

Abstract
Spastin is a microtubule-severing AAA (ATPases associated with diverse cellular activities) protein needed for cell division and intracellular vesicle transport. Currently, we lack chemical inhibitors to probe spastin function in such dynamic cellular processes. To design a chemical inhibitor of spastin we tested selected heterocyclic-scaffolds against wildtype protein and constructs with engineered mutations in the nucleotide-binding site that do not substantially disrupt ATPase activity. These data, along with computational docking, guided improvements in compound potency and selectivity and led to spastazoline, a pyrazolyl-pyrrolopyrimidine-based cell-permeable probe for spastin. These studies also identified spastazoline resistance-conferring point mutations in spastin. Spastazoline, along with matched inhibitor-sensitive and inhibitor-resistant cell lines we generated, were used in parallel experiments to dissect spastin-specific phenotypes in dividing cells. Together, our findings suggest how chemical probes for AAA proteins, along with inhibitor resistance-conferring mutations, can be designed and used to dissect dynamic cellular processes.
Keywords: chemical probes, AAA proteins, resistance mutations, spastin, cell division, nuclear envelope
Introduction
In humans the AAA superfamily (ATPases associated with diverse cellular activities) has ~100 proteins whose functions have been linked to a wide range of cellular processes, including cell division, cytoskeleton organization, and organelle biogenesis1. As AAA proteins carry out functions that can occur within minutes to seconds, chemical probes that inhibit their activities in cells on similarly fast timescales can be valuable tools to dissect dynamic mechanisms2. Currently we have well-characterized selective chemical probes for only a handful of AAA proteins such as dynein, a microtubule-based motor protein, midasin, a ribosome biogenesis factor, and valosin-containing protein (VCP), a regulator of ubiquitin-dependent proteolysis3–6. However, chemical inhibitors of most AAA proteins are not available.
Spastin, fidgetin, and katanin are AAA proteins closely related at the level of sequence and are collectively referred to as microtubule-severing enzymes7. Biochemical assays have shown that spastin and katanin can sever microtubules into smaller filaments in an ATP hydrolysis-dependent manner8,9. Based on these findings, models have been proposed for how microtubule-severing enzymes contribute to the assembly, disassembly, or maintenance of microtubule-based structures in axons and dendrites, primary cilia, and dividing cells10. In addition, recent studies have suggested a role for spastin in the biogenesis and function of the nuclear envelope and membrane organelles such as lysosomes and endosomes11–13. Thus far, the only chemical known to target a microtubule-severing enzyme in cells is a purine-based compound that can pull-down katanin from cellular lysates14. However, the direct and specific inhibition of katanin by this compound has not been demonstrated14, and it is unclear how useful chemical probes for microtubule-severing proteins can be developed.
In principle, chemical inhibitors of AAA proteins can be identified that target either the nucleotide-binding site or an allosteric site. An allosteric inhibitor-binding site has been characterized for VCP and the vacuolar protein sorting-associated protein 4 (VPS4)15,16. However, it is unclear if an equivalent site exists in other AAA proteins. Targeting the nucleotide-binding site could represent a more general strategy; however designing nucleotide-competitive inhibitors of AAA proteins is challenging for at least three reasons. First, the active site is conserved across the AAA superfamily17. Second, this site in AAA proteins can undergo activity-associated conformational transitions that substantially alter its overall shape18. Third, we lack structural data for any eukaryotic AAA protein bound to a nucleotide-competitive inhibitor. These data can be crucial for structure-based inhibitor design19.
It is now emerging that analyses of resistance-conferring mutations can help establish the direct physiological targets of chemical inhibitors and provide insights into how compounds interact with their targets20–22. When these mutations do not disrupt protein activity they can also be introduced in cells to systematically examine on-target and off-target activities of chemical inhibitors22. On-target inhibitor phenotypes can be identified as those observed in the cells expressing the wildtype protein but not in those expressing the allele with the resistance-conferring mutation22. For these reasons, resistance-conferring mutations could also be useful during the early steps of inhibitor development when un-optimized compounds (e.g. screening hits) without robust models for inhibitor-target interactions or data on off-target effects need to be evaluated. Thus far, resistance-conferring mutations have been typically identified using genetic-based screens that rely on selection of growth in tractable systems, as has been the case for cytotoxic chemical inhibitors of AAA proteins5,23. However, to identify mutations in AAA proteins that do not impair function but confer inhibitor resistance can be challenging.
Here, we use protein structure and sequence analyses to design mutant alleles of spastin that retain ATPase activity. Testing selected heterocyclic scaffolds against wildtype protein and constructs with these mutations, along with computational docking, helped design spastazoline, a pyrazolyl-pyrrolopyrimidine-based inhibitor of spastin. In addition, we identified a spastazoline resistance-conferring point mutation in spastin. Spastazoline-induced phenotypes were examined in cells expressing either wildtype spastin or an allele with a resistance-conferring point mutation. Together, these studies reveal how spastin-specific cell division phenotypes can be analyzed using chemical probes and cognate resistance-conferring mutations.
Results
Mutant alleles of spastin that retain ATPase activity
To design mutant alleles of spastin we focused on the nucleotide-binding pocket. We selected D. melanogaster spastin (hereafter, Dm-spastin), as it is a biochemically well-characterized microtubule-severing enzyme24. We first compared the sequences of the AAA domains in Dm-spastin and four related AAA proteins: human FIGL1 (Hs-FIGL1), human pachytene homology-like protein 2 (Hs-PCH2), mouse VCP (Mm-VCP, which has two AAA domains and is identical to the human protein), and X. laevis katanin (Xl-katanin) (Supplementary Figure 1a, 1b). These proteins were selected as recombinant forms of either their AAA domains or constructs with ATPase activity had been previously reported25–27. Using available structural data (Dm-spastin, Hs-FIGL1, and Mm-VCP)15,24,28 or homology-based models (Hs-PCH2 and Xl-katanin, see methods for details) we identified residues that were likely to be within ~6 Å of the adenine (Fig. 1a, 1b). These residues are located in five structural motifs of the AAA domain, which are named the N-terminal loop (N-loop), the phosphate-binding loop (P-loop), the hinge motif, helix-4, and the sensor-II motif29 (Supplementary Figure 1c).
Figure 1. Identifying and characterizing ‘variability hot-spot’ mutations in spastin.
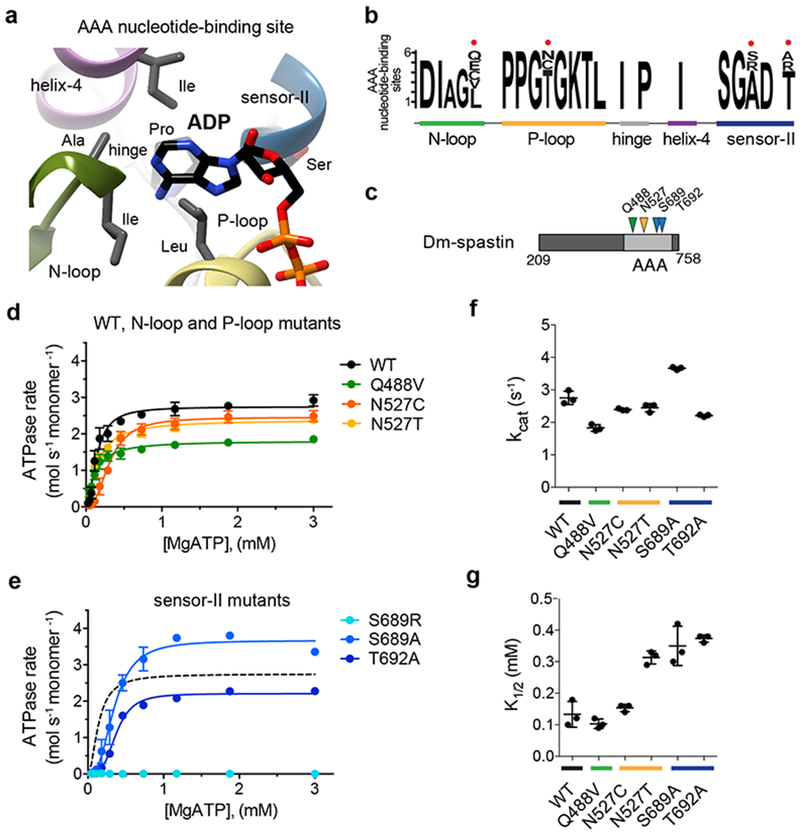
(a) Structural model (ribbon diagram) of the nucleotide-binding site in FIGL1 (PDB: 3D8B) showing adenosine-5’ diphosphate (ADP, stick representation), structural motifs (N-loop: green, P-loop: yellow, helix 4: magenta, sensor-II: blue, hinge: gray), and selected residues within ~6 Å of the adenine. (b) A modified “sequence logo” diagram for residues in the nucleotide-binding site of six AAA domains. The variability hot-spot residues for proteins in the AAA superfamily are indicated (red circles). The relevant structural motifs in the nucleotide-binding site are also highlighted. (c) Schematic shows the AAA domain (light gray box), the first and last residues of the Dm-spastin construct (not to scale), and the residues that were mutated in Dm-spastin’s AAA domain (colored arrows). (d-e) ATP concentration-dependence of the steady-state activity of wildtype (WT) and mutant Dm-spastin constructs, analyzed using an NADH-coupled assay. Rates were fit to the Michaelis-Menten equation for cooperative enzymes (average ± s.d., n = 3). For comparison data for the WT protein (from d) are also shown in e. (f, g) Catalytic turnover number (kcat; f), and the ATP concentration required for half-maximal velocity (K1/2; g) of recombinant Dm-spastin constructs. Lines represent average ± s.d. (n = 3). Measured values for these parameters are provided in Supplementary Figure 1h. The corresponding structural motifs for the mutated residues are color-coded as in a.
As expected for a conserved active site, most of these residues are invariant across the five AAA proteins examined. However, we noted four amino acid positions – one in the N-loop, one in the P-loop, and two in the sensor-II motif – that have diverged significantly (Fig. 1b). These residues, which we name “variability hot-spot” residues, contribute in large part to the sequence variation in these nucleotide-binding motifs across these AAA proteins (Supplementary Figure 1d). Sequence alignment of the five structural motifs of the nucleotide-binding site in 24 AAA domains indicates that these variability hot-spot residues can be identified across this protein superfamily (Supplementary Figures 1e, 1f). We hypothesized that a residue at a variability hot-spot position could be replaced with a residue from the equivalent position in a related AAA protein, to obtain an active site mutant that retains enzymatic activity.
To test this hypothesis, we generated recombinant constructs of Dm-spastin wildtype and Dm-spastin carrying mutations at the variability hot-spot residues and characterized their steady-state ATP-hydrolysis activity (Fig. 1c–1e and Supplementary Figures 1g, 1h). In addition to the wildtype protein, five of the six mutant constructs were active (Fig. 1d, 1e and Supplementary Figure 1h provides additional enzyme activity parameters). Only one construct, with a mutation at a sensor-II variability hot-spot residue (S689R), yielded an inactive recombinant protein (Fig. 1e). Dm-spastin had a catalytic activity (kcat) of ~3 s−1 at saturating ATP concentrations (Fig. 1f). The five active mutants had kcat values within ~1.5-fold of the wildtype protein, ranging from ~2 s−1 for the N527T mutant to ~4 s−1 for the S689A mutant (Fig. 1f). The ATP concentrations required for half-maximal enzymatic velocity (K1/2) were also within a narrow range, from ~0.1 mM (wildtype, Q488V, and N527T) to ~0.4 mM (S689A) (Fig. 1g). Together, these data indicate that swapping variability hot-spot residues can yield AAA protein alleles that retain catalytic activity.
A starting chemical scaffold to design spastin inhibitors
To find chemical starting points for designing spastin inhibitors we examined conserved features of the nucleotide-binding site. In Dm-spastin, as well as other AAA proteins, we noted a pattern of two hydrogen bonds between the adenine and the backbone of a residue in the N-loop (e.g. A404 in FIGL1 (PDB:3D8B); G481 in VCP (PDB:5FTK); Supplementary Figure 2a). Therefore, we selected a collection of 33 chemically diverse kinase inhibitors that could mimic the adenine H-bonding interactions to test against AAA proteins (Supplementary Table 1).
To profile the inhibitory activity and selectivity of these compounds we generated recombinant forms of Hs-FIGL1, Hs-PCH2, Mm-VCP, and Xl-katanin, and characterized their steady-state ATPase activities (Fig. 2a, 2b, and Supplementary Figures 2b–2f). Consistent with other reports, we found K1/2 values for ATP hydrolysis to be in the high micromolar range3,26,29. While the kcat varied >50-fold (Mm-VCP: ~0.1 s−1; Xl-katanin: ~6 s−1; Supplementary Figure 2d), the K1/2 values varied only ~3-fold (Hs-PCH2: ~150 µM; Xl-katanin and Mm-VCP: ~450 µM; Fig. 2b). For these enzymes, the range of K1/2 was comparable to what we found for the active Dm-spastin mutants, suggesting that variation in the hot-spot residues can account for differences in how these proteins interact with the nucleotide.
Figure 2. Identifying chemical scaffolds that inhibit Dm-spastin.
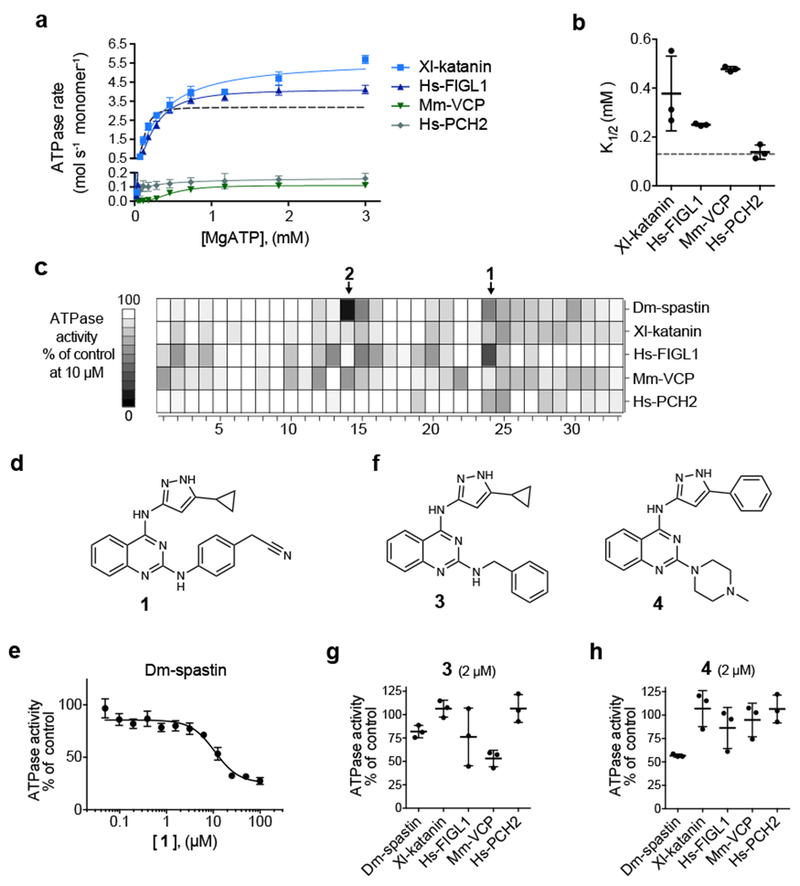
(a) ATP concentration-dependence of the steady-state activity of four AAA protein constructs, analyzed using an NADH-coupled assay. Schematics for these constructs along with SDS-PAGE gels are shown in Supplementary Figures 2b–2c. Rates were fit to the Michaelis-Menten equation for cooperative enzymes (average ± s.d., n = 3). (b) ATP concentration required for half-maximal velocity (K1/2) of four AAA proteins. Lines represent average ± s.d. (n = 3, for comparison the corresponding data for Dm-spastin are also shown in panels a and b, dashed line). Additional enzyme activity parameters are provided in Supplementary Figures 2d–2f. (c) ATPase activity of five AAA proteins in the presence of selected compounds (10 µM). Heat map corresponds to percent ATPase activity relative to DMSO control (average, n = 2, bins: 10%). Chemical structures of all compounds tested are provided in Supplementary Table 1. (d) Chemical structure of compound 1 (structure of compound 2 is shown in Supplementary Table 1). (e) Concentration-dependent inhibition of the steady-state ATPase activity of Dm-spastin by compound 1. Graph shows percent residual ATPase activity values relative to DMSO control (average ± s.d., n = 3) fit to a sigmoidal dose-response equation, IC50 was not determined as complete inhibition at the highest concentration tested was not observed. (f) Chemical structures of two analogs of compound 1. (g, h) Percent steady-state ATPase activity of five AAA proteins in the presence of compounds 3 or 4 (2 µM). Lines represent average ± s.d. (n = 3). (c, e-h) In these assays an MgATP concentration of 0.5 mM was used.
We next tested the selected compounds (10 µM) against Dm-spastin and the other four AAA proteins (Fig. 2c). While most compounds did not show appreciable inhibition of any of these enzymes, we identified two compounds, one with a 4-aminopyrazolylquinazoline core (1) and the other with a 3,5-diamino-triazole core (2) that inhibited at least one AAA protein by >50% (0.5 mM ATP, Fig. 2d and Supplementary Table 1). Both compounds 1 and 2 substantially inhibited the ATPase activity of Dm-spastin at 10 µM and compound 1 also inhibited Hs-FIGL1 (Fig. 2c). We prioritized compound 1 for further studies here, and will examine compound 2 in subsequent work.
Dose-dependent analysis revealed that compound 1 inhibited Dm-spastin with potency in the micromolar range (Fig. 2e). To examine the structure-activity relationship against AAA proteins we synthesized and tested compound 1 analogs with modifications of the substituents on the pyrazolylquinazoline core (10 µM, Fig. 2f–2h, and Supplementary Table 2 shows additional synthesized analogs). We found that compound 3, in which a benzyl group replaces the phenylacetonitrile, more potently inhibited the activity of VCP compared to compound 1 (Fig. 2f, 2g). In contrast, compound 4 in which a N-methyl piperazine replaces the phenylacetonitrile and a phenyl replaces the cyclopropyl in the pyrazole ring, showed increased selectivity for Dm-spastin (Fig. 2f, 2h). Testing Dm-spastin inhibition by compound 4 across a range of ATP concentrations revealed increases in K1/2 at higher compound concentrations, but not substantial changes in kcat (Supplementary Figure 2g–h), consistent with compound 4 binding to the Dm-spastin nucleotide-binding site. Together, these data suggest that the pyrazolylquinazoline scaffold could provide a useful starting point to develop ATP-competitive inhibitors of spastin.
A model for how compound 4 binds spastin
To examine how compound 4 binds spastin we tested it against Dm-spastin constructs with mutations at the variability hot-spot residues. We found that the mutation in the N-loop (Q488V) reduced the potency of 4 by ~20-fold (Fig. 3a), while the mutations in the P-loop reduced potency by ~3-fold (N527C) or >35-fold (N527T) (Fig. 3b). In contrast, mutations in the sensor-II motif led to ~2-fold reduction (T692A) or no substantial difference (S689A) in the potency of this inhibitor (Fig. 3c). These data suggested that the P-loop and N-loop variability hot-spot residues may interact with the inhibitor, and that this information could be used to confirm or reject predictions from computational docking solutions.
Figure 3. Testing mutant constructs to build a model for the chemical inhibition of spastin.
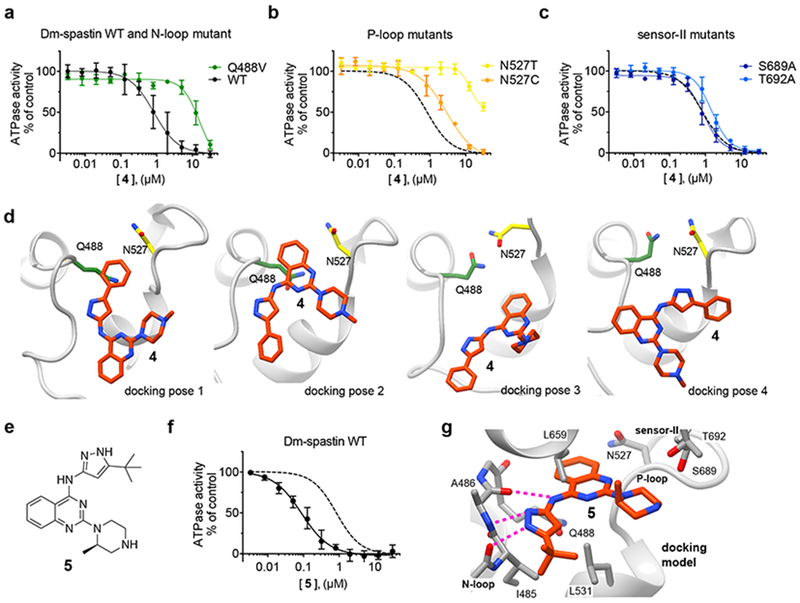
(a-c) Compound 4 concentration-dependent inhibition of the steady-state ATPase activity of Dm-spastin constructs with mutations at variability hot-spot residues and Dm-spastin WT. Data were fit to a sigmoidal dose-response equation and IC50 were calculated (Dm-spastin constructs, WT: 1.0 ± 0.6 µM; Q488V: ~12.5 µM; N527C: 2.9 ± 0.5 µM; N527T: >30 µM; S689A: 0.9 ± 0.2 µM; and T692A: 1.5 ± 0.4 µM; 0.5 mM MgATP, average ± s.d., n = 3). For comparison, analysis of the inhibition of WT (data from a) is also shown in b and c (dashed line). (d) Four computational docking poses showing compound 4 (stick representation, carbon atoms in orange) bound to Dm-spastin (ribbon-and-stick representation, gray ribbons) along with the Q488 (green) and N527 (yellow) variability hot-spot residues. (e) Chemical structure of compound 5. (f) Compound 5 concentration-dependent inhibition of the steady-state ATPase activity of Dm-spastin WT. Graph shows values fit to a sigmoidal dose-response equation. IC50 = 106 ± 35 nM, 0.5 mM MgATP, average ± s.d., n = 3. For comparison, data for compound 4 are shown (dashed line). (g) Computational docking model for compound 5 bound to Dm-spastin. Potential hydrogen-bonding interactions between the aminopyrazole and the protein backbone are highlighted (dashed lines). Other key amino acids in Dm-spastin nucleotide-binding site are also shown. All images were generated using UCSF Chimera. Additional details of the models are provided in Supplementary Figure 3a and 3f.
Next we used available Dm-spastin structural models to dock compound 4 into the spastin nucleotide-binding site24. Spastin structures reported to date do not have nucleotide bound, suggesting the crystallography models may not match the conformation that binds nucleotide-competitive inhibitors24,30. To generate additional spastin conformations we used molecular dynamics simulations and computationally docked compound 4 in the nucleotide-binding site of each spastin conformer31 (see Online Methods for details). This method yielded four models with similar docking scores for how compound 4 and spastin may interact (Fig. 3d). In three of the four models, we noted adenine-like hydrogen-bonding interactions between the inhibitor amino-pyrazole group and the backbone of N-loop A486 (Supplementary Figure 3a). However, only two of these docking poses had the inhibitor in close proximity to the variability hot-spot residues in the N-loop and the P-loop, and were therefore consistent with the potency shifts observed in the mutant spastin constructs (poses 1 and 2, Fig. 3d and Supplementary Figure 3a).
We reasoned that the two different docking solutions could arise due to the chemical equivalence of the benzene rings in the quinazoline and the pyrazolyl moieties. To distinguish between these two potential binding modes we designed modifications to effectively disrupt the symmetry of the inhibitor. In particular, we synthesized compound 5, in which the phenyl group in the pyrazolyl was replaced by a non-aromatic tert-butyl group, and the piperazine N-methyl group was shifted to the 2-position of the ring (Fig. 3e). Compound 5 inhibited Dm-spastin steady-state ATPase activity (~10-fold more potently than compound 4, Fig. 3f), and also blocked Dm-spastin dependent microtubule-severing (2 µM inhibitor, Supplementary Figures. 3b–3e).
Gratifyingly, docking analysis revealed only one favored pose for compound 5 bound to spastin (Fig. 3g). This model was also consistent with the data obtained by testing mutant constructs. In this compound 5-spastin model, the quinazoline core is buried in the adenine-binding pocket within van der Waals distance to the N-loop and P-loop variability hot-spot residues, the piperazine group is proximal to the sensor-II helix, and the aminopyrazole group establishes a network of hydrogen-bonding interactions with the backbone of the N-loop (Fig. 3g and Supplementary Figure 3f).
Designing a selective inhibitor of human spastin
To develop a chemical probe for human spastin we purified a recombinant form of human spastin from E. coli (hereafter, Hs-spastin, Supplementary Figures 4a, 4b). Hs-spastin had similar ATPase activity relative to Dm-spastin (Supplementary Figure 4c), and was inhibited by compound 5, albeit with reduced potency (IC50 = 4.4 ± 2.1 µM, 1 mM ATP, average ± s.d., n = 3, Fig. 4a).
Figure 4. Developing a potent and selective inhibitor of human spastin.
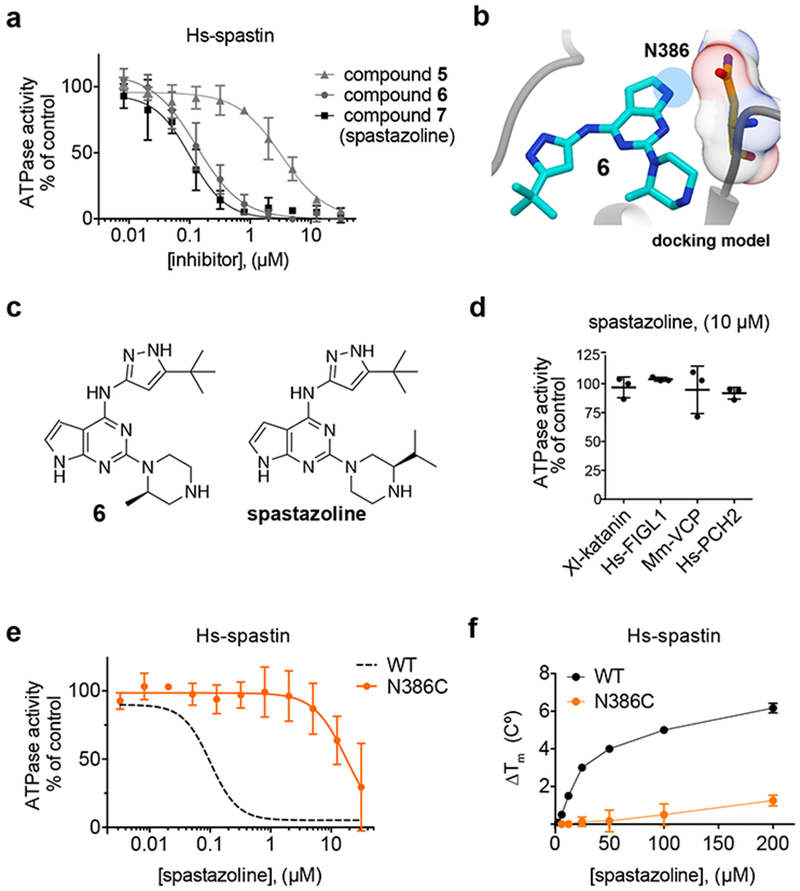
(a) Concentration-dependent inhibition of the steady-state ATPase activity of the Hs-spastin construct by compound 5, 6 and spastazoline. Graph shows values fit to a sigmoidal dose-response equation (average ± s.d., n = 3, 1 mM MgATP). IC50 = compound 5: 4.4 ± 2.1 µM, compound 6: 132 ± 55 nM; spastazoline: 99 ± 18 nM, average ± s.d.; n = 3. (b) The predicted model for compound 6 (blue, stick representation) bound to spastin (gray, ribbon representation) is shown. The N386 residue (stick and surface representation), and the region occupied by inhibitor atoms in close proximity to the N386 residue are also highlighted (blue circle). Additional details of the model are shown in Supplementary Figure 4d. (c) Chemical structures of compounds 6 and 7 (spastazoline). (d) Percent steady-state ATPase activity of four AAA proteins in the presence of spastazoline (10 µM, 0.5 mM MgATP, line represents average ± s.d., n = 3). (e) Concentration-dependent inhibition of the steady-state ATPase activity of an Hs-spastin mutant construct (N386C). Graph shows values fit to a sigmoidal dose-response equation (average ± s.d., n = 3, 1 mM MgATP). For comparison the curve corresponding to the data for the wildtype Hs-spastin is shown (WT, dashed line, data from c). Characterization of the Hs-spastin WT and N386C constructs is shown in Supplementary Figures 4a–4c and Supplementary Figures 4g–4i. (f) Concentration-dependent effect of spastazoline on the heat-induced unfolding (∆Tm) of Hs-spastin WT and N386C mutant constructs analyzed used differential scanning fluorimetry (average ± s.d., n = 3). Supplementary Figures 4j and 4k show representative differential scanning fluorimetry experiments.
The inhibitor-Dm-spastin model suggests that the hydrogen at the 8-position of compound 5’s quinazoline ring would be in close proximity to the N386 side chain in human spastin (equivalent to Drosophila spastin N527) (Fig. 3g). An asparagine residue is rarely found at the P-loop variability hot-spot position of AAA proteins (Supplementary Table 3). Therefore, we hypothesized that inhibitor potency and selectivity for spastin could be improved by optimizing the interaction with the N386 residue in Hs-spastin. We designed and synthesized an analog in which a pyrrolopyrimidine replaced compound’s 5 quinazoline core, as this would introduce an electropositive group in the position predicted to be proximal to the N386 side chain (compound 6, Fig. 4b, 4c). We found that compound 6 inhibited Hs-spastin with ~30-fold improved potency with respect to compound 5 (Fig. 4a, IC50 = 132 ± 55 nM, 1 mM ATP, average ± s.d., n = 3). Computational docking analysis suggested two possible poses for compound 6 bound to spastin, but a hydrogen-bonding interaction with the P-loop asparagine was possible only in one pose (Supplementary Figure 4d).
As kinases are known targets of these chemical scaffolds32, we examined the activity of compound 6 against 64 human kinases. We found that compound 6 inhibited only four of the 64 kinases >50% at ~15-fold the IC50 for Hs-spastin (2 µM, Supplementary Table 4). To reduce off-target activity we compared the binding modes of compound 6 to spastin and to kinases, using available inhibitor-kinase structural data (Supplementary Figure 4d and 4e). We observed that compound 6’s piperazine ring is likely to be buried in the interior of the kinase pocket33, but is facing the bulk solvent in our inhibitor-spastin model (Supplementary Figure 4f). Therefore, we synthesized compound 7 (Fig 4c), hereafter named spastazoline, in which the 2-methyl in the piperazine group was modified to a 3-isopropyl group so as to increase steric hindrance when interacting with the kinase pocket without disrupting spastin binding. We found that spastazoline only inhibited one of the 64 kinases tested (NTRK1) >50% (Supplementary Table 4). Importantly, spastazoline potently inhibited Hs-spastin (Fig. 4a, IC50 = 99 ± 18 nM, average ± s.d.; n = 3, 1 mM MgATP) and did not appreciably inhibit any of the four related AAA proteins that we have characterized (10 µM, Fig. 4d). Together these data suggest that spastazoline could be a useful chemical probe for human spastin, if potential off-target effects can be systematically addressed.
We hypothesized that a mutation at N386 would disrupt the spastin-inhibitor interaction and confer resistance to spastazoline. We purified a recombinant Hs-spastin construct with a N386C mutation (equivalent to the N527C mutation in Dm-spastin) and found that the ATPase activity of this mutant construct was comparable to that of wildtype Hs-spastin (Supplementary Figures 4a–4c). As predicted by our model, spastazoline inhibited Hs-spastin-N386C >100-fold less potently than the wildtype protein (Fig. 4e). Additionally, we found that spastazoline did not appreciably stabilize the Hs-spastin N386C mutant against thermal induced denaturation (ΔTm: ~1.5 ºC at 200 µM, Fig. 4f and Supplementary Figure 4g), while it significantly stabilized the WT construct (ΔTm: ~6.0 ºC, Fig. 4f and Supplementary Figure 4h). Together, these data indicate that our approach not only led to a potent chemical inhibitor of spastin but also could identify resistance-conferring mutations.
Probing spastin function during cell division
To probe spastin cellular functions using spastazoline (Fig. 5), we focused on the M87e4 spastin (containing the alternatively spliced exon 4), the most abundant spastin isoform in human cells33. As the N386C mutation suppresses spastazoline-binding in biochemical assays, we generated two HeLa cell lines in which green fluorescent protein (GFP)-tagged spastin M87e4, either wildtype (HeLa-WT) or mutant allele (HeLa-N386C), were introduced using the Flp-In T-Rex system. We found that these transgenes were expressed at comparable levels (Supplementary Figures 5a–5c).
Figure 5. Using spastazoline and a cognate resistance-conferring mutation to probe spastin function in cell division.
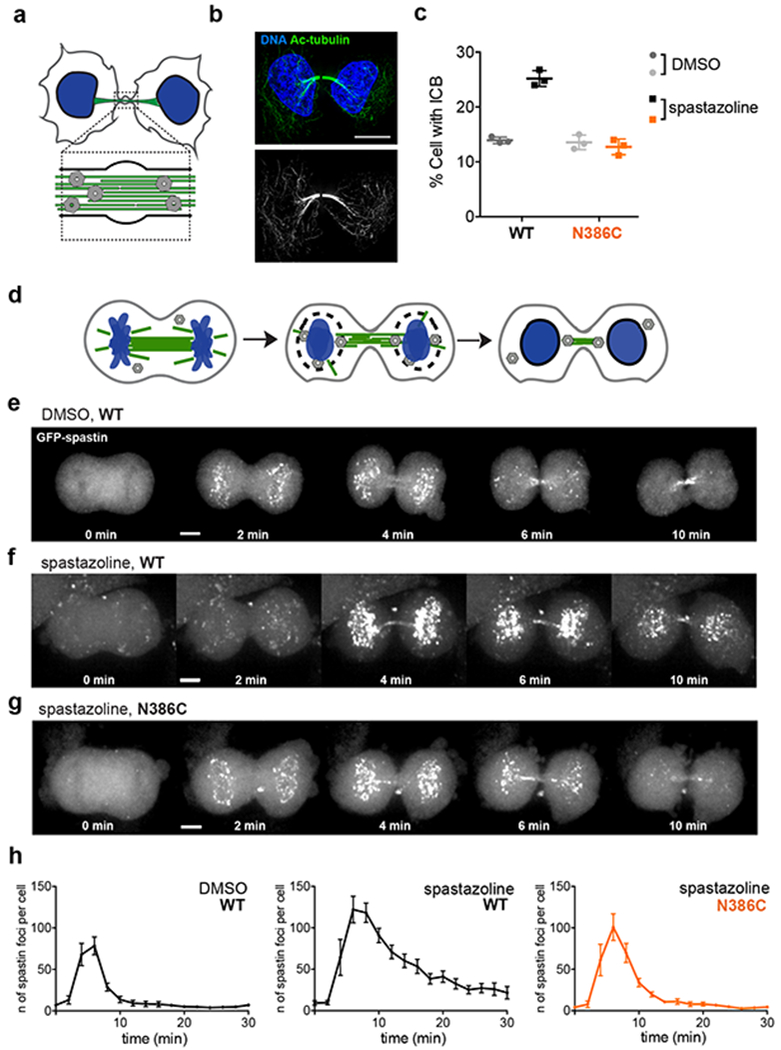
(a) Schematic for an intercellular bridge (DNA: blue, microtubules: green, spastin: gray). (b) Immunofluorescence images of fixed HeLa-WT cells stained for acetylated tubulin. Two-color overlay (DNA: blue; acetylated tubulin: green), along with the corresponding gray scale image of acetylated tubulin staining, are shown. Scale bar = 10 µm. (c) Percent of cell with an intercellular bridge observed in fixed HeLa-WT and -N386C cells treated with DMSO (0.1%) or spastazoline (10 µM) for 4.5 hrs (lines represents average ± s.d., n = 3, >2500 cells analyzed per condition). Representative images for fixed HeLa-WT and HeLa-N386C cells treated with spastazoline and stained for acetylated tubulin are shown in Supplementary Figures 5d–e. (d) Schematic of nuclear envelope reformation in a dividing cell, highlighting spastin (gray) localization at the intersection points between microtubules (green) and the reassembling nuclear envelope (black) around chromosomes (blue). (e-g) Effect of spastazoline treatment on spastin localization in dividing cells. Maximum-intensity confocal projections show distributions of GFP at the indicated times after ingression of the cleavage furrow is first observed (t=0) in HeLa-WT cells treated with DMSO control (e) and HeLa-WT or -N386C cells treated with spastazoline (10 µM for 1 hour, f and g). Scale bar = 5 µm. Representative images from three independent experiments are shown. h) Number of spastin foci observed in anaphase HeLa-WT cells treated with DMSO, and HeLa-WT or –N386 cells treated with spastazoline (n = 8 cells per condition, average ± s.e.m., three independent experiments, values for HeLa-N386C cells treated with DMSO are shown in Supplementary Figure 5j).
To examine spastin-dependent inhibitor-induced phenotypes we focused on cell division. RNAi-mediated spastin knockdown has been shown to disrupt disassembly of the intercellular bridge, a membrane- and microtubule-based structure that connects daughter cells during the final stage of cell division34 (Fig. 5a). To readily identify intercellular bridges, which are enriched in tubulin post-translational modifications associated with stable microtubules, we imaged acetylated tubulin using immunofluorescence microscopy (Fig. 5b). Treating HeLa-WT cells with spastazoline for 4.5 hours (10 µM) resulted in a ~2-fold increase in the number of cells with intercellular bridges compared to DMSO control (25.2 ± 1.4% vs. 13.9 ± 0.6%, n=3, average ± s.d. Fig. 5c and Supplementary Figure 5d). Importantly, in HeLa-N386C, the number of cells with an intercellular bridge was similar to that observed in the DMSO control (13.6 ± 1.4% vs. 12.7 ± 1.3%, n=3, average ± s.d., Fig. 5c and Supplementary Figure 5e). Spastazoline treatment (10 µM) did not impact the viability of HeLa cells (Supplementary Figure 5f), and also did not result in overt changes in the organization of microtubules (Supplementary Figure 5g). Furthermore, spastazoline did not inhibit ATPase activity of a recombinant human VPS4 (Supplementary Figures 5h, 5i), another AAA protein required for intercellular bridge disassembly35. Together, these data indicate that spastazoline inhibits intercellular bridge disassembly by interfering with spastin activity, as the inhibitor-dependent phenotype was observed in wildtype cells, but not cells expressing the mutant allele of spastin.
During anaphase the nuclear envelope is rapidly, within minutes, reformed around daughter nuclei36 (Fig. 5d). RNAi knockdown studies have revealed a role for spastin in nuclear envelope reformation13. To examine the effects of spastazoline during nuclear envelope reformation, we tracked EGFP-spastin dynamics in dividing HeLa-WT cells. We found that EGFP-spastin accumulated at chromatin within 2–4 min after cleavage furrow ingression initiation (Fig. 5e, 5h). This signal was lost within ~10 min. These localization dynamics suggest that spastin dissociates as the nuclear envelope reassembles.
Imaging HeLa-WT cells in anaphase treated with spastazoline (10 µM for 1 hour) also revealed GFP-spastin puncta on chromosomes (Fig. 5f). Remarkably, spastin puncta persisted for several minutes compared to DMSO controls (Fig. 5f). Even 20 min after the initiation of cleavage furrow ingression the spastazoline treated cells had a higher number of GFP-spastin puncta than controls (44 ± 9 vs. 5 ± 2; average ± s.e.m. Fig. 5h). Imaging HeLa-N386C cells, which express spastazoline-resistant spastin, revealed the recruitment of mutant spastin to anaphase chromosomes with similar kinetics as wildtype (Fig. 5g and Supplementary Figure 5j). However, treatment with spastazoline did not substantially alter spastin dynamics in anaphase Hela-N386C cells. In particular, the number of observed GFP-spastin dots 20 min after cleavage furrow ingression was comparable to controls (8 ± 3; 1.6-fold increase with respect to wildtype, average ± s.e.m., Fig. 5g, 5h). Together, these data are consistent with a model in which spastin activity and localization dynamics contribute to nuclear envelope reformation during anaphase.
Discussion
Here we report the design of spastazoline, an ATP-competitive chemical inhibitor of human spastin (Fig. 6). We also identify a cognate resistance-conferring mutation in human spastin that together with spastazoline can be used to probe spastin’s function in dynamic cellular processes.
Figure 6. Engineering “silent” mutations to generate models for inhibitor-target interactions and to identify inhibitor resistance-conferring mutations.
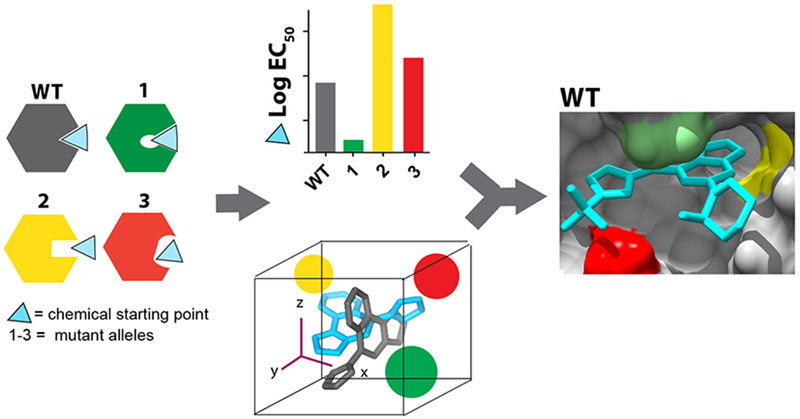
For a selected site in a target protein, constructs with mutations that retain activity are generated to alter its shape and electrostatics. Potency of a chemical scaffold is tested against wildtype and mutant constructs. These data, along with computational docking, can guide improvements in inhibitor potency and specificity and help identify inhibitor resistance-conferring mutations.
Current models indicate that spastin and the endosomal sorting complex required for transport (ESCRT)-III, to which spastin binds37, coordinate microtubule disassembly with membrane constriction and fusion events during nuclear envelope reformation13,38,39. Our data suggest that inhibition of spastin activity likely suppresses microtubule severing and thereby impairs spastin dissociation and the loss of ESCRT-III from the sites of membrane constriction. RNAi-based knockdown of VPS4 or the ESCRT-III protein CHMP2A results in DNA damage associated with compromised nuclear envelope integrity13. Consistent with these observations, we suggest that disrupting spastin activity could also result in DNA damage due to defects in nuclear envelope reformation.
Cells depleted of spastin also show defects in the assembly, maintenance and dynamics of intracellular membrane organelles such as endosomes and lysosomes11,12. As the effects of chronic spastin depletion are likely to accumulate over several hours, the ability to control spastin activity on much faster timescales with spastazoline would be useful to examine intracellular trafficking of membrane organelles. Mutations in the spastin gene are linked to inherited neurodegenerative conditions called hereditary spastic paraplegias39, and targeting spastin has been proposed as a therapeutic strategy for Alzheimer’s disease40. In post-mitotic neurons, DNA damage associated with spastin inhibition is not likely and therefore, pharmacological inhibition of spastin in models for neurodegenerative diseases should be further explored. Spastazoline is also likely to be a valuable tool for these studies.
A critical step in our approach is to identify amino acids in the target protein that can be mutated to alter the shape and electrostatics of the inhibitor-binding site without substantially disrupting the protein’s activity (Fig. 6). In the case of spastin we find that variability hot-spot residues in the nucleotide-binding pocket can be swapped to introduce mutations to generate alleles that retain biochemical activity. Variability hot-spots mutations that reduce inhibitor potency are likely to identify direct inhibitor-target interactions, and can be used to optimize compound potency and selectivity. Testing compounds against wildtype and mutant alleles can help select the most likely inhibitor-target binding mode from the solutions yielded by computational docking methods (Fig. 6).
We speculate that selective chemical probes for AAA proteins could be designed by engineering interactions with unique combinations of variability hot-spot residues. The core structures of compounds that bind conserved structural features of ATPases can be used as starting scaffolds to which different chemical groups may be added to contact the variability hot-spot residues in the target AAA protein. For instance, pyrazolyl-heterocycle-based scaffolds are likely to be valuable starting points for developing inhibitors of katanin, a microtubule-severing AAA protein closely related to spastin. In katanin the P-loop and N-loop variability hot-spot residues are threonine and leucine respectively, compared to asparagine and glutamine at equivalent sites in spastin, and engineering contacts with these residues could lead to katanin-specific chemical probes. For AAA proteins in which the shape of the nucleotide-binding site and the variability hot-spot residues diverge more substantially from spastin, other core scaffolds may be needed. Mutant AAA protein alleles can be generated and their inhibition by these scaffolds could be tested to develop models for inhibitor-target binding and guide compound optimization. These analyses would also help identify cognate inhibitor resistance-conferring mutations that can be used along with the inhibitor to decipher target-specific phenotypes in cells (Fig. 6).
In principle, the mutagenesis-based approach we used to develop spastazoline could help design inhibitors of targets for which high-resolution structural data for inhibitor-protein interactions are not readily available, or the inhibitor-bound conformation states are not known. Additional studies will be needed to determine if variability hot-spot residues can be identified and swapped to design functionally silent mutations for protein-protein interaction or other non-enzymatic sites that can be targeted by chemical inhibitors. Identifying and analyzing resistance-conferring mutations at an early stage of inhibitor design may also help develop chemical therapeutics against which resistance is less likely to arise.
Supplementary Material
Acknowledegments
We thank J. Steinman and P. Verma for help with AAA protein purification, and M. Grasso for the purification of human VPS4B. We thank F. Glickman and C. Adura of the Rockefeller University High-Throughput and Spectroscopy Resource Center for assistance with assay development and A. North of the Rockefeller Bioimaging Resource Center. We are especially grateful to members of the Kapitein Lab (Utrecht University): L. Kapitein, K. Jansen and W. Nijenhuis, for testing our spastin inhibitors in cellular assays. We are also grateful to R. Heald (University of California, Berkeley) and C. Campsteijn (Oslo University Hospital) for plasmids, and to A. Roll-Mecak (NIH) for plasmid and a protein sample for the initial assay validation. T.C. was supported by the EMBO Long-Term Fellowship for post-doctoral studies and by the Kestenbaum award for research in neurodegenerative diseases. M.E.K. acknowledges support from an NIH training grant (GM066699). T.M.K. is grateful to the NIH (GM98578) for supporting this research.
Footnotes
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Erzberger JP & Berger JM Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu Rev Biophys Biomol Struct 35, 93–114 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Lampson MA & Kapoor TM Unraveling cell division mechanisms with small-molecule inhibitors. Nat Chem Biol 2, 19–27 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Chou TF et al. Reversible inhibitor of p97, DBeQ, impairs both ubiquitin-dependent and autophagic protein clearance pathways. Proc Natl Acad Sci U S A 108, 4834–9 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Firestone AJ et al. Small-molecule inhibitors of the AAA+ ATPase motor cytoplasmic dynein. Nature 484, 125–9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawashima SA et al. Potent, Reversible, and Specific Chemical Inhibitors of Eukaryotic Ribosome Biogenesis. Cell 167, 512–524 e14 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Magnaghi P et al. Covalent and allosteric inhibitors of the ATPase VCP/p97 induce cancer cell death. Nat Chem Biol 9, 548–56 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Roll-Mecak A & McNally FJ Microtubule-severing enzymes. Curr Opin Cell Biol 22, 96–103 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roll-Mecak A & Vale RD The Drosophila homologue of the hereditary spastic paraplegia protein, spastin, severs and disassembles microtubules. Curr Biol 15, 650–5 (2005). [DOI] [PubMed] [Google Scholar]
- 9.McNally FJ & Vale RD Identification of katanin, an ATPase that severs and disassembles stable microtubules. Cell 75, 419–29 (1993). [DOI] [PubMed] [Google Scholar]
- 10.Sharp DJ & Ross JL Microtubule-severing enzymes at the cutting edge. J Cell Sci 125, 2561–9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Allison R et al. An ESCRT-spastin interaction promotes fission of recycling tubules from the endosome. J Cell Biol 202, 527–43 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Allison R et al. Defects in ER-endosome contacts impact lysosome function in hereditary spastic paraplegia. J Cell Biol 216, 1337–1355 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vietri M et al. Spastin and ESCRT-III coordinate mitotic spindle disassembly and nuclear envelope sealing. Nature 522, 231–5 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Kuo TC et al. Purine-Type Compounds Induce Microtubule Fragmentation and Lung Cancer Cell Death through Interaction with Katanin. J Med Chem 59, 8521–34 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Banerjee S et al. 2.3 A resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition. Science 351, 871–5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pohler R et al. A Non-Competitive Inhibitor of VCP/p97 and VPS4 Reveals Conserved Allosteric Circuits in Type I and II AAA ATPases. Angew Chem Int Ed Engl 57, 1576–1580 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Beyer A Sequence analysis of the AAA protein family. Protein Sci 6, 2043–58 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Puchades C et al. Structure of the mitochondrial inner membrane AAA+ protease YME1 gives insight into substrate processing. Science 358(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davis AM, Teague SJ & Kleywegt GJ Application and limitations of X-ray crystallographic data in structure-based ligand and drug design. Angew Chem Int Ed Engl 42, 2718–36 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Han T et al. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 356(2017). [DOI] [PubMed] [Google Scholar]
- 21.Wacker SA, Houghtaling BR, Elemento O & Kapoor TM Using transcriptome sequencing to identify mechanisms of drug action and resistance. Nat Chem Biol 8, 235–7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kapoor TM & Miller RM Leveraging Chemotype-Specific Resistance for Drug Target Identification and Chemical Biology. Trends Pharmacol Sci 38, 1100–1109 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anderson DJ et al. Targeting the AAA ATPase p97 as an Approach to Treat Cancer through Disruption of Protein Homeostasis. Cancer Cell 28, 653–665 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roll-Mecak A & Vale RD Structural basis of microtubule severing by the hereditary spastic paraplegia protein spastin. Nature 451, 363–7 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rouiller I et al. Conformational changes of the multifunction p97 AAA ATPase during its ATPase cycle. Nat Struct Biol 9, 950–7 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Ye Q et al. TRIP13 is a protein-remodeling AAA+ ATPase that catalyzes MAD2 conformation switching. Elife 4(2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loughlin R, Wilbur JD, McNally FJ, Nedelec FJ & Heald R Katanin contributes to interspecies spindle length scaling in Xenopus. Cell 147, 1397–407 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karlberg T et al. Crystal structure of human fidgetin-like protein 1 in complex with ADP. DOI:1–2210/pdb3D8B/pdb. [Google Scholar]
- 29.Evans KJ, Gomes ER, Reisenweber SM, Gundersen GG & Lauring BP Linking axonal degeneration to microtubule remodeling by Spastin-mediated microtubule severing. J Cell Biol 168, 599–606 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taylor JL, White SR, Lauring B & Kull FJ Crystal structure of the human spastin AAA domain. J Struct Biol 179, 133–7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Amaro RE et al. Ensemble Docking in Drug Discovery. Biophys J 114, 2271–2278 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Statsuk AV et al. Tuning a three-component reaction for trapping kinase substrate complexes. J Am Chem Soc 130, 17568–74 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Claudiani P, Riano E, Errico A, Andolfi G & Rugarli EI Spastin subcellular localization is regulated through usage of different translation start sites and active export from the nucleus. Exp Cell Res 309, 358–69 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Connell JW, Lindon C, Luzio JP & Reid E Spastin couples microtubule severing to membrane traffic in completion of cytokinesis and secretion. Traffic 10, 42–56 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mierzwa BE, Chiaruttini N, Redondo-Morata L, von Filseck JM, König J, Larios J, Poser I, Müller-Reichert T, Scheuring S, Roux A, Gerlich DW. Dynamic subunit turnover in ESCRT-III assemblies is regulated by Vps4 to mediate membrane remodelling during cytokinesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schellhaus AK, De Magistris P & Antonin W Nuclear Reformation at the End of Mitosis. J Mol Biol 428, 1962–85 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Yang D et al. Structural basis for midbody targeting of spastin by the ESCRT-III protein CHMP1B. Nat Struct Mol Biol 15, 1278–86 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Olmos Y, Hodgson L, Mantell J, Verkade P & Carlton JG ESCRT-III controls nuclear envelope reformation. Nature 522, 236–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blackstone C, O’Kane CJ & Reid E Hereditary spastic paraplegias: membrane traffic and the motor pathway. Nat Rev Neurosci 12, 31–42 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zempel H, Luedtke J, Kumar Y, Biernat J, Dawson H, Mandelkow E, and Mandelkow EM Amyloid-beta oligomers induce synaptic damage via Tau-dependent microtubule severing by TTLL6 and spastin. The EMBO J 32, 2920–2937, (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mancuso G & Rugarli EI A cryptic promoter in the first exon of the SPG4 gene directs the synthesis of the 60-kDa spastin isoform. BMC Biol 6, 31 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Uphoff CC & Drexler HG Detection of mycoplasma contaminations. Methods Mol Biol 946, 1–13 (2013). [DOI] [PubMed] [Google Scholar]
- 43.Schindelin J et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–82 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hartman JJ et al. Katanin, a microtubule-severing protein, is a novel AAA ATPase that targets to the centrosome using a WD40-containing subunit. Cell 93, 277–87 (1998). [DOI] [PubMed] [Google Scholar]
- 45.Feng BY & Shoichet BK A detergent-based assay for the detection of promiscuous inhibitors. Nat Protoc 1, 550–3 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ziolkowska NE & Roll-Mecak A In vitro microtubule severing assays. Methods Mol Biol 1046, 323–34 (2013). [DOI] [PubMed] [Google Scholar]
- 47.Pettersen EF et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–12 (2004). [DOI] [PubMed] [Google Scholar]
- 48.Friesner RA et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem 49, 6177–96 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.