

Summary
Resistance to the last-resort antibiotic colistin is now widespread and new therapeutics are urgently required. We report the first in toto chemical synthesis and pre-clinical evaluation of octapeptins, a class of lipopeptides structurally related to colistin. The octapeptin biosynthetic cluster consisted of three non-ribosomal peptide synthetases (OctA, OctB, OctC) that produced an amphiphilic antibiotic, octapeptin C4, which was shown to bind to and depolarize membranes. Whilst active against MDR strains in vitro, octapeptin C4 displayed poor in vivo efficacy, most likely due to high plasma protein binding. NMR solution structures, empirical structure-activity and structure-toxicity models were used to design synthetic octapeptins active against MDR and extensively-drug resistance (XDR) bacteria. The scaffold was then subtly altered to reduce plasma protein binding, while maintaining activity against MDR and XDR bacteria. In vivo efficacy was demonstrated in a murine bacteremia model with a colistin-resistant P. aeruginosa clinical isolate.
Introduction
The increasing incidence of bacterial resistance in the clinic, coupled with a barren antibiotic development pipeline has been dubbed ‘the perfect storm’ (Gould, 2009). The World Health Organization (WHO) has identified antimicrobial resistance as one of the three greatest threats to human health. The situation is more dire for Gram-negative ‘superbugs’ (notably Pseudomonas aeruginosa, Acinetobacter baumannii and Klebsiella pneumoniae), against which no novel classes of antibiotics are available. The emergence of polymyxin resistance means in essence that there will be no effective antibiotics available for treatment of infections caused by these Gram-negative ‘superbugs’.
The octapeptins are a family of lipooctapeptide antibiotics with a broad antimicrobial spectrum (Meyers, et al., 1976; Storm, et al., 1977). They are structurally similar to the polymyxin lipodecapeptides, and can be considered as a sub-class that retain the cyclic heptapeptide core, but have a truncated linear peptide tail (Figure 1, Table 2). Both octapeptins and polymyxins contain multiple cationic diaminobutyric acid (Dab) residues at the same positions within the heptapeptide core. The major differences between the polymyxins and octapeptins are that the octapeptins contain: (i) a hydrophobic Leu residue at position 8 instead of a polar Thr residue at the corresponding position 10 in the polymyxins; (ii) a D-Dab at position 1 in the truncated exocyclic tail instead of L-Dab at the corresponding position 3 in the polymyxins; and (iii) N-terminal 3(R)-hydroxy fatty acids rather than a mixture of alkyl fatty acids seen at the N-terminal of the polymyxins (Table 2).
Figure 1. Overview of the octapeptin biosynthetic gene cluster.
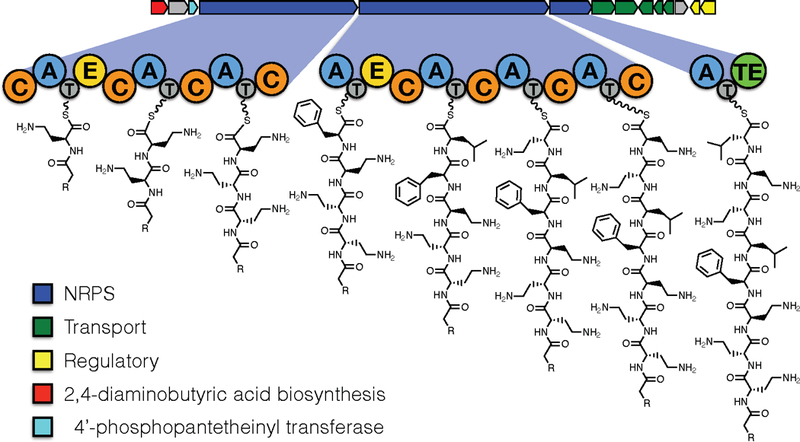
The exploded panel shows the domain arrangement and predicted biosynthetic intermediate for each NRPS module in the cluster. The key indicates predicted functions for the open reading frames within the cluster as deduced by blast search against functionally characterised BGCs in the MiBig repository (Medema et al. 2015).
