

Summary
Bacteriophages draw scientific attention in medicine and biotechnology, including phage engineering, widely used to shape biological properties of bacteriophages. We developed engineered T4‐derived bacteriophages presenting seven types of tissue‐homing peptides. We evaluated phage accumulation in targeted tissues, spleen, liver and phage circulation in blood (in mice). Contrary to expectations, accumulation of engineered bacteriophages in targeted organs was not observed, but instead, three engineered phages achieved tissue titres up to 2 orders of magnitude lower than unmodified T4. This correlated with impaired survival of these phages in the circulation. Thus, engineering of T4 phage resulted in the short‐circulating phage phenotype. We found that the complement system inactivated engineered phages significantly more strongly than unmodified T4, while no significant differences in phages’ susceptibility to phagocytosis or immunogenicity were found. The short‐circulating phage phenotype of the engineered phages suggests that natural phages, at least those propagating on commensal bacteria of animals and humans, are naturally optimized to escape rapid neutralization by the immune system. In this way, phages remain active for longer when inside mammalian bodies, thus increasing their chance of propagating on commensal bacteria. The effect of phage engineering on phage pharmacokinetics should be considered in phage design for medical purposes.
Introduction
Bacteriophages (phages), which are among the most abundant biological entities on earth, are extensively studied as important tools for medicine and biotechnology. Natural phages are applicable for antibacterial therapies that make use of the intrinsic ability of phages to kill bacteria. Phage therapy is considered a hope to help overcome the antibiotic‐resistance crisis that we currently face (Kutter et al., 2010; Abedon et al., 2011; Pirnay et al., 2011; Miedzybrodzki et al., 2012; Kazmierczak et al., 2014; Gorski et al., 2016). Biotechnology, in turn, explores phage potential for diverse modifications. This field has been emerging during recent years (Pires et al., 2016). Modified phage virions can be versatile and universal nanocarriers, suitable for the delivery of biologically active elements (Jiang et al., 1997; Sathaliyawala et al., 2006; Yacoby and Benhar, 2008; Yao et al., 2016), as well as biosensors capable of detecting and labelling biological targets (Lee et al., 2017; Yue et al., 2017). Phage engineering has also been proposed to enhance, expand or target biological activity of therapeutic phages in vivo when used as antibacterials (Dabrowska et al., 2014b; Gorski et al., 2015; Pires et al., 2016).
Administration of phages to humans or to animals exposes phages to interactions with the immune system (Gorski et al., 2012) that eventually determine phage pharmacokinetics (Hodyra‐Stefaniak et al., 2015; Van Belleghem et al., 2017). Phage pharmacokinetics, in turn, determines phage therapeutic efficacy and outcomes of the treatment (Payne et al., 2000; Levin and Bull, 2004; Cairns et al., 2009). Although induction of phage‐specific antibodies is by far the most widely investigated aspect of immune reactions to phages (Uhr et al., 1962a,b; Ochs et al., 1971; Smith et al., 1987; Fogelman et al., 2000; Huff et al., 2010; Dabrowska et al., 2014a; Majewska et al., 2015; Lusiak‐Szelachowska et al., 2017), it is the innate immune response that plays the key role in phage clearance from animal and human bodies in non‐immunized individuals. Innate immunity is non‐specific and it removes phage particles even when no specific response to bacteriophages has yet developed. Non‐specific removal of phages is executed mainly by the mononuclear phagocytic system (MPS, previously: reticulo‐endothelial system or RES), whereby phages are filtered and inactivated by phagocytosis. The spleen and liver, which are key elements of the MPS, have been demonstrated as major ‘phage traps’ inside bodies (Keller and Engley, 1958; Inchley, 1969; Geier et al., 1973; Hodyra‐Stefaniak et al., 2015). To some extent, bacteriophages can also be neutralized by the serum complement system in a way similar to inactivation of other viruses (Sulkin et al., 1957; Hajek and Mandel, 1966; Dabrowska et al., 2014a; Hodyra‐Stefaniak et al., 2015). Bacteriophages can also affect the innate part of immune response. In general, phage effect on the immune response seems to be anti‐inflammatory. Phage, that naturally binds bacterial products, may moderate signals from bacterial PAMPs (Pathogen‐Associated Molecular Patterns) and thus decrease inflammation (Miedzybrodzki et al., 2008; Gorski et al., 2012; Miernikiewicz et al., 2016; Zhang et al., 2018). Recently, direct anti‐inflammatory phage effect on mammalian cells has been detected by gene expression profiling of peripheral blood monocytes. This effect may further affect outcomes of phage therapeutic application (Van Belleghem et al., 2017).
Differences in phage susceptibility to neutralization by the innate immune system may be responsible for differences in phage ability to remain active in vivo. This was first reported by Merril et al. (Merril et al., 1996), who isolated long‐circulating phages. Long‐circulating phages are mutants or variants capable of maintaining their antibacterial activity for longer (than parental strains) when they circulate in mammalian blood. Bacteriophages present in the circulation immediately disseminate throughout the body (Dabrowska et al., 2005); thus, prolonged circulation indicates maintenance of higher titres in the whole body. Long‐circulating phages have been identified as more efficient in phage therapy of experimental septicaemia (Merril et al., 1996; Vitiello et al., 2005; Capparelli et al., 2006, 2007).
As an alternative to searching for phage mutants able to circulate longer in the system, we have previously proposed phage engineering with small peptides that promote phage accumulation in selected tissues (Gorski et al., 2015). It is unclear if the modified phages, by adsorption to selected cell types, would leave the circulation and thus result in a short‐circulating phenotype. Nevertheless, increasing phage concentration at the site of infection may be essential for achieving the ‘inundation threshold’, which is the minimum phage density that can prevent a bacterial infection from progressing (Abedon, 2011). In localized infections, this might greatly increase the success of treatment. The targeting (homing) peptides can be presented on the phage surface by phage display technology (Gorski et al., 2015). The idea relates to the fundamental studies of Ruoslahti, who identified homing peptides for brain and kidney (Pasqualini and Ruoslahti, 1996). Peptides facilitating delivery and homing to many tissues were further identified by the use of phage display libraries (Arap et al., 2002a,b; Duerr et al., 2004; Kang et al., 2008; Giordano et al., 2009; Budynek et al., 2010; Li et al., 2011, 2012; Teesalu et al., 2013), thus demonstrating broad potential to target drugs to selected organs, potentially improving outcomes of many kinds of treatment (Ruoslahti, 2012).
Here, we report a study of seven T4‐derived phages, each presenting targeting peptides on the head surface. Phage virions were engineered by phage display as previously described (Oslizlo et al., 2011; Ceglarek et al., 2013). They presented peptides targeting the lung (2 peptides), prostate (2 peptides) or brain (1 peptide), or facilitating translocation from the intestine lumen to the circulation (2 peptides). All seven types of engineered phages were investigated in vivo for their pharmacokinetics and compared to non‐modified T4 phage. Phage titres were tested in blood and in selected organs including the spleen and liver. To understand individual pharmacokinetics of engineered phages, immune responses elicited by the engineered phages were identified in terms of both the innate immune response (phagocytosis, serum complement activity) and the adaptive immune response (antibodies).
Results
Circulation of engineered bacteriophages in targeted tissues, spleen, liver and blood
The following seven types of engineered bacteriophage T4 were constructed; these phages displayed peptides targeting the lungs (T4‐L1 and T4‐L2), the prostate (T4‐P1 and T4‐P2), the brain (T4‐B), and facilitating translocation from the gut lumen to the circulation (T4‐G1 and T4‐G2) (for sequences and references see Table 1 in the Material and Methods section). All engineered phages presented the peptides as N‐terminal fusions to surface protein Hoc. We tested by anti‐Hoc antibody reaction relative saturation of phage particles with Hoc fusions (Fig. S1). We confirmed that Hoc fusions were present on all types of engineered phages. The ability of displayed peptides to target selected cells was confirmed in vitro in representative phages; these were demonstrated to bind (T4‐P1, T4‐B) or to translocate across targeted cells (Fig. S2). Investigated bacteriophages were injected i.v. into mice (T4‐L1, T4‐L2, T4‐P1, T4‐P2, T4‐B) or added to drinking water (T4‐G1, T4‐G2). Unmodified T4 phage served as a control in each case and it was applied by identical route and schedule as engineered phages. Eventually, phages disseminated in the whole body, since active phages were detected in all targeted organs as well as in the spleen and liver (Figs 1 and 2). However, expected accumulation of engineered phages in targeted organs was not observed in any case (Fig. 1). We did not observe any cross‐reactivity between types of modified phages (data not shown). Further, phages T4‐B and T4‐G2 achieved approximately 2 orders of magnitude lower titres in targeted organs than the parental strain (Fig. 1), which was opposite to expected outcomes of phage modifications. Concordant results were observed longer after the administration (up to 24 hours after administration) (data not shown).
Table 1.
Targeting peptides presented on T4 phage
| Targeted organ | Designation of phage displaying this peptide | Sequence of targeting peptide | Reference |
|---|---|---|---|
| Lungs | T4‐L1 | CGFECVRQCPERC | Rajotte and Ruoslahti (1999) |
| Lungs | T4‐L2 | CGSPGWVRC | Giordano et al. (2008) |
| Brain | T4‐B | TGNYKALHPHNG | Li et al. (2011) |
| Prostate | T4‐P1 | RRAGGS | Arap et al. (2002b) |
| Prostate | T4‐P2 | SMSIARL | Arap et al. (2002a) |
| Translocation from gut lumen to circulation | T4‐G1 | YPRLLTP | Duerr et al. (2004) |
| Translocation from gut lumen to circulation | T4‐G2 | CSKSSDYQC | Kang et al. (2008) |
Figure 1.
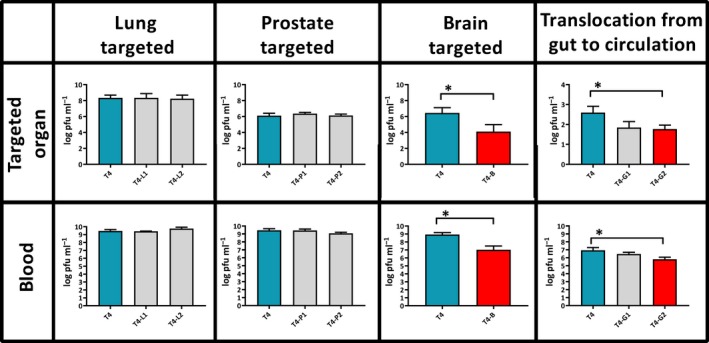
Phage titres in targeted organs and in blood of mice after administration of T4 phage presenting peptides targeting lungs (T4‐L1 and T4‐L2), prostate (T4‐P1 and T4‐P2), brain (T4‐B) and facilitating translocation from gut to circulation (T4‐G1 and T4‐G2). Targeted organs: Mice (N = 5 to 6) were injected intravenously with a phage dose of 5 × 109 pfu per mouse (each phage) and phage titre was measured in tissues 2 h later (T4‐L1, T4‐L2, T4‐P1, T4‐P2, T4‐B) or phage was added to drinking water at 5 × 1010 pfu per ml and phage titre was measured 10 h later in blood (T4‐G1 and T4‐G2). In each case, non‐modified T4 phage (blue bars) was used as a control. Log10 of mean phage concentrations in selected organs (pfu per gram of tissue or per ml of blood) is presented (bars) with standard deviation (whiskers). Differences statistically significant in comparison to control phage are indicated with asterisks and red colour of a relevant bar (Mann–Whitney U‐test, P < 0.05). Blood: in all cases, mice (N = 5 to 6) were injected intravenously with tested phages 1 × 109–1 × 1010 pfu per mouse to compare phage circulation in blood, phage titre in blood was measured 2 h later. In each case non‐modified T4 phage (T4, 1 × 1010 pfu per mouse) was used as a control. Log10 of mean phage concentrations is presented (bars) with standard deviation (whiskers). Differences statistically significant in comparison to control phage are indicated with asterisks and a red bar (Mann–Whitney U‐test, P < 0.05).
Figure 2.
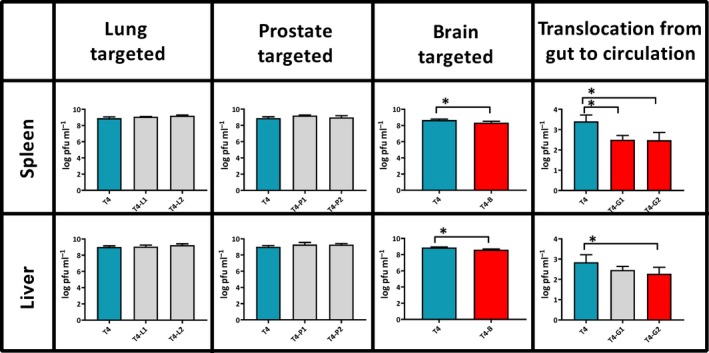
Phage titres in murine spleen and liver after administration of T4 phage presenting peptides targeting lungs (T4‐L1 and T4‐L2), prostate (T4‐P1 and T4‐P2), brain (T4‐B) and facilitating translocation from gut to circulation (T4‐G1 and T4‐G2). Mice (N = 5 to 6) were injected intraperitoneally with a phage dose of 5 × 109 pfu per mouse (each phage) and phage titre was measured in tissues 2 h later (T4‐L1, T4‐L2, T4‐P1, T4‐P2, T4‐B) or phage was added to drinking water at 5 × 1010 pfu per ml and phage titre was measured 10 h later in blood (T4‐G1 and T4‐G2). In each case, non‐modified T4 phage (blue bars) was used as a control. Log10 of mean phage concentrations in selected organs (pfu per gram of tissue or per ml of blood) is presented (bars) with standard deviation (whiskers). Differences statistically significant in comparison to control phage are indicated with asterisks and red colour of a relevant bar (Mann–Whitney U‐test, P < 0.05).
Intravenous (i.v.) administration of modified bacteriophages was used to assess their ability to survive in the circulation. All investigated bacteriophages were administered i.v. to mice, and unmodified T4 phage served as the control. Two hours after administration, phage titre was determined in blood. No differences were observed between the blood titre of control T4 phage and modified phages T4‐L1, T4‐L2, T4‐P1, T4‐P2 and T4‐G1. However, blood titres of T4‐B and T4‐G2 were from 1 to 2 orders of magnitude lower than those of the unmodified control (P = 0.04164 and P = 0.007382, respectively) (Fig. 1). Consistent results were observed longer up to 24 h after administration (data not shown). Notably, concentrations of T4‐B and T4‐G2 bacteriophages in the spleen and liver were also significantly lower than concentrations of the unmodified T4 phage (Fig. 2). Lower titres of T4‐B and T4‐G2 in all investigated tissues suggested that those engineered phages had a generally weaker ability to survive in living animal bodies.
Interactions of engineered bacteriophages with innate immunity
Phage ability to survive in the circulation may depend on phage susceptibility to neutralization by the innate immune system (Merril et al., 1996; Hodyra‐Stefaniak et al., 2015). Thus, we investigated ex vivo interactions of the phages T4‐B, T4‐G1, T4‐G2 and unmodified T4 (control) with two major parts of the innate immunity response: complement system and phagocytes. Phages were incubated with blood sera as the source of complement, and with isolated phagocytic cells which were polymorphonuclear cells (PMNs) or peripheral blood mononuclear cells (PBMCs). Human blood was used in this part of the study to make the observations more useful for therapeutic and other medical solutions in humans.
Exposure of phages to the complement system significantly decreased phage activity: phage titre remaining after incubation with active sera ranged from 4.7% (T4‐B) to 43.7% (T4) of initial phage activity, while it was not decreased in the same phages incubated with inactivated sera (P < 0.001) (Fig. 3). Unmodified T4 phage was less susceptible to neutralization by the complement system than phages displaying targeting peptides. This difference was significant when compared to T4‐B and T4‐G2 (P = 0.007 and P = 0.0485, respectively) (Fig. 3). Incubation of phages with phagocytes, in turn, did not result in significant differences between titres of remaining engineered bacteriophages and control phage T4; the overall decrease of phage titre ranged from 20 to 40% of the initial phage titre (control) (Fig. 4). Thus, individual phage susceptibility to phagocytosis by immune cells does not seem to contribute to differences between engineered and non‐engineered bacteriophages in their ability to survive in circulation in vivo, while the complement system plays an important role in inactivation of bacteriophages, having a stronger effect on the engineered ones.
Figure 3.
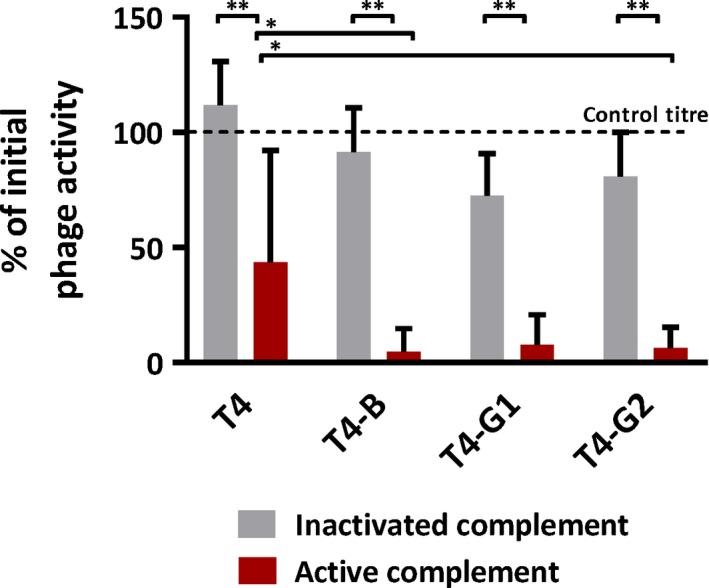
Effect of human complement system ex vivo on non‐modified bacteriophage T4 and on T4 presenting peptides targeting brain (T4‐B), and facilitating translocation from gut to circulation (T4‐G1 and T4‐G2). Blood samples from six healthy human volunteers were use. Individuals defective for the serum complement activity were excluded from the study. Serum was isolated from blood samples and incubated 1:1 with phage preparations (107 pfu ml−1) for 1 h at 37°C, either active (red bars) or after heat inactivation for 1.5 h at 56°C (grey bars). After incubation, phage activity was tested by the double‐layer plate method. Phage titre was compared to control titre without incubation with serum (initial phage activity) and presented as % of the control. **Statistically significant P < 0.001, *statistically significant P < 0.05.
Figure 4.
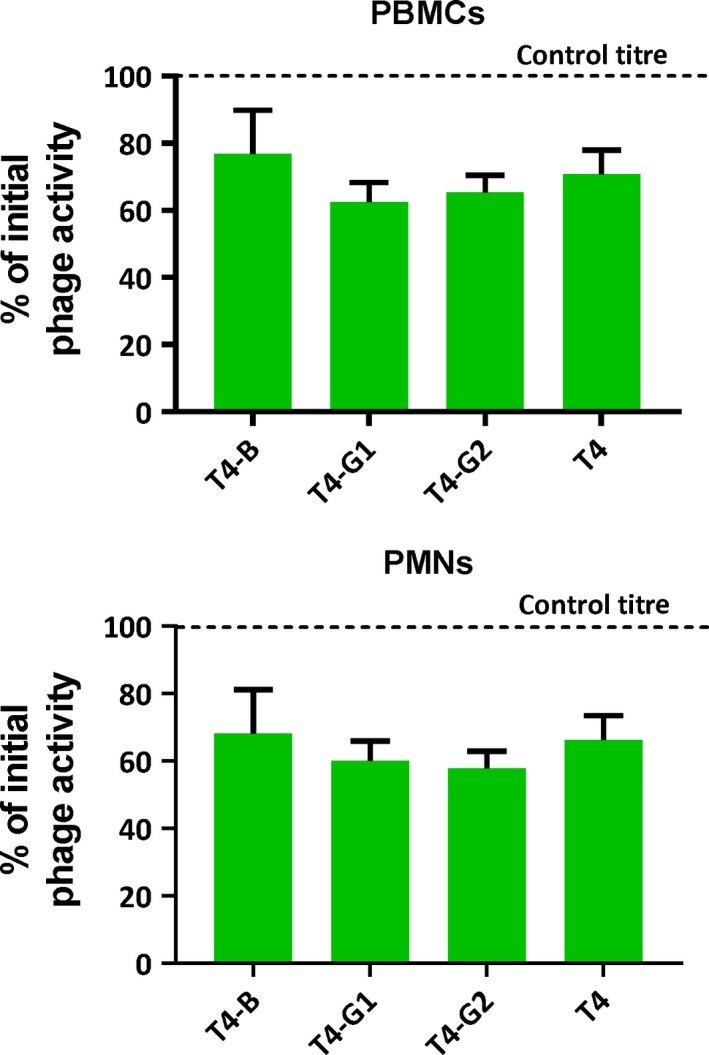
Effect of human phagocytes ex vivo on non‐modified bacteriophage T4 and on T4 presenting peptides targeting brain (T4‐B), and facilitating translocation from gut to circulation (T4‐G1 and T4‐G2). Polymorphonuclear neutrophils (PMNs) and mononuclear blood cells (PBMCs) were used in a final density of 106 cells ml−1. Phages T4‐B1, T4‐G1, T4‐G2 and T4 as the control were added to a final count of 105/ml (volume: 1 ml) and incubated for 1.5 h at 37°C, viable phage titre detected after incubation was determined by RTD in the culture supernatant, results were presented as the per cent of initial phage titre (control titre).
Induction of antibodies by engineered bacteriophages
Innate immunity and adaptive immunity are linked by many pathways and they collaboratively contribute to resulting immune responses to foreign antigens. Complement‐conjugated antigens are effective in rapid development of the specific immune response (Dempsey et al., 1996). Thus, we investigated whether differences in phage abilities to interact with innate immunity eventually affected the adaptive immunity response, specifically phage‐specific antibody production. However, we did not observe significant differences between levels of specific IgM and IgG elicited in mice in response to treatment with engineered phages T4‐B, T4‐G1, T4‐G2 and unmodified T4. The following typical pattern of induction was observed: the IgM peak was observed around day 7, IgG increased gradually from the beginning of the experiment, a major rise of IgG was noted approximately from day 7, and the IgG level remained high until the end of the experiment on day 50 (Fig. 5). This is in line with antibody induction patterns observed in murine models for other Myoviridae bacteriophage F8 (Hodyra‐Stefaniak et al., 2015).
Figure 5.
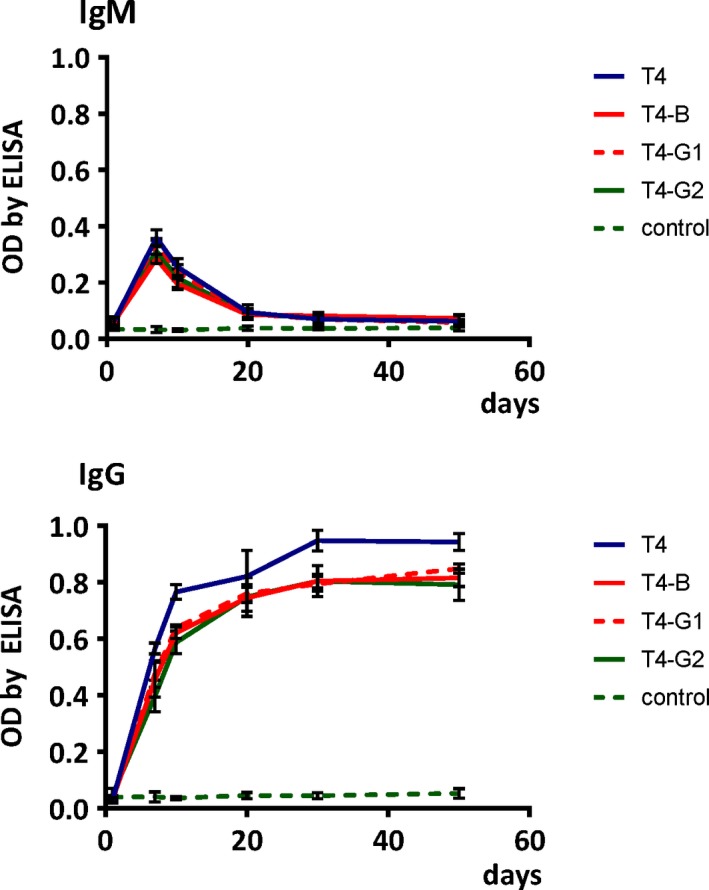
Kinetics of phage‐specific antibody production in mice treated with T4 phages presenting targeting peptides. Upper panel: induction of IgM specific to phages targeting brain (T4‐B) and facilitating translocation from gut to circulation (T4‐G1, T4‐G2), days 1–50 after injection, is presented; lower panel: induction of IgG specific to phages targeting brain (T4‐B) and facilitating translocation from gut to circulation (T4‐G1, T4‐G2). Mice (N = 5) were injected with modified phages i.p. at 1 × 109 pfu per mouse; in each case, non‐modified T4 phage (T4, 1 × 109 pfu per mouse) was used as a control. Blood was collected from tail veins on days 1, 7, 10, 20, 30, 50 and specific antibodies were assessed by ELISA; optical density (OD) by ELISA is presented (points with trend line) with standard deviation (whiskers).
Discussion
In this study, we studied engineered bacteriophages presenting tissue‐homing peptides, by evaluating their tissue accumulation and blood circulation in vivo in an animal model. Although this study was inspired by very encouraging data from selection of tissue‐homing peptides by phage display (Pasqualini and Ruoslahti, 1996; Arap et al., 2002a,b; Duerr et al., 2004; Kang et al., 2008; Giordano et al., 2009; Li et al., 2011, 2012; Teesalu et al., 2013; Gorski et al., 2015), we have not observed the expected concentration of engineered bacteriophages in targeted organs, as a result of any phage modifications tested (Fig. 1). Probably, the major difference between the aforementioned studies that allowed for selection of targeting peptides and the study presented herein is the phage display system that was used. Here, we used T4, which is a tailed phage with a large icosahedral head (Caudovirales), while those previous studies applied filamentous phages such as fd, M13 or related phages (Inoviridae). Caudovirales and Inoviridae substantially differ in their morphology and in the way that foreign peptides are exposed on the phage, including peptide‐neighbouring and linking elements (Fig. 6). Thus, these differences are probably important enough to impair the targeting properties of peptides when presented on T4.
Figure 6.
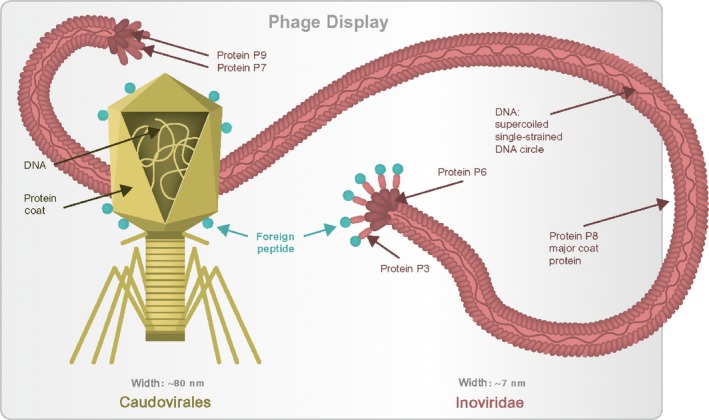
Structural differences between T4 phage‐derived and filamentous phage‐derived vectors presenting tissue targeting peptides.
In some cases (phage T4‐B and T4‐G2), we did not observe phage accumulation in targeted organs, in fact phage titres were significantly lower than that of unmodified phage. This was opposite to the expected results. However, lower phage titres in targeted tissues correlated with lower titres in spleens and livers (Fig. 1), which suggested that phage titre in the whole body was generally lower. The range of this decrease varied from only 2‐fold up to 100‐fold 2 hours after application. Such a situation may result from impaired phage survival in a living system. Impaired phage survival in the circulation was indeed demonstrated for T4‐B and T4‐G2, when compared to unmodified T4 (Fig. 2). Thus, in addition to long‐circulating phages (Merril et al., 1996), we have observed short‐circulating phages.
Seeking the mechanisms underlying the short‐circulating phenotype, we investigated the immunity reactions to phages. Pharmacokinetics in naïve animals is determined by interactions with innate immunity, where the major roles can be played by phagocytes and possibly by the complement system. We have not found any important effect of phagocytosis (Fig. 4), but the effect of complement on phages was significant. Phage activity decreased after phage exposure to complement by almost 2 orders of magnitude (Fig. 3). Engineered phages T4‐B and T4‐G2 were inactivated by the complement system significantly more strongly than non‐modified T4 (Fig. 3). Although we did not find an important role of phagocytes in the observed phenotype, one should note that in a living system complement and phagocytes cooperate. Foreign objects when opsonized by complement proteins are more readily engulfed by phagocytes. This means that in more natural situation, phagocytosis of these short‐circulating phages can also be more effective, even though the primary reason is their reactivity to the complement system.
Complement proteins (C3b) form covalent bond to surface of targeted object (e.g. a virus) and they recruit further components of the cascade, eventually forming a large structure (with a major contribution of C6, C7, C8 and C9). This complex measures 30.5 nm in its longest dimension, and the upper rim width of 24 nm (Serna et al., 2016). This means that each complex, while many can be formed at each phage, have considerable dimensions comparing to phage (approx. head 111 × 78 nm, tail 113 × 18 nm, baseplate 52 × 27 nm). We hypothesize that protein complexes formed by the complement system are at least able to block normal functions of phage proteins, including their steric rearrangements and their adhesion to bacterial surfaces. It is unclear if phage particles can be disrupted by complement system complexes, but natural activity of these complexes is to perforate targeted objects.
Sokoloff et al. (2000, 2001, 2004) observed that interactions of T7‐based phage vectors with the complement system were mediated by natural IgM antibodies. Natural IgM antibodies are considered to contribute to important immunoregulatory and housekeeping functions in mammals by recognition of apoptotic cells and enhancing their phagocytic clearance, but their full repertoire of function has not been determined yet (Gronwall et al., 2012). In T7 phage display vectors, natural IgM recognized and bound phage particles, eventually activating the complement cascade. Importantly, binding of IgM to phage vectors depended on C‐terminal sequences of peptides presented on capsids. A long‐circulating phenotype in rats was generated by presenting peptides with C‐terminal lysine or arginine (Sokoloff et al., 2000).
In our study reported herein, foreign peptides presented on the phage capsid did not expose their C‐terminus, since the fusion to Hoc protein on T4 phage was N‐terminal. However, Vitiello et al. (2005) reported that in phage lambda, the long‐circulating phenotype (in mice) was mediated by a mutation located inside the gene coding major capsid protein; this mutation resulted in substitution of glutamic acid for lysine inside the relevant protein (not at the C‐terminus) (Vitiello et al., 2005). Although the authors proposed lower susceptibility of mutated phage to capture by the mononuclear phagocytic system (referred by them as reticulo‐endothelial system), it is possible that the long‐circulating phenotype might instead result from lower phage susceptibility to inactivation by the complement system. Thus, studies of Sokoloff et al. (2000) and Vitiello et al. (2005) suggest that exposure of lysine (K) or arginine (R) on the phage head surface helps the phage to escape the immune response (long‐circulating phenotype), while lack of these two makes the phage more sensitive to inactivation in vivo. We have not found exactly the same correlation among the investigated targeting peptides, since lysine (K) was present in two of the peptides mediating the short‐circulating phenotype. However, all peptides making T4 phage short‐circulating lacked arginine (R), which was present in all others (Fig. 7). This may suggest that the lack of arginine contributes to the short‐circulating phenotype. We propose the amino acid composition of phage capsids as the factor strongly affecting phage interaction with the complement system. This is in line with the fact, that an important step of complement action is formation of a covalent bond between C3b complement proteins and surface carbohydrate or protein of targeted object (e.g. a virus). Thus, changes in biochemical characteristics of these surfaces may change vulnerability to complement action. Nevertheless, this mechanism needs further investigation and so far should be regarded as a hypothesis.
Figure 7.

Amino acid sequences of peptides presented on engineered bacteriophages. Arginine (R, blue) was previously identified as attenuating phage interactions with complement system, bold – peptides mediating short‐circulating phenotype of peptide‐presenting bacteriophages.
One should consider that modifications in the phage particle that make it a phage display platform, not the foreign peptides, may affect phage circulation in a living animal. The engineering itself may affect the phage. In this study, a phage deprived of its surface protein Hoc served as the platform for presentation of the peptides; the phage display system was used to put Hoc fused to the tested peptides on the platform. Thus, differences between the platform and the wild‐type phage may play a role in the observed effects. We also observed differences between relative saturation of phage particles with the fusions, but no correlation between this saturation and impaired phage pharmacokinetics was found. This suggests that the observed results did not derive from differences in the saturation of phage particle with fusions. However, in our opinion, some effect of differences between wild phage particles and those deprived of the surface proteins cannot be fully excluded and they may contribute to the resulting effect of phage engineering on phage pharmacokinetics. The problem should be a subject of further investigations.
The short‐circulating phenotype seems unfavourable in therapeutic application of phages. Rapid elimination of phages results in a low overall phage titre in the body, which makes it difficult to achieve the ‘inundation threshold’ that is necessary to control infection (Abedon, 2011). Poor accumulation of phages in tissues is not a good predictor of therapeutic outcomes. Obviously, more studies are needed to understand if and how our results on phage circulation and organ penetration are relevant for clinical phage therapy. Phage penetration may be species‐specific, and other routes of phage administration should also be tested to draw conclusions which may be applicable to treatment of patients. However, changes of phage pharmacokinetics that result from phage engineering should be taken into account in attempts to engineer phages or phage‐derived delivery vectors for medical purposes (e.g. in design of nanocarriers). Designed modifications need to be tested for their effect on phage pharmacokinetics, which may differ between different types of bacteriophages and different types of modifications. Specifically, some coliphages, such as T4, may have developed mechanisms for escaping the immune response in mammals, since mammals are a natural environment for these phages. Coliphages can be easily modified due to the abundance of Escherichia coli laboratory strains, coli‐related vectors and well‐established systems for phage display, but also other types of phages may offer a good platform for modifications. Thus, careful selection and individual testing of phage strains designed for particular applications seem to be crucial for their successful use in vivo.
Conclusions
Natural bacteriophages seem to be naturally optimized (to some extent) to circulate in mammalian bodies escaping rapid neutralization, at least those phages that propagate on commensal bacteria of animals and humans. This is in line with evolutionary selection pressure: circulating inside mammalian bodies, phages benefit from evading activity of the immune system. They remain active for longer, thus increasing their chance of encountering sensitive bacteria among mammalian commensals. The complement system contributes to phage neutralization in vivo, and thus phages less prone to its activity benefit from longer survival. Phage engineering, especially presenting foreign elements on the phage surface, can destroy optimized biochemical properties of a phage particle and make the phage more visible to the immune system. Eventually, pharmacokinetics of engineered phages differ from that of natural ones, and it can be unfavourable for therapeutic use: the phage may acquire the short‐circulating phenotype. However, even when more prone to innate immunity action, short‐circulating phages do not induce a stronger specific immune response. Unfavourable effects of phage engineering can manifest in one type of bacteriophage (e.g. Caudovirales), while not being observed in others (e.g. filamentous phages). Thus, careful selection and individual testing of phage strains designed for particular applications seem to be crucial for their optimal use in vivo.
Experimental procedure
Bacteriophages
The wild‐type T4 phage was purchased from American Type Culture Collection (ATCC) (Rockville, Maryland, USA). Phage display of targeting peptides on T4 was completed as previously described with minor modifications (Oslizlo et al., 2011). Briefly, a phage variant without the protein Hoc was cultured on E. coli transformed with expression vectors coding Hoc‐targeting peptide fusions (N‐terminal), that is, fusions were incorporated into phage capsid during natural phage assembly inside bacteria. Expression vectors were constructed in pCDFDuet‐1 (Novagen) by cloning of PCR products coding Hoc‐targeting peptide fusions into the XhoI/BglIII restriction site. The sequences coding for targeting peptides were introduced in a PCR reaction from the forward primer. Peptides fused to Hoc and displayed on T4 phage are listed in Table 1. All cloning vectors were directly sequenced for control of construction accuracy and tested for effective expression of the Hoc fusions in E. coli B834 (OverExpress). Appropriate clones were used for the phage display cultures.
Lysates were purified by filtration through polysulfone membranes and by chromatography: gel filtration on Sepharose 4B (Sigma‐Aldrich, Poland). The preparation was dialysed using 1000 kDa‐pore membranes against sterile and pure PBS and filtered with 0.22 μm PVDF filters (Millipore, Europe). Phage concentrations were measured by the double‐layer method of Adams or by routine test dilution (RTD), and presence of Hoc fusions was confirmed by ELISA with anti‐Hoc murine serum (Supporting Information) (Dabrowska et al., 2014a). LPS concentration was controlled by EndoLISA (Hyglos GmbH, Germany), according to the manufacturer's instructions. Diluted samples or standard dilution with binding buffer were incubated overnight at room temperature with shaking. Subsequently, the plate was washed and assay reagent was added. The fluorescent signal was detected immediately in a fluorescence reader (Synergy H4 H4MLFPTAD BioTek Instruments USA). Effective concentration of LPS was less than 1 EU per mouse or per ml of solutions used for ex vivo complement and phagocytosis assays.
Animal model of phage circulation in vivo
The male BALB/c (6–10 weeks) mice were purchased from the Center of Experimental Medicine, Medical University of Bialystok, Poland, or Mossakowski Medical Research Centre, Polish Academy of Sciences, Warsaw, Poland and bred under specific pathogen‐free (SPF) conditions in the Animal Breeding Center of the Institute of Immunology and Experimental Therapy (IIET).
For the study of engineered phages in targeted organs, spleen and liver, mice (N = 5 to 6) were (i) injected intraperitoneally with 5 × 109 pfu per mouse of phages targeting lungs, brain or prostate (T4‐L1, T4‐L2, T4‐P1, T4‐P2, T4‐B), phage titre being measured in the tissues 2 h later, or (ii) treated with phage added to drinking water 5 × 1010 pfu per ml of phages modified with peptides facilitating translocation from gut to circulation (T4‐G1, T4‐G2), and phage titre was measured 4 h later in blood. In each case, non‐modified T4 phage that was cultured and purified identically to modified phages was used as a control. Animals were sacrificed by cervical dislocation, liver and spleen were excised in all animals, and targeted organs were excised according to the tested phage type. Organs were homogenized and weighed, and the homogenates were serially diluted with PBS (1 g equalling 1 ml). The phage titre in each tissue/organ was determined by the RTD method. In the case of phages modified with peptides facilitating translocation from gut to circulation, murine blood was collected from the tail vein into heparinized tubes, under local anaesthesia (lidocaine) and phage titre was determined by the RTD method.
For the study of engineered phages’ survival in circulation, mice (N = 5 to 6) were injected intravenously 1 × 109–1 × 1010 pfu per mouse and their titre in blood was measured 2 hours later. In each case, non‐modified T4 phage that was cultured and purified identically to modified phages was used as a control. Blood was collected from the orbital plexus vein into heparinized tubes, under anaesthesia, and phage titre was determined by the RTD method.
Animal model of antibody induction
The male C57Bl6/J (6–10 weeks) mice were purchased from the Center of Experimental Medicine, Medical University of Bialystok, Poland, and bred under SPF conditions in the Animal Breeding Center of the IIET.
Mice (N = 5 to 6) were injected intraperitoneally (i.p.) with modified phages T4‐B, T4‐G1 or T4‐G2; the dose was 1 × 1010 pfu per mouse. Non‐modified T4 phage that was cultured and purified identically to modified phages was used as a control. Blood was collected from tail veins on days 1, 7, 10, 20, 30 and 50; blood was collected into clotting tubes. Serum was separated from the blood by double centrifugation at 2250 × g and used for the ELISA assay.
ELISA was conducted on MaxiSorp flat‐bottom 96‐well plates (Nunc, Thermo Scientific, Europe) that were covered overnight with phages (5 × 109 pfu ml−1). Subsequently, wells were washed with PBS and blocked with 1% SuperBlock Blocking Buffer (Thermo Scientific, Europe). Diluted serum (1/100 in PBS) was added (100 μl of diluted serum per well). The plate was incubated at 37°C for 2 h and washed with 0.05% Tween 20 in PBS (Serva, Europe) five times. Diluted detection secondary antibody was applied (100 μl per well): peroxidase‐conjugated AffiniPure goat anti‐mouse IgM (Jackson ImmunoResearch Laboratories) or peroxidase‐conjugated AffiniPure goat anti‐mouse IgG (Jackson ImmunoResearch Laboratories). The plate was incubated for 1 h at room temperature in the dark. TMB substrate reagents for peroxidase were used according to the manufacturer's instructions (DY999, R&D Systems, Europe) and incubated for 20 min. Twenty‐five μl of 2N H2SO4 was added, and absorbance was measured at 450 nm (main reading) and 570 nm (background).
Phage inactivation by the complement system
The human complement effect on engineered bacteriophages was evaluated in sera of 8 healthy volunteers, with their written consent, and in accordance with the local Commission of Bioethics, Wroclaw Medical University (approval no. 503/2015). Potential deficiencies of the complement activity in the donors were tested by the diagnostic Complement System Screen (WIESLAB, Euro Diagnostica AB, Sweden) before the experiment, and samples with any abnormal complement system activity were excluded. Blood was collected into clotting tubes, and serum was separated from the blood by double centrifugation at 2250 g. Half of each serum sample was heat‐inactivated by incubation at 56°C for 1 h to inhibit the serum complement system. Each donor's serum was further tested as a complement‐inactivated or non‐inactivated serum. To test the inhibitory effect of human sera on bacteriophages, each engineered bacteriophage – T4‐L1, T4‐L2, T4‐P1, T4‐P2, T4‐B, T4‐G1, T4‐G2 (107 pfu ml−1) – and T4 as the control (107 pfu ml−1) was mixed with serum samples (1:1) and incubated at 37°C for 1 h. After incubation, phage activity was tested by the double‐layer plate method.
Phage inactivation by phagocytes
Human phagocytes’ effect on engineered bacteriophages was evaluated in sera of 6 healthy volunteers, with their written consent, and in accordance with the local Commission of Bioethics, Wroclaw Medical University (approval no. 503/2015). Fractionation and isolation of human peripheral blood phagocytes from heparinized blood samples (polymorphonuclear neutrophils (PMNs) and mononuclear blood cells (PBMCs)) were conducted according to Boyum (Boyum, 1968) with modifications. Briefly, PBMCs and PMNs were isolated using a density gradient (Histopaque 1119 and Histopaque 1077 (Sigma‐Aldrich)) by centrifugation (700g, 30 min, 20°C). The PMN‐rich layer of Histopaque 1119 and mononuclear‐cell‐rich layer of Histopaque 1077 were collected, and the cells were washed (5 min, 870 × g, 4°C) in PBS three times. Morphology, quantity and viability of the cells were verified by optical microscopy in the histological Bürker chamber. The cells were then diluted in Hanks solution supplemented with 5% fetal calf serum (FCS) to achieve the required cell density for the further experiments.
Phagocytosis was tested in 24‐well plates. Cells were used in a final density of 106 cells per ml. Phages T4‐B1, T4‐G1, T4‐G2 and T4 as the control were added to a final count of 105/ml (volume: 1 ml). Cultures were incubated for 1.5 h at 37°C, in 5% CO2 with occasional gentle mixing. Samples were centrifuged (200 × g, 4°C, 5 min) and viable phage titre was determined by RTD in the culture supernatant.
Ethics statements
All experiments with human samples were approved by the local Commission of Bioethics, Wroclaw Medical University (no. 503/2015), and they were conducted only with the written consent of a healthy volunteer who entered the study as a blood donor.
All animal experiments were performed according to EU Directive 2010/63/EU for animal experimentations and were approved by the 1st Local Committee for Experiments with the Use of Laboratory Animals, Wroclaw, Poland (no. 07/2017 and no. 71/2015). The authors followed the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines (Kilkenny et al., 2012).
Statistics
All experiments were repeated 2–3 times; they were not summarized; exemplary experiments with their individual N values and statistical significance were presented. Statistical analysis was performed using the Kruskal–Wallis ANOVA or Mann–Whitney U‐test with the Statistica 8.0 software package (www.statsoft.pl).
Conflict of interest
None declared.
Supporting information
Fig. S1. Saturation of phage particles with Hoc‐peptide fusions.
Fig. S2. Comparison of engineered phages and T4 phage affinity to targeted cells.
Acknowledgements
Project supported by the National Science Centre in Poland (grant no. UMO‐2014/13/N/NZ6/03985) and by Wroclaw Centre of Biotechnology, The Leading National Research Centre (KNOW) program for years 2014‐2018.
Microbial Biotechnology (2019) 12(4), 730–741
Funding Information
Project supported by the National Science Centre in Poland (grant no. UMO‐2014/13/N/NZ6/03985) and by Wroclaw Centre of Biotechnology, The Leading National Research Centre (KNOW) program for years 2014–2018.
References
- Abedon, S. (2011) Phage therapy pharmacology: calculating phage dosing. Adv Appl Microbiol 77: 730–40. [DOI] [PubMed] [Google Scholar]
- Abedon, S.T. , Kuhl, S.J. , Blasdel, B.G. , and Kutter, E.M. (2011) Phage treatment of human infections. Bacteriophage 1: 66–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arap, W. , Haedicke, W. , Bernasconi, M. , Kain, R. , Rajotte, D. , Krajewski, S. , et al (2002a) Targeting the prostate for destruction through a vascular address. Proc Natl Acad Sci USA 99: 1527–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arap, W. , Kolonin, M.G. , Trepel, M. , Lahdenranta, J. , Cardo‐Vila, M. , Giordano, R.J. , et al (2002b) Steps toward mapping the human vasculature by phage display. Nat Med 8: 121–127. [DOI] [PubMed] [Google Scholar]
- Boyum, A. (1968) Isolation of mononuclear cells and granulocytes from human blood. Isolation of monuclear cells by one centrifugation, and of granulocytes by combining centrifugation and sedimentation at 1 g. Scand J Clin Lab Invest Suppl 97: 77–89. [PubMed] [Google Scholar]
- Budynek, P. , Dabrowska, K. , Skaradzinski, G. , and Gorski, A. (2010) Bacteriophages and cancer. Arch Microbiol 192: 315–320. [DOI] [PubMed] [Google Scholar]
- Cairns, B.J. , Timms, A.R. , Jansen, V.A. , Connerton, I.F. , and Payne, R.J. (2009) Quantitative models of in vitro bacteriophage‐host dynamics and their application to phage therapy. PLoS Pathog 5: e1000253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capparelli, R. , Ventimiglia, I. , Roperto, S. , Fenizia, D. , and Iannelli, D. (2006) Selection of an Escherichia coli O157:H7 bacteriophage for persistence in the circulatory system of mice infected experimentally. Clin Microbiol Infect 12: 248–253. [DOI] [PubMed] [Google Scholar]
- Capparelli, R. , Parlato, M. , Borriello, G. , Salvatore, P. , and Iannelli, D. (2007) Experimental phage therapy against Staphylococcus aureus in mice. Antimicrob Agents Chemother 51: 2765–2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceglarek, I. , Piotrowicz, A. , Lecion, D. , Miernikiewicz, P. , Owczarek, B. , Hodyra, K. , et al (2013) A novel approach for separating bacteriophages from other bacteriophages using affinity chromatography and phage display. Sci Rep 3: 3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabrowska, K. , Switala‐Jelen, K. , Opolski, A. , Weber‐Dabrowska, B. , and Gorski, A. (2005) Bacteriophage penetration in vertebrates. J Appl Microbiol 98: 7–13. [DOI] [PubMed] [Google Scholar]
- Dabrowska, K. , Miernikiewicz, P. , Piotrowicz, A. , Hodyra, K. , Owczarek, B. , Lecion, D. , et al (2014a) Immunogenicity studies of proteins forming the T4 phage head surface. J Virol 88: 12551–12557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabrowska, K. , Kazmierczak, Z. , Majewska, J. , Miernikiewicz, P. , Piotrowicz, A. , Wietrzyk, J. , et al (2014b) Bacteriophages displaying anticancer peptides in combined antibacterial and anticancer treatment. Future Microbiol 9: 861–869. [DOI] [PubMed] [Google Scholar]
- Dempsey, P.W. , Allison, M.E. , Akkaraju, S. , Goodnow, C.C. , and Fearon, D.T. (1996) C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science 271: 348–350. [DOI] [PubMed] [Google Scholar]
- Duerr, D.M. , White, S.J. , and Schluesener, H.J. (2004) Identification of peptide sequences that induce the transport of phage across the gastrointestinal mucosal barrier. J Virol Methods 116: 177–180. [DOI] [PubMed] [Google Scholar]
- Fogelman, I. , Davey, V. , Ochs, H.D. , Elashoff, M. , Feinberg, M.B. , Mican, J. , et al (2000) Evaluation of CD4 + T cell function In vivo in HIV‐infected patients as measured by bacteriophage phiX174 immunization. J Infect Dis 182: 435–441. [DOI] [PubMed] [Google Scholar]
- Geier, M.R. , Trigg, M.E. , and Merril, C.R. (1973) Fate of bacteriophage lambda in non‐immune germ‐free mice. Nature 246: 221–223. [DOI] [PubMed] [Google Scholar]
- Giordano, R.J. , Lahdenranta, J. , Zhen, L. , Chukwueke, U. , Petrache, I. , Langley, R.R. , et al (2008) Targeted induction of lung endothelial cell apoptosis causes emphysema‐like changes in the mouse. J Biol Chem 283: 29447–29460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano, R.J. , Edwards, J.K. , Tuder, R.M. , Arap, W. , and Pasqualini, R. (2009) Combinatorial ligand‐directed lung targeting. Proc Am Thorac Soc 6: 411–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski, A. , Miedzybrodzki, R. , Borysowski, J. , Dabrowska, K. , Wierzbicki, P. , Ohams, M. , et al (2012) Phage as a modulator of immune responses: practical implications for phage therapy. Adv Virus Res 83: 41–71. [DOI] [PubMed] [Google Scholar]
- Gorski, A. , Dabrowska, K. , Hodyra‐Stefaniak, K. , Borysowski, J. , Miedzybrodzki, R. , and Weber‐Dabrowska, B. (2015) Phages targeting infected tissues: novel approach to phage therapy. Future Microbiol 10: 199–204. [DOI] [PubMed] [Google Scholar]
- Gorski, A. , Miedzybrodzki, R. , Weber‐Dabrowska, B. , Fortuna, W. , Letkiewicz, S. , Rogoz, P. , et al (2016) Phage therapy: combating infections with potential for evolving from merely a treatment for complications to targeting diseases. Front Microbiol 7: 1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronwall, C. , Vas, J. , and Silverman, G.J. (2012) Protective roles of natural IgM antibodies. Front Immunol 3: 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajek, P. , and Mandel, L. (1966) Antibody response of young animals to bacteriophages of different immunological behaviour: phi X 174 and T2. Folia Microbiol (Praha) 11: 282–289. [DOI] [PubMed] [Google Scholar]
- Hodyra‐Stefaniak, K. , Miernikiewicz, P. , Drapala, J. , Drab, M. , Jonczyk‐Matysiak, E. , Lecion, D. , et al (2015) Mammalian host‐versus‐phage immune response determines phage fate in vivo. Sci Rep 5: 14802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huff, W.E. , Huff, G.R. , Rath, N.C. , and Donoghue, A.M. (2010) Immune interference of bacteriophage efficacy when treating colibacillosis in poultry. Poult Sci 89: 895–900. [DOI] [PubMed] [Google Scholar]
- Inchley, C.J. (1969) The actvity of mouse Kupffer cells following intravenous injection of T4 bacteriophage. Clin Exp Immunol 5: 173–187. [PMC free article] [PubMed] [Google Scholar]
- Jiang, J. , Abu‐Shilbayeh, L. , and Rao, V.B. (1997) Display of a PorA peptide from Neisseria meningitidis on the bacteriophage T4 capsid surface. Infect Immun 65: 4770–4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, S.K. , Woo, J.H. , Kim, M.K. , Woo, S.S. , Choi, J.H. , Lee, H.G. , et al (2008) Identification of a peptide sequence that improves transport of macromolecules across the intestinal mucosal barrier targeting goblet cells. J Biotechnol 135: 210–216. [DOI] [PubMed] [Google Scholar]
- Kazmierczak, Z. , Gorski, A. , and Dabrowska, K. (2014) Facing antibiotic resistance: Staphylococcus aureus phages as a medical tool. Viruses 6: 2551–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller, R. , and Engley, F.B. Jr (1958) Fate of bacteriophage particles introduced into mice by various routes. Proc Soc Exp Biol Med 98: 577–580. [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W.J. , Cuthill, I.C. , Emerson, M. , and Altman, D.G. (2012) Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. Osteoarthritis Cartilage 20: 256–260. [DOI] [PubMed] [Google Scholar]
- Kutter, E. , De Vos, D. , Gvasalia, G. , Alavidze, Z. , Gogokhia, L. , Kuhl, S. , and Abedon, S.T. (2010) Phage therapy in clinical practice: treatment of human infections. Curr Pharm Biotechnol 11: 69–86. [DOI] [PubMed] [Google Scholar]
- Lee, J.H. , Fan, B. , Samdin, T.D. , Monteiro, D.A. , Desai, M.S. , Scheideler, O. , et al (2017) Phage‐based structural color sensors and their pattern recognition sensing system. ACS Nano 11: 3632–3641. [DOI] [PubMed] [Google Scholar]
- Levin, B.R. , and Bull, J.J. (2004) Population and evolutionary dynamics of phage therapy. Nat Rev Microbiol 2: 166–173. [DOI] [PubMed] [Google Scholar]
- Li, J. , Feng, L. , Fan, L. , Zha, Y. , Guo, L. , Zhang, Q. , et al (2011) Targeting the brain with PEG‐PLGA nanoparticles modified with phage‐displayed peptides. Biomaterials 32: 4943–4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. , Zhang, Q. , Pang, Z. , Wang, Y. , Liu, Q. , Guo, L. , and Jiang, X. (2012) Identification of peptide sequences that target to the brain using in vivo phage display. Amino Acids 42: 2373–2381. [DOI] [PubMed] [Google Scholar]
- Lusiak‐Szelachowska, M. , Zaczek, M. , Weber‐Dabrowska, B. , Miedzybrodzki, R. , Letkiewicz, S. , Fortuna, W. , et al (2017) Antiphage activity of sera during phage therapy in relation to its outcome. Future Microbiol 12: 109–117. [DOI] [PubMed] [Google Scholar]
- Majewska, J. , Beta, W. , Lecion, D. , Hodyra‐Stefaniak, K. , Klopot, A. , Kazmierczak, Z. , et al (2015) Oral application of T4 phage induces weak antibody production in the gut and in the blood. Viruses 7: 4783–4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merril, C.R. , Biswas, B. , Carlton, R. , Jensen, N.C. , Creed, G.J. , Zullo, S. , and Adhya, S. (1996) Long‐circulating bacteriophage as antibacterial agents. Proc Natl Acad Sci USA 93: 3188–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miedzybrodzki, R. , Switala‐Jelen, K. , Fortuna, W. , Weber‐Dabrowska, B. , Przerwa, A. , Lusiak‐Szelachowska, M. , et al (2008) Bacteriophage preparation inhibition of reactive oxygen species generation by endotoxin‐stimulated polymorphonuclear leukocytes. Virus Res 131: 233–242. [DOI] [PubMed] [Google Scholar]
- Miedzybrodzki, R. , Borysowski, J. , Weber‐Dabrowska, B. , Fortuna, W. , Letkiewicz, S. , Szufnarowski, K. , et al (2012) Clinical aspects of phage therapy. Adv Virus Res 83: 73–121. [DOI] [PubMed] [Google Scholar]
- Miernikiewicz, P. , Klopot, A. , Soluch, R. , Szkuta, P. , Keska, W. , Hodyra‐Stefaniak, K. , et al (2016) T4 phage tail adhesin Gp12 counteracts LPS‐induced inflammation in vivo. Front Microbiol 7: 1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochs, H.D. , Davis, S.D. , and Wedgwood, R.J. (1971) Immunologic responses to bacteriophage phi‐X 174 in immunodeficiency diseases. J Clin Invest 50: 2559–2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oslizlo, A. , Miernikiewicz, P. , Piotrowicz, A. , Owczarek, B. , Kopciuch, A. , Figura, G. , and Dabrowska, K. (2011) Purification of phage display‐modified bacteriophage T4 by affinity chromatography. BMC Biotechnol 11: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualini, R. , and Ruoslahti, E. (1996) Organ targeting in vivo using phage display peptide libraries. Nature 380: 364–366. [DOI] [PubMed] [Google Scholar]
- Payne, R.J. , Phil, D. , and Jansen, V.A. (2000) Phage therapy: the peculiar kinetics of self‐replicating pharmaceuticals. Clin Pharmacol Ther 68: 225–230. [DOI] [PubMed] [Google Scholar]
- Pires, D.P. , Cleto, S. , Sillankorva, S. , Azeredo, J. , and Lu, T.K. (2016) Genetically engineered phages: a review of advances over the last decade. Microbiol Mol Biol Rev 80: 523–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirnay, J.P. , De Vos, D. , Verbeken, G. , Merabishvili, M. , Chanishvili, N. , Vaneechoutte, M. , et al (2011) The phage therapy paradigm: pret‐a‐porter or sur‐mesure? Pharm Res 28: 934–937. [DOI] [PubMed] [Google Scholar]
- Rajotte, D. , and Ruoslahti, E. (1999) Membrane dipeptidase is the receptor for a lung‐targeting peptide identified by in vivo phage display. J Biol Chem 274: 11593–11598. [DOI] [PubMed] [Google Scholar]
- Ruoslahti, E. (2012) Peptides as targeting elements and tissue penetration devices for nanoparticles. Adv Mater 24: 3747–3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathaliyawala, T. , Rao, M. , Maclean, D.M. , Birx, D.L. , Alving, C.R. , and Rao, V.B. (2006) Assembly of human immunodeficiency virus (HIV) antigens on bacteriophage T4: a novel in vitro approach to construct multicomponent HIV vaccines. J Virol 80: 7688–7698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serna, M. , Giles, J.L. , Morgan, B.P. , and Bubeck, D. (2016) Structural basis of complement membrane attack complex formation. Nat Commun 7: 10587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, H.W. , Huggins, M.B. , and Shaw, K.M. (1987) Factors influencing the survival and multiplication of bacteriophages in calves and in their environment. J Gen Microbiol 133: 1127–1135. [DOI] [PubMed] [Google Scholar]
- Sokoloff, A.V. , Bock, I. , Zhang, G. , Sebestyen, M.G. , and Wolff, J.A. (2000) The interactions of peptides with the innate immune system studied with use of T7 phage peptide display. Mol Ther 2: 131–139. [DOI] [PubMed] [Google Scholar]
- Sokoloff, A.V. , Bock, I. , Zhang, G. , Hoffman, S. , Dama, J. , Ludtke, J.J. , et al (2001) Specific recognition of protein carboxy‐terminal sequences by natural IgM antibodies in normal serum. Mol Ther 3: 821–830. [DOI] [PubMed] [Google Scholar]
- Sokoloff, A.V. , Puckett, M. , Ludtke, J.J. , and Fetterly, B. (2004) Sequence‐specific binding of normal serum immunoglobulin M to exposed protein C‐termini. Immunology 112: 237–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulkin, S.E. , Finkelstein, R.A. , and Rosenblum, E.D. (1957) Effect of zymosan on bacteriophage clearance. Science 125: 742–743. [DOI] [PubMed] [Google Scholar]
- Teesalu, T. , Sugahara, K.N. , and Ruoslahti, E. (2013) Tumor‐penetrating peptides. Front Oncol 3: 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhr, J.W. , Finkelstein, M.S. , and Baumann, J.B. (1962a) Antibody formation. III. The primary and secondary antibody response to bacteriophage phi X 174 in guinea pigs. J Exp Med 115: 655–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhr, J.W. , Finkelstein, S. , and Franklin, E.C. (1962b) Antibody response to bacteriophage phi‐X‐174 in non‐mammalian vertebrates. Proc Soc Exp Biol Med 111: 13–15. [DOI] [PubMed] [Google Scholar]
- Van Belleghem, J.D. , Clement, F. , Merabishvili, M. , Lavigne, R. , and Vaneechoutte, M. (2017) Pro‐ and anti‐inflammatory responses of peripheral blood mononuclear cells induced by Staphylococcus aureus and Pseudomonas aeruginosa phages. Sci Rep 7: 8004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitiello, C.L. , Merril, C.R. , and Adhya, S. (2005) An amino acid substitution in a capsid protein enhances phage survival in mouse circulatory system more than a 1000‐fold. Virus Res 114: 101–103. [DOI] [PubMed] [Google Scholar]
- Yacoby, I. , and Benhar, I. (2008) Targeted filamentous bacteriophages as therapeutic agents. Expert Opin Drug Deliv 5: 321–329. [DOI] [PubMed] [Google Scholar]
- Yao, V.J. , D'Angelo, S. , Butler, K.S. , Theron, C. , Smith, T.L. , Marchio, S. , et al (2016) Ligand‐targeted theranostic nanomedicines against cancer. J Control Release 240: 267–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue, H. , He, Y. , Fan, E. , Wang, L. , Lu, S. , and Fu, Z. (2017) Label‐free electrochemiluminescent biosensor for rapid and sensitive detection of Pseudomonas aeruginosa using phage as highly specific recognition agent. Biosens Bioelectron 94: 429–432. [DOI] [PubMed] [Google Scholar]
- Zhang, L. , Hou, X. , Sun, L. , He, T. , Wei, R. , Pang, M. , and Wang, R. (2018) Staphylococcus aureus bacteriophage suppresses LPS‐induced inflammation in MAC‐T bovine mammary epithelial cells. Front Microbiol 9: 1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Saturation of phage particles with Hoc‐peptide fusions.
Fig. S2. Comparison of engineered phages and T4 phage affinity to targeted cells.