

Abstract
Purpose of review
This review will examine advances in our understanding of the association between high-density lipoprotein (HDL) function and cardiovascular disease (CVD) in patients with chronic kidney disease (CKD).
Recent findings
Large randomized statin trials and related meta-analyses confirm that lipid-lowering therapy benefits patients with mild to moderate CKD, leaving a degree of residual cardiovascular risk similar to that documented in the general population. However, patients with advanced CKD on dialysis show little to no cardiovascular benefits from lipid-lowering therapy and have an exaggerated residual cardiovascular risk. HDL quantity and functionality may explain some of the residual risk. CKD modulates the level, composition and functionality of HDL, including impaired cholesterol acceptor function and pro-inflammatory effects. Although these abnormalities prevail in CKD, they do not track together and thus support the idea of separate and distinct mechanistic pathways for each of these critical functions of HDL.
Summary
CKD-induced perturbations in HDL composition, metabolism and functionality may contribute to the excess CVD in patients with CKD and present new therapeutic targets for intervention in this population.
Keywords: cardiovascular disease, cholesterol efflux, chronic kidney disease, high-density lipoprotein
INTRODUCTION
The dramatic reduction in cardiovascular events is, in large part, due to lipid-lowering therapy (statins) targeting low-density lipoprotein cholesterol (LDL-C). Despite the success of statins, a 60– 75% residual cardiovascular disease (CVD) risk remains, and thus there is the potential for significant further risk reduction beyond that achieved by lipid-lowering treatment. CKD patients have long been recognized as having increased CVD, although factors underlying the heightened risk are unknown. Further, as CKD patients have historically been excluded from lipid-lowering trials, the therapeutic effectiveness in this patient population was also unknown. Recent retrospective analyses of patients with CKD recruited into clinical trials with cardiovascular endpoints studying hypertensive and dyslipidemic cohorts reveal that lipid-lowering is effective in preventing cardiovascular events in patients with mild to moderate CKD. This conclusion is now firmly supported by the Study of Heart and Renal Protection (SHARP) that specifically evaluated therapeutic intervention in CKD patients. Interestingly, the residual cardiovascular risk observed in CKD patients is similar to that seen in the general population. On the other hand, little to no benefit has been observed in trials of patients whose CKD has progressed to end-stage renal disease (ESRD) requiring dialysis, despite robust reductions in LDL-C. Further, although a therapeutic benefit was observed over a range of CKD in SHARP [1■■], patients with ESRD on dialysis had much less risk reduction compared with patients with moderate CKD. Thus, patients with advanced CKD on dialysis bear an exaggerated residual cardiovascular risk. Understanding the mechanisms underlying the residual risk despite achieving target LDL-C is critical, as additional strategies different and complementary to lipid-lowering agents may be necessary that are especially relevant to CKD. One such strategy may involve high-density lipoprotein (HDL).
CARDIOVASCULAR DISEASE IN CHRONIC KIDNEY DISEASE: INCIDENCE
Chronic kidney disease increases the risk of CVD [2–4]. Individuals with early CKD have a natural history that predicts death from CVD is a more likely consequence than progression to ESRD and distinguishes the CKD patients as among the highest risk cohorts for cardiovascular events of any studied population [5]. These observations are significant because early CKD now affects some 10–16% of the population worldwide, a figure that is projected to rise [6,7]. Although cardiovascular mortality in CKD is due to many causes, atherosclerotic coronary artery disease (CAD) is abundant across the entire spectrum of CKD. A recent analysis of 1010 consecutive patients undergoing coronary angiography revealed that individuals with CAD have significantly increased rate of CKD compared with persons without CAD (18.8% versus 5.4%) [8]. Although the exact prevalence of CAD across all stages of CKD has not yet been firmly established, it is nonetheless widely accepted that atherosclerotic CAD is significantly higher in CKD patients than in the general population [9,10].
TRADITIONAL, NONTRADITIONAL AND RESIDUAL RISKS
Traditional risk factors, including hypertension and diabetes mellitus, are common across the entire CKD spectrum. However, even after statistical adjustments for these risks, CKD independently predicts CVD morbidity and mortality. A large-scale meta-analysis that included more than a million participants encompassing the general population as well as high-risk and chronic disease populations reiterated the concept that CKD [assessed by estimated glomerular filtration rate (eGFR) and albumin:creatinine ratio (ACR)] increases the relative risk of mortality and progression to ESRD in individuals with hypertension [7]. Importantly, the association was even stronger in individuals without hypertension. Additional analysis of this database also revealed that eGFR and ACR predict mortality irrespective of the presence or absence of diabetes [11]. In a separate study, Tonelli et al. [12] compared coronary artery events in individuals with diabetes and individuals with CKD. The incidence of myocardial infarction was similar in diabetic individuals and persons with CKD stages 1–4 but without diabetes. Individuals with more advanced CKD, especially those with more severe proteinuria, had markedly heightened cardiovascular risk compared with diabetic individuals without CKD. Together, these studies emphasize that in addition to the traditional risk factors, CKD per se is a powerful independent risk for future coronary events and mortality.
Hyperlipidemia, specifically elevated level of LDL-C, the traditional risk regarded as the primary driver of CVD in the general population, is not consistent in CKD. The divergence between LDL-C levels and CVD becomes especially apparent as the decline in renal function progresses to ESRD [1■■,13–16]. Other risk factors relevant in the general population, for example hypertension and increased BMI, also lose their prognostic value in the setting of CKD [14,17]. Such observations have led to a search for nontraditional risks specific to CKD, including malnutrition, low albumin, inflammation, oxidant stress, anemia, hyperhomocysteinemia and dysregulation of calcium/phosphorus metabolism. Although experimental and clinical support for each of these potential hazards exist (especially malnutrition/inflammation), none have been definitively proven as causal in the accelerated CVD occurring in CKD.
There is abundant evidence confirming that LDL-C lowering by various HMG-CoA reductase inhibitors (statins) reduces CVD; nonetheless, the potential for sizable additional risk reduction exists. Meta-analysis of more than 90 000 patients with a mean follow-up time of 5 years reported that for every 40 mg/dl reduction in LDL-C, cardiovascular event rates diminished by 21% [18]. In the PROVE-IT trial, aggressive lipid lowering was associated with a residual risk (fatal or no-fatal CHD) of 22.4% after a 2-year follow-up [19]. Review of the major studies with therapies based on lowering of LDL-C by statins found the relative risk reduction in CAD to be 15–37%, which predicts a residual risk in the range of 63–85% [20]. The therapeutic response and, therefore, the residual risk observed in individuals with predialysis CKD is very similar to that remaining in the general population. By contrast, CKD patients who progress to ESRD requiring dialysis are unique in their apparent recalcitrance to lessening the residual risk. This suggests that the uremic environment limits responsiveness to lipid-lowering therapy and reflects greater contribution of factors underlying the residual risk, for example insulin resistance, procoagulable state, other dyslipidemias, for example elevated triglycerides, preponderance of atherogenic LDL particles, accumulation of cholesterol-rich remnant particles and HDL-C. Low HDL-C level, and more recently, reduced HDL function, may explain some of the residual risk and has become a target to further decrease CVD [18,21,22].
HIGH-DENSITY LIPOPROTEIN: LEVEL, STRUCTURE AND FUNCTION
Epidemiologic studies have established that decreased levels of HDL-C are associated with increased CVD, even in individuals on lipid-lowering therapies [18,22–24]. However, the value of HDL-C as a biomarker has been questioned by the increasing appreciation of exceptions to the relationship. Thus, unlike LDL-C, genome-wide association studies have not found that genetic factors regulating HDL-C levels are associated with CAD [25]. Further, genetic variations in the HDL metabolic pathway that decrease or increase the concentration of HDL-C [i.e. apolipoprotein (apo)A-1Milano and cholesteryl ester transfer protein (CETP) deficiency, lecithin/cholesterol acyltransferase (LCAT), hepatic lipase deficiency] do not follow the inverse relationship between the level and CVD events or atherosclerosis [26–28]. Also, the recent disappointing clinical trials showing that significantly raised HDL-C levels do not provide atheroprotection (inhibition of proatherogenic CETP inhibitor torcetrapib in ILLUMINATE and dalcetrapib in dal-OUTCOMES as well as niacin treatment in AIM-HIGH) further underscore that, in isolation, levels of HDL-C may be insufficient as a marker of antiatherogenic effects or therapeutic target [21,29,30■■]. Instead, the studies raise the possibility that not all HDL particles are equally protective and that clinical assays that measure the total quantity of HDL-C may not reflect important qualitative and functional differences. HDL is a very complex lipoprotein and global measures of the HDL-C may fail to capture functionality that relate to changes in the structure, composition and biochemical characteristics of its components. Thus, aside from the level of HDL-C, changes in HDL functions may reflect the relative impact of particular subfractions, alterations in its particle composition or modifications of lipids or proteins [31■].
A key mechanism by which HDL exerts its antiatherogenic effects is reverse cholesterol transport (RCT), a multistep, multiorgan process to remove excess cholesterol from peripheral cells, transport it in plasma for hepatic delivery, followed by excretion in bile and intestine [32]. Modulation of the first step in RCT, namely, cholesterol efflux, can affect atherosclerotic plaques in animal models, whereas impairment in HDL’s capacity to promote cholesterol efflux from cultured macrophage foam cells has been shown to predict subclinical atherosclerosis (increased carotid artery intima–media thickness in healthy volunteers) and likelihood of CAD (angiographic CAD in a case– control study) in a non-CKD population [33■■]. Importantly, these results occurred independently of HDL-C or apoA-I levels. In the atherosclerosis-prone setting of familial hypercholesterolemia, HDL2 particles have also been shown to have reduced cholesterol efflux capacity as well as impaired ability of the HDL to deliver cholesterol esters to the liver [34].
Aside from transport of excess cholesterol, HDL is a key modulator in other processes relevant to atherogenesis such as inflammation and oxidant stress. HDL also protects and supports the endothelium through inhibition of monocyte chemotaxis, adhesion molecule expression, enhanced nitric oxide and prostacyclin production. HDL promotes anticoagulation, augments urokinase-dependent fibrinolysis while inhibiting platelet activation/aggregation [35]. Experimental and clinical examples of these processes include reports that HDL of patients with diabetes failed to stimulate endothelial cell nitric oxide production and promote endothelial repair in a carotid artery injury model in mice [36]. HDL of patients with CAD, rather than stimulating, actually inhibited endothelial cell nitric oxide production and lost the capacity to limit endothelial inflammatory activation and endothelial repair in vivo [37■]. The functional diversity of HDL complements proteomic analyses that indicate that only one-third of HDL’s protein cargo relates to lipid metabolism, whereas the majority are proteins involved in acute-phase response [serum amyloid A (SAA), fibronectin], complement activation and protease inhibition (serine protease inhibitors) [38]. The protein composition of HDL has been shown to differ between healthy individuals and patients with CAD [39]. Treatment with statin and niacin therapy altered the HDL proteome to more closely resemble that of healthy age and sex-matched controls [40]. These observations underscore that the antiatherogenic capacity of HDL particles encompasses a variety of antiatherogenic effects that reflect a dynamic variability in particle size, structure, composition as well as modifications in the proteome or lipidome.
CHRONIC KIDNEY DISEASE AFFECTS CIRCULATING LEVEL, MATURATION AND COMPOSITION OF HIGH-DENSITY LIPOPROTEIN-CHOLESTEROL
CKD reduces circulating HDL-C reflecting depressed synthesis as well as increased degradation and abnormal clearance of its major protein, apoA-I. CKD also affects HDL-C levels by impairing its normal maturation. Although decreased HDL-C is seen across all stages of CKD, the precise biological impact of reduced HDL-C concentrations remains to be determined. For example, a recent study [41] found a linear association between HDL-C levels and carotid intima– media thickness (CIMT) in patients with moderately depressed levels of eGFR. However, studies in dialysis patients found no association between HDL-C concentration and cardiovascular or all-cause mortality [42,43].
In addition to reduced circulating HDL-C, CKD, specifically ESRD, affects the composition of HDL particles, including increasing triglycerides, decreasing cholesterol content, lowering apoA-I/apoA-II and increasing apoC-II/apoC-III levels. Holzer et al. [44■■] showed that compared with HDL isolated from normal controls, the proteome of HDL from dialysis patients had increased acute phase protein, SAA1, albumin, lipoprotein-associated phospholipase A2 and apoC-III. The lipidome of uremic HDL was also different, including reduced phospholipid but increased triglyceride and lysophospholipid. Using shotgun proteomics, Weichhart et al. [45■] identified 49 HDL-associated proteins in hemodialysis patients. Compared with control, uremic HDL was enriched in surfactant protein B (SP-B), apoC-II, SAA1 and α−1-microglobulin/bikunin precursor. This specific set of proteins is distinct from those enriched in HDL of non-CKD patients with CAD, that is apoC-IV, paraoxonase 1 (PON1), C3, apoA-IV and apoE [39]. Such differences suggest that a unique HDL proteome may contribute to CVD susceptibility of the ESRD populations.
It is also notable that SP-B, SAA and pigment epithelium derived factor were elevated in HDL of predialysis patients with moderate CKD and became enriched from CKD4 to ESRD. This study found that although HDL of controls lessened production of inflammatory cytokines, uremic HDL did not have an anti-inflammatory effect and was rather proinflammatory, an effect that was linked to SAA1. Thus, the SAA levels in HDL of ESRD patients significantly correlated with impaired anti-inflammatory ability. Similar observation was made by Tolle et al. [46■], who also found the HDL of dialysis patients to be enriched in SAA1 and have lower anti-inflammatory capacity. This study linked the effect to activation of formyl-peptide receptor 2. Clearly, alterations in the HDL composition observed in ESRD patients may well affect a host’s HDL functions (discussed below). The extent to which more modest renal impairment modifies the proteome or lipidome and the functional consequences of such modifications remains to be clarified.
CHRONIC KIDNEY DISEASE AFFECTS HIGH-DENSITY LIPOPROTEIN FUNCTION
We examined the inflammatory and lipid-handling functions of HDL from patients with ESRD on maintenance hemodialysis (ESRD-HD) [47■■]. HDL from ESRD-HD was dramatically less effective than normal HDL in accepting cholesterol from macrophages [median 6.9% with interquartile range (IQR) 1.4–10.2] vs. control 14.9% (9.8– 17.8), P < 0.001]. The profound efflux impairment was also seen in ESRD-HD diabetic individuals compared with diabetic individuals without kidney disease [median 8.1% (IQR 3.3–12.9) vs. control 13.6% (11.0–15.9), P=.009]. The efflux remained significantly lower in ESRD-HD patients after adjustment for age, sex, race, BMI, high-sensitivity C-reactive protein (CRP), total cholesterol, triglycerides, serum HDL level, serum LDL level, CVD and use of angiotensin-converting enzyme inhibitor/angiotensin II receptor blocker (ARB) [difference, 10.2% with 95% confidence interval (CI) 5.8–14.5, P < 0.0001]. These data fit well with results of Holzer et al. [44■■], who also found reduced efflux ability of HDL in dialysis patients compared with HDL from controls, although that data did not specifically examine whether coexisting comorbidities such as diabetes, obesity, underlying CVD and demographic characteristics contributed to the observed differences. Statin use did not improve efflux capacity to HDL from ESRD-HD [6.8% (1.8–8.8) vs. 6.8% (1.4–10.5) for statin users and nonusers, respectively]. The results echo findings in non-CKD individuals that intervention with statins does not improve cholesterol efflux, which is consistent with the concept that the therapeutic benefit of statins is through mechanisms that are distinct from promoting cholesterol efflux [33■■]. The results complement the confounding data that although lipid-lowering therapies are beneficial in predialysis patients [1■■,48–50], interventions with statins provide only limited benefits against acute events in the ESRD-HD population even in the face of robust reduction in LDL-C [1■■,15,16]. These findings raise the possibility that HDL function may explain the unremitting residual risk in patients with advanced CKD receiving the standard of care.
Strikingly, HDL-C acceptor capacity impairment in ESRD-HD patients persisted irrespective of the serum HDL-C level (Fig. 1). These results illustrate that HDL-C levels may be a poor surrogate for cholesterol efflux capacity in the ESRD-HD. Similar to results reported by Khera et al. [33■■], HDL-C’s efflux capacity in normal controls was also not dependent on concentration of HDL-C. Overall, these observations reiterate the possible importance of alterations in the HDL particle, size, charge and composition on its function, and therefore atherogenesis.
FIGURE 1.
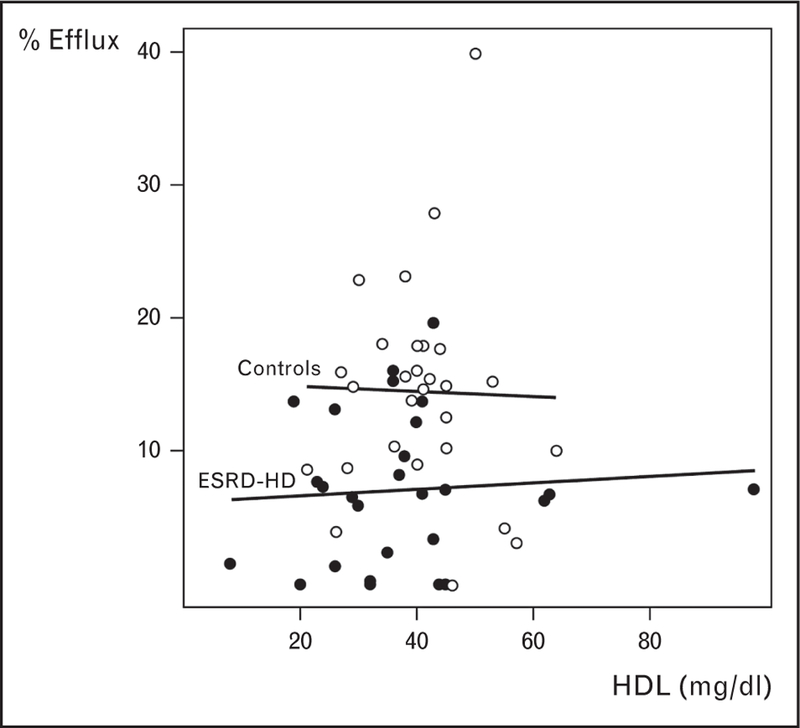
Cholesterol efflux capacity and high-density lipoprotein-cholesterol levels showing no correlation across the range of high-density lipoprotein-cholesterol in end-stage renal disease patients on maintenance hemodialysis (ESRD-HD) or in normal controls.
To determine whether the cholesterol acceptor capacity of HDL can be modulated by activation of cellular transporters, we treated human monocyte leukemia cells (THP-1) cells with a liver X receptor (LXR) agonist that increased expression of the cellular transporter, ATP-binding cassette A1 (ABCA1) and ATP-binding cassette G1 (ABCG1). Upregulation in ABCA1/G1 significantly increased cholesterol efflux to control HDL. LXR-activation of macrophage ABCA1/G1 also increased cholesterol efflux to HDL from ESRD-HD. These observations complement our experimental study that uninephrectomy-induced amplification in atherosclerosis and foam cell formation is due to impaired cholesterol efflux that reflects repression of macrophage ABCA1 [51]. Together, these findings raise the novel possibility that, even in the face of dysfunctional HDL that characterizes advanced CKD, activation of cellular transporters may abrogate pro-atherogenic pathways. Whether such intervention can lessen CVD risk in this population remains to be clarified.
Using the same HDL samples, we examined the cellular inflammatory response in macrophages exposed to HDL of ESRD-HD or non-CKD controls. Compared with the cytokine response in THP-1 cells treated with control HDL, HDL isolated from ESRD-HD caused a significantly greater inflammatory response in interleukin-1 β (IL-1β), interleukin-6 (IL-6) and tumour necrosis factor-α (TNF-α). In addition to enhancing cytokine gene expression, the HDL of ESRD-HD had impaired antichemotactic function. These observations are in line with the understanding that CKD is a proinflammatory/prooxidant state, with various systemic markers of inflammation and oxidant stress increasing as renal function declines [52] as well as the studies showing that CKD specifically impairs the anti-inflammatory functions of HDL [45■,46■]. Weichhart et al. [45■] examined Staphylococcus aureus induced inflammation of monocytes pretreated with HDL from ESRD patients on dialysis, and found that HDL from dialysed individuals had defective anti-inflammatory potency as compared with that from control individuals. These results complement previous findings showing that uremic HDL has an impaired ability to inhibit LDL oxidation [43,53]. Even within the dialysis population, individuals with prooxidant HDL had more comorbidities and increased risk of cardiovascular and all-cause mortalities than individuals whose HDL had better antioxidant ability, although not circulating levels of HDL-C [43]. A prospective study in more than 400 dialysis patients by Honda et al. [54] showed that high levels of oxidized HDL were associated with increased CIMT. Patients with a combination of high ox-HDL and high IL-6 have a significantly greater increase in CIMT at 3 years of follow up as well as an increased risk for CVD events and CVD-related mortality. Together, these findings suggest the functionality of the HDL particle may be an important determinant for antiatherogenic processes in the CKD setting, and may present a new therapeutic target.
It is interesting that in contrast to its effects on cholesterol efflux, therapy with statin abolished the difference in the cytokine response between HDL isolated from dialysis patients and non-CKD control individuals. Thus, IL-1β, IL-6 and TNF-α response to HDL from the subgroup of ESRD-HD patients on statin therapy was not different from that of controls taking statins. These results underscore the possibility that the inflammatory effects of uremic HDL respond to statin therapy, but that this effect is not linked with HDL’s effects on cellular HDL’s cholesterol handling. The divergence in HDL’s inflammatory and lipid-handling response to statin therapy prompted us to examine the association between anti-inflammatory and lipid-handling properties of a given HDL sample across the entire study population. We found no correlation between individual macrophage cholesterol efflux capacity of HDL and markers of systemic inflammation, namely, circulating high-sensitivity CRP. There was also no association between individual macrophage cholesterol efflux capacity of HDL and IL-1β, IL-6 and TNF-α response of macrophages within ESRD-HD individuals. Although the correlation between lipid handling and inflammation was closer in control individuals, this did not achieve statistical significance. It is important to underscore that the results do not negate the importance of either inflammation or abnormal lipid handling in ESRD. Instead, the findings raise the intriguing possibility of separate and distinct mechanistic pathways for different functions of HDL. These findings also suggest that reduced HDL ability to mediate cholesterol efflux may be the key driver for excess CVD among ESRD, and that correction of both cholesterol handling and inflammation must be remedied to achieve a therapeutic benefit in this population. This concept may explain why statins have a limited benefit in this population and supports the idea that both cholesterol efflux and inflammation may have to be targeted with more specific therapies to achieve clinical improvement in this population.
CONCLUSION
Causes underlying the enormous CVD burden in CKD remain unclear. Evidence has accumulated that lipid-lowering therapy in persons with mild to moderate CKD provides similar benefit as in the general population. Nonetheless, a similarly large residual cardiovascular risk unresponsive to the lipid-lowering therapy remains. This residual risk is larger in ESRD patients who appear resistant to the benefits of lipid-lowering therapy. A potential contributor to the residual risk is HDL. Although HDL level is a recognized negative risk for CVD, the concept of HDL dysfunction provides an additional mechanism by which HDL may affect biological processes influencing CVD. CKD decreases the level of circulating HDL and also alters the composition of the HDL particles, rendering HDL dysfunctional. Specific changes in the composition of particles within HDL as well as modifications in the proteome or lipidome of the HDL may cause one or more dysfunctions, and raising the levels of a dysfunctional HDL may not provide benefit. Therefore, therapy should be aimed at improving HDL function, possibly by targeting specific compositional moieties within the HDL particle.
KEY POINTS.
CKD increases cardiovascular disease.
Lipid-lowering therapy in patients with mild to moderate CKD is beneficial and leaves a residual cardiovascular risk that is similar to the general population; lipid-lowering therapy does not lessen residual risk in patients with advanced CKD on dialysis.
HDL quality may be an important contributor to residual cardiovascular risk.
CKD affects the quantity and composition of HDL particles, which in turn impairs their functionality.
Therapies affecting lipoprotein levels should be complemented by interventions that specifically target the composition and modification of HDL particles that impair processes relevant to CVD (cellular lipid homeostasis, anti-inflammatory/antioxidant activity, support of endothelium).
Footnotes
Conflicts of interest
There are no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
■ of special interest
■■ of outstanding interest
Additional references related to this topic can also be found in the Current World Literature section in this issue (pp. 357–358).
- 1.■■.Baigent C, Landray MJ, Reith C, et al. The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (Study of Heart and Renal Protection): a randomised placebo-controlled trial. Lancet 2011; 377:2181–2192.A large, randomized, placebo-controlled trial that focused on patients with moderate to severe CKD as well as those on dilaysis showing that low-dose statin therapy is well tolerated and reduces CVD events in patients with stage 3–4 CKD. The study was inconclusive about CVD benefit in patients on dialysis.
- 2.Go AS, Chertow GM, Fan D, et al. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med 2004; 351:1296–1305. [DOI] [PubMed] [Google Scholar]
- 3.Klausen KP, Scharling H, Jensen JS. Very low level of microalbuminuria is associated with increased risk of death in subjects with cardiovascular or cerebrovascular diseases. J Intern Med 2006; 260:231–237. [DOI] [PubMed] [Google Scholar]
- 4.Schmieder RE, Mann JF, Schumacher H, et al. Changes in albuminuria predict mortality and morbidity in patients with vascular disease. J Am Soc Nephrol 2011; 22:1353–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keith DS, Nichols GA, Gullion CM, et al. Longitudinal follow-up and outcomes among a population with chronic kidney disease in a large managed care organization. Arch Intern Med 2004; 164:659–663. [DOI] [PubMed] [Google Scholar]
- 6.Levey AS, de Jong PE, Coresh J, et al. The definition, classification, and prognosis of chronic kidney disease: a KDIGO Controversies Conference report. Kidney Int 2011; 80:17–28. [DOI] [PubMed] [Google Scholar]
- 7.Mahmoodi BK, Matsushita K, Woodward M, et al. Associations of kidney disease measures with mortality and end-stage renal disease in individuals with and without hypertension: a meta-analysis. Lancet 2012; 380:1662–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu H, Yan L, Ma GS, et al. Association of chronic kidney disease and coronary artery disease in 1010 consecutive patients undergoing coronary angiography. J Nephrol 2012; 25:219–224. [DOI] [PubMed] [Google Scholar]
- 9.Baber U, Stone GW, Weisz G, et al. Coronary plaque composition, morphology, and outcomes in patients with and without chronic kidney disease presenting with acute coronary syndromes. JACC Cardiovasc Imag 2012; 5:S53–S61. [DOI] [PubMed] [Google Scholar]
- 10.Weiner DE, Tighiouart H, Elsayed EF, et al. The relationship between non-traditional risk factors and outcomes in individuals with stage 3 to 4 CKD. Am J Kidney Dis 2008; 51:212–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fox CS, Matsushita K, Woodward M, et al. Associations of kidney disease measures with mortality and end-stage renal disease in individuals with and without diabetes: a meta-analysis. Lancet 2012; 380:1662–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tonelli M, Muntner P, Lloyd A, et al. Risk of coronary events in people with chronic kidney disease compared with those with diabetes: a population-level cohort study. Lancet 2012; 380:807–814. [DOI] [PubMed] [Google Scholar]
- 13.Chawla V, Greene T, Beck GJ, et al. Hyperlipidemia and long-term outcomes in nondiabetic chronic kidney disease. Clin J Am Soc Nephrol 2010; 5:1582–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kovesdy CP, Anderson JE, Kalantar-Zadeh K. Inverse association between lipid levels and mortality in men with chronic kidney disease who are not yet on dialysis: effects of case mix and the malnutrition-inflammation-cachexia syndrome. J Am Soc Nephrol 2007; 18:304–311. [DOI] [PubMed] [Google Scholar]
- 15.Fellstrom BC, Jardine AG, Schmieder RE, et al. Rosuvastatin and cardiovascular events in patients undergoing hemodialysis. N Engl J Med 2009; 360:1395–1407. [DOI] [PubMed] [Google Scholar]
- 16.Wanner C, Krane V, Marz W, et al. Atorvastatin in patients with type 2 diabetes mellitus undergoing hemodialysis. N Engl J Med 2005; 353:238–248. [DOI] [PubMed] [Google Scholar]
- 17.Shah DS, Polkinghorne KR, Pellicano R, Kerr PG. Are traditional risk factors valid for assessing cardiovascular risk in end-stage renal failure patients? Nephrology (Carlton) 2008; 13:667–671. [DOI] [PubMed] [Google Scholar]
- 18.Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90 056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278. [DOI] [PubMed] [Google Scholar]
- 19.Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–504. [DOI] [PubMed] [Google Scholar]
- 20.Rader DJ. Reducing residual cardiovascular risk: the role of raising HDL-C. Medscape Education Article 569095 http://www.medscape.org/viewarticle/569095
- 21.Barter PJ, Caulfield M, Eriksson M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med 2007; 357:2109–2122. [DOI] [PubMed] [Google Scholar]
- 22.Briel M, Ferreira-Gonzalez I, You JJ, et al. Association between change in high density lipoprotein cholesterol and cardiovascular disease morbidity and mortality: systematic review and meta-regression analysis. BMJ 2009; 338:b92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barter P, Gotto AM, LaRosa JC, et al. HDL cholesterol, very low levels of LDL cholesterol, and cardiovascular events. N Engl J Med 2007; 357:1301–1310. [DOI] [PubMed] [Google Scholar]
- 24.Di Angelantonio E, Sarwar N, Perry P, et al. Major lipids, apolipoproteins, and risk of vascular disease. JAMA 2009; 302:1993–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Voight BF, Peloso GM, Orho-Melander M, et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet 2012; 380:572–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johannsen TH, Kamstrup PR, Andersen RV, et al. Hepatic lipase, genetically elevated high-density lipoprotein, and risk of ischemic cardiovascular disease. J Clin Endocrinol Metab 2009; 94:1264–1273. [DOI] [PubMed] [Google Scholar]
- 27.Calabresi L, Baldassarre D, Simonelli S, et al. Plasma lecithin:cholesterol acyltransferase and carotid intima-media thickness in European individuals at high cardiovascular risk. J Lipid Res 2011; 52:1569–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sirtori CR, Calabresi L, Franceschini G, et al. Cardiovascular status of carriers of the apolipoprotein A-I(Milano) mutant: the Limone sul Garda study. Circulation 2001; 103:1949–1954. [DOI] [PubMed] [Google Scholar]
- 29.Roche: Roche provided update on Phase III study of dalcetrapid Investor Update; 2012. http://www.roche.com/investors/ir_update/inv-update-2012-05-07.htm
- 30.■■.Boden WE, Probstfield JL, Anderson T, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med 2011; 365:2255–2267.A large clinical trial that casts doubt on niacin treatment finding of absence of treatment benefits with the addition of modified-release niacin in patients with low baseline HDL-C and intensively treated LDL-C.
- 31.■.de la Llera Moya M, McGillicuddy FC, Hinkle CC, et al. Inflammation modulates human HDL composition and function in vivo. Atherosclerosis 2012; 222:390–394.This study in humans supports the concept that inflammation promotes atherogenic HDL dysfunction with impaired RCT independently of changes in plasma HDL-C and apoA-I.
- 32.Wang X, Rader DJ. Molecular regulation of macrophage reverse cholesterol transport. Curr Opin Cardiol 2007; 22:368–372. [DOI] [PubMed] [Google Scholar]
- 33.■■.Khera AV, Cuchel M, de la Llera-Moya M, et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med 2011; 364:127–135.This study provided evidence that HDL can promote cholesterol efflux from cultured macrophage foam cells and that efflux may be a key process in explaining the inverse relationship between HDL and risk of atherosclerotic CAD in humans.
- 34.Bellanger N, Orsoni A, Julia Z, et al. Atheroprotective reverse cholesterol transport pathway is defective in familial hypercholesterolemia. Arterioscler Thromb Vasc Biol 2011; 31:1675–1681. [DOI] [PubMed] [Google Scholar]
- 35.Soran H, Hama S, Yadav R, Durrington PN. HDL functionality. Curr Opin Lipidol 2012; 23:353–366. [DOI] [PubMed] [Google Scholar]
- 36.Sorrentino SA, Besler C, Rohrer L, et al. Endothelial-vasoprotective effects of high-density lipoprotein are impaired in patients with type 2 diabetes mellitus but are improved after extended-release niacin therapy. Circulation 2010; 121:110–122. [DOI] [PubMed] [Google Scholar]
- 37.■.Besler C, Heinrich K, Rohrer L, et al. Mechanisms underlying adverse effects of HDL on eNOS-activating pathways in patients with coronary artery disease. J Clin Invest 2011; 121:2693–2708.This study revealed that the capacity of HDL to stimulate endothelial nitric oxide production is important for endothelial anti-inflammatory effects of HDL.
- 38.Davidsson P, Hulthe J, Fagerberg B, Camejo G. Proteomics of apolipoproteins and associated proteins from plasma high-density lipoproteins. Arterioscler Thromb Vasc Biol 2010; 30:156–163. [DOI] [PubMed] [Google Scholar]
- 39.Vaisar T, Pennathur S, Green PS, et al. Shotgun proteomics implicates protease inhibition and complement activation in the antiinflammatory properties of HDL. J Clin Invest 2007; 117:746–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Green PS, Vaisar T, Pennathur S, et al. Combined statin and niacin therapy remodels the high-density lipoprotein proteome. Circulation 2008; 118: 1259–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lamprea-Montealegre JA, Astor BC, McClelland RL, et al. CKD, plasma lipids, and common carotid intima-media thickness: results from the Multi-Ethnic Study of Atherosclerosis. Clin J Am Soc Nephrol 2012; 11:1777–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kilpatrick RD, McAllister CJ, Kovesdy CP, et al. Association between serum lipids and survival in hemodialysis patients and impact of race. J Am Soc Nephrol 2007; 18:293–303. [DOI] [PubMed] [Google Scholar]
- 43.Kalantar-Zadeh K, Kopple JD, Kamranpour N, et al. HDL-inflammatory index correlates with poor outcome in hemodialysis patients. Kidney Int 2007; 72:1149–1156. [DOI] [PubMed] [Google Scholar]
- 44.■■.Holzer M, Birner-Gruenberger R, Stojakovic T, et al. Uremia alters HDL composition and function. J Am Soc Nephrol 2011; 22:1631–1641.This study used mass spectrometry and biochemical analysis to show uremic alterations in HDL proteome and lipidome affect cholesterol efflux ability of HDL.
- 45.■.Weichhart T, Kopecky C, Kubicek M, et al. Serum amyloid A in uremic HDL promotes inflammation. J Am Soc Nephrol 2012; 23:934–947.This study showed specific changes in the composition of HDL of dialysis patients with some proteins, for example surfactant protein, increased in HDL of predialysis patients.
- 46.■.Tolle M, Huang T, Schuchardt M, et al. High-density lipoprotein loses its anti-inflammatory capacity by accumulation of pro-inflammatory-serum amyloid A. Cardiovasc Res 2012; 94:154–162.This study shows enrichment in SAA in HDL of patients with ESRD on dialysis.
- 47.■■.Yamamoto S, Yancey PG, Ikizler TA, et al. Dysfunctional high-density lipoprotein in patients on chronic hemodialysis. J Am Coll Cardiol 2012; 60:2372–2379.This study suggests that cholesterol efflux is a key driver of excess CVD in dialysis patients and suggests cellular cholesterol transporters as potential therapeutic targets in this population.
- 48.Ridker PM, MacFadyen J, Cressman M, Glynn RJ. Efficacy of rosuvastatin among men and women with moderate chronic kidney disease and elevated high-sensitivity C-reactive protein: a secondary analysis from the JUPITER (Justification for the Use of Statins in Prevention-an Intervention Trial Evaluating Rosuvastatin) trial. J Am Coll Cardiol 2010; 55:1266–1273. [DOI] [PubMed] [Google Scholar]
- 49.Navaneethan SD, Nigwekar SU, Perkovic V, et al. HMG CoA reductase inhibitors (statins) for dialysis patients. Cochrane Database Syst Rev 2009:CD004289. [DOI] [PubMed]
- 50.Palmer SC, Craig JC, Navaneethan SD, et al. Benefits and harms of statin therapy for persons with chronic kidney disease: a systematic review and meta-analysis. Ann Intern Med 2012; 157:263–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zuo Y, Yancey P, Castro I, et al. Renal dysfunction potentiates foam cell formation by repressing ABCA1. Arterioscler Thromb Vasc Biol 2009; 29:1277–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krane V, Wanner C. Statins, inflammation and kidney disease. Nat Rev Nephrol 2011; 7:385–397. [DOI] [PubMed] [Google Scholar]
- 53.Moradi H, Pahl MV, Elahimehr R, Vaziri ND. Impaired antioxidant activity of high-density lipoprotein in chronic kidney disease. Transl Res 2009; 153:77–85. [DOI] [PubMed] [Google Scholar]
- 54.Honda H, Ueda M, Kojima S, et al. Oxidized high-density lipoprotein as a risk factor for cardiovascular events in prevalent hemodialysis patients. Atherosclerosis 2012; 220:493–501. [DOI] [PubMed] [Google Scholar]