

Abstract
Nemaline myopathy, which is characterized by the accumulation of ‘’rod’’ bodies in muscle fibers is a very rare inherited muscle disease. According to the underlying mutation, the disease has varying severity of clinical outcomes. Patients with severe forms of the disease die because of hypotonia, feeding difficulties, aspiration pneumonia, and respiratory failure in the neonatal or infancy period. Mild forms of the disease present with walking-swallowing difficulties and respiratory distress in late childhood or adulthood. A two-and-a-half-month-old boy was monitored in our Pediatric Intensive Care Unit with hypotonia, pneumonia, and respiratory distress. Nemaline myopathy was diagnosed as the result of a muscle biopsy. An advanced molecular examination revealed heterozygous mutations in the skeletal muscle α-actin (ACTA1) gene, which is the second most common cause of this disease. Nemaline myopathy should be kept in mind in patients of all age groups with respiratory failure and walking difficulty secondary to muscle weakness.
Keywords: ACTA1 gene, hypotonia, muscle biopsy, nemaline myopathy, respiratory failure
Abstract
Nemalin miyopatisi oldukça nadir görülen kalıtımsal bir kas hastalığı olup kas liflerinde ‘’rod’’(nemalin) cisimciği birikimi ile tanımlanmaktadır. Hastalık altta yatan mutasyona ve mutasyonun kalıtım biçimine göre değişen ağırlıkta klinik gidişe sahiptir. Ağır şekillerinde olgular yutma ve solunum kaslarının etkilenmesi sonucu beslenme yetersizliği, aspirasyon pnömonisi ve solunum yetmezliği nedeni ile yenidoğan ya da süt çocukluğu döneminde kaybedilmektedir. Geç başlangıçlı hafif olgular yaşam kalitesini bozan yürüme-yutma zorluğu ve solunum sıkıntısı ile geç çocukluk ya da erişkin yaşta bulgu verebilmektedir. Hipotoni, pnömoni ve solunum sıkıntısı ile Çocuk Yoğun Bakım Birimi’nde izlenen iki buçuk aylık erkek bebeğe kas biyopsisi sonucu nemalin miyopatisi tanısı koyuldu. İleri moleküler inceleme sonucu hastalığın ikinci en sık nedeni olan “Skeletal Muscle α-Actin” (ACTA1) geninde heterozigot mutasyon saptandı. Yenidoğan döneminden erişkin döneme kadar kas güçsüzlüğüne bağlı solunum yetmezliği ve yutma-yürüme güçlüğü varlığında yapısal miyopatiler içinde nemalin miyopatisi akılda bulundurulmalı, şüphenilen olgulara kas biyopsisi ya/ya da genetik inceleme yapılmalıdır.
Anahtar sözcükler: ACTA1 geni, hipotoni, kas biyopsisi, nemalin miyopatisi, solunum yetmezliği
Introduction
Nemaline myopathy (NM) is a rare genetic muscle disorder characterized by the accumulation of ‘rod’ bodies in muscle fibers. It is estimated that NM has an incidence of 1/50 000 and is responsible of 17% of all congenital myopathies (1). The disease causes variable clinical prognosis according to the underlying mutation. Patients whose symptoms start from birth are lost in the neonatal period or infancy because of severe hypotonia, malnutrition, aspiration pneumonia, and respiratory failure. Mild cases with late onset may be manifested in late childhood or adulthood with walking-swallowing difficulty, which disrupts quality of life, and respiratory distress occuring because of weakness in the respiratory muscles (1, 2).
In this article, a pediatric patient who was followed up in the Pediatric Intensive Care Unit (PICU) with hypotonia, pneumonia, and respiratory distress, and was diagnosed as having NM with muscle biopsy and gene mutation testing, is presented with the objective of drawing attention to this rare muscle disease.
Case
A two-and-a-half-month-old male baby who had been hospitalized in the Pediatrics Ward for one month because of hypotonia, pneumonia, and respiratory distress was internalized in th PICU with the same symptoms. It was learned that he was born from the first pregnancy of a 28-year-old mother in the 38th gestational week by cesarean section because of polyhydramniosis and non-progressive labor with a birth weight of 2960 g. There was no consanguinity between the parents. It was stated that there was a considerable decrease in the baby’s movements during pregnancy. On physical examination, the height was found as 59 cm (50th percentile), the weight was found as 4550 g (3–10th percentile) and the head circumference was found as 38 cm (10–25th percentile). His consciousness was open and he was interested in the surroundings. The Glasgow Coma Score (GCS) was found as 15. His body temperature was measured as 37.2°C, apical heart beat was 145/min, respiratory rate was 62/min, and blood pressure was found 88/49 mm Hg. Marked hypotonia and dismorphic findings including small chin, long face, and highly arched palate were found. On examination of the respiratory system, bilateral crepitant rales were found. Examination of the other systems was found to be normal. Biochemical measurements were found to be normal except for a slight elevation in transaminases (AST: 82 IU/L, ALT: 84 IU/L) and respiratory acidosis was found (pH: 7.09, pCO2: 85 mm Hg, HCO3: 24 mmol/L). A complete bood count was found as normal. Among the infection markers, C-reactive protein was found as 0.53 mg/dL (0.01–0.5) and procalcitonin was found as 0.48 ng/mL (<0.5). On lung radiography, diffuse infiltration was observed in the right lung. The patient was intubated and mechanical ventilation was initiated. The antibiotics he was receiving (intravenous ampicillin and cefotaxim) were switched to intravenous vancomycin and meropenem.
In further investigations, the blood lactate, amino acid, carnitine, acylcarnitine, very-long-chain fatty acid levels, and urine organic acids were found as normal. Echocardiography, fundoscopic examination, and bone radiographs were found as normal. Mild laryngomalacia was found on bronchoscopy and a sweat test was within the normal limits. Upper gastrointestinal endoscopy was normal. No pathology was found on brain magnetic resonance imaging except for a choroid plexus cyst. Chromosomal analysis, spinal muscular atrophy gene examination, and fluoresence in-situ hybridization (FISH), which was performed in terms of Prader-Willi syndrome, were found to be normal. In the microscopic examination of the muscle biopsy, which was performed after all these results were obtained, a ‘nemaline body’ (rod body) was observed mostly in the sarcolemmal regions (Fig. 1a–c). A diagnosis of NM was made with these findings and further molecular examination was planned. NM_001100.3: c.1075A>C (p.I359L) (heterozygous) mutation was found in skeletal muscle α-actin” (ACTA1) gene whole genome sequencing, which was performed with the new-generation sequence analysis method (Fig. 2). The parents’ genetic tests directed to the disease were found to be normal.
Figure 1.
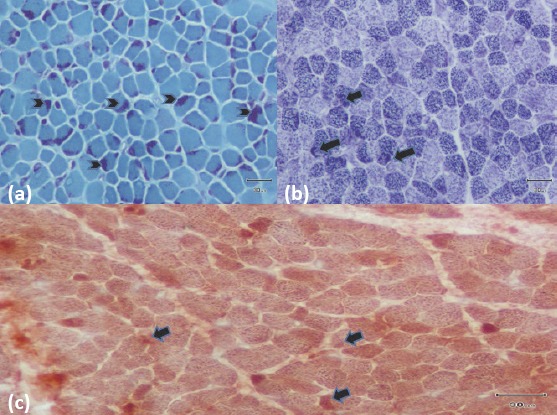
(a) Difference in diameter between both types of muscle fibers with modified Gomori-trichrome staining (more prominent in the type 1 muscle fibers), increased endomisial connective tissue and intracytoplasmic dark red-blue tubular structures (“rods”) with a small diameter located in the periphery (more intensive and prominent in the atrophic type 1 muscle fibers) on light microscopic examination on muscle biopsy. (b) Activity with NADH-TR histochemical staining in the areas compatible with the intracytoplasmic dark red-blue tubular structures (“rods”) with a small diameter located in the periphery (arrows). (c) Activity with COX enzyme histochemical staining in the areas compatible with the intracytoplasmic dark red-blue tubular structures (“rods”) with a small diameter located in the periphery (arrows)
Figure 2.
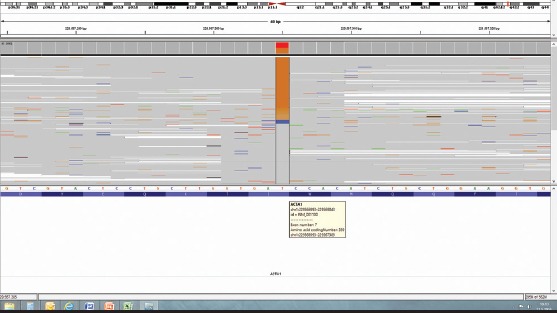
YNM_001100.3: c.1075A>C (p.I359L) (heterozygous) mutation in the ACTA1 gene whole genome sequencing performed using new-generation sequence analysis
Tracheostomy was performed on the 23rd day of hospitalization in the patient who failed extubation preparation tests because of hypotonia and insufficient respiratory effort. Subsequenly, the patient was discharged on the 45th day of hospitalization, education was given to his family, and he was followed up with a house ventilator. Our patient is aged 18 months at the present time, he is being connected to house ventilator at night and his respiration in room air is sufficent in the daytime. He can walk with assistance, he is being fed with a nasogastric tube because of swallowing difficulty, and gastrostomy is being planned. Written informed consent was received from the parents of this patient for this case presentation.
Discussion
Nemaline myopathy is a genetic myopathy. It was first described by Shy et al. (3) in 1963. Actin, which is a round structural protein, constitutes polymerized helix-shaped actin filaments. It provides mechanical support for cells, determines the shape of cells, and enables cells to move. In nemaline myopathy, especially the structure of alpha actin, is disrupted and intracellular accumulation and dysfunction occur as a result of disruption in the nuclear structure of the fine filament in the sarcomere (1).
Autosomal dominant (OD) and autosomal recessive (AR) types have been reported. Frequently, facial, axial, and proximal limb muscles are involved (1, 2, 4). It is characterized by muscle weakness, respiratory distress, feeding-swallowing problems, aspiration pneumonia, hypotonia, and dismorphc findings including highly arched palate, retrognatia, and a long and expressionless face (1, 2). It is divided into six types acccording to the mutation and inheritance. The severe congenital (newborn) type has a fatal prognosis generally in the first year because of respiratory failure. In typical (mild prognosis) congenital NM, weakness, hypotonia and feeding problems occur frequently in the neonatal period or in the first year of life. In this mild congenital type, muscle weakness and hypotonia are milder compared with severe and moderate congenital type. Movement against gravity is present and the respiratory muscles are involved less or the disease progresses more slowly (1). Polyhydramniosis may be observed during pregnancies in mothers of affected babies, especially in congenital NM types (4). Compatible with the literature, polyhydramniosis was present in the mother of our patient whose parents did not have consanguinity, and our patient had a long face and highly arched palate. Feeding-swallowing problems, which occured from the age of one and a half months and progressed slowly, respiratory distress, and ability to move against gravity suggested the mild congential type of NM in our patient.
The diagnosis of NM is made with muscle biopsy and mutation analysis (1–4). The level of creatinine kinase is normal or slightly elevated. Electromyography (EMG) may be normal or show nonspecific myopathic characteristics especially at advanced ages. Most of the time, performing EMG is technically difficult in the first months (1, 4). Rod bodies are observed, especially below the sarcolemmal region and around the nucleus on light microscopic examination with the Gomori-trichrome stain. With this staining, the involved striated muscle fibers are stained blue-green, and the ‘rod’ bodies are stained red (1–4). The creatinine kinase level was found to be normal in our patient and EMG could not be performed because of technical difficultiies. Muscle biopsy revealed a difference in diameter between both types of muscle fibers with modified Gomori-trichrome staining (more prominent in the type 1 muscle fibers), increased endomisial connective tissue and intracytoplasmic dark red-blue tubular structures (“rod”) with a small diameter located in the periphery (more intensive and prominent in the atrophic type 1 muscle fibers) (Fig. 1a). In the areas compatible with the intracytoplasmic dark red-blue tubular structures (rods) with a small diameter located in the periphery, activity was observed with NADH-TR and COX enzyme histochemical stainings (Fig. 1b, Fig. 1c).
Current literature reports that mutations in 10 different genes including ACTA1, NEB, TPM3, TPM2, TNNT1, CFL2, KBTBD13, KLHL40, KLHL41 and LMOD3 cause NM disease. Among these mutations, the most common mutation is NEB mutation and the second most common mutation is ACTA1 mutation (1, 5). It is difficult to predict the phenotype according to genotype because clinical findings in different types of the disease are frequently similar to each other and it is not possible to perform mutation analysis in all subjects (1). In our patient, we found the ACTA1 mutation, which is the second most common mutation in NM, described by Ilkowski et al. (6) for the first time (Fig. 2). ACTA1, which can be inherited both autosomal dominantly and autosomal recessively, is found in 15–25% of patients with NM. It is the cause in 50% of severe and fatal cases and may also be responsible for childhood- and adult-onset mild forms (1, 7). Our case was evaluated to be quite likely compatible with the OD form because a heterozygous mutation was observed in our patient and there was no consanguinity between the parents. Absence of mutation in the parents on genetic analysis suggested that this mutation occured de novo. De novo mutations are common mechanisms in diseases that show OD inheritance (8). The absence of a second mutation in our patient and emergence of clinical manifestations when the mutation was sporadic and heterozygous, suggested that it was an OD mutation. There was also a possibility that a de novo mutation occured in the second allele with a second mutation that had deep intronic location inherited from one of the parents or that we could not find in the regulator regions. The fact that the ACTA1 mutations are observed with a rate of 25% among all mutations (a prevalence of aproximately 0,5/100,000) in nemaline myopathy shows that this possibility is very low (1, 8).
Muscle weakness and respiratory insufficiency did not progress on clinical follow-up at the age of 18 months in our patient, who we thought was compatible with mild congenital NM. He is using a house ventilator only at night, he does not need oxygen support in the daytime, and his own respiratory effort is sufficient. Levesque et al. (9) reported that the clinical picture was relieved at the end of a 6-month follow-up period in a female patient who was found to have an ACTA1 mutation and who was diagnosed in infancy with the same cinical findings as our patient.
In conclusion, muscle biopsy should be considered in the differential diagnosis of newborns and infants who have hypotonia, respiratory distress, feeding difficulty, dysmorphic appearance, and joint anomalies, even if the creatinine kinase levels are found to be normal. Nemaline myopathy, which was found in our patient, is a rare muscle disease that can be diagnosed by muscle biopsy and/or genetic analysis, and characterized by varying degrees of muscle weakness from the neonatal period to adulthood.
Footnotes
Informed Consent: Written informed consent was obtained from patient’s parent.
Peer-review: Externally peer-reviewed.
Author Contributions: Consept - O.Y., M.E.; Design - Ö.Ş., S.C., O.Y.; Supervision - E.Ş., H.S.K.; Funding - Ö.Ş., S.C.; Data Collection and/or Processing - O.Y., E.Ş., H.S.K., C.H.A; Analysis and/or Interpretation - E.Ş., H.S.K., M.E.; Literature Review - O.Y., M.T.P., C.H.A.; Writing - O.Y., E.Ş., H.S.K.; Critical Review - E.Ş., M.E.
Conflict of Interest: No conflict of interest was declared by the authors.
Financial Disclosure: The authors declared that this study has received no financial support.
Hasta Onamı: Yazılı hasta onamı hastanın ebeveynlerinden alınmıştır.
Hakem Değerlendirmesi: Dış bağımsız.
Yazar Katkıları: Fikir - O.Y., M.E.; Tasarım - Ö.Ş., S.C., O.Y.; Denetleme - E.Ş., H.S.K.; Kaynaklar - Ö.Ş., S.C.; Veri Toplanması ve İşlemesi - O.Y., E.Ş., H.S.K., C.H.A; Analiz ve Yorum - E.Ş., H.S.K., M.E.; Literatür Taraması - O.Y., M.T.P., C.H.A.; Yazıyı Yazan - O.Y., E.Ş., H.S.K.; Eleştirel İnceleme - E.Ş., M.E.
Çıkar Çatışması: Yazarlar çıkar çatışması bildirmemişlerdir.
Mali Destek: Yazarlar bu çalışma için mali destek almadıklarını beyan etmişlerdir.
References
- 1.North KN, Ryan MM. In: Nemaline myopathy. GeneReviews. Pagon RA, Adam MP, Ardinger HH, editors. Seattle: University of Washington, Seattle; 2015. pp. 1993–2019. [PubMed] [Google Scholar]
- 2.Yin X, Pu CQ, Wang Q, Liu JX, Mao YL. Clinical and pathological features of patients with nemaline myopathy. Mol Med Rep. 2014;10:175–82. doi: 10.3892/mmr.2014.2184. [DOI] [PubMed] [Google Scholar]
- 3.Shy GM, Engel WK, Somers JE, Wanko T. Nemaline Myopathy. A New Congenital Myopathy. Brain. 1963;86:793–810. doi: 10.1093/brain/86.4.793. [DOI] [PubMed] [Google Scholar]
- 4.Ryan MM, Schnell C, Strickland CD, et al. Nemaline myopathy:a clinical study of 143 cases. Ann Neurol. 2001;50:312–20. doi: 10.1002/ana.1080. [DOI] [PubMed] [Google Scholar]
- 5.Nowak KJ, Davis MR, Wallgren-Pettersson C, Lamont PJ, Laing NG. Clinical utility gene card for:Nemaline myopathy - update 2015. Eur J Hum Genet. 2015;23 doi: 10.1038/ejhg.2015.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ilkovski B, Cooper ST, Nowak K, et al. Nemaline myopathy caused by mutations in the muscle alpha-skeletal-actin gene. Am J Hum Genet. 2001;68:1333–43. doi: 10.1086/320605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gineste C, Le Fur Y, Vilmen C, et al. Combined MRI and ³¹P-MRS investigations of the ACTA1(H40Y) mouse model of nemaline myopathy show impaired muscle function and altered energy metabolism. PLoS One. 2013;8:e61517. doi: 10.1371/journal.pone.0061517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prevalence of rare diseases:Bibliographic data- Orphanet Report Series, Rare Diseases collection. November 2016, Number 1:Diseases listed in alphabetical order. Available from:URL: http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_diseases.pdf .
- 9.Levesque L, Del Bigio MR, Krawitz S, Mhanni AA. A de novo dominant mutation in ACTA1 causing congenital nemaline myopathy associated with a milder phenotype:expanding the spectrum of dominant ACTA1 mutations. Neuromuscul Disord. 2013;23:239–42. doi: 10.1016/j.nmd.2012.12.004. [DOI] [PubMed] [Google Scholar]