

Abstract
Pancreatic cancer (PC) is insidious with a high mortality rate due to the lack of symptomology prior to diagnosis. Mitochondrial involvement in PC development is becoming accepted, and exposure to cadmium (Cd) is suspected of being a risk factor for the development of PC; however, the mechanisms involved remain unclear. In this study, we examined the role of Cd as a mitochondrial toxicant and whether alterations in mitochondrial function may be an underlying cause for the development of PC. In this study, cadmium chloride (CdCl2)-mediated toxicity in hTERT-HPNE and AsPC-1 pancreatic cell lines was determined by MTT assay. We also investigated the release of LDH and the generation of free radicals. Mitochondrial toxicity assays were performed in media containing glucose (25 mM) or galactose (10 mM) and following exposure to CdCl2 (0-100 μM) followed by MTT assay. For the confirmation of mitochondrial toxicity, we measured the release of ATP following exposure to CdCl2. Initial experiments confirmed that exposure to CdCl2 did not reduce the viability of either cell line until a concentration of >10 μM was used. Non-linear analysis of the response curves revealed lethal concentration 50% (LC50) values for CdCl2 in the HPNE cells of 77 μM compared to 42 μM in the AsPC-1 cells (P<0.01). The CdCl2-mediated mitochondrial toxic effects were greater in the HPNE cells, suggesting a heightened sensitivity to the effects of CdCl2, not due to elevated oxidative stress. Increased mitochondrial toxic sensitivity was indicated by a 73.4% reduction in IC50 values in the HPNE cells cultured in galactose compared to culture in glucose media, whereas the AsPC-1 cells exhibited a 58.8% reduction in IC50 values. In addition, the higher concentration of CdCl2 elicited a significant cell-dependent effect on ATP release in both cell lines, suggestive of CdCl2 being a mitochondrial toxicant. Cell survival was unaffected following exposure to low concentrations of CdCl2; however, exposure did alter mitochondrial function (control cells > tumor cells). Therefore, the findings of this study indicate that the mitochondria may be a site of action for cadmium in promoting tumor development.
Keywords: Warburg effect, oxidative stress, cadmium, glycolysis, oxidative phosphorylation, cytotoxicity
Introduction
The effects of cadmium (Cd) on biological systems have been extensively studied; however, they remain relatively enigmatic. Cd has an exceptionally long half-life in biological systems (years) which may underlie the physiological changes associated with exposure to Cd. Cd is found widely throughout the environment and is one of the more abundant toxicants to which humans are exposed in both occupational and environmental settings (1-3). Cadmium exerts toxic effects on many organs and systems of organs (4,5). Similarly, its endocrine disruptive activities have been described (6-9), as well as its hormetic effects (10). Cadmium, as well as Cd-containing compounds, have been classified as carcinogenic by the International Agency for Research on Cancer (11,12) based on epidemiological studies showing a causal connection with the development of lung cancer. Epidemiological studies have also implicated exposure to Cd to be involved in the development of other types of cancer, such as kidney cancer (13-15), prostate cancer (14,16,17), cancer of the testis (17), bladder cancer (18), breast cancer (19) and overall cancer mortality (20).
The latest report on the global burden of cancer worldwide using GLOBOCAN 2018 estimated pancreatic cancer (PC) as the seventh leading cause of cancer-related mortality among both males and females with the projection that in the 28 countries of the European Union, this type of cancer will surpass breast cancer as the third leading cause of cancer-related mortality in the future (21). The prognosis of this type of cancer is very poor, mainly due to late-stage diagnosis. Hence, the elucidation of the etiology of this type of cancer and the identification of prognostic factors for it are of utmost importance. The quest for a legitimate biomarker to identify PC in the early stages is ongoing (22,23). A recent review article suggested the possible role of exposure to Cd in the development of PC (24). Some of the mechanisms involved in the carcinogenetic effects of Cd on the pancreas may be the induction of oxidative stress and interaction with bioelements, which have been observed and discussed in a number of studies (25-32). The development of PC is likely to be associated with the silencing of certain tumor suppressor genes and/or the activation of oncogenes, although the underlying molecular mechanisms remain undefined (22,23,33,34). However, interactions between certain environmental chemicals, with Cd being one of them, and these genes can be expected. Furthermore, the micro-RNA-mediated regulation of pancreatic cell proliferation has been demonstrated by Basu et al (35). Subsequently, microRNAs (miRNAs or miRs) were found to be present specifically in the mitochondria, originating from mitochondrial DNA and regulating genes coding for mitochondrial proteins in a direct manner, and consequently mitochondrial function (36). Concerning the role of Cd as a mitochondrial toxin, it has recently been studied, but with mixed results, suggesting that Cd may exert direct and indirect effects on the mitochondria (37-40). This ability of Cd to exert toxic effects on the mitochondria could certainly be one of the mechanisms of its carcinogenic effects on cells.
The nature of Cd-related mitochondrial toxicity has led to speculation that the toxicity observed can be mediated either by direct or indirect mechanisms. The aim of this study was to enhance our understanding of the effects of Cd on the mitochondria and to determine whether Cd is a direct or an indirect mitochondrial toxin. Current literature has been unclear and at times, conflicting (37). One concept that is becoming more accepted is that there is an organ-specificity regarding Cd toxicity. There is overlap between some of the major organs examined, although there are also differences in Cd-mediated responses. In the kidneys, there is speculation that Cd interferes with normal mitochondrial function, leading to increased oxidative stress and apoptosis (40), which is further supported by in vitro studies utilizing human kidney cell lines showing that Cd directly alters mitochondrial membrane permeability and potential (39). Li et al extended these findings to isolated and purified mitochondria (38). In hepatic cells, exposure to Cd has been shown to decrease mitochondrial membrane permeability, but also to directly alter apoptotic pathways by increasing the activity of both caspase-3 and caspase-9, as well as increasing the activity of p53 (41). Recently, the effects of CdCl2 on the pancreas and pancreatic cells have been further described (24). In pancreatic β-cells (RIN-m5F cells), exposure to CdCl2 has been shown to result in the death of β-cells following increased oxidative stress followed by a downstream-mediated activation of apoptotic pathways regulated by the mitochondria (42). Some researchers have postulated that environmental or occupational exposure to Cd, and the resulting Cd-mediated death or damage to β-cells is an underlying cause of diabetes and obesity (43).
In this study, we examined the effects of cadmium chloride (CdCl2) on mitochondrial function due to the involvement of mitochondria in the 'Warburg effect'. The Warburg effect describes how tumor cells bypass oxidative phosphorylation [responsible for the generation of 38 adenosine triphosphates (ATPs)] and instead utilize aerobic glycolysis [generating only 2 ATPs, but other metabolites which could be useful in cellular proliferation (44)]. An advancement in our understanding of the peculiar phenomenon that was identified by Otto Warburg almost 100 years ago has progressed very little until the last 5-10 years when a significant surge in interest has taken place (45,46). Although one single theory has not come to the forefront, numerous hypotheses have been presented, suggesting that the switch from oxidative phosphorylation to aerobic glycolysis signals the transition from a normal cell to a tumor cell (46). Another hypothesis is that switching to aerobic glycolysis can protect the tumor cell from sugar-induced death when glucose uptake is high, and by changing to a high production of lactate, pro-proliferative intermediates can be produced (47). Compounds, such as Cd, that damage the mitochondria promote a shift towards aerobic glycolysis, thus leading to tumor formation. Researchers that have examined toxicant [1-methyl-4-phenyl-1,2,3,6-tetrahydro-pyridine (MPTP)] damage to the mitochondria have reported that oxidative phosphorylation is decreased, that apoptotic pathways are altered and there is a bioenergetic shift in glucose utilization (48). Indirect genetic damage to the mitochondria that will result in epigenetic alterations has also been demonstrated in tumor cells, as well as inherited genetic abnormalities that predispose or facilitate the progression of the Warburg effect in tumor cells (49). The apparent importance of the mitochondria and the Warburg effect has led researchers to postulate that either 'turning' oxidative phosphorylation 'back on' or shunting away from lactate production/utilization may be an effective means of controlling tumor growth (45,50).
Recently, we performed a literature search for 'cadmium', 'Warburg Effect', or 'Crabtree Effect' and the references that we obtained were more specific for the effects of Cd on sugar utilization in yeast, which is what the Crabtree effect initially described (51,52). The effect of Cd on cellular energy utilization is an area of research that needs to be addressed for a better understanding of the effects of Cd on the pancreas and on the development of PC, diabetes, obesity and other pancreatic dysfunctions. In this study, we designed a series of assays that would systematically address the Cd-mediated effects on pancreatic cells using a control epithelial cell line (HPNE) and the pancreatic tumor cell line, AsPC-1. The goals of these experiments were to first determine the level of toxicity following the exposure of both cell lines to Cd, and to then use subtoxic concentrations to determine mitochondrial toxicity. Our initial concentrations were determined using Cd concentrations obtained from pancreatic tissue, from a pancreatic tumor, the area surrounding the tumor and from 'healthy' pancreatic tissue (53). After determining the threshold for Cd-mediated toxicity, we examined the production of free radicals following exposure to Cd and then used the glucose-galactose assay to assess the toxic potential of Cd on the mitochondria by measuring the shift in the toxicity curve and change in IC50 values comparing the glucose curve to the galactose curve. From these experiments, we aim to improve our understanding of Cd-mediated intracellular changes in pancreatic cells.
Materials and methods
Cell lines and cell culture maintenance
All cell culture experiments were performed using standard Biosafety Level 2 (BSL-2) guidelines. The study was approved by the Oklahoma State University Center for Health Science Institutional Biosafety committee in compliance with all National Institutes of Health regulations. Cell lines purchased from the American Type Culture Collection (ATCC). Pancreatic hTERT-HPNE ('human pancreatic Nestin-expressing' cells; ATCC® CRL-4023™, immortalized pancreatic control cells, referred to as 'HPNE') and AsPC-1 (ATCC® CRL-1682™, pancreatic tumor cells) cells were grown and maintained as described in the ATCC-suggested protocols. HPNE cell growth media consisted of DMEM base media with 2 mM glutamine, and 1.5 g/l sodium bicarbonate supplemented with M3 Base media supplement (M300F-500; Incell Corp.) at a ratio of 3:1. To produce complete growth media, 5% fetal bovine serum, 10 ng/ml human epidermal growth factor, 1 mg/ml glucose and 750 ng/ml Puromycin (Sigma-Aldrich and Corning) were added to the DMEM:M3 base. Base AsPC-1 media was RPMI-1640 with 2 mM glutamine, and complete growth media consisted of supplementation with 10% fetal bovine serum, and 1% penicillin/streptomycin (Sigma-Aldrich and Corning). Assay media used in the cytotoxicity assays was a base MEM supplemented with 2 mM glutamine and 1% fetal bovine serum. Assay media was phenol-free and low serum to minimize the confounding factors associated with the influence of protective growth factors in serum. Cells were allowed to adhere for at least 24 h before exposure. Following adherence, growth media were removed and replaced with assay media containing the appropriate treatment(s).
Cell viability and cell growth assays
All lactate dehydrogenase (LDH) assays (Cytotox-ONE™ kit; G7891, Promega) were performed essentially as previously described (54). The HPNE and AsPC-1 cell lines (20-50,000 cells/well) were plated and allowed to adhere for at least 24 h prior to the assay. LDH assays determined i) the number of viable cells within the culture; ii) the percentage of the total number of cells that were viable. Initial serum-comparison assays experiments examined the effects of various serum concentrations on cell growth and viability. Following the removal of the growth media, MEM supplemented with 0% (control), or 1, 2.5, 5 and 10% fetal bovine serum was added to appropriate wells followed by incubation at 37°C in 5% CO2 for 48 h. Following exposure, the LDH assay was carried out according to the manufacturer's instructions. The ability to quantify dead and live cells is described in the following formula: {[(total RFU) - (Media RFU)] ÷ (total RFU)} ×100. The amount of fluorescence produced was quantified using a Biotek plate reader (Biotek Instruments) set to an excitation wavelength of 485 nm and an emission wavelength of 530 nm.
Once we determined whether or not changing the serum concentration would affect cell viability and cell number, we exposed both the HPNE and AsPC-1 cells to 2 ppm (18 μM), 6 ppm (54 μM) and 14 ppm (126 μM) CdCl2 for 48 h. We have previously analyzed human pancreatic tissue and measured the content of Cd (53). The highest concentration was found within the tumor, the intermediate concentration was in the region immediately surrounding the tumor zone, and the lowest concentration was found at a site distant from the tumor. We used the molecular contribution of Cd to CdCl2 and calculated the concentration of Cd alone. This assay was performed to aid in the identification of the threshold of toxicity associated with exposure to CdCl2. Following exposure for 48 h, both cell viability and the total number of cells were determined as described in the serum-comparison assays described above. The rationale for using CdCl2 as our form of Cd is based on existing literature and published studies using CdCl2 both in in vitro and in vivo studies (24,25,27,31,53). The use of CdCl2 will strengthen conclusions comparing our findings to the existing literature using CdCl2.
Measurement of oxidative stress
Both cell lines were grown in 25 cm2 flasks and seeded into a 96-well (black/clear bottom) plate at an initial density of 10−5 cells/ml (or 10−4 cells/well). The cells were allowed to adhere for 24 h and to initiate the assay, the growth media was removed followed by washing the cells (twice) with warmed PBS. The cells were then loaded with 100 μM DCFH-diacetate (DCFH-DA) for 30 min at 37°C in 5% CO2, while being protected from light. Following the removal and washing with PBS, the cells were exposed to one of three treatments; control (PBS only; 0 μM CdCl2), 50 μM CdCl2 or 100 μM CdCl2. Baseline fluorescence was determined in the PBS-only group prior to the addition of CdCl2. The cells were returned to the incubator to allow for the continuation of the reaction. The plates were removed after 30, 60, 90 and 120 min and the fluorescence resulting from the conversion of dichlorofluorescin (DCFH) to dichlorofluorescein (DCF) was measured using a Biotek plate reader set to an excitation wavelength of 485 nm and an emission wavelength of 530 nm. The amount of fluorescence detected is directly correlated to the amount of oxidative stress occurring within the cell. Data were then expressed as the means ± SEM of n=8 assayed in duplicate.
Determination of lethal concentration 50% (LC50) values for CdCl2
The HPNE and AsPC-1 cell lines were maintained as described above. The cells were plated at a final density of approximately 105 cells/ml in clear 96-well plates. The cells were allowed to adhere for at least 24 h and the media were carefully removed and assay media with the appropriate concentrations of CdCl2 (0-10−3 M) were added to the wells. Following 48 h of exposure, 12 mM stock (1.2 mM final concentration) MTT (Molecular Probes) was added to each well and the plates were returned to the incubator (37°C and 5% CO2) for 4 h followed by the addition of detergent for another 3-4 h. The amount of insoluble formazan was determined by measuring the absorbance at 565 nm using the BioTek plate reader. Data were then expressed as the means ± SEM of n=6 in duplicate.
Mitochondrial toxicity assays
HPNE and AsPC-1 cell lines were plated at a final density of 20,000-50,000 cells/well and allowed to adhere for at least 24 h prior to the assay. The initiation of toxicant exposure begins by removing the growth media and replacing with DMEM supplemented with either 25 mM glucose or 10 mM galactose. This method of determining mitochondrial toxicity is similar to that described in the study by Sanuki et al (55) and Dott et al (56). The treatment groups consisted of cells exposed to Cd (0.1-100 μM) or control (media only) for 4 h followed by the addition of 12 mM MTT dye (1.2 mM final concentration). The absorbance was measured at 565 nm, and the data were then expressed as the means ± SEM of n=12 in duplicate.
To establish the association between cell death and the loss of ATP, we used a luciferin-based detection system (Mitochondrial ToxGlo™; Promega) (57). The principle of the mitochondrial toxicity tests is that substituting 10 mM galactose for 25 mM glucose will increase susceptibility to mitochondrial toxins (58-60). The cells were exposed to CdCl2 (1 or 50 μM) for 48 h prior to the initiation of the assay. These two concentrations were selected based on 50 μM approximating 6 ppm, and 1 μM was significantly lower than our threshold of 10 μM. Following 30 min of incubation at 37°C in a CO2 incubator, viability was determined by measuring the fluorescence (485 nmex/525 nmem) emitted. The second step in the multiplex was to quantify the amount of ATP present by directly adding the luciferin-based detection system into the wells that have already been quantified for cell viability with the amount of luminescence being directly proportional to the amount of ATP present. Collectively, by comparing the cellular responses in the two datasets, a better understanding of CdCl2 can be obtained as a cellular toxicant that results in mitochondrial dysfunction or non-mitochondrial related cytotoxic mechanisms.
Effect of CdCl2 on mitochondrial permeability
To assess cell health, we utilized a dye that is permeable to the mitochondrial and will fluoresce red or green depending on the membrane potential of the mitochondria. A high mitochondrial membrane potential (ΔψM) is representative of a healthy cell, whereas a low ΔψM is indicative of an unhealthy cell. The higher the ratio, the greater the polarization of the mitochondrial membrane. We utilized the JC-1 kit (Cayman Chemical Co.) for this purpose. The cells were plated at approximately 105 cells/well and allowed to adhere for 24 h. Exposure was initiated by removing the growth media from each well and adding the treatment media with CdCl2 (1 μM), or the vehicle (water). The cells were returned to the incubator for 48 h at 37°C and 5% CO2. Following exposure, the cells were prepared according to manufacturer instructions; staining was initiated by the addition of 10 μ JC-1 dye and the cells were returned to the incubator for 20 min. The fluorescence intensity was measured using the BioTek plate reader at 535ex/595em to measure JC-1 aggregates (healthy cells; green) and then a second reading using 485ex/535em which measures JC-1 monomers (apoptotic/unhealthy cells; red).
Statistical analysis: All data were analyzed using GraphPad Prism software (version 7.04)
Data from initial CdCl2 exposure assays were analyzed by one-way ANOVA followed by Dunnett's post hoc comparison to control values. In the oxidative stress and LDH assays, data were analyzed by two-way ANOVA (2×3 or cell line x concentration) using Sidak's or Tukey's test for multiple comparisons, respectively. All data are expressed as the means ± SEM of n=4 (ATP assay), n=6 (MTT LC50 assay), n=8 (DCFH oxidative stress assay) or n=12 (mitochondrial toxicity assay) performed in either duplicate or triplicate. In the LC50 and mitochondrial toxicity assays, the data were first fit by non-linear single-site nonlinear regression inhibition curve (single-site) to determine the IC50 value for CdCl2 in each media. A ≥3-fold leftward shift in LC50 or IC50 values in the galactose group was indicative of mitochondrial toxicity. When curve-fitting, we utilized the D'Agostino and Pearson Test for Normality (omnibus K2) to assure that the curves followed a Gaussian distribution. One-way analysis of variance was performed with Dunnett's test for post hoc comparison to control LC50 values. For individual comparisons of LC50 values, a Student's t-test was used to compared cell lines and treatments. For analyzing the mitochondrial toxicity data using dual-labeling, we utilized a three-way ANOVA (2×2×2; cell type, concentration, media) design with Sidak's test for post hoc multiple comparisons. Data are presented first as the result of the ANOVA where appropriate (F-value) representing whether there was a significant association within the group, such as a concentration effect. Post-hoc multiple comparisons were then performed, usually comparing to the control value; however, the value obtained from the post-hoc comparison is labeled in the figures by a symbol. A significance level was set at P<0.05.
Results
Cell viability: Effect of exposure to Cd and low serum concentration: There is a need to perform assays in reduced serum
Fetal bovine serum contains multiple factors that have been known to scavenge free radicals and afford protection to the cells when exposed to a toxic insult (61-63). These assays were performed to assess changes in cell viability in the presence of reducing concentrations of serum (fetal bovine serum), as well as a change in basal media from the standard optimized growth media to MEM with minimal supplementation. In the HPNE cells (Fig. 1A), no marked differences (P=0.61) were observed in either viability between the serum groups or between the growth media and corresponding MEM. In the AsPC-1 cells, there was a robust increase (F5,41=27.34; P<0.0001) in viability in the presence of reduced serum (Fig. 1B). From 0 to 5% the AsPC-1 cells exhibited a 20-25% greater viability compared to the growth media. It was noteworthy that reduced serum improved the viability of AsPC-1 cells. Previous studies have suggested that reducing the serum concentration will reduce viability and cellular proliferation in a variety of cell lines (64,65). This study, at least to the best of our knowledge, is the first to examine the effects of a reduced serum concentration on AsPC-1 viability and growth.
Figure 1.

Effects of reduced serum (fetal bovine serum) concentration on the viability of (A) HPNE cells; and (B) AsPC-1 cells. Reducing the fetal bovine serum concentration from 10 to 0% did not markedly alter HPNE viability compared to the growth media control. In the AsPC-1 cells, there was a generalized increase in cell viability from 20-25%, apart from the 10% FBS group. The data presented represent n=4 performed in duplicate and are expressed as the means ± SEM. Data were analyzed by one-way ANOVA followed by Sidak post hoc test for the comparisons of the treatment (fetal bovine serum) to the growth media cell viability. **P<0.01 compared to growth media control values.
Initial experiments examined the effects of CdCl2 on the viability of HPNE and AsPC-1 cells by exposing the cells to CdCl2 at final concentrations of 0, 2, 6 and 14 ppm (Table I). These concentrations were selected based on data from human studies showing 14 ppm CdCl2 in the core of a pancreatic tumor, with 6 ppm on the tumor margin and 2 ppm associated with 'healthy' tissue distant from the tumor center (53). As evidenced by the data presented in Table I, we observed only minimal changes in viability or cell number in cells exposed to CdCl2. In the HPNE cells, we observed an effect of treatment, (approximately 5%) in both cell number and viability compared to the control values (F4,15=37.59; P<0.0001). Post hoc comparisons revealed no differences between the treatment and control groups. In the AsPC-1 cells, exposure to CdCl2 (treatment effect) resulted in a significant change in both viability (F4,15=66.69; P<0.0001) and in cell number (F4,15=10.91; P=0.0002). Dunnett's post hoc analysis was used to compare changes in the treatment groups to the control values. Only exposure to the highest concentration of CdCl2, 14 ppm, elicited significant reductions in both cellular viability (51%) and cell number (54%). Collectively, these data suggest that the control HPNE cells are relatively resistant following changes in serum concentration, as well as CdCl2 exposure. We observed minimal changes in both cell viability, as well as in cell number which would be indicative of proliferation/growth changes. The AsPC-1 cells, on the other hand, were more sensitive to the changes in serum concentration and exposure to CdCl2. Generally, only the highest concentration of CdCl2 elicited significant reductions in both cell viability and number. Subsequent experiments assessed CdCl2-mediated changes in oxidative stress and mitochondrial toxicity as a secondary means for CdCl2 exposure resulting in cellular damage.
Table I.
Effects of exposure to CdCl2 for 48 h on the live cell number and viability of HPNE and AsPC-1 cells.
| CdCl2 concentration
|
|||||
|---|---|---|---|---|---|
| Growth media | Low-serum MEM | 2 ppm | 6 ppm | 14 ppm | |
| HPNE | |||||
| Live cell no. (RFU) | 3,711±100 | 3,591±71 | 3,478±70 | 3,585±52 | 3,518±87 |
| Viability (%) | 84.2±0.3 | 81.9±0.2 | 80.7±0.4 | 80.6±0.2 | 78.6±0.5 |
| AsPC-1 | |||||
| Live cell no. (RFU) | 15,501±1,518 | 15,247±1,206 | 12,318±231 | 12,048±1,033 | 7,054±678a |
| Viability (%) | 62.4±2.3 | 71.1±1.1 | 58.9±1.4 | 56.9±6.6 | 30.7±4.1a |
P<0.0001 compared to the growth media. Data are expressed as the means ± SEM, n=4. CdCl2, cadmium chloride.
To determine the LC50 value of CdCl2 in the HPNE and AsPC-1 cells, each cell line was incubated with 7 concentrations of CdCl2 (1 to 1 mM) for 48 h, and cell viability was determined by MTT staining (n=6). Curves for both cell lines were best fit to a single-site model (Fig. 2). Individual response curves were analyzed to assess the LC50 for CdCl2 in each assay run. LC50 data were compared for the HPNE (77.3±6.8 μM) and AsPC-1 (42.1±4.0 μM) cells by Student's t-test and we observed a significant (P=0.0013) 45.5% reduction in LC50 values in the AsPC-1 group (Fig. 2, inset). We also compared our means at each concentration to their corresponding control values to determine the threshold at which we observed a significant decrease in cell viability. This threshold provides a value where CdCl2 effects are significantly different from the control and compliments our LC50 values. Using a one-way ANOVA with Dunnett's test to compare treatment groups to their corresponding control, we were able to identify the threshold at which the concentration yielded a significant reduction in cell number. The viability of both the HPNE (F6,35=92.27; P<0.0001) and AsPC-1 (F6,35=115; P<0.0001) cells was significantly affected by the increasing concentrations of CdCl2. The maximum concentration where we observed no reduction in viability was 10 μM in both cell lines. Therefore, although there was a slight rightward shift in LC50 values in the AsPC-1 group, the threshold for viability reduction was unaltered between the cell lines.
Figure 2.
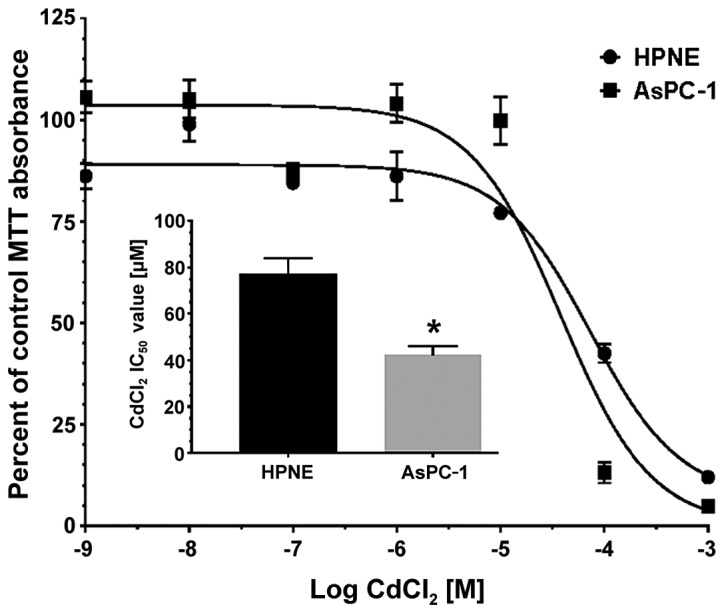
Determination of LC50 values for CdCl2 in HPNE and AsPC-1 cells. Cells were plated in clear 96-well plate at approximately 10−5 cells/ml and allowed to adhere for 24 h. before the addition of assay media containing CdCl2 (1 to 1 mM). Cells were returned to the incubator for 48 h. MTT assay was performed as described in the Materials and methods and the data were best fit to a single-site model using nonlinear iterative curve fitting (GraphPad Prism). Individual LC50 values were calculated for each curve (inset) and AsPC-1 cells exhibited a lower LC50 value compared to HPNE LC50 values (P<0.01). Data are expressed as the means ± SEM of 6 assays performed in duplicate (n=6). *P<0.01 compared to the HPNE LC50 value. LC50, lethal concentration 50%; CdCl2, cadmium chloride.
Cd-mediated induction of oxidative stress
The precise role of CdCl2 in the generation of free radicals has been a topic of considerable discussion. It is clear that transition metals have been implicated in the generation of free radicals (66,67). There is also evidence to suggest that oxidative stress and associated inflammation are underlying causes of cancer (68,69). The role of CdCl2 is not as clear as with other metals such as iron, lead, mercury. In this study, the ability of 50 or 100 μM CdCl2 to increase oxidative stress in the HPNE and AsPC-1 cells (Fig. 3A and B, respectively) was assessed by measuring DCF fluorescence. Prior to expsoure to CdCl2, basal DCF fluorescence was measured to yield a baseline to which subsequent treatment values would be compared. The basal levels of DCF fluorescence were 57.7% higher in the AsPC-1 cells compared to the HPNE values (555.8 and 352.4 RFU respectively; P<0.0001) at the initial 0-time-point. When comparing the means between the AsPC-1 and HPNE cells at 60 min of incubation, the trend was similar with the AsPC-1 cells, demonstrating a 48.4% increase in oxidative stress (595.2 vs. 401.2 RFU respectively; P<0.0001). Data were analyzed using a three-way ANOVA (cell type x time x treatment) and there was a clear association regarding cell type (F1,56=531.2; P<0.0001) and time (F4,105=162.6; P<0.0001) responses to CdCl2. In both cell lines, increasing the time of exposure resulted in an increased fluorescence. The trend for these increases was similar between the HPNE (F4,105=26.75; P<0.0001) and AsPC-1 (F4,105=59.71; P<0.0001) cells with no significant association between time and CdCl2 concentration in either the HPNE (F8,105=1.766; P=0.09) or AsPC-1 (F8,105=0.2748; P=0.92) cells.
Figure 3.

Oxidative stress in (A) HPNE and (B) AsPC-1 cells following exposure to CdCl2. Cells were grown and plated as described in the methods section. After loading with 100 μM DCFH, cells were washed twice with warm PBS and baseline fluorescence was measured (time 0). Treatment groups of (0 μM, PBS-only), and 50 or 100 μM CdCl2 were added to the appropriate treatment wells. DCF fluorescence was quantified at 30, 60, 90 and 120 min using a BioTek plate reader set at 485 nm(ex)/530 nm(em). The RFU values expressed were corrected for background (35-48 RFU) for each group. There was a significant effect of treatment-concentration in both HPNE (F2,105=10.45; P<0.0001) and AsPC-1 (F2,105=16.45; P<0.0001) cells. Data are expressed as the means ± SEM of 8 assays (n=8) performed in duplicate. CdCl2, cadmium chloride. *P<0.01 compared to the corresponding 0-time-point.
Mitochondrial toxicity elicited by exposure to Cd
The next series of experiments examined the effects of increasing concentrations of CdCl2 on cellular toxicity in the HPNE (Fig. 4A) and AsPC-1 (Fig. 4B) cells. Following 4 h of exposure to CdCl2, cellular viability was measured by MTT assay. This assay examined the shift in IC50 value for CdCl2 following exposure to the metal in the presence of 25 mM glucose or 10 mM galactose-containing media. A rightward shift in IC50 for the galactose curve of 2.5-3.0-fold would be indicative of a mitochondrial toxin (55,56). All curves where fit to a single-site model with a r2 values of 0.72, 0.73, 0.75 and 0.86 for HPNE-glucose, HPNE-galactose, AsPC-1-glucose and AsPC-1-galactose, respectively. After assessing the normality values, all curves passed as fitting normal Gaussian distribution (P-values of 0.07, 0.56, 0.17 and 0.92, respectively).
Figure 4.

Mitochondrial toxicity following exposure to various concentrations of CdCl2 in the presence of 25 mM glucose or 10 mM galactose. Cells were maintained in regular growth media until assayed. Cells were exposed to 0.01-100 μM CdCl2 for 4 h and the number of viable cells was estimated by MTT assay in the (A) HPNE and (B) AsPC-1 cells. Note the slight leftward shift in the curves for the 10 mM galactose group indicating a greater sensitivity to CdCl2 toxicity. Data are expressed as the means ± SEM of 12 assays (n=12) performed in duplicate and represent a composite of all assays analyzed by nonlinear curve fitting - single site inhibition analysis. CdCl2, cadmium chloride.
Individual IC50 values were obtained for each sample using non-linear curve fitting and graphically represented as the means ± SEM for each group (Fig. 5A). There was a significant effect of media on the IC50 values (glucose vs. galactose; F1,44=25.38; P<0.0001) that was observed in both cell lines. Sidak post hoc analysis revealed that only the IC50 value in the HPNE-galactose group was significantly (P<0.01) lower than that in the HPNE-glucose group (Fig. 5A). When examining the shift in IC50 values (Fig. 5B), we observed that there was almost a 2- to 4-fold rightward shift in IC50 values in both cell lines. The IC50 shift suggests that CdCl2 does act as a mitochondrial toxin in the HPNE and AsPC-1 cells.
Figure 5.

Comparison of CdCl2 IC50 values in HPNE and AsPC-1 cells following 4 h of exposure to CdCl2. Media contained either 25 mM glucose or 10 mM galactose. Individual IC50 values were obtained from the data in Fig. 4 by analyzing each curve independently and then grouped by cell type. IC50 values (A) are expressed as μM and the rightward fold-shift in mean IC50 values (B) are expressed as the means ± SEM of 12 assays (n=12) performed in duplicate. CdCl2, cadmium chloride. *P<0.01 compared to HPNE IC50 value with 25 mM glucose.
Using a dual-labeling assay kit, we measured both cell viability (Fig. 6A) and ATP release from the mitochondria, which is indicative of mitochondrial toxicity (Fig. 6B). The use of the more sensitive dual-labeling system revealed a complex response pattern to the action of CdCl2 in both cell lines. The four possible outcomes from this assay are: i) No change in ATP signal + no change in membrane integrity = not a mitochondrial toxicant; ii) reduced ATP + increased membrane integrity = primary necrosis; iii) reduced ATP + no change in membrane integrity = mitochondrial toxin; and iv) very low ATP at all concentrations + discordant change in membrane integrity = mitochondrial toxicant. Exposure to CdCl2 significantly (F1,24=214.5; P<0.0001) affected membrane integrity (Fig. 6A). The cytotoxicity response was also dependent on the type of media, glucose vs. galactose (F1,24=13.54; P=0.0012) and cell type, HPNE vs. AsPC-1 (F1,24=173.4; P<0.0001). There was a significant (F1,24=37.11; P<0.0001) association between the media and CdCl2 concentration. In each group, exposure to 50 μM CdCl2 resulted in a significant reduction (P<0.05) in viability compared to the corresponding 1 μM CdCl2 values in each of the groups. Viability in both the 1 and 50 μM CdCl2 groups in the galactose/AsPC-1 group was significantly (P<0.05) lower than the corresponding 25 mM glucose viability values. Across the CdCl2 concentrations and glucose/galactose supplementation, the viability values were significantly (P<0.05) reduced in the AsPC-1 cells compared to the corresponding viability values in the HPNE cells.
Figure 6.

Determination of membrane integrity and ATP release following exposure to 1 or 50 μM CdCl2 in the HPNE and AsPC-1 cells. (A) Effects of exposure to CdCl2 on membrane integrity following incubation in the presence of 25 mM glucose, or 10 mM galactose. AsPC-1 cells displayed a greater sensitivity to CdCl2 compared to the HPNE cell responses, and this great sensitivity was most evident when cells were incubated in the presence of 10 mM galactose. (B) Measurement of ATP release from damaged cells demonstrated a small ATP reduction in the AsPC-1 50 μM CdCl2 group. Incubation in the presence of 10 mM galactose revealed that AsPC-1 cells were most sensitive to the actions of CdCl2. Data represent the means ± SEM of 4 assays (n=4) performed in duplicate (aP<0.05 compared to the corresponding 25 mM glucose values; bP<0.05 compared to the corresponding HPNE group values; *P<0.05 compared to the corresponding 1 μM CdCl2 values). CdCl2, cadmium chloride.
When examining the levels of ATP, we observed a less robust change across he treatment groups (Fig. 6B), yet the trends were similar compared to the data observed from the cytotoxicity experiments. Exposure to CdCl2 significantly (F1,24=55.58; P<0.0001) affected membrane integrity (Fig. 6A). The cytotoxicity response was also dependent on the type of media, glucose vs. galactose (F1,24=22.67; P<0.0001) and cell type, HPNE vs. AsPC-1 (F1,24=9.10; P=0.006). There were two interactions that were significantly associated and these were media type x CdCl2 concentration (F1,24=21.03; P=0.0001) and cell type x CdCl2 concentration (F1,24=10.53; P=0.0034), suggesting that the CdCl2 concentration had a significant impact on both membrane integrity and ATP quantity, depending on the media (glucose vs. galactose) type or cell type (HPNE vs. AsPC-1). The AsPC-1 cells exhibited a slightly higher sensitivity to the effects of CdCl2, as indicated in Fig. 6B. The sensitivity of the AsPC-1 cells was particularly visible at the higher concentration of 50 μM of CdCl2, with the reduction in ATP was more pronounced in the 10 mM galactose group. If there is a change in membrane integrity associated with the changes in ATP, then that would be indicative of a necrotic toxin, leading to a reduction in ATP. We observed two associated interactions, with both the media and the cell type being significantly associated with the concentration of CdCl2.
An additional measure of mitochondrial function is the measure of mitochondrial permeability (referred to as ΔψM). We used the JC-1 dye due to its ability to permeate the mitochondria, and to either form aggregates (healthy cells) or monomers (unhealthy cells) which fluoresce at different wavelengths. We observed that CdCl2 (1 μM) had a significant effect on the health of the cells (F1,16=7.35; P=0.0184), slightly reducing the ratio of health/unhealthy cells in both cell lines (Fig. 7). Neither treatment group differed significantly from its corresponding control value.
Figure 7.
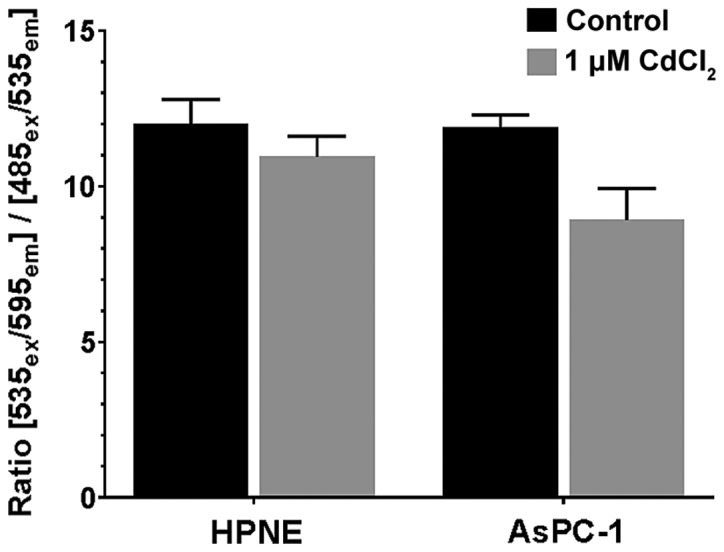
Measurement of mitochondrial permeability using JC-1 dye following exposure to 1 μM CdCl2 in HPNE and AsPC-1 cells. Fluorescence intensity was measured at 535ex/595em to measure JC-1 aggregates (healthy cells; green) and then a second reading using 485ex/535em which measures JC-1 monomers (apoptotic/unhealthy cells; red). The ratio of green/red represents a relative number of cells which are 'healthy'. There was a significant effect of treatment (F1,16=7.349; P=0.0154) with reductions in the ratio following exposure to 1 μM CdCl2 in both groups. Individual comparisons to control values did not reveal any difference in the fluorescent ratio in either cell line. Data represent the means ± SEM of 5 assays (n=5) performed in duplicate. CdCl2, cadmium chloride.
Discussion
On the periodic table, some of the most toxic metals reside in the d-block or transition metal block. Cd is part of this group; however, Cd is somewhat different in that it is a fifth-period element, unlike chromium, manganese, iron, and cobalt which are fourth-period elements. In addition, Cd is at the end of the transitional metal fifth-period row which makes it a 'post-translational element'. Therefore, there are similarities between Cd and other metals that are known toxicants, carcinogens, and free radical generators, such as mercury, iron, chromium, cobalt, nickel and manganese, although additional studies are required to improve our understanding of the cellular and intracellular actions of Cd. In this study, we aimed to work towards that end. Initial experiments examined the effects of CdCl2 on viability and cell number in media containing decreasing concentrations of fetal bovine serum. We did this to confirm the lack of serum effect for short exposure times. Other studies have reported that increasing the serum concentration up to 10% can affect the response of different cell types to toxicants (64,70). In this study, we reported that following exposure conducted in a low-serum environment, neither the growth or viability of neither cell line was significantly affected. The lack of effect aids in further in vitro studies and we can control the influence and impact that serum has on toxicity studies as a possible protectant. The lack of cytotoxicity observed following the exposure of HPNE cells to CdCl2 is not surprising since, control cells should have better defenses to protect against CdCl2-mediated damage. In addition, the 48-h exposure period may not be long enough for overt cytotoxic damage to be observed. Assay condition parameters are a consideration for further studies to avoid obscuring potential necrotic or apoptotic changes. Subsequent assays used a reduced serum to remove as many confounds as possible from additional toxicology testing. One variable we did not address in our studies is the possibility that low-level CdCl2 exposure may alter cellular responses, such as apoptosis.
The ability of elemental Cd to bioaccumulate in humans and concentrate in organs, such as the liver and kidneys can increase Cd-mediated toxicity over time (71). Exposure to CdCl2 can result in apoptotic, as well as necrotic changes (72-74). The results of this study indicated that exposure to only the higher concentration of CdCl2 resulted in a reduction in cell viability and cell number. This reduction was only observed in the AsPC-1 cells. This study intended to establish 'threshold' levels of exposure (concentration and duration) for use in subsequent mitochondrial toxicity experiments. Since we were attempting to find a threshold concentration for toxicity, it was not unexpected that we observed a lack of robust changes in cell viability, proliferation or cell number after a 48-h exposure. Our understanding of Cd as an environmental toxicant is expanding, and its role in the development of cancer has become widely accepted (24,75-77). Health organizations have classified Cd as a carcinogen (76). However, the exact mechanism(s) through which Cd exerts these carcinogenic effects remain largely unknown. Questions have been put forth as to whether it is a 'direct vs. indirect' effect, or potentially an 'upstream vs. downstream' effect. Further studies are required to describe the intracellular effects of Cd. In this study, we chose to target the mitochondria as an intracellular organelle that needs to function normally to aid the cell in providing the necessary energy requirements. When the mitochondria fail to function normally, tumor formation can follow (46).
The determination of a CdCl2 LC50 value in cell cultures is hard to find. It has previously been shown that the LC50 for CdCl2 is approximately 70-80 μM in various aquatic organisms (77). Other researchers have indicated that the lethality of CdCl2 is dependent on both the concentration and the duration of the exposure (78). In hepatic and pituitary cultures, the reported LC50 values for CdCl2 range from 40-50 μM (78,79). In this study, our findings of 40-70 μM fall within the LC50 range that has been reported elsewhere. The concentrations selected in the present study should represent concentrations that are: i) Below; ii) above; and iii) at the LC50 value. Caution was needed not to use concentrations that were too high which would prove lethal to a large percentage number of cells. That would skew our findings and conclusions since we would primarily be examining a subpopulation of cells that was resilient enough to guard against CdCl2 toxicity. Yet a concentration that was too low would not elicit a change in cellular function, and a change in apoptotic function in particular.
The secondary aim of this study was to establish and utilize 'subtoxic' concentrations of CdCl2 to assess oxidative stress elicited by exposure to CdCl2. Using approximately the mid-concentration, 6 ppm or 54 μM, of CdCl2, we can see that CdCl2 was only weakly a pro-oxidant, leading to a 15-20% increase in free radicals over 120 min of exposure. Measuring DCF fluorescence relies on the direct generation of free radicals by CdCl2. In vivo studies have shown that the administration of CdCl2 does increase biomarker expression commonly associated with increased oxidative stress (80,81). Therefore, changes in the oxidative status of the cell alone cannot be sufficient alone to elicit intracellular changes leading to apoptotic changes (82). In the kidneys, researchers have postulated that Cd interferes with normal mitochondrial function, leading to increased oxidative stress and apoptosis (38-40). In hepatic cells, CdCl2 exposure has been shown to decrease ΔψM, but also to directly alter apoptotic pathways by increasing the activity of both caspase-3 and caspase-9, as well as increasing the activity of p53 (41). In the pancreas, CdCl2 exposure has been shown to result in the death of β-cells following increased oxidative stress followed by a downstream-mediated activation of apoptotic pathways regulated by mitochondria (42). Other researchers have postulated that environment or occupational exposure to Cd, and the resulting Cd-mediated death or damage to β-cells is an underlying cause of diabetes and obesity (43). It is apparent that the Cd-mediated effects in the pancreas can alter β-cell function, hormonal responses, and may lead to the development of PC.
Healthy or 'normal' cells prefer oxidative phosphorylation (generation of 38 ATPs) to derive their energy (Crabtree), but immortalized cells lines may be adapted to using high glucose, generating ATP via glycolysis (only generates 2 ATPs), which will decrease the sensitivity of the cells to mitochondrial toxins (44). In this study, we examined the effects of CdCl2 on mitochondrial function and the Warburg effect, which involves the differential production of ATP in media containing glucose or galactose. Subsequently, by comparing the toxicity of a compound in the presence of glucose media, or the presence of galactose media, we can determine direct mitochondrial toxicity compared to other indirect mechanisms (55,57,58). The use of galactose-containing (10 mM) media will force the cell to increase their reliance on oxidative phosphorylation to generate ATP. The direct assessment of CdCl2 as a mitochondrial toxin suggested that CdCl2 is a weak mitochondrial toxicant. Comparisons of the glucose/galactose MTT curves in the presence of increasing concentrations of CdCl2 revealed an almost 3-fold shift in IC50 values with glucose IC50 values > galactose IC50 values.
In support of this finding was our observation that ATP levels were reduced, primarily in AsPC-1 cells, with little concomitant changes in viability, suggestive of mitochondrial toxicity (57,59). These data suggest that the CdCl2 concentration has a significant impact on both membrane integrity and ATP quantity depending on media (glucose vs. galactose) type or cell type (HPNE vs. AsPC-1). When examining the data to determine whether CdCl2 is a mitochondrial toxin, or whether it elicits toxicity through another mechanism, the amount of ATP present needs to be quantified. We would have expected a reduction in ATP in the presence of CdCl2 compared to control values suggesting a mitochondrial toxin. The cytotoxic response must then be examined, with no change in membrane integrity + reduction in ATP being indicative of being a mitochondrial toxin.
Following the addition of the JC-1 dye data, we observed only a small decrease in the ratio of healthy to unhealthy cells in both cell lines. A caveat of these findings is that we used 1 μM CdCl2. The rationale for this concentration was that our threshold was approximately 10 μM CdCl2 when we began to see a reduction in cellular viability. We decided that a 10-fold lower concentration would be sufficient to induce changes in cellular function without resulting in a loss of viability, which would make our data difficult to interpret since the mitochondrial information would be from cells that were able to survive CdCl2 exposure. From our data, it can be hypothesized that CdCl2 is a weak mitochondrial toxin, with adverse effects evident at the highest CdCl2 concentrations of 10 μM or higher.
Although one single theory has not come to the forefront, numerous hypotheses have been presented suggesting that the switch from oxidative phosphorylation to aerobic glycolysis signals the transition from a normal cell to a tumor cell (46). Another hypothesis is that switching to aerobic glycolysis can protect the tumor cell from a sugar-induced death when glucose uptake is high, and by switching to a high production of lactate, pro-proliferative intermediates can be produced (47). Compounds, such as CdCl2, that damage the mitochondria promote a shift towards aerobic glycolysis, thus leading to tumor formation. Studies that have examined toxicant (MPTP) damage to the mitochondria have reported that oxidative phosphorylation is decreased, that apoptotic pathways are altered and there is a bioenergetic shift in glucose utilization (48). Indirect genetic damage to the mitochondria that will result in epigenetic alterations have has also been demonstrated in tumor cells, as well as inherited genetic abnormalities that predispose or facilitate the progression of the Warburg effect in tumor cells (49). The apparent importance of the mitochondria and the Warburg effect has led researchers to postulate that either 'turning' oxidative phosphorylation 'back on' or shunting away from lactate production/utilization may be an effective means of controlling tumor growth (45,50). In this study, we demonstrated a rightward shift in IC50 values for both the HPNE and AsPC-1 cells, suggesting that exposure to CdCl2 plays a role in mitochondrial dysfunction. Additional experiments supporting the finding of CdCl2 as a weak mitochondrial toxicant, include the investigation of the aspects of mitochondrial toxicity, such as changes in mitochondrial respiration, transition pore permeability, and the activation of apoptotic peptides. The ability of Cd to accumulate within mammalian tissues is a variable that needs to be addressed due to the long biological half-life. This characteristic of Cd may result in toxicity when exposure concentrations are low, but exposure duration is extended. Additional studies are underway to examine the effects of CdCl2 exposure on intracellular function when subtoxic concentrations of CdCl2 are added to the growth media for extended periods of time (weeks). Exposure in this manner would better represent long-term chronic exposure that many individuals are subjected to in the environment.
Collectively, these data enhance our understanding of Cd-mediated intracellular toxicity. The results also lead us to additional questions. Unlike other well-studied metals, such as mercury, iron, nickel and manganese, CdCl2 appears to exert multiple weak effects, such as on oxidative stress, viability, ATP loss and oxidative phosphorylation as an energy source. CdCl2 does behave as a weak mitochondrial toxicant as indicated by the 2- to 4-fold shift in IC50 values comparing the glucose and galactose curves. Since we observed only modest changes in viability, cell number and ATP release, it can be hypothesized that changes following low concentrations of CdCl2 are most likely due to a complex and cumulative set of small changes as opposed to substantial changes. The assessment of the direct vs. indirect effects of CdCl2 on cellular and mitochondrial function is essential. Potential CdCl2-mediated changes which occur upstream or downstream from the mitochondria will be pivotal in determining where and how CdCl2 is eliciting its cellular effects. Additional studies are underway to examine the molecular alterations in intracellular organelles associated with long-term CdCl2 exposure addressing our larger question regarding CdCl2 as a mitochondrial toxicant.
Acknowledgments
Not applicable.
Funding
The authors received support for this research in part from OSU intramural funding [#154333-7] and Ministry of Education, Science and Technological Development of Serbia (Project III 46009). The funders had no role in the design of the study, or in the collection, analyses, or interpretation of the data, in the writing of the manuscript, and in the decision to publish the results.
Availability of data and materials
All data generated or analyzed during this study are included in this published article or are available from the corresponding author on reasonable request.
Authors' contributions
DRW and ABD were responsible for the conceptualization, methodology, formal data analysis and interpretation, data curation, writing/original draft preparation, writing-review and editing, project administration, and funding acquisition. DAS and AT were involved in consultation regarding the conceptualization and interpretation of the data, data curation, composition of the draft manuscript, as well as the final manuscript and post-review editing. VD was responsible for earlier clinical studies that established the pancreatic concentrations of Cd and working to quantify in vivo Cd concentrations. VD and AB aided in the design of the experiments and the establishment of appropriate/relevant concentrations of Cd to be used in the study. AB worked with the overall design and interpretation of the data and assisted in the writing of the final manuscript. AS worked with the design of the viability studies, performance of viability assays and the corresponding data acquisition and analysis for those assays. All authors have read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
DAS is the Editor-in-Chief for the journal, but had no personal involvement in the reviewing process, or any influence in terms of adjudicating on the final decision, for this article. The other authors declare that they have no competing interests regarding this work.
References
- 1.Faroon O, Ashizawa A, Wright S, Tucker P, Jenkins K, Ingerman L, Rudisill C. Toxicological Profile for Cadmium. Agency for Toxic Substances and Disease Registry. 2012:1–487. [PubMed] [Google Scholar]
- 2.Renieri E, Alegakis A, Kiriakakis M, Vinceti M, Ozcagli E, Wilks M, Tsatsakis A. Cd, Pb and Hg Biomonitoring in Fish of the Mediterranean Region and Risk Estimations on Fish Consumption. Toxics. 2014;2:417–442. doi: 10.3390/toxics2030417. [DOI] [Google Scholar]
- 3.Renieri EA, Safenkova IV, Alegakis AK, Slutskaya ES, Kokaraki V, Kentouri M, Dzantiev BB, Tsatsakis AM. Cadmium, lead and mercury in muscle tissue of gilthead seabream and seabass: Risk evaluation for consumers. Food Chem Toxicol. 2019;124:439–449. doi: 10.1016/j.fct.2018.12.020. [DOI] [PubMed] [Google Scholar]
- 4.Mezynska M, Brzóska MM. Environmental exposure to cadmium-a risk for health of the general population in industrialized countries and preventive strategies. Environ Sci Pollut Res Int. 2018;25:3211–3232. doi: 10.1007/s11356-017-0827-z. [DOI] [PubMed] [Google Scholar]
- 5.Satarug S. Dietary Cadmium Intake and Its Effects on Kidneys. Toxics. 2018;6:15. doi: 10.3390/toxics6010015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buha A, Antonijević B, Bulat Z, Jaćević V, Milovanović V, Matović V. The impact of prolonged cadmium exposure and co-exposure with polychlorinated biphenyls on thyroid function in rats. Toxicol Lett. 2013;221:83–90. doi: 10.1016/j.toxlet.2013.06.216. [DOI] [PubMed] [Google Scholar]
- 7.Buha A, Matovic V, Antonijevic B, Bulat Z, Curcic M, Renieri EA, Tsatsakis AM, Schweitzer A, Wallace D. Overview of cadmium thyroid disrupting effects and mechanisms. Int J Mol Sci. 2018;19:1501. doi: 10.3390/ijms19051501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Silva N, Peiris-John R, Wickremasinghe R, Senanayake H, Sathiakumar N. Cadmium a metalloestrogen: Are we convinced? J Appl Toxicol. 2012;32:318–332. doi: 10.1002/jat.1771. [DOI] [PubMed] [Google Scholar]
- 9.Xing Y, Xia W, Zhang B, Zhou A, Huang Z, Zhang H, Liu H, Jiang Y, Hu C, Chen X, et al. Relation between cadmium exposure and gestational diabetes mellitus. Environ Int. 2018;113:300–305. doi: 10.1016/j.envint.2018.01.001. [DOI] [PubMed] [Google Scholar]
- 10.Renieri EA, Sfakianakis DG, Alegakis AA, Safenkova IV, Buha A, Matović V, Tzardi M, Dzantiev BB, Divanach P, Kentouri M, et al. Nonlinear responses to waterborne cadmium exposure in zebrafish. An in vivo study. Environ Res. 2017;157:173–181. doi: 10.1016/j.envres.2017.05.021. [DOI] [PubMed] [Google Scholar]
- 11.IARC . IARC monographs on the evaluation of the carcinogenic risk of chemicals to man. 1993. [Google Scholar]
- 12.Iarc and IARC Working Group on the Evaluation of Carcinogenic Risks to Humans International Agency for Research on Cancer Iarc Monographs on the Evaluation of Carcinogenic Risks to Humans. Personal habits and indoor combustions. IARC Monogr Eval Carcinog Risks Hum. 2012;100E [PMC free article] [PubMed] [Google Scholar]
- 13.Huff J, Lunn RM, Waalkes MP, Tomatis L, Infante PF. Cadmium-induced cancers in animals and in humans. Int J Occup Environ Health. 2007;13:202–212. doi: 10.1179/oeh.2007.13.2.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartwig A. Cadmium and cancer. In: Sigel A, Sigel H, Sigel RK, editors. Cadmium: From Toxicity to Essentiality. Springer; Netherlands, Dordrecht: 2013. pp. 491–507. [Google Scholar]
- 15.Il'yasova D, Schwartz GG. Cadmium and renal cancer. Toxicol Appl Pharmacol. 2005;207:179–186. doi: 10.1016/j.taap.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 16.Vinceti M, Venturelli M, Sighinolfi C, Trerotoli P, Bonvicini F, Ferrari A, Bianchi G, Serio G, Bergomi M, Vivoli G. Case-control study of toenail cadmium and prostate cancer risk in Italy. Sci Total Environ. 2007;373:77–81. doi: 10.1016/j.scitotenv.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 17.Goyer RA, Liu J, Waalkes MP. Cadmium and cancer of prostate and testis. Biometals. 2004;17:555–558. doi: 10.1023/B:BIOM.0000045738.59708.20. [DOI] [PubMed] [Google Scholar]
- 18.Feki-Tounsi M, Hamza-Chaffai A. Cadmium as a possible cause of bladder cancer: A review of accumulated evidence. Environ Sci Pollut Res Int. 2014;21:10561–10573. doi: 10.1007/s11356-014-2970-0. [DOI] [PubMed] [Google Scholar]
- 19.Van Maele-Fabry G, Lombaert N, Lison D. Dietary exposure to cadmium and risk of breast cancer in postmenopausal women: A systematic review and meta-analysis. Environ Int. 2016;86:1–13. doi: 10.1016/j.envint.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 20.Cho YA, Kim J, Woo HD, Kang M. Dietary cadmium intake and the risk of cancer: A meta-analysis. PLoS One. 2013;8:e75087. doi: 10.1371/journal.pone.0075087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 22.Mytilinaiou M, Nikitovic D, Berdiaki A, Kostouras A, Papoutsidakis A, Tsatsakis AM, Tzanakakis GN. Emerging roles of syndecan 2 in epithelial and mesenchymal cancer progression. IUBMB Life. 2017;69:824–833. doi: 10.1002/iub.1678. [DOI] [PubMed] [Google Scholar]
- 23.Tzanakakis GN, Margioris AN, Tsatsakis AM, Vezeridis MP. The metastatic potential of human pancreatic cell lines in the liver of nude mice correlates well with cathepsin B activity. Int J Gastrointest Cancer. 2003;34:27–38. doi: 10.1385/IJGC:34:1:27. [DOI] [PubMed] [Google Scholar]
- 24.Buha A, Wallace D, Matovic V, Schweitzer A, Oluic B, Micic D, Djordjevic V. Cadmium Exposure as a Putative Risk Factor for the Development of Pancreatic Cancer: Three Different Lines of Evidence. BioMed Res Int. 2017;2017:1981837. doi: 10.1155/2017/1981837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andjelkovic M, Buha Djordjevic A, Antonijevic E, Antonijevic B, Stanic M, Kotur-Stevuljevic J, Spasojevic-Kalimanovska V, Jovanovic M, Boricic N, Wallace D, et al. Toxic effect of acute cadmium and lead exposure in rat blood, liver, and kidney. Int J Environ Res Public Health. 2019;16:E274. doi: 10.3390/ijerph16020274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buha A, Bulat Z, Dukić-Ćosić D, Matović V. Effects of oral and intraperitoneal magnesium treatment against cadmium-induced oxidative stress in plasma of rats. Arh Hig Rada Toksikol. 2012;63:247–254. doi: 10.2478/10004-1254-63-2012-2217. [DOI] [PubMed] [Google Scholar]
- 27.Matović V, Buha A, Bulat Z, Ðukić-Ćosić D, Miljković M, Ivanišević J, Kotur-Stevuljević J. Route-dependent effects of cadmium/cadmium and magnesium acute treatment on parameters of oxidative stress in rat liver. Food Chem Toxicol. 2012;50:552–557. doi: 10.1016/j.fct.2011.12.035. [DOI] [PubMed] [Google Scholar]
- 28.Djukić-Cosić D, Curcić Jovanović M, Plamenac Bulat Z, Ninković M, Malicević Z, Matović V. Relation between lipid peroxidation and iron concentration in mouse liver after acute and subacute cadmium intoxication. J Trace Elem Med Biol. 2008;22:66–72. doi: 10.1016/j.jtemb.2007.09.024. [DOI] [PubMed] [Google Scholar]
- 29.Bulat Z, Dukić-Osi-Ćosić D, Antonijević B, Bulat P, Vujanović D, Buha A, Matović V. Effect of magnesium supplementation on the distribution patterns of zinc, copper, and magnesium in rabbits exposed to prolonged cadmium intoxication. Sci World J. 2012;2012 doi: 10.1100/2012/572514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Djukić-Cosić D, Ninković M, Malicević Z, Plamenac-Bulat Z, Matović V. Effect of supplemental magnesium on the kidney levels of cadmium, zinc, and copper of mice exposed to toxic levels of cadmium. Biol Trace Elem Res. 2006;114:281–291. doi: 10.1385/BTER:114:1:281. [DOI] [PubMed] [Google Scholar]
- 31.Matović V, Buha A, Ðukić-Ćosić D, Bulat Z. Insight into the oxidative stress induced by lead and/or cadmium in blood, liver and kidneys. Food Chem Toxicol. 2015;78:130–140. doi: 10.1016/j.fct.2015.02.011. [DOI] [PubMed] [Google Scholar]
- 32.Bulat Z, Đukić-Ćosić D, Antonijević B, Buha A, Bulat P, Pavlović Z, Matović V. Can zinc supplementation ameliorate cadmium-induced alterations in the bioelement content in rabbits? Arh Hig Rada Toksikol. 2017;68:38–45. doi: 10.1515/aiht-2017-68-2919. [DOI] [PubMed] [Google Scholar]
- 33.Zhang DX, Dai YD, Yuan SX, Tao L. Prognostic factors in patients with pancreatic cancer. Exp Ther Med. 2012;3:423–432. doi: 10.3892/etm.2011.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu J, Ji S, Liang C, Qin Y, Jin K, Liang D, Xu W, Shi S, Zhang B, Liu L, et al. Critical role of oncogenic KRAS in pancreatic cancer (Review) Mol Med Rep. 2016;13:4943–4949. doi: 10.3892/mmr.2016.5196. [DOI] [PubMed] [Google Scholar]
- 35.Basu A, Jiang X, Negrini M, Haldar S. MicroRNA-mediated regulation of pancreatic cancer cell proliferation. Oncol Lett. 2010;1:565–568. doi: 10.3892/ol_00000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duarte FV, Palmeira CM, Rolo AP. The role of microRNAs in mitochondria: Small players acting wide. Genes (Basel) 2014;5:865–886. doi: 10.3390/genes5040865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cannino G, Ferruggia E, Luparello C, Rinaldi AM. Cadmium and mitochondria. Mitochondrion. 2009;9:377–384. doi: 10.1016/j.mito.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 38.Li M, Xia T, Jiang CS, Li LJ, Fu JL, Zhou ZC. Cadmium directly induced the opening of membrane permeability pore of mitochondria which possibly involved in cadmium-triggered apoptosis. Toxicology. 2003;194:19–33. doi: 10.1016/S0300-483X(03)00327-5. [DOI] [PubMed] [Google Scholar]
- 39.Mao WP, Zhang NN, Zhou FY, Li WX, Liu HY, Feng J, Zhou L, Wei CJ, Pan YB, He ZJ. Cadmium directly induced mitochondrial dysfunction of human embryonic kidney cells. Hum Exp Toxicol. 2011;30:920–929. doi: 10.1177/0960327110384286. [DOI] [PubMed] [Google Scholar]
- 40.Gobe G, Crane D. Mitochondria, reactive oxygen species and cadmium toxicity in the kidney. Toxicol Lett. 2010;198:49–55. doi: 10.1016/j.toxlet.2010.04.013. [DOI] [PubMed] [Google Scholar]
- 41.Lasfer M, Vadrot N, Aoudjehane L, Conti F, Bringuier AF, Feldmann G, Reyl-Desmars F. Cadmium induces mitochondria-dependent apoptosis of normal human hepatocytes. Cell Biol Toxicol. 2008;24:55–62. doi: 10.1007/s10565-007-9015-0. [DOI] [PubMed] [Google Scholar]
- 42.Chang KC, Hsu CC, Liu SH, Su CC, Yen CC, Lee MJ, Chen KL, Ho TJ, Hung DZ, Wu CC, et al. Cadmium induces apoptosis in pancreatic β-cells through a mitochondria-dependent pathway: The role of oxidative stress-mediated c-Jun N-terminal kinase activation. PLoS One. 2013;8:e54374. doi: 10.1371/journal.pone.0054374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tinkov AA, Filippini T, Ajsuvakova OP, Aaseth J, Gluhcheva YG, Ivanova JM, Bjørklund G, Skalnaya MG, Gatiatulina ER, Popova EV, et al. The role of cadmium in obesity and diabetes. Sci Total Environ. 2017;601-602:741–755. doi: 10.1016/j.scitotenv.2017.05.224. [DOI] [PubMed] [Google Scholar]
- 44.Hammad N, Rosas-Lemus M, Uribe-Carvajal S, Rigoulet M, Devin A. The Crabtree and Warburg effects: Do metabolite-induced regulations participate in their induction? Biochim Biophys Acta. 2016;1857:1139–1146. doi: 10.1016/j.bbabio.2016.03.034. [DOI] [PubMed] [Google Scholar]
- 45.Diaz-Ruiz R, Rigoulet M, Devin A. The Warburg and Crabtree effects: On the origin of cancer cell energy metabolism and of yeast glucose repression. Biochim Biophys Acta. 2011;1807:568–576. doi: 10.1016/j.bbabio.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 46.Devic S. Warburg effect - a consequence or the cause of carcinogenesis? J Cancer. 2016;7:817–822. doi: 10.7150/jca.14274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de Alteriis E, Cartenì F, Parascandola P, Serpa J, Mazzoleni S. Revisiting the Crabtree/Warburg effect in a dynamic perspective: A fitness advantage against sugar-induced cell death. Cell Cycle. 2018;17:688–701. doi: 10.1080/15384101.2018.1442622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Solaini G, Sgarbi G, Baracca A. Oxidative phosphorylation in cancer cells. Biochim Biophys Acta. 2011;1807:534–542. doi: 10.1016/j.bbabio.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 49.Chandra D, Singh KK. Genetic insights into OXPHOS defect and its role in cancer. Biochim Biophys Acta. 2011;1807:620–625. doi: 10.1016/j.bbabio.2010.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mot AI, Liddell JR, White AR, Crouch PJ. Circumventing the Crabtree Effect: A method to induce lactate consumption and increase oxidative phosphorylation in cell culture. Int J Biochem Cell Biol. 2016;79:128–138. doi: 10.1016/j.biocel.2016.08.029. [DOI] [PubMed] [Google Scholar]
- 51.Lamas-Maceiras M, Rodríguez-Belmonte E, Becerra M, González-Siso MI, Cerdán ME. KlGcr1 controls glucose-6-phosphate dehydrogenase activity and responses to H O cadmium and arsenate in Kluyveromyces lactis. Fungal Genet 2 2, Biol. 2015;82:95–103. doi: 10.1016/j.fgb.2015.07.004. [DOI] [PubMed] [Google Scholar]
- 52.Torres AM, Maceiras ML, Belmonte ER, Naveira LN, Calvo MB, Cerdán ME. KlRox1 pcontributes to yeast resistance to metals and is necessary for KlYCF1 expression in the presence of cadmium. Gene. 2012;497:27–37. doi: 10.1016/j.gene.2012.01.050. [DOI] [PubMed] [Google Scholar]
- 53.Djordjevic VR, Wallace DR, Schweitzer A, Boricic N, Knezevic D, Matic S, Grubor N, Kerkez M, Radenkovic D, Bulat Z, et al. Environmental cadmium exposure and pancreatic cancer: Evidence from case control, animal and in vitro studies. Environ Int. 2019;128:353–361. doi: 10.1016/j.envint.2019.04.048. [DOI] [PubMed] [Google Scholar]
- 54.Wallace DR, Bates E. Differential effects of organic and inorganic mercury on phenotypically variant breast cancer cell lines. J Clin Toxicol. 2015;5:1000273. doi: 10.4172/2161-0495.1000273. [DOI] [Google Scholar]
- 55.Sanuki Y, Araki T, Nakazono O, Tsurui K. A rapid mitochondrial toxicity assay utilizing rapidly changing cell energy metabolism. J Toxicol Sci. 2017;42:349–358. doi: 10.2131/jts.42.349. [DOI] [PubMed] [Google Scholar]
- 56.Dott W, Mistry P, Wright J, Cain K, Herbert KE. Modulation of mitochondrial bioenergetics in a skeletal muscle cell line model of mitochondrial toxicity. Redox Biol. 2014;2:224–233. doi: 10.1016/j.redox.2013.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Niles AL, Moravec RA, Eric Hesselberth P, Scurria MA, Daily WJ, Riss TL. A homogeneous assay to measure live and dead cells in the same sample by detecting different protease markers. Anal Biochem. 2007;366:197–206. doi: 10.1016/j.ab.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 58.Marroquin LD, Hynes J, Dykens JA, Jamieson JD, Will Y. Circumventing the Crabtree effect: Replacing media glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants. Toxicol Sci. 2007;97:539–547. doi: 10.1093/toxsci/kfm052. [DOI] [PubMed] [Google Scholar]
- 59.Rossignol R, Gilkerson R, Aggeler R, Yamagata K, Remington SJ, Capaldi RA. Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res. 2004;64:985–993. doi: 10.1158/0008-5472.CAN-03-1101. [DOI] [PubMed] [Google Scholar]
- 60.Rodríguez-Enríquez S, Juárez O, Rodríguez-Zavala JS, Moreno-Sánchez R. Multisite control of the Crabtree effect in ascites hepatoma cells. Eur J Biochem. 2001;268:2512–2519. doi: 10.1046/j.1432-1327.2001.02140.x. [DOI] [PubMed] [Google Scholar]
- 61.Taverna M, Marie AL, Mira JP, Guidet B. Specific antioxidant properties of human serum albumin. Ann Intensive Care. 2013;3:4. doi: 10.1186/2110-5820-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hsiao IL, Huang YJ. Effects of serum on cytotoxicity of nano- and micro-sized ZnO particles. J Nanopart Res. 2013;15:1829. doi: 10.1007/s11051-013-1829-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Leelakanok N, Fischer CL, Bates AM, Guthmiller JM, Johnson GK, Salem AK, Brogden KA, Brogden NK. Cytotoxicity of HBD3 for dendritic cells, normal human epidermal keratinocytes, hTERT keratinocytes, and primary oral gingival epithelial keratinocytes in cell culture conditions. Toxicol Lett. 2015;239:90–96. doi: 10.1016/j.toxlet.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Umebayashi C, Oyama Y, Chikahisa-Muramastu L, Nakao H, Nishizaki Y, Nakata M, Soeda F, Takahama K. Tri-n-butyltin-induced cytotoxicity on rat thymocytes in presence and absence of serum. Toxicol In Vitro. 2004;18:55–61. doi: 10.1016/j.tiv.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 65.Qiao D, Seidler FJ, Slotkin TA. Developmental neurotoxicity of chlorpyrifos modeled in vitro: Comparative effects of metabolites and other cholinesterase inhibitors on DNA synthesis in PC12 and C6 cells. Environ Health Perspect. 2001;109:909–913. doi: 10.1289/ehp.01109909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stohs SJJ, Bagchi D. Oxidative mechanisms in the toxicity of metal ions. Free Radic Biol Med. 1995;18:321–336. doi: 10.1016/0891-5849(94)00159-H. [DOI] [PubMed] [Google Scholar]
- 67.Ercal N, Gurer-Orhan H, Aykin-Burns N. Toxic metals and oxidative stress part I: Mechanisms involved in metal-induced oxidative damage. Curr Top Med Chem. 2001;1:529–539. doi: 10.2174/1568026013394831. [DOI] [PubMed] [Google Scholar]
- 68.Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160:1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 69.Galaris D, Evangelou A. The role of oxidative stress in mechanisms of metal-induced carcinogenesis. Crit Rev Oncol Hematol. 2002;42:93–103. doi: 10.1016/S1040-8428(01)00212-8. [DOI] [PubMed] [Google Scholar]
- 70.Thomas MG, Marwood RM, Parsons AE, Parsons RB. The effect of foetal bovine serum supplementation upon the lactate dehydrogenase cytotoxicity assay: Important considerations for in vitro toxicity analysis. Toxicol In Vitro. 2015;30:300–308. doi: 10.1016/j.tiv.2015.10.007. [DOI] [PubMed] [Google Scholar]
- 71.Cadm ium toxicity and treatment. ScientificWorldJournal. 2013;2013:394652. [Google Scholar]
- 72.Franco R, Sánchez-Olea R, Reyes-Reyes EM, Panayiotidis MI. Environmental toxicity, oxidative stress and apoptosis: Ménage à trois. Mutat Res. 2009;674:3–22. doi: 10.1016/j.mrgentox.2008.11.012. [DOI] [PubMed] [Google Scholar]
- 73.Chen C, Xun P, Nishijo M, Sekikawa A, He K. Cadmium exposure and risk of pancreatic cancer: A meta-analysis of prospective cohort studies and case-control studies among individuals without occupational exposure history. Environ Sci Pollut Res Int. 2015;22:17465–17474. doi: 10.1007/s11356-015-5464-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen P, Duan X, Li M, Huang C, Li J, Chu R, Ying H, Song H, Jia X, Ba Q, et al. Systematic network assessment of the carcinogenic activities of cadmium. Toxicol Appl Pharmacol. 2016;310:150–158. doi: 10.1016/j.taap.2016.09.006. [DOI] [PubMed] [Google Scholar]
- 75.Waalkes MP. Cadmium carcinogenesis. Mutat Res. 2003;533:107–120. doi: 10.1016/j.mrfmmm.2003.07.011. [DOI] [PubMed] [Google Scholar]
- 76.WHO Exposure to Cadmium: A Major Public Health Concern. Geneva. 2010 [Google Scholar]
- 77.Muthukumaravel K, Kumarasamy P, Amsath A, Paulraj MG. Toxic effect of cadmium on the electrophoretic protein patterns of gill and muscle of Oreochromis mossambicus. E-J Chem. 2007;4:284–286. doi: 10.1155/2007/834538. [DOI] [Google Scholar]
- 78.Fotakis G, Timbrell JA. In vitro cytotoxicity assays: Comparison of LDH, neutral red, MTT and protein assay in hepatoma cell lines following exposure to cadmium chloride. Toxicol Lett. 2006;160:171–177. doi: 10.1016/j.toxlet.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 79.Hinkle PM, Kinsella PA, Osterhoudt KC. Cadmium uptake and toxicity via voltage-sensitive calcium channels. J Biol Chem. 1987;262:16333–16337. [PubMed] [Google Scholar]
- 80.Bertin G, Averbeck D. Cadmium: Cellular effects, modifications of biomolecules, modulation of DNA repair and genotoxic consequences (a review) Biochimie. 2006;88:1549–1559. doi: 10.1016/j.biochi.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 81.Cuypers A, Plusquin M, Remans T, Jozefczak M, Keunen E, Gielen H, Opdenakker K, Nair AR, Munters E, Artois TJ, et al. Cadmium stress: An oxidative challenge. Biometals. 2010;23:927–940. doi: 10.1007/s10534-010-9329-x. [DOI] [PubMed] [Google Scholar]
- 82.Tzanakakis GN, Krambovitis E, Tsatsakis AM, Vezeridis MP. The preventive effect of ketoconazole on experimental metastasis from a human pancreatic carcinoma may be related to its effect on prostaglandin synthesis. Int J Gastrointest Cancer. 2002;32:23–30. doi: 10.1385/IJGC:32:1:23. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article or are available from the corresponding author on reasonable request.