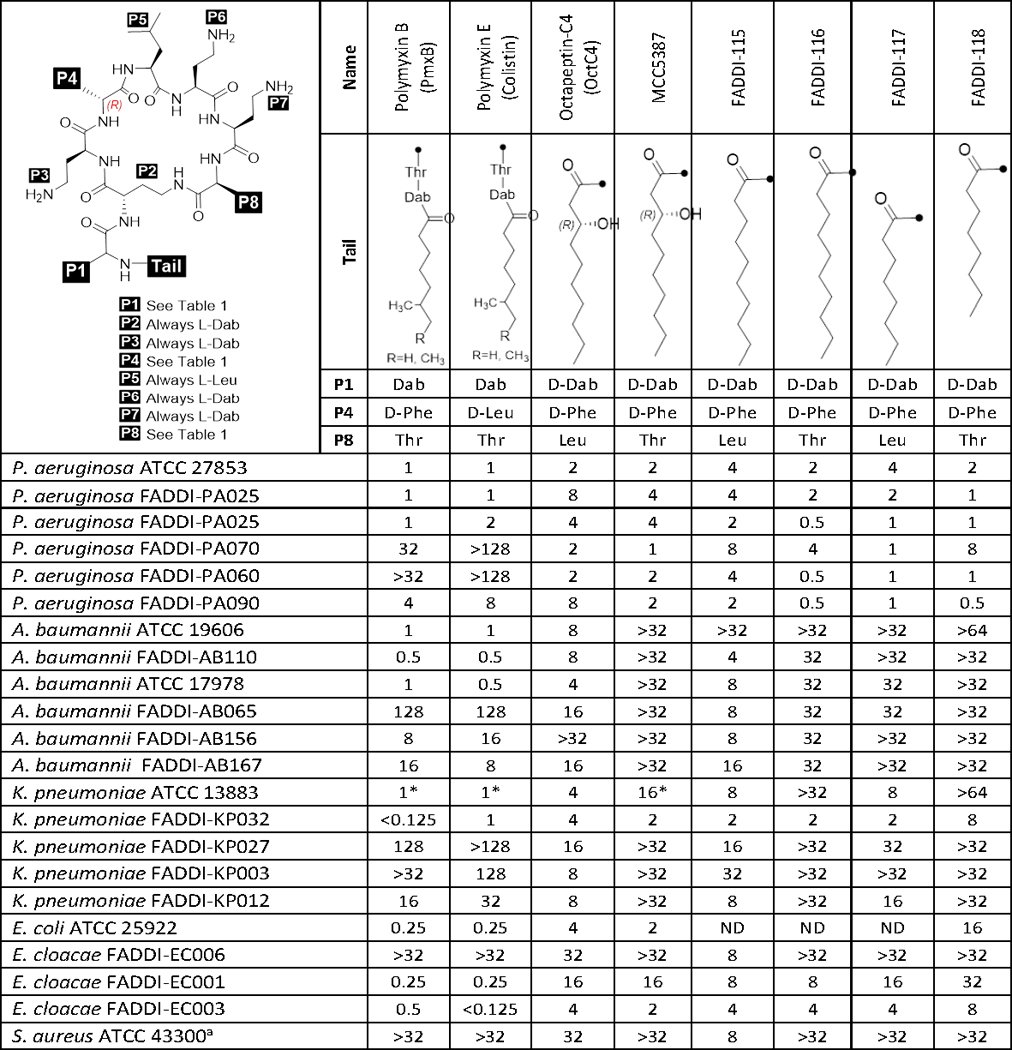
Table 2.
Structures of octapeptin~C4, polymyxin B, coiistin and novel octapeptin analogues; and their minimum inhibitory concentrations (MICs) against Gram- negative and Gram-positive bacteria.
 |
The octapeptins were discovