

Abstract
Microglial activation has been recognized as a major contributor to inflammation of the epileptic brain. Seizures are commonly accompanied by remarkable microgliosis and loss of neurons. In this study we utilize the CX3CR1GFP/+ CCR2RFP/+ genetic mouse model, in which CX3CR1+ resident microglia and CCR2+ monocytes are labeled with GFP and RFP, respectively. Using a combination of time-lapse two-photon imaging and whole-cell patch clamp recording, we determined the distinct morphological, dynamic, and electrophysiological characteristics of infiltrated monocytes and resident microglia, and the evolution of their behavior at different time points following kainic acid-induced seizures. Seizure activated microglia presented enlarged somas with less ramified processes, whereas, infiltrated monocytes were smaller, highly motile cells that lacked processes. Moreover, resident microglia, but not infiltrated monocytes, proliferate locally in the hippocampus after seizure. Microglial proliferation was dependent on the colony stimulating factor 1 receptor (CSF-1R) pathway. Pharmacological inhibition of CSF-1R reduced seizure-induced microglial proliferation, which correlated with attenuation of neuronal death without altering acute seizure behaviors. Taken together, we demonstrated that proliferation of activated resident microglia contributes to neuronal death in the hippocampus via CSF-1R after status epilepticus, providing potential therapeutic targets for neuroprotection in epilepsy.
Keywords: microglia, monocytes, epilepsy, proliferation, infiltration, colony stimulating factor 1 receptor
Introduction
Epilepsy is one of the most common neurological disorders, affecting approximately 50 million people, and is characterized by abnormal hypersynchrony of neurons. Currently available anti-epileptic drugs preferentially target neuronal mechanisms, but are only effective in two-third of the patients. Hence, there is an urgent need for alternative therapeutic strategies. An increasing body of clinical and experimental evidence suggests that neuroinflammation in the brain is a crucial mechanism underlying the pathology of epilepsy (Eyo et al. 2017; Vezzani 2014). Microglia are the innate immune cells of the brain and major contributors to neuroinflammation and neuronal loss following status epilepticus (Avignone et al. 2008; Bosco et al. 2018; Matsuda et al. 2015; Tian et al. 2017). Within animal models of epilepsy, microglia become chronically activated with increased cell numbers in the hippocampus (Drage et al. 2002; Shapiro et al. 2008). A similar increase in microglial reactivity has also been reported in human patients with intractable seizures (Beach et al. 1995). Although well studied, the source of microgliosis remains controversial. Some studies indicate that circulating cells from the bloodstream can infiltrate the CNS and contribute to the microglial pool (Djukic et al. 2006; Flügel et al. 2001; Simard and Rivest 2004), while other studies suggest that local proliferation of reactive microglia is the sole source (Ajami et al. 2007; Gu et al. 2016b). Recent studies however, showed that in addition to resident microglia, infiltrating immune cells (i.e., monocytes, leukocytes) invade the hippocampus after seizures and contribute significantly to pathogenesis (Tian et al. 2017; Varvel et al. 2016; Zattoni et al. 2011). In this study, we used complimentary techniques to investigate the source of microgliosis, particularly the respective contribution of monocyte infiltration and local proliferation in epilepsy.
Although microglia and monocytes share common macrophage properties and can act synergistically (London et al. 2013; Peng et al. 2016), their functions can be highly divergent within different pathological conditions (Mildner et al. 2011; Prinz et al. 2011; Yamasaki et al. 2014). A recent study showed that microglia were highly inflammatory, while infiltrated monocytes contributed to phagocytosis after ischemic injury (Ritzel et al. 2015). Conversely, monocytes were proposed to be inflammatory in experimental autoimmune encephalomyelitis (Yamasaki et al. 2014) and AD mouse models (Hohsfield and Humpel 2015). Under epileptic conditions, infiltration of monocytes is believed to contribute to the worsening of inflammation following seizures (Tian et al. 2017; Varvel et al. 2016). Hence, understanding the different functions and properties of infiltrating monocytes and resident microglia will help development of drugs that can modulate these cells to improve prognosis post-seizure.
Microglial proliferation is the main source for microgliosis in several disease conditions. Studies have shown a link between microglia numbers and seizure outcomes (Kim et al. 2015). One study showed that toll-like receptor 2 was involved in microglial proliferation after kainic acid (KA)-induced seizures (Hong et al. 2010). Importantly, microglial activation and proliferation was found to be prevalent in epilepsy patients and immunomodulatory therapy resulted in greater than 90% reduction in seizure activity (Najjar et al. 2011). These reports suggest that microglial proliferation may profoundly impact prognosis following epilepsy. However, the mechanism underlying seizure-induced microglial proliferation remains unclear. Here, we investigated the functional implication and molecular mechanism underlying microglial proliferation in a mouse model of KA-induced seizures.
Materials and Methods:
Animals
Six- to twelve-week-old age-matched male mice were used in accordance with institutional guidelines, and all experiments were approved by the animal care and use committees at the Mayo Clinic, Rutgers University, and Anhui Medical University. C57BL/6J mice (wildtype, WT), CX3CR1GFP/+: CCR2RFP/+mice, and CX3CR1creER/+:Rosa26tdTomato/+ reporter mice were used in this study. C57BL/6J mice were purchased from Charles River Laboratories, Inc. CX3CR1GFP/+:CCR2RFP/+ mice were generated by crossbreeding CCR2RFP/RFP mice (Saederup et al. 2010) with CX3CR1GFP/GFP mice (Jung et al., 2000). CX3CR1creER/+:Rosa26tdTomato/+ reporter mice were generated by crossbreeding CX3CR1creER/creER mice with Rosa26tdTomato/tdTomato mice. All transgenic mice were purchased from Jackson Laboratories, Inc., except CX3CR1creER/creER mice (gift from Dr. Wen-Biao Gan at New York University). All transgenic mice were on a C57BL/6J background to avoid strain-related variations in KA susceptibility to seizures (McKhann et al. 2003) and showed no detectable developmental defects. Animals were housed in standard cages and maintained under controlled room temperature and humidity with 12/12-hour light-dark cycles.
Inducible expression of tdTomato in brain resident microglia
To selective label brain resident microglia, CX3CR1creER/+:Rosa26tdTomato/+ reporter mice were used. Tamoxifen (TM, Sigma) was dissolved in corn oil (Sigma) and administered intraperitoneally (i.p., 150 mg/kg, 20 mg/ml) into adult CX3CR1creER/+: Rosa26tdTomato/+ mice twice in 48 h intervals. TM induced the expression of tdTomato in resident microglial and infiltrating monocytes. Since infiltrated monocytes have a shorter lifespan due to rapid turnover, tdTomato-expressing monocytes are replaced by monocytes lacking tdTomato at 4 weeks following TM injection, while resident microglia still expressed tdTomato (Parkhurst et al. 2013; Peng et al. 2016). Thus, to distinguish infiltrated monocytes from resident microglia, KA-induced seizures were induced 4 weeks following TM induction.
KA-induced seizure model
The KA-induced seizure models were described previously (Eyo et al. 2015; Eyo et al. 2014). Briefly, mice were anesthetized with isoflurane, stereotaxically implanted with a 26-gauge stainless steel guide cannula aimed towards the lateral cerebral ventricle (from the bregma: −0.2 mm anteroposterior (AP), +0.95 mm mediolateral (ML), −2.0 mm dorsoventral (DV)), and then were intracerebroventricularly (i.c.v.) injected with KA at 0.15 μg in 5 μl of artificial cerebrospinal fluid (ACSF). Sham controls were injected with 5 μl of ACSF. Seizure behavior was monitored under a modified Racine scale as follows (Eyo et al. 2015; Eyo et al. 2014): (1) freezing behavior; (2) rigid posture with raised tail; (3) continuous head bobbing and forepaws shaking; (4) rearing, falling, and jumping; (5) continuous level 4; and (6) loss of posture and generalized convulsion activity. Mice that progressed to at least stage 3 were killed at different time points after seizure. The mice with seizure score more than 4 in the first one hour after KA injection was regarded as severe seizure, while mice with scores lesser than 4 were considered to be mild/moderate.
Acute brain slices preparation
Acute coronal brain slices containing the hippocampus were prepared from 6–8-week-old CX3CR1GFP/+ CCR2RFP/+ mice that received either KA or sham treatment. Briefly, mice were anesthetized and swiftly decapitated. Brains from decapitated mice were carefully removed and placed in ice-cold oxygenated (95% O2 and 5% CO2) ACSF with the following composition (in mM): NaCl, 124; NaHCO3, 25; KCl, 2.5; KH2PO4, 1; CaCl2, 2; MgSO4, 2; glucose, 10 and sucrose added to make 300–320 mOsmol. Coronal slices (300 μm) were prepared and transferred to a recovery chamber for 30 mins with oxygenated ACSF with the same composition as above at room temperature before two-photon imaging or electrophysiological studies.
Two-photon Imaging
Resident microglia (CX3CR1GFP/+) and infiltrated monocytes (CCR2RFP/+) were imaged using a two-photon microscope (Scientifica Inc, UK) with a Ti:Sapphire laser (Mai Tai; Spectra Physics) tuned to 1040 nm with a 40X water immersion lens (0.8 NA; Olympus). Fluorescence was detected using two photomultiplier tubes in whole-field detection mode and a 565 nm dichroic mirror with 525/50 nm (green /red channel) emission filters. Laser power was maintained at 25 mW or below. Typically, 15 consecutive z stack images were collected at 1.5 μm intervals every min. To perform a general laser injury, we focused the laser 66X and parked it at ~250 mW at 900 nm for 3 s. The lesion site was induced in a 15 × 15 pixel frame and the size of the resulting laser burn was estimated to vary between 8–15 μm (25–50 pixels) in diameter. For imaging experiments, a minimum of two to three slices from different mice from the same litter were randomly selected for imaging per treatment group/condition. Images were obtained between 50–100 μm from the slice surface. 20 μm thick sections were made from projection z-stack images taken at 1.5 μm intervals. Pixel size was 1024 × 1024 and field of view was 325 μm × 325 μm × 20 μm. For number of cells (per field of view), soma size (cross-sectional area), process number (per microglia) and process length analysis of microglia and monocytes in the processed image was manually evaluated from the maximum projected images using Image J. For responding process velocity toward laser-induced injury, time-lapse movies were first registered using the StackReg plugin to eliminate any x-y drift. For process velocity analysis, individual processes were then tracked using the Manual Tracking plugin. For number of responding processes, only process from cell soma located within 200 μm were analyzed. For peak process velocity, the maximum velocity of each process was averaged within each treatment group. Migrating processes were selected at random but only processes that were maintained through at least five frames were used. The average process velocity through the tracked period was determined and averaged from at least eight processes per experiment for 5–7 experiments.
Slice electrophysiology
Whole cell patch-clamp recordings were made on GFP+ microglia and RFP+ monocytes from cortical slices (~50 μm deep) at 1d after icv KA. Recording electrodes (4–5 MΩ) contained a K-based internal solution composed of (in mM): 120 K-gluconate, 5 NaCl, 1 MgCl2, 0.5 EGTA, 10 Na2Phosphocreatine, and 10 HEPES (pH 7.2; 280–300 mOsmol. The membrane potential was held at −60 mV for microglia/monocytes. Data were amplified and filtered at 2 kHz by a patch-clamp amplifier (Multiclamp 700B), digitalized (DIGIDATA 1440A), stored, and analyzed by pCLAMP (Molecular Devices, Union City, CA). The intrinsic membrane properties of microglia and monocytes are listed in Table 1. Data were discarded when the input resistance changed >20% during recording. The voltage ramp test was performed from −140 to 60 mV in 500 ms. For electrophysiology, a minimum of five cells from at least three different mice from the same litter were randomly selected for recording per condition.
Table1.
Intrinsic membrane properties of microglia and monocytes.
| Membrane capacitance Cm (pF) |
Membrane resistance (GΩ) |
Resting membrane potential (mV) |
|
|---|---|---|---|
|
Microglia GFP+ 1d PBS |
21.23 ± 2.72 | 1.47 ± 0.26 | −20.86 ± 1.54 |
|
Microglia GFP+ 1d KA |
31.75 ± 5.45 ** | 1.17 ± 0.11 | −32.69 ± 4.21** |
|
Monocytes RFP+ 1d KA |
17.97 ± 3.28 | 1.89 ± 1.05 | −15.46 ± 1.37 |
Compared with GFP+ 1d PBS microglia, p < 0.01
Loss of activity of CSF-1
For loss of function experiments, CSF-1 neutralized antibody (200 ng in 5 μl ACSF, R&D Catalog# AF416) or an isotype control antibody (Rat IgG) were injected into ventricles 2 h before KA injection and 2 h after KA injection and twice per day, for additional 3 consecutive days through a guide cannula (from the bregma: −0.2 mm anteroposterior (AP), +0.95 mm mediolateral (ML), −2.0 mm dorsoventral (DV)) (n=6). The effect of neutralizing CSF-1 was detected by Western blot with anti-CSF-1 antibody (1:500, R&D, Catalog# AF416). Inhibition of the tyrosine kinase activity of CSF-1R was achieved by the administration of GW2580 (Millipore, Catalog#344036), as previously described (Conway et al. 2005; Yan et al. 2017). GW2580 (200 μM in 5 μl ACSF) was injected 2 h before KA injection and 8 h after KA injection and once per day for an additional 3 consecutive days through a guide cannula placed in the lateral ventricles and comparing to vehicle control mice. The animals assigned for seizure scoring were assigned randomly for each treatment group.
Fluoro-Jade B staining
Fluoro-Jade B (FJB) is an anionic fluorochrome capable of selectively staining neurons with compromised membrane integrity. Briefly, tissue sections were air-dried, and then immersed in 0.06% potassium permanganate solution for 10 mins. Slides were washed briefly in ddH2O and then placed in a solution of 0.001% FJB dye dissolved in 0.1% acetic acid for 10 mins, followed by washing in ddH2O, drying, clearing with xylene, and mounting with a cover slip. To quantify FJB-positive cells, three sections per mouse were selected. The numbers of FJB-positive cells from 9 locations per mouse (3 fields per section × 3 sections per mouse) were averaged and results are reported as the number of FJB-positive neurons per field of view and expressed as mean ± SEM.
Fluorescent immunostaining
Mice were deeply anaesthetized with isoflurane (5% in O2) and perfused with 20 ml PBS followed by 20 ml of pre-cold 4% paraformaldehyde (PFA) in PBS containing 1.5% picric acid. The brains were removed and post-fixed with the same 4% PFA for 6–8 h at 4 oC. The samples were then transferred to 30% sucrose in PBS for 48 h. Sample sections (15 μm in thickness) were prepared on gelatin-coated glass slide with a cryostat (Leica). The sections were blocked with 5% goat or 5% donkey serum and 0.3% TritonX-100 (Sigma) in TBS for 60 mins, and then incubated overnight at 4 oC with primary antibody for rabbit-anti-Iba1 (1:500, Wako Chemicals, Catalog# 019–19741), goat-anti-Iba1 (1:200, Abcam, Catalog# ab107159), rat-anti-CD11b (1:500, Biolegend, Catalog# 101202), Rat anti-CD169 (1:400, AbD serotec, Catalog# MCA884), rabbit anti-GFAP (1:500, Chemicon, Catalog# AB5804), rabbit anti-NeuN (1:100, Abcam, Catalog# ab177487), rabbit-anti-Ki67 (1:500, Abcam, Catalog# ab16667), rabbit anti-BrdU (1:500, Sigma, Catalog# B8434), or rabbit anti-cleaved casapase-3 (1:300, Cell signaling, Catalog# 9664). The sections were then incubated for 90 mins at room temperature, with corresponding secondary antibodies (1:500, Life Technologies). The sections were mounted with fluoromount-G (Southern Biotech) and images were collected with a fluorescent microscope and confocal microscope (LSM510, Zeiss). Cell counting and fluorescent signal intensity was quantified using Image J. Quantitative measurement of protein-protein immunofluorescent co-localization was performed in Image J with “Co-localization Finder” plugin.
Nissl staining
For Nissl staining, hippocampal tissue sections were mounted and stained with 0.5% cresyl violet. The number of CA1 and CA3 pyramidal neurons in a defined area (1.0 mm × 0.25 mm) was counted in at least three sections per mouse brain. All assessments of histological sections were blindly performed.
BrdU for microglia proliferation
The thymidine analog bromodeoxyuridine (BrdU) (Sigma) was used to label proliferating and recently post-mitotic cells in the seizure mice brain. The BrdU solution was diluted in 1M PBS just before use and intraperitoneally (i.p. 100 mg/kg) injected twice per day for different periods after KA injection. Tissue sections were pretreated with 50% formamide in 2 × standard saline citrate (SSC) for 2 h at 65 °C, followed in 2 × SSC (15 mins, RT), 2N HCl (30 mins, 37 °C), 0.1 M borate buffer (10 mins, RT) and rinsed in Tris-buffered saline (TBS, pH 7.3) for three times at room temperature. After then, BrdU immunostaining was performed as described in our previous studies (Gu et al. 2016b).
Western blot
Hippocampus samples were extracted and homogenized in lysis buffer containing protease and phosphatase inhibitors, and the homogenates were centrifuged at 7000 g for 15 mins at 4°C as described previously (Wu et al. 2012). The supernatants were collected and the protein concentration was determined by Bradford method, and 30 μg of proteins were loaded for each lane and separated by SDS-PAGE. After the transfer, the blots were incubated overnight at 4°C with one of the primary antibodies listed below: CSF-1 (1:500, R&D, AF416), β-actin (1:5000, rabbit; Santa Cruz). These blots were then washed, incubated with HRP-conjugated secondary antibody and developed in an enhanced chemiluminescent solution. Specific bands were evaluated by apparent molecular size. Finally, images were captured using a Fuji-Film LAS-300 (Fuji, Sheffield, UK) and the intensity of the selected bands was analyzed using Image J software.
Statistical Analysis
All data are expressed as mean ± SEM. Quantification of cells was done with Image J software (NIH Image). Changes of values of each experimental group were tested using Student’s t-test or one way ANOVA, followed by post-hoc Tukey’s test to establish significance. Differences were considered significant for p < 0.05.
Results
Increased neuronal death and microgliosis after KA induced epilepsy.
Neuronal death and microgliosis have been noted in the hippocampus in both human patients and experimental animal models of epilepsy (Vezzani 2014). In order to characterize seizure-induced microgliosis, we performed i.c.v. injection of KA to induce both mild and severe seizures in mice (Figure 1A). A Racine seizure score greater than 4 in the first one hour after KA injection was considered to be a severe seizure. Not surprisingly, we found the degree of neuronal damage determined by FJB staining after KA injection was dependent on the severity of seizures (Figure 1B–C). For instance, both CA3 and CA1 areas show dramatic FJB staining in severe seizures while only CA3 area of the hippocampus shows the cell death in mild seizure group. In addition, we found that increased neuronal loss was accompanied by increased number of Iba1+ cells, defined as microgliosis, after severe seizures (Figure 1D, Supplementary Figure 1A–B). The number of Iba1+ cells increased dramatically following seizure, peaked at 3d, and returned to control levels by 14d after i.c.v. KA (Figure 1D–E, Supplementary Figure 1A–B). These results indicate that the degree of seizure is associated with the level of microgliosis and microglial activation. In this study, we focus on severe seizures in order to examine the source and significance of microgliosis in both the CA1 and CA3 following seizures.
Figure 1. Increased neuronal cell death and Iba1+ cell population in the hippocampus following KA-induced seizures.
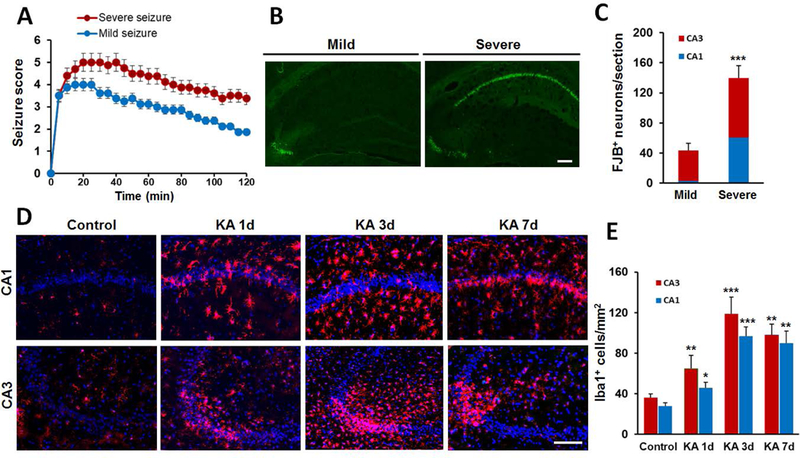
A, Following KA (i.c.v, 0.15 μg in 5 μl ACSF) injection, seizure scores were used to identify the extent of mild and severe seizure. B, Representative images of FJB staining from mild and severe seizure mice at 3d post i.c.v. KA. C, Quantitative data showing neuronal injury in mild and severe seizure. D, Increased Iba1+ cells in the hippocampus at different times after severe seizure. E, Quantitative data of D showing the number of Iba1+ cells in the hippocampus. Data are presented as mean ± S.E.M. n = 6 mice for each group. *p<0.05, **p<0.01, ***p<0.001, versus control. Scale bar = 100 μm.
Heterogeneous Iba1+ population is made up of resident microglia and infiltrated monocytes.
Next, we investigated the composition of microgliosis, which is the expanded Iba1+ population after seizures. Using the CX3CR1GFP/+ CCR2RFP/+ genetic mouse model, we were able to distinguish activated resident microglia (GFP+) from the infiltrating monocytes (RFP+) in the hippocampus at 1d, 3d, and 7d after i.c.v. KA (Figure 2A–B). In the CA1 and CA3 regions, the number of GFP+ microglia increased significantly after seizures and reached a peak at 7d after i.c.v. KA (Figure 2A–B). We observed that the infiltration of RFP+ monocytes was also increased in both the CA1 and CA3 after severe seizures with peak infiltration 3d after KA (Figure 2A–B). Under conditions of mild seizures, monocyte infiltration was only observed in the CA3 but not CA1 hippocampal region (Data not shown). Monocyte infiltration after seizures was further confirmed using CD169 staining, a marker to distinguish the infiltrating monocytes from resident microglia (Gordon et al. 2014; Gu et al. 2016b). We note that CD169 (red) marked a subset of Iba1+ cells (green) corresponding to the monocyte population that peaked at 1d-3d following KA and declined by 7d (Figure 2C–D). Taken together, our results indicate that both infiltrated monocytes and activated resident microglia contribute to the expanded microglial Iba1+ population, namely microgliosis, following severe KA-induced seizures.
Figure 2: Iba1+ population is heterogeneous and is made up of activated resident microglia and infiltrated monocytes.
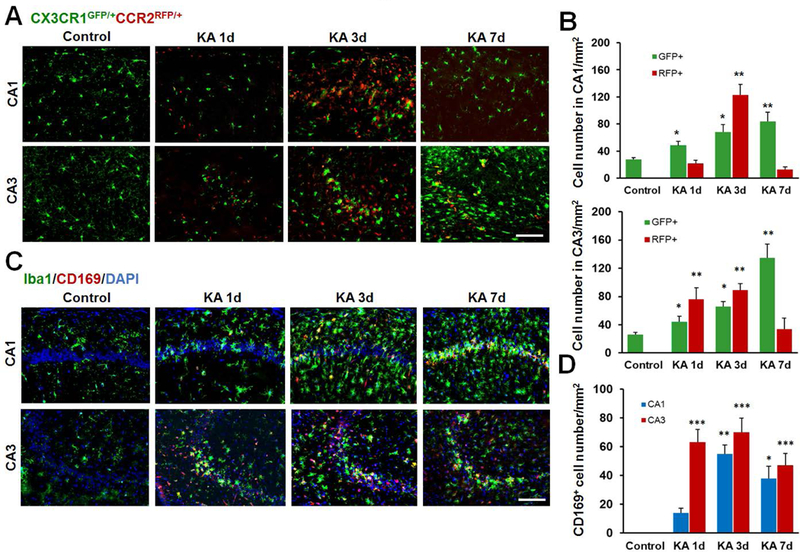
A, Representative confocal images of the hippocampus at different time points following KA-induced seizures in double transgenic CX3CR1GFP/+:CCR2RFP/+ mice. Resident GFP+ microglia (CX3CR1GFP/+) are in green and infiltrated RFP+ monocytes (CCR2RFP/+) are in red. Note the obvious appearance of CCR2RFP/+ cells in the hippocampus at day 3 after KA injection. B, Quantitative data showing increased GFP+ and RFP+ cells in the hippocampus CA1 and CA3 following KA injection. C, Representative images showing that the Iba1+ cell population is made up of two distinct sub-populations-CD169 (red) positive infiltrated monocytes and Iba1 (green) positive cells 1d, 3d, and 7d following KA i.c.v. injection. D, Summarized data depicting the number of CD169+ cells in the hippocampus CA1 and CA3 at different time points following i.c.v. KA. n = 5 mice for each group. *p<0.05, **p<0.01, ***p<0.001, versus control. Scale bar = 100 μm.
Activated resident microglia and infiltrated monocytes display distinct morphological and electrophysiological properties.
We next characterized the morphological and electrophysiological properties of GFP+ microglia and RFP+ monocytes in CX3CR1GFP/+ CCR2RFP/+ mice at 1d, 3d, and 7d after i.c.v. KA using two-photon microscopy and whole-cell patch clamp recordings in live hippocampal slices. We noted that the seizure-induced activated resident microglia were enlarged with stationery soma and fewer ramified dynamic processes, whereas, infiltrated monocytes had smaller somas lacking processes (Figure 3A–B). The soma size of microglia increased dramatically, reaching a maximum at 1d and returned to normal levels at 3d and 7d following i.c.v. KA (Figure 3B). However, the number of microglial processes and the average process length remained significantly reduced at 1d, 3d, and 7d following KA, suggesting that the microglia are still in a state of mild activation (Figure 3B). Interestingly, we also identified the emergence of a new population of GFP+:RFP+ cells that shared characteristics of both microglia and monocytes (i.e., smaller soma size like monocytes, but processes like microglia). This data suggests that a sub-population of cells may evolve into CX3CR1GFP/+:CCR2RFP/+ expressing cells with morphological behavior indicative of a transitionary state at later time points (3d and 7d) following seizures (Figure 3A–B).
Figure 3. Characterization of morphological and electrophysiological properties of resident microglia and infiltrated monocytes following KA-induced seizures in CA3.
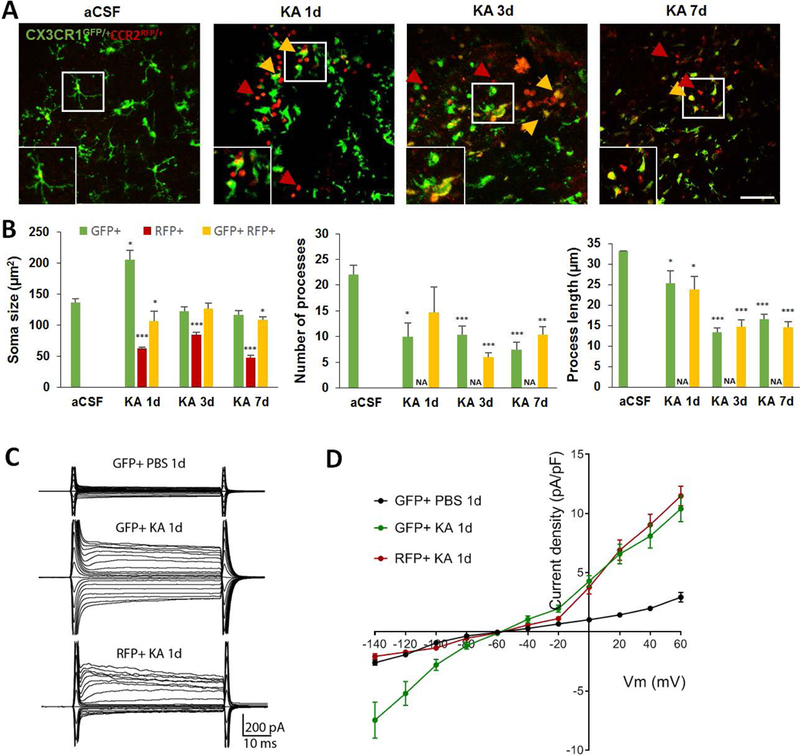
A, Representative two-photon images showing resident CX3CR1GFP/+ microglia, CCR2RFP/+ monocytes (red arrow) and double positive CX3CR1GFP/+:CCR2RFP/+ (yellow arrow) cells in the CA3 1d, 3d, and 7d following i.c.v. KA. Inset shows higher magnification image of the area highlighted in the white box. Scale bar = 50 μm. B, Summarized data showing soma size (μm2), number of processes and process length of GFP only, RFP only, and GFP+:RFP+ cells at different time points after KA-induced seizures. Data are presented as mean ± S.E.M. n = 3 mice per each group. *p<0.05, **p<0.01, ***p<0.001, versus GFP+ cells in aCSF condition. Since RFP+ cells lacked processes, the comparisons of process number and length was not applicable (NA). C, Membrane currents of CX3CR1GFP/+ microglia and CCR2RFP/+ monocytes at 1d after KA. D, Summarized plot of current density (pA/pF) versus voltage (mV) of CX3CR1GFP/+ microglia and CCR2RFP/+ monocytes in CA3 hippocampus at 1d after KA-induced seizures.
Next, we compared the electrophysiological properties of resident microglia and infiltrated monocytes after seizures. To this end, we performed whole-cell patch clamp recordings in GFP+ and RFP+ cells from hippocampal slices of mice. A minimum of five cells from at least three different mice were randomly selected for recording per condition. The resting microglia was recorded from 1d PBS injected mice. The GFP+ activated microglia and RFP+ monocytes were recorded from the CA3 at 1d after KA. We found that resting resident microglia exhibited small currents with linear current-voltage relationship at resting states, whereas there are more outward and inward K currents in activated microglia (Figure 3C–D). Interestingly, infiltrating monocytes show mostly outward K currents (Figure 3C–D). These results along with the intrinsic membrane properties (Table 1) indicate that the two cell sources of seizure-induced microgliosis are unique in their electrophysiological properties. Moreover, the significant increase in cell capacitance and hyperpolarized resting membrane potential of activated (i.e., GFP+ 1d KA microglia) versus the resting microglia (i.e., GFP+ 1d PBS microglia) is further indicative of an increase to soma size and microglia activation (Table 1).
Dynamic properties of activated resident microglia and infiltrated monocytes.
In addition to their morphological and electrophysiological properties, we also delineated the dynamic differences between the resident microglia and infiltrated monocytes after KA induced seizures. We found that both GFP+ only and GFP+:RFP+ populations responded to laserburn injury, with their cellular processes moving towards the injured site (Figure 4A, Supplementary Movie 1). However, the RFP+ only population was indifferent to laser-induced injury and did not show any directed movement towards or away from the injury location (Figure 4A). Hence, we first characterized the basal dynamic behavior of infiltrated monocytes in hippocampal slices after KA-induced seizure. Such characterization of monocyte behavior in infiltrated tissue has not been reported previously. Interestingly, in contrast to the stationary cell body of resident microglia, the infiltrated monocytes showed amoeboid movement, which we characterized in terms of velocity and distance travelled in the neuronal parenchyma. The average velocity of monocytes peaked at 1d and decreased at 3d and 7d after seizures (Figure 4B). However, the distance travelled by the monocytes in the monitored field of view during the observation period of 30 mins was maximum at 3d after KA-induced seizures (Figure 4B). This suggests that the infiltrated monocytes possess migratory properties that are more prominent at early time points following infiltration and reduce as they spend more time in the neuronal milieu.
Figure 4. Characterization of dynamic properties of resident microglia and infiltrated monocytes following KA-induced seizures in CA3.
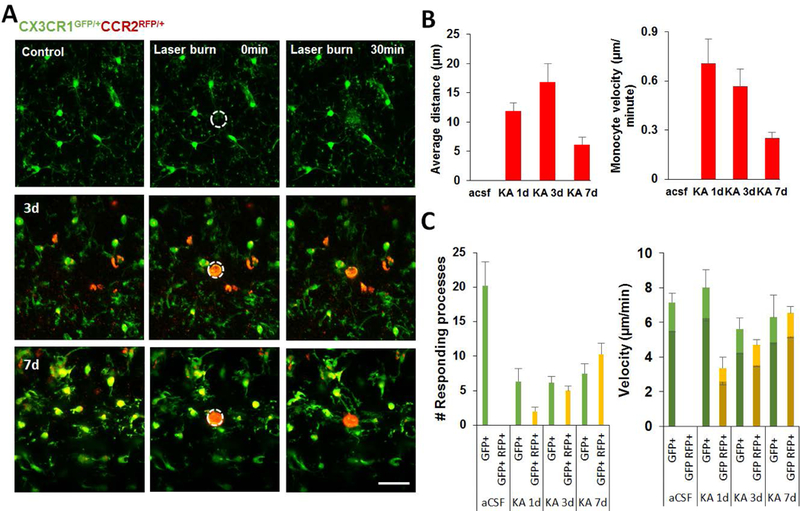
A, Representative images showing the response of activated resident microglia and infiltrated monocytes to laser burn injury at 1d, 3d, and 7d following i.c.v. KA. Scale bar = 50 μm. B, Summarized data showing characteristics of infiltrated monocyte motility such as mean distance travelled per min and average velocity in the CA3 region following KA-induced infiltration. C. Bar graphs showing number of responding processes towards laser burn and the microglial process velocity. The velocities of GFP+ cells (green) and GFP+:RFP+ cells are denoted as light green/yellow for peak velocity and dark green/yellow for average velocity.
In response to laser burn, we identified that the number of responding processes drastically reduced in the CA3 region at 1d, 3d, and 7d after KA administration (Figure 4C, Supplementary Movie 1). Moreover, GFP+ only microglia possessed faster response to laser injury at 1d after seizures but both peak and average process response velocity declined at later time points (Figure 4C). In contrast, the GFP+:RFP+ transitioning cells, showed significantly slower response velocity at 1d, but progressively increased 3d and 7d after seizures (Figure 4C).
Microglial proliferation contributes to expansion of the Iba1+ population.
While cell migration and proliferation may contribute to the increased number of resident microglia specifically, the source of the expanded population of total Iba1+ cells after seizures remains unclear. Hence, we investigated the contribution of proliferation via Ki67 and BrdU, known measures of cellular proliferation. We observed a significant amount of Ki67 staining co-localized with Iba1+ positive cells in the hippocampus at 1d, 2d, 3d, and 7d after KA seizures (Figure 5A–C). This result was further confirmed using BrdU (Figure 5D–E). We next used several strategies to identify whether resident microglia or infiltrated monocytes were proliferating. First, we determined that only GFP+ but not RFP+ cells were positive for Ki67 in CX3CR1GFP/+:CCR2RFP/+ mice (Figure 6A). We quantified the microglial proliferation and found that ~59.2% and ~45.1% of microglia were Ki67+:GFP+ in the CA1 and CA3, respectively at 3d after KA (Figure 6C). Secondly, we performed CD11b staining in CX3CR1creER/+: R26tdTomato/+ reporter mice, in which resident microglia are labeled by both CD11b and Tdtomato while infiltrated monocytes are CD11b+:Tdtomato-. Consistently, we found that Ki67 was localized only in CD11b+:Tdtomato+ resident microglia, but not in CD11b+:Tdtomato- monocytes (Figure 6B, D). We also determined that ~72.7% and 74.3% of Tdtomato+ microglial cells contained Ki67 at 3d following seizures in the CA1 and CA3, respectively (Figure 6D). Lastly, we found no co-labeling of CD169, a known monocyte marker, with Ki67 (Figure 6E). Taken together, these data suggest that only resident microglia and not infiltrating monocytes proliferate following seizures in the hippocampus. Among proliferating cells, we noted that astroglial proliferation occurred as early as 1d after KA seizure (Supplementary Figure 2), although astrocyte proliferation was not as prominent as microglia. These results suggest that local proliferation of resident microglia is the predominant source of microgliosis after status epilepticus.
Figure 5. Microglia, but not monocytes, contribute to seizure-induced increase to the proliferating Iba1+ population.
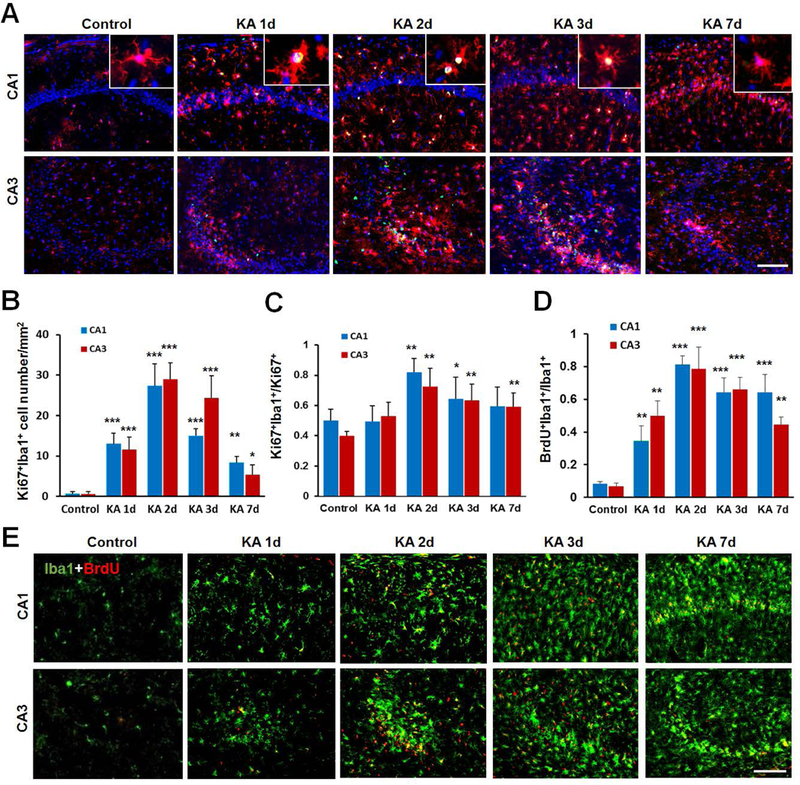
A, Representative images of Ki67 (green) and Iba1 (red) immunostaining in the hippocampus CA1 and CA3 at different time points after KA injection. The enlarged panel shows co-localization of Ki67 immunostaining with Iba1+ microglia. B, The time course of Ki67 and Iba1 co-expressing cells, which peaks at day 2 after KA injection. n = 5 mice for each group. C, Shown is the percentage of Ki67+:Iba1+ cells among total Ki67+ cells at different time points after KA injection. n = 5 mice for each time point. D, E, Representative images and quantification showing co-localization of BrdU (red) and Iba1 (green) in the hippocampus at different time following KA injection. BrdU (i.p., 100 mg/kg, 2 pulses/day) was applied immediately after KA treatment. n =5 mice for each time point. *p<0.05, **p<0.01, ***p<0.001, versus control. Scale bar = 100 μm.
Figure 6. Microglial proliferation predominates seizure induced microgliosis.
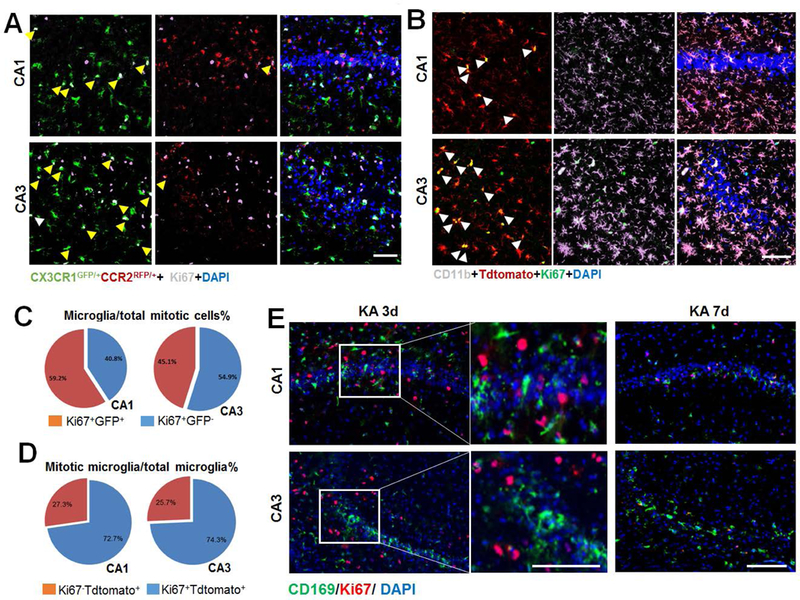
A, Representative images of Ki67 immunostaining in the hippocampus at 3d after KA injection in double transgenic CX3CR1GFP/+:CCR2RFP/+ mice. Arrows denote co-localization of Ki67 (white) immunostaining with GFP+ resident microglia. Note, no Ki67 immunostaining co-localized with RFP+:GFP- infiltrated monocyte. Scale bar = 50 μm. B, Representative images of the hippocampus showing the co-localization of Ki67 (green) and tdTomato+ (red)(white arrow) at day 2 following KA treatment in CX3CR1creER/+:R26tdTomato/+ reporter mice. Note, tdTomato-CD11b+ cells do not express Ki76. Scale bar = 50 μm. C, The percentage of Ki67+:GFP+ cells among total Ki67+ cells per mm2 at KA 3d in CX3CR1GFP/+:CCR2RFP/+ mice. n = 4. D, The percentage of Ki67+:tdTomato+ cells among total tdTomato+ cells per mm2 at KA 2d in CX3CR1creER/+:R26tdTomato/+ reporter mice. n= 4. E, Representative images of Ki67 (red) and CD169 (green) immunostaining in the hippocampus at 3d and 7d after KA treatment in WT mice. n = 5 mice for each time point. Scale bar = 100 μm.
Microglial proliferation is CSF-1R dependent.
We next investigated the molecular pathway underlying microglial proliferation in the hippocampus and its implication in KA-induced seizures. To this end, we tested colony stimulating factor-1 receptor (CSF-1R), a known regulator of microglial survival, as a potential target for reducing microglial proliferation (Elmore et al. 2014). Consistent with the role of CSF-1 pathway in the acute responses of microglia, we found that the protein expression of CSF-1 was significantly increased at 1d but not at 3d following KA-induced seizures (Figure 7A,B). To block the CSF-1 pathway, we treated mice with either anti-CSF-1 antibody (CSF-1Ab) or CSF-1R antagonist GW2580 and measured the outcomes at different time points after KA treatment (Figure 7C). Anti-CSF-1 antibody, but not corresponding control IgG, was able to largely reduce CSF-1 upregulation in the hippocampus at 1d after KA seizure, confirming that anti-CSF-1 antibody is an effective way to neutralize CSF-1 (Figure 7D,E).
Figure 7. Microglial proliferation is CSF-1 Receptor dependent.
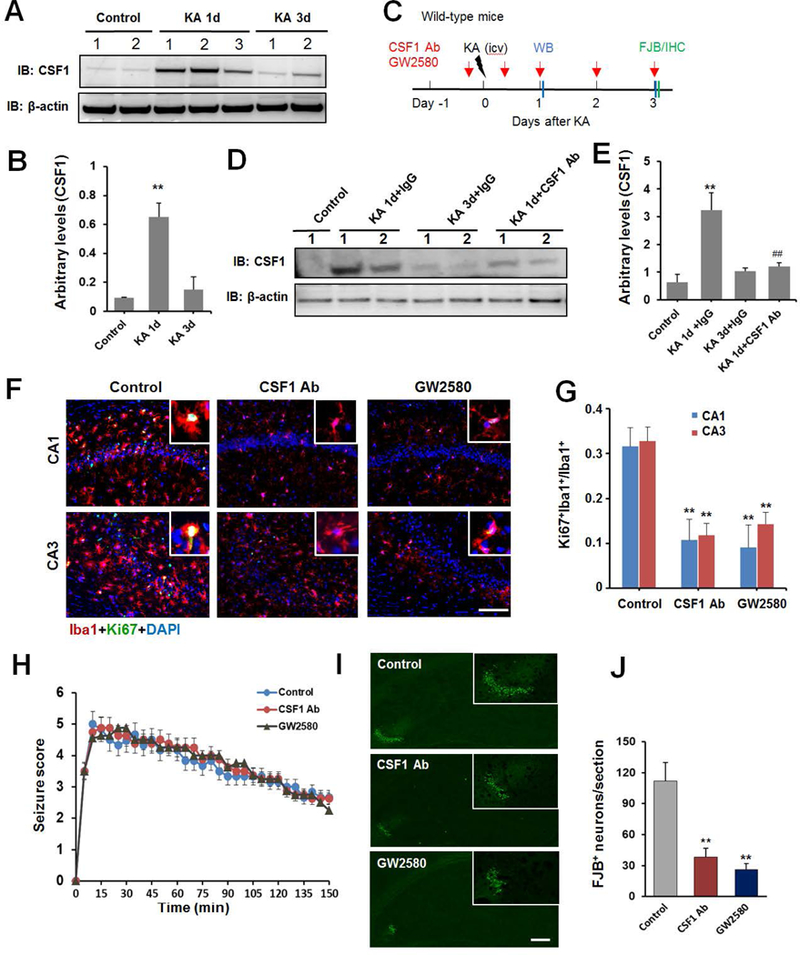
A, Western blot assay shows expression of CSF-1 in the hippocampus of mice in control, 1d and 3d after i.c.v. KA and the corresponding β-actin loading control. B, Summarized data showing increase in CSF-1 expression at 1d, but not 3d, after i.c.v. KA. n = 5 mice for each group. C. An experimental diagram showing the timeline of drug treatments, KA injection, immunostaining, and Western blot assay. D, E CSF-1 neutralizing antibody (Ab) reduces CSF-1 expression in Western blot assay. n = 5 mice for each group. F, Representative images of Ki67 (green) with Iba1 (red) immunostaining in hippocampus at day 3 after KA injection in CSF-1Ab or GW2580 treated mice. G, Quantitative data is shown the ratio of Ki67+:Iba1+ to total Iba1+ cells per mm2 in the hippocampus from control, CSF-1Ab or GW2580 treated seizure mice. n=5 for each group. H, Seizure scores following KA injection in the presence or absence of CSF-1Ab or CSF-1R inhibitor GW2580. n=8 for each group. I, Representative images of FJB staining at day 3 after KA delivery with or without CSF-1 neutralizing Ab or GW2580 treatment. J, Quantitative data showing CSF-1Ab or GW2580 treatment reduces seizure-induced neuronal injury. n =5 mice for each group. **p<0.01, versus control. #p<0.05 versus IgG control at 1d after icv KA. Scale bar = 100 μm.
To test CSF-1 signaling in KA-induced microglial proliferation, we evaluated the Ki67 staining after the treatment of CSF-1 antibody or GW5280 at 1d after KA seizure. We were able to confirm that microglial proliferation at 3d following KA was dramatically reduced by treatment with either CSF-1Ab or GW2580 (Figure 7F–G). In addition to affecting proliferation, treatment with GW2580 affected microglia morphology, with reduced primary microglial branch numbers and microglia soma size in GW2580-treated mice compared to untreated mice after KA treatment (Supplemental Figure 3A–B). Interestingly, we noted that monocyte (CD169+ cells) infiltration was also significantly reduced in GW2580-treated mice following KA-induced seizures (Supplemental Figure 3C–D). Moreover, we ensured that the concentration of the drug GW2580 and CSF-1 antibody was only sufficient to reduce proliferation but not affect microglial survival (Figure 7F–G). Indeed, we did not observe microglia apoptosis with cleaved caspase-3 staining after CSF-1Ab treatment (Supplementary Figure 4A), and we confirmed that GW2580 and CSF-1Ab reduce microglial proliferation via BrdU labeling (Supplementary Figure 4B). Additionally, although the seizures score remained unaffected after CSF-1Ab or GW2580 treatment (Figure 7H), FJB staining found that seizure-induced neuronal cell death was significantly reduced in experimental groups treated with CSF-1Ab and GW2580 (Figure 7I,J). Using Nissl staining, our results further confirmed that the hippocampus structure was preserved to a certain extent following treatment with CSF-1Ab and GW2580 (Supplementary Figure 5). Taken together, these results indicate that blocking microglial proliferation did not alter acute seizure behaviors, but is sufficient to reduce neuronal death following KA-induced seizures.
Discussion
The present study shows that both activated resident microglia and infiltrated monocytes contribute to the expanded microgliosis population in the hippocampus after KA-induced seizures. Notably, microglia and monocytes possess very distinct morphological, dynamic, and electrophysiological properties. We further show that microglia but not monocytes proliferate in the hippocampus following seizures and blocking microglial proliferation can reduce seizure-induced neuronal loss (Figure 8).
Figure 8. Seizure induced microgliosis in the hippocampus.
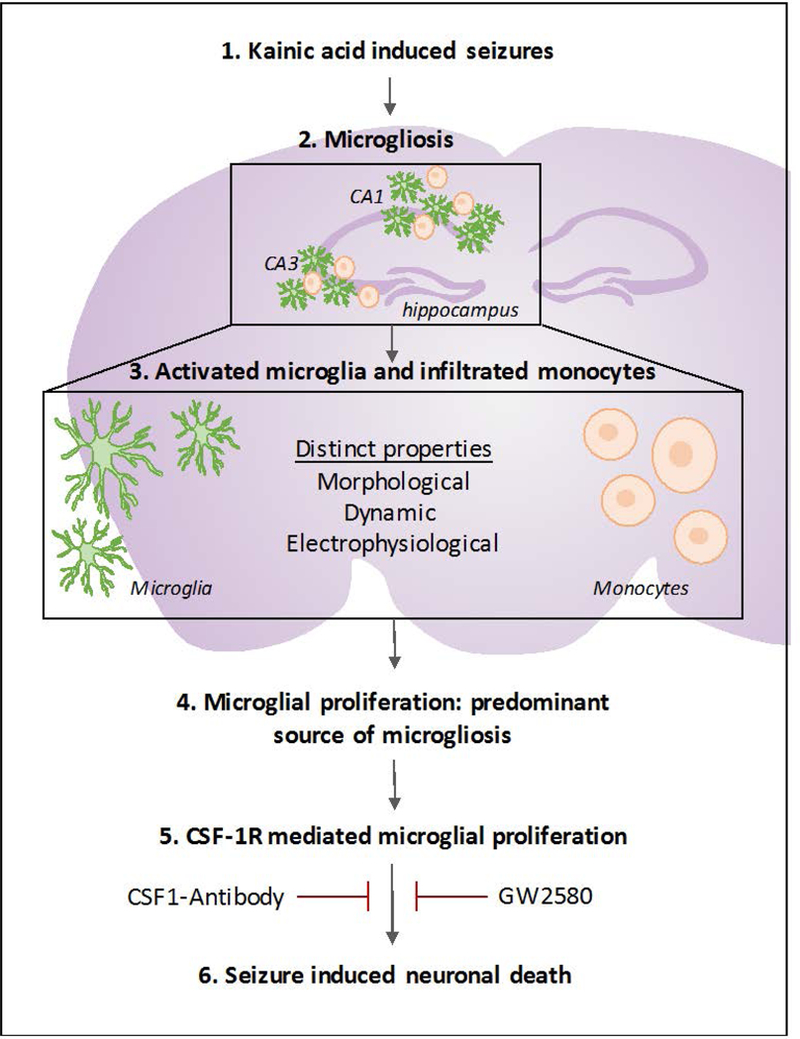
Schematic showing seizure induced microgliosis is comprised of morphologically and physiologically distinct activated resident microglia and infiltrated monocytes. Moreover, it illustrates that microglia, and not monocytes, proliferate in the hippocampus following seizures and that blocking microglial proliferation via antagonism of the colony stimulating factor 1 receptor may help reduce seizure-induced neuronal loss.
Microgliosis is a common pathological feature following status epilepticus. Research studies have shown a positive correlation between increased microglial numbers and neuronal death and seizure severity (Avignone et al. 2008). Here, we found that the degree of microgliosis occurring within the CA1 and CA3 regions of the hippocampus corresponded to the level of neuronal loss and seizure severity. In our previous study, we showed microglial activation was localized to the CA3 region following moderate seizures (Tian et al. 2017). This is consistent with the notion that the degree of microgliosis directly correlated with seizure activity (Kim et al. 2015). However, the source and heterogeneity of the seizure-induced expansion in microglial populations still remains unclear and the lack of proper technical tools has made this question controversial (Ajami et al. 2007; Mildner et al. 2011). Here, using a transgenic mouse model, we show that the observed seizure-induced expansion of microglial population is actually comprised of both local microglial proliferation and monocytes infiltration. This is in line with our recent study as well as another study showing the infiltration of monocytes following chemically-induced epilepsy pathogenesis (Tian et al. 2017; Varvel et al. 2016; Zattoni et al. 2011). The infiltration of monocytes was shown to be dependent on the CCL2-CCR2 chemokine signaling pathway and reduced infiltration in CCR2 knockout mice corresponded to reduced neuronal loss, reduced neuroinflammation, and better functional recovery (Tian et al. 2017; Varvel et al. 2016). This suggests that in addition to activating resident microglia, infiltrated monocytes also could contribute significantly to pathological progression of epilepsy.
The diversity of the CNS-resident microglia and infiltrated monocytes, has largely been overlooked. Only recently have their differential roles in various pathological conditions been acknowledged (London et al. 2013). Following ischemic injury, resident microglia were found to be pro-inflammatory, whereas, infiltrated monocytes predominantly contributed to phagocytosis (Ritzel et al., 2015). Similarly, infiltrated monocytes were noted to possess distinct roles for amyloid clearance in Alzheimer’s disease (Mildner et al. 2011) and demyelination in multiple sclerosis (Yamasaki et al. 2014). In this study, we characterized the properties of microglia and monocytes following seizures, in order to better understand and manipulate them for therapeutic purposes. Seizure-induced activation of microglia was, expectedly, characterized by time-dependent changes in the morphological state including increase in microglial cell number, increased soma size and reduction in number of processes/process length (Avignone et al. 2008; Eyo et al. 2017). However, we found that infiltrated monocytes after status epilepticus lacked cellular processes and were highly motile cells when compared to the relatively stationary microglial (Eyo et al. 2018b). Interestingly, microglia and monocytes showed distinct electrophysiological properties as well. In the resting state, microglia express a unique membrane channel pattern consisting of a an inward rectifying Kir and outward rectifying KV channel components (Kettenmann et al. 1990). However, following seizures, we observed an additional outward delayed rectifying K+ conductance, similar to the upregulated Kv1.3 currents seen in lipopolysaccharide treated microglia (Nguyen et al. 2017), as well as in microglia activated after kainate-induced status epilepticus (Menteyne et al. 2009). Interestingly, the infiltrated monocytes also showed a strong outward K current following seizure indicative of activation. This is in line with a recent report showing increased K currents in human monocytes following treatment with lipopolysaccharide (Kim et al. 2015). Further, biophysical and pharmacological characterization confirm the involvement of potassium channels containing Kv1.3 subunits in activated microglia following status epilepticus (Menteyne et al. 2009). Although microglia and monocytes express a repertoire of potassium channels, the Kv1.3 channel is of particular interest because it is highly expressed in activated microglia (Rangaraju et al. 2015) and implicated in proliferation, activation, migration as well as cytokine release (Charolidi et al. 2015; Kotecha and Schlichter 1999; Vicente et al. 2003). The increase in Kv1.3-like currents in microglia and monocytes following KA administration is suggestive of activation. However, the role of the K channel in seizure-induced microglial proliferation and monocyte infiltration needs to be investigated.
We also characterized the dynamic properties of the resident microglia and infiltrated monocytes. We noted a reduction in the basal velocity of microglial processes towards laser injury at 1d after KA, but returned to control levels at later time points. This is in contrast with the elevated purinergic receptor-mediated directional velocities noted in activated microglia after status epilepticus (Avignone et al. 2015). This discrepancy may be due to the differential response of microglia in CA1 (Avignone et al. 2015) versus CA3 (our investigation) regions of the hippocampus after KA-induced seizures. Consistently, we noted regional difference of microglial response to NMDA-induced neuronal hyperactivities in mouse hippocampus (Eyo et al. 2018a). In addition, we noted an increase in expression of P2Y12 in the CA1, whereas the expression was down-regulated in the CA3 after kainic acid induced seizures (data not shown). We also found that infiltrated monocytes did not respond to laser injury. This likely due to monocytes lacking purinergic receptors P2Y12 expression (Gu et al. 2016a; Hickman et al. 2013; Sasaki et al. 2003). Surprisingly, our study identified the emergence of a new population of cells that resembled the conversion of infiltrated monocytes into resident microglia at late time points after KA-indued seizures. We noted that the CX3CR1GFP/+:CCR2RFP/+ transitioning monocytes evolved more microglia-like characteristics and eventually responded to laser injury. Similar transitions were observed in brain engrafts where circulating monocytes differentiated into microglia following bacterial infection (Djukic et al. 2006). Depending on stimuli, monocytes have been shown to differentiate into perivascular macrophages, microglia, or even dendritic cells based on their surface marker expression (Hohsfield and Humpel 2015; Krutzik et al. 2005; Schmidtmayer et al. 1994; Ziegler-Heitbrock 2007). It is unclear whether the transitioned monocytes remain in the neuronal parenchyma as permanent resident microglia or have shorter life spans. Since, infiltrated monocytes have shown to assist microglia in phagocytosis, it would be of clinical relevance to investigate their fate in the milieu following infiltration. Alternatively, a subpopulation of peripheral monocytes is known to express CX3CR1 receptor (Auffray et al. 2007; Jung et al. 2000) and may be part of the infiltrating monocyte population following seizures. In the current scenario, it is unclear whether the monocytes are transitioning into microglia or microglia under certain conditions may start expressing CCR2 receptors and thus the nature of such transformation needs to be clarified.
Our study further showed that seizure-induced microgliosis is driven primarily through local proliferation of resident microglia in the hippocampus. The correlation between excitotoxicity and microgliosis make it plausible to assume that increased neuronal excitability might be the major trigger for microglial proliferation following seizures (Zhu et al. 2010). Recent studies suggest that colony-stimulating factor 1 (CSF-1) is critical for microglial proliferation under physiological conditions (Elmore et al., 2014) and microglia solely express CSF-1R in the normal brain (Erblich et al. 2011; Sierra et al. 2007). In addition, CSF-1R is reported to be major driver for microglial proliferation in the spinal dorsal horn after peripheral nerve injury (Gu et al. 2016b; Guan et al. 2016). Although microglial activation and proliferation have been reported following epilepsy, the underlying molecular mechanisms have remained unclear. A previous report showed that toll-like receptor 2 signaling was important for seizure-induced microglial proliferation (Hong et al. 2010). Here, we show that CSF-1R signaling affected seizure-induced changes in microglia morphology, microglia proliferation, monocyte infiltration and resultant neuronal death. Interestingly, our results suggest that microglial proliferation may promote monocyte infiltration after seizures. In line with this idea, our previous results have demonstrated that CCL2 is upregulated activated microglia (Tian et al. 2017). Although, blockage of CSF-1R prevented neuronal death and preserved hippocampal structure, the direct effect on neuronal activity and hippocampus function needs to be investigated. Microglial proliferation has been associated with seizure activity and immunomodulation has been shown to improve seizure outcomes (Najjar et al. 2011). Therefore, targeting microglial proliferation, specifically CSF-1R signaling, might be a good therapeutic strategy for the treatment of patients with epilepsy. The current study also furthers our understanding of the unique properties of microglia and monocyte which can be exploited in order to develop novel therapeutic solutions for epileptic patients.
Supplementary Material
Time-lapse video from two-photon images showing response of GFP+ only and GFP+:RFP+ cells towards laser burn and lack of response by RFP+ only cells at 7d after KA and corresponding control.
Supplementary Figure 1. Changes in morphology and cell number of Iba1+ cells following KA-induced seizures.
A, Representative images of Iba1-labled hippocampal microglia from control and KA (i.c.v., 0.15 μg in 5 μl ACSF) treated mice. Scale bar = 100 μm. The lowest panel showing microglial morphology in representative slices from control and KA-treated mice. Scale bar = 10 μm. B, Quantitative data showing the number and the fluorescence intensity of Iba1+ cells following KA-induced seizures. n = 6 mice for each group. *p<0.05, **p<0.01, ***p<0.001, versus control.
Supplementary Figure 2. The time course of astrocyte proliferation in the hippocampus of KA-induced seizure mice.
A, Representative images of Ki67 (green) and GFAP (red) immunostaining in the hippocampus at different time points in KA treated mice. Scale bar =100 μm. The enlarged panel showing co-localization of Ki67 immunostaining with GFAP+ astrocytes. B, Quantitative data is shown the percentage of Ki67+:GFAP+ cells among total Ki67+ cells per mm2 in the hippocampus, which peaks at 3d after KA injection. n = 6 mice for each group. *p<0.05, **p<0.01 versus control.
Supplementary Figure 3. CSF-1R inhibitor GW2580 treatment attenuates microglial activation and monocyte recruitment in KA-induced seizures mice.
A, Representative images of Iba1 immunostaining in the hippocampus from control and GW2580-treated seizure mice at 3d after KA injection. Inset shows outline of microglial morphologies in the white box of the representative slices from control and GW2580-treated mice used to determine microglial area. Scale bar = 20 μm. B, Quantitative data showing GW2580 decreases primary microglial branch numbers and microglia soma size in representative slices from control and GW2580-treated mice. n = 5 mice for each group. C. Representative images of Iba1 (green) and CD169 (red) immunostaining in the hippocampus CA1 and CA3 at day 3 after KA injection in the presence or absence of GW2580. Scale bar = 100 μm. D, Quantitative data showing GW2580 treatment reduces seizure-induced CD169+ monocytes recruitment in the hippocampus. n = 5 mice for each group. *p < 0.05, ** p< 0.01, versus control.
Supplementary Figure 4. CSF-1Ab treatment did not induce microglial apoptosis in KA-induced seizure mice.
A, Representative images of cleaved casapase-3 (green) and Iba1 (red) immunostaining in the hippocampus at day 3 after KA injection in control and CSF-1Ab treated mice. B, Representative images of BrdU (red) with Iba1 (green) immunostaining in the hippocampus at day 3 after KA injection in CSF-1Ab or GW2580 treated mice. C, Quantitative data is shown the number of BrdU+:Iba1+ cells per mm2 in the hippocampus from control, CSF-1Ab, or GW2580 treated seizure mice. n =5 mice for each group. ***P < 0.001 versus control. Scale bar = 100 μm.
Supplementary Figure 5. CSF-1R inhibitor GW2580 treatment reverses the neuron loss in KA induced seizure mice.
A, Representative images of Nissl staining in the hippocampus at day 3 after KA injection from control, CSF-1Ab, or GW2580 treated mice. B, The number of surviving neurons in Nissl stained sections was counted in the CA3 area of ipsilateral side of the hippocampus in control, CSF-1Ab, or GW2580 treated mice 3d after KA injection. n = 5 for each group, ***p < 0.001 versus control. C. Representative images of NeuN staining in the hippocampus from control, CSF-1Ab, or GW2580 treated seizure mice. Note the more severe neuron loss occurred in the CA3 from control seizure mice compared with that from the GW2580 or CSF-1Ab treated seizure mice. n =5 mice per group. Scale bar = 100 μm.
Main Points.
Microglia and monocytes contribute to observed hippocampal microgliosis after seizures.
Microglia, but not monocytes, proliferate in the hippocampus after seizures.
Inhibiting microglial proliferation does not affect seizure severity but attenuated seizure-induced neuronal loss.
Acknowledgements:
This work is supported by National Institute of Health (R01NS088627 and R21DE025689 to W.L-J.), National Natural Science Foundation of China (81302755 to F.L.) and Key University Science Research Project of Anhui Province (KJ2017A167 to F.L.)
Footnotes
Conflict of Interest: the authors declare no competing financial interests.
Reference
- Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. 2007. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci 10:1538–43. [DOI] [PubMed] [Google Scholar]
- Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, Sarnacki S, Cumano A, Lauvau G, Geissmann F. 2007. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science 317:666–70. [DOI] [PubMed] [Google Scholar]
- Avignone E, Lepleux M, Angibaud J, Nagerl UV. 2015. Altered morphological dynamics of activated microglia after induction of status epilepticus. J Neuroinflammation 12:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avignone E, Ulmann L, Levavasseur F, Rassendren F, Audinat E. 2008. Status epilepticus induces a particular microglial activation state characterized by enhanced purinergic signaling. J Neurosci 28:9133–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach TG, Woodhurst WB, MacDonald DB, Jones MW. 1995. Reactive microglia in hippocampal sclerosis associated with human temporal lobe epilepsy. Neurosci Lett 191:27–30. [DOI] [PubMed] [Google Scholar]
- Bosco DB, Zheng J, Xu Z, Peng J, Eyo UB, Tang K, Yan C, Huang J, Feng L, Wu G and others. 2018. RNAseq analysis of hippocampal microglia after kainic acid-induced seizures. Mol Brain 11:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charolidi N, Schilling T, Eder C. 2015. Microglial Kv1.3 Channels and P2Y12 Receptors Differentially Regulate Cytokine and Chemokine Release from Brain Slices of Young Adult and Aged Mice. PLoS One 10:e0128463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway JG, McDonald B, Parham J, Keith B, Rusnak DW, Shaw E, Jansen M, Lin P, Payne A, Crosby RM and others. 2005. Inhibition of colony-stimulating-factor-1 signaling in vivo with the orally bioavailable cFMS kinase inhibitor GW2580. Proc Natl Acad Sci U S A 102:16078–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djukic M, Mildner A, Schmidt H, Czesnik D, Bruck W, Priller J, Nau R, Prinz M. 2006. Circulating monocytes engraft in the brain, differentiate into microglia and contribute to the pathology following meningitis in mice. Brain 129:2394–403. [DOI] [PubMed] [Google Scholar]
- Drage MG, Holmes GL, Seyfried TN. 2002. Hippocampal neurons and glia in epileptic EL mice. J Neurocytol 31:681–92. [DOI] [PubMed] [Google Scholar]
- Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, Kitazawa M, Matusow B, Nguyen H, West BL and others. 2014. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 82:380–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erblich B, Zhu L, Etgen AM, Dobrenis K, Pollard JW. 2011. Absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PLoS One 6:e26317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyo UB, Bispo A, Liu J, Sabu S, Wu R, DiBona VL, Zheng J, Murugan M, Zhang H, Tang Y and others. 2018a. The GluN2A Subunit Regulates Neuronal NMDA receptor-Induced Microglia-Neuron Physical Interactions. Sci Rep 8:828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyo UB, Gu N, De S, Dong H, Richardson JR, Wu LJ. 2015. Modulation of microglial process convergence toward neuronal dendrites by extracellular calcium. J Neurosci 35:2417–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyo UB, Mo M, Yi MH, Murugan M, Liu J, Yarlagadda R, Margolis DJ, Xu P, Wu LJ. 2018b. P2Y12R-Dependent Translocation Mechanisms Gate the Changing Microglial Landscape. Cell Rep 23:959–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyo UB, Murugan M, Wu LJ. 2017. Microglia-Neuron Communication in Epilepsy. Glia 65:5–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyo UB, Peng J, Swiatkowski P, Mukherjee A, Bispo A, Wu LJ. 2014. Neuronal Hyperactivity Recruits Microglial Processes via Neuronal NMDA Receptors and Microglial P2Y12 Receptors after Status Epilepticus. J Neurosci 34:10528–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flügel A, Bradl M, Kreutzberg GW, Graeber MB. 2001. Transformation of donor-derived bone marrow precursors into host microglia during autoimmune CNS inflammation and during the retrograde response to axotomy. J Neurosci Res 66:74–82. [DOI] [PubMed] [Google Scholar]
- Gordon S, Plüddemann A, Martinez Estrada F. 2014. Macrophage heterogeneity in tissues: phenotypic diversity and functions. Immunol Rev 262:36–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu N, Eyo UB, Murugan M, Peng J, Matta S, Dong H, Wu LJ. 2016a. Microglial P2Y12 receptors regulate microglial activation and surveillance during neuropathic pain. Brain Behav Immun 55:82–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu N, Peng J, Murugan M, Wang X, Eyo UB, Sun D, Ren Y, DiCicco-Bloom E, Young W, Dong H and others. 2016b. Spinal Microgliosis Due to Resident Microglial Proliferation Is Required for Pain Hypersensitivity after Peripheral Nerve Injury. Cell Rep 16:605–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Z, Kuhn JA, Wang X, Colquitt B, Solorzano C, Vaman S, Guan AK, Evans-Reinsch Z, Braz J, Devor M and others. 2016. Injured sensory neuron-derived CSF1 induces microglial proliferation and DAP12-dependent pain. Nat Neurosci 19:94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, El Khoury J. 2013. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci 16:1896–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohsfield LA, Humpel C. 2015. Migration of blood cells to β-amyloid plaques in Alzheimer’s disease. Exp Gerontol 65:8–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong J, Cho IH, Kwak KI, Suh EC, Seo J, Min HJ, Choi SY, Kim CH, Park SH, Jo EK and others. 2010. Microglial Toll-like receptor 2 contributes to kainic acid-induced glial activation and hippocampal neuronal cell death. J Biol Chem 285:39447–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR. 2000. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol 20:4106–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenmann H, Hoppe D, Gottmann K, Banati R, Kreutzberg G. 1990. Cultured microglial cells have a distinct pattern of membrane channels different from peritoneal macrophages. J Neurosci Res 26:278–87. [DOI] [PubMed] [Google Scholar]
- Kim KS, Jang JH, Lin H, Choi SW, Kim HR, Shin DH, Nam JH, Zhang YH, Kim SJ. 2015. Rise and Fall of Kir2.2 Current by TLR4 Signaling in Human Monocytes: PKC-Dependent Trafficking and PI3K-Mediated PIP2 Decrease. J Immunol 195:3345–54. [DOI] [PubMed] [Google Scholar]
- Kotecha SA, Schlichter LC. 1999. A Kv1.5 to Kv1.3 switch in endogenous hippocampal microglia and a role in proliferation. J Neurosci 19:10680–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krutzik SR, Tan B, Li H, Ochoa MT, Liu PT, Sharfstein SE, Graeber TG, Sieling PA, Liu YJ, Rea TH and others. 2005. TLR activation triggers the rapid differentiation of monocytes into macrophages and dendritic cells. Nat Med 11:653–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London A, Cohen M, Schwartz M. 2013. Microglia and monocyte-derived macrophages: functionally distinct populations that act in concert in CNS plasticity and repair. Front Cell Neurosci 7:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda T, Murao N, Katano Y, Juliandi B, Kohyama J, Akira S, Kawai T, Nakashima K. 2015. TLR9 signalling in microglia attenuates seizure-induced aberrant neurogenesis in the adult hippocampus. Nat Commun 6:6514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann GM, Wenzel HJ, Robbins CA, Sosunov AA, Schwartzkroin PA. 2003. Mouse strain differences in kainic acid sensitivity, seizure behavior, mortality, and hippocampal pathology. Neuroscience 122:551–61. [DOI] [PubMed] [Google Scholar]
- Menteyne A, Levavasseur F, Audinat E, Avignone E. 2009. Predominant functional expression of Kv1.3 by activated microglia of the hippocampus after Status epilepticus. PLoS One 4:e6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mildner A, Schlevogt B, Kierdorf K, Böttcher C, Erny D, Kummer MP, Quinn M, Brück W, Bechmann I, Heneka MT and others. 2011. Distinct and non-redundant roles of microglia and myeloid subsets in mouse models of Alzheimer’s disease. J Neurosci 31:11159–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najjar S, Pearlman D, Miller DC, Devinsky O. 2011. Refractory epilepsy associated with microglial activation. Neurologist 17:249–54. [DOI] [PubMed] [Google Scholar]
- Nguyen HM, Grössinger EM, Horiuchi M, Davis KW, Jin LW, Maezawa I, Wulff H. 2017. Differential Kv1.3, KCa3.1, and Kir2.1 expression in “classically” and “alternatively” activated microglia. Glia 65:106–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR, Lafaille JJ, Hempstead BL, Littman DR, Gan WB. 2013. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155:1596–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Gu N, Zhou L, B Eyo U, Murugan M, Gan WB, Wu LJ. 2016. Microglia and monocytes synergistically promote the transition from acute to chronic pain after nerve injury. Nat Commun 7:12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz M, Priller J, Sisodia SS, Ransohoff RM. 2011. Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat Neurosci 14:1227–35. [DOI] [PubMed] [Google Scholar]
- Rangaraju S, Gearing M, Jin LW, Levey A. 2015. Potassium channel Kv1.3 is highly expressed by microglia in human Alzheimer’s disease. J Alzheimers Dis 44:797–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritzel RM, Patel AR, Grenier JM, Crapser J, Verma R, Jellison ER, McCullough LD. 2015. Functional differences between microglia and monocytes after ischemic stroke. J Neuroinflammation 12:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saederup N, Cardona AE, Croft K, Mizutani M, Cotleur AC, Tsou CL, Ransohoff RM, Charo IF. 2010. Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PLoS One 5:e13693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Hoshi M, Akazawa C, Nakamura Y, Tsuzuki H, Inoue K, Kohsaka S. 2003. Selective expression of Gi/o-coupled ATP receptor P2Y12 in microglia in rat brain. Glia 44:242–50. [DOI] [PubMed] [Google Scholar]
- Schmidtmayer J, Jacobsen C, Miksch G, Sievers J. 1994. Blood monocytes and spleen macrophages differentiate into microglia-like cells on monolayers of astrocytes: membrane currents. Glia 12:259–67. [DOI] [PubMed] [Google Scholar]
- Shapiro LA, Wang L, Ribak CE. 2008. Rapid astrocyte and microglial activation following pilocarpine-induced seizures in rats. Epilepsia 49 Suppl 2:33–41. [DOI] [PubMed] [Google Scholar]
- Sierra A, Gottfried-Blackmore AC, McEwen BS, Bulloch K. 2007. Microglia derived from aging mice exhibit an altered inflammatory profile. Glia 55:412–24. [DOI] [PubMed] [Google Scholar]
- Simard AR, Rivest S. 2004. Bone marrow stem cells have the ability to populate the entire central nervous system into fully differentiated parenchymal microglia. FASEB J 18:998–1000. [DOI] [PubMed] [Google Scholar]
- Tian DS, Peng J, Murugan M, Feng LJ, Liu JL, Eyo UB, Zhou LJ, Mogilevsky R, Wang W, Wu LJ. 2017. Chemokine CCL2-CCR2 Signaling Induces Neuronal Cell Death via STAT3 Activation and IL-1beta Production after Status Epilepticus. J Neurosci 37:7878–7892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varvel NH, Neher JJ, Bosch A, Wang W, Ransohoff RM, Miller RJ, Dingledine R. 2016. Infiltrating monocytes promote brain inflammation and exacerbate neuronal damage after status epilepticus. Proc Natl Acad Sci U S A 113:E5665–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A 2014. Epilepsy and inflammation in the brain: overview and pathophysiology. Epilepsy Curr 14:3–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente R, Escalada A, Coma M, Fuster G, Sánchez-Tilló E, López-Iglesias C, Soler C, Solsona C, Celada A, Felipe A. 2003. Differential voltage-dependent K+ channel responses during proliferation and activation in macrophages. J Biol Chem 278:46307–20. [DOI] [PubMed] [Google Scholar]
- Wu LJ, Wu G, Akhavan Sharif MR, Baker A, Jia Y, Fahey FH, Luo HR, Feener EP, Clapham DE. 2012. The voltage-gated proton channel Hv1 enhances brain damage from ischemic stroke. Nat Neurosci 15:565–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki R, Lu H, Butovsky O, Ohno N, Rietsch AM, Cialic R, Wu PM, Doykan CE, Lin J, Cotleur AC and others. 2014. Differential roles of microglia and monocytes in the inflamed central nervous system. J Exp Med 211:1533–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X, Maixner DW, Li F, Weng HR. 2017. Chronic pain and impaired glial glutamate transporter function in lupus-prone mice are ameliorated by blocking macrophage colony-stimulating factor-1 receptors. J Neurochem 140:963–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zattoni M, Mura ML, Deprez F, Schwendener RA, Engelhardt B, Frei K, Fritschy JM. 2011. Brain infiltration of leukocytes contributes to the pathophysiology of temporal lobe epilepsy. J Neurosci 31:4037–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Zheng H, Shao X, Wang W, Yao Q, Li Z. 2010. Excitotoxicity of TNFalpha derived from KA activated microglia on hippocampal neurons in vitro and in vivo. J Neurochem 114:386–96. [DOI] [PubMed] [Google Scholar]
- Ziegler-Heitbrock L 2007. The CD14+ CD16+ blood monocytes: their role in infection and inflammation. J Leukoc Biol 81:584–92. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Time-lapse video from two-photon images showing response of GFP+ only and GFP+:RFP+ cells towards laser burn and lack of response by RFP+ only cells at 7d after KA and corresponding control.
Supplementary Figure 1. Changes in morphology and cell number of Iba1+ cells following KA-induced seizures.
A, Representative images of Iba1-labled hippocampal microglia from control and KA (i.c.v., 0.15 μg in 5 μl ACSF) treated mice. Scale bar = 100 μm. The lowest panel showing microglial morphology in representative slices from control and KA-treated mice. Scale bar = 10 μm. B, Quantitative data showing the number and the fluorescence intensity of Iba1+ cells following KA-induced seizures. n = 6 mice for each group. *p<0.05, **p<0.01, ***p<0.001, versus control.
Supplementary Figure 2. The time course of astrocyte proliferation in the hippocampus of KA-induced seizure mice.
A, Representative images of Ki67 (green) and GFAP (red) immunostaining in the hippocampus at different time points in KA treated mice. Scale bar =100 μm. The enlarged panel showing co-localization of Ki67 immunostaining with GFAP+ astrocytes. B, Quantitative data is shown the percentage of Ki67+:GFAP+ cells among total Ki67+ cells per mm2 in the hippocampus, which peaks at 3d after KA injection. n = 6 mice for each group. *p<0.05, **p<0.01 versus control.
Supplementary Figure 3. CSF-1R inhibitor GW2580 treatment attenuates microglial activation and monocyte recruitment in KA-induced seizures mice.
A, Representative images of Iba1 immunostaining in the hippocampus from control and GW2580-treated seizure mice at 3d after KA injection. Inset shows outline of microglial morphologies in the white box of the representative slices from control and GW2580-treated mice used to determine microglial area. Scale bar = 20 μm. B, Quantitative data showing GW2580 decreases primary microglial branch numbers and microglia soma size in representative slices from control and GW2580-treated mice. n = 5 mice for each group. C. Representative images of Iba1 (green) and CD169 (red) immunostaining in the hippocampus CA1 and CA3 at day 3 after KA injection in the presence or absence of GW2580. Scale bar = 100 μm. D, Quantitative data showing GW2580 treatment reduces seizure-induced CD169+ monocytes recruitment in the hippocampus. n = 5 mice for each group. *p < 0.05, ** p< 0.01, versus control.
Supplementary Figure 4. CSF-1Ab treatment did not induce microglial apoptosis in KA-induced seizure mice.
A, Representative images of cleaved casapase-3 (green) and Iba1 (red) immunostaining in the hippocampus at day 3 after KA injection in control and CSF-1Ab treated mice. B, Representative images of BrdU (red) with Iba1 (green) immunostaining in the hippocampus at day 3 after KA injection in CSF-1Ab or GW2580 treated mice. C, Quantitative data is shown the number of BrdU+:Iba1+ cells per mm2 in the hippocampus from control, CSF-1Ab, or GW2580 treated seizure mice. n =5 mice for each group. ***P < 0.001 versus control. Scale bar = 100 μm.
Supplementary Figure 5. CSF-1R inhibitor GW2580 treatment reverses the neuron loss in KA induced seizure mice.
A, Representative images of Nissl staining in the hippocampus at day 3 after KA injection from control, CSF-1Ab, or GW2580 treated mice. B, The number of surviving neurons in Nissl stained sections was counted in the CA3 area of ipsilateral side of the hippocampus in control, CSF-1Ab, or GW2580 treated mice 3d after KA injection. n = 5 for each group, ***p < 0.001 versus control. C. Representative images of NeuN staining in the hippocampus from control, CSF-1Ab, or GW2580 treated seizure mice. Note the more severe neuron loss occurred in the CA3 from control seizure mice compared with that from the GW2580 or CSF-1Ab treated seizure mice. n =5 mice per group. Scale bar = 100 μm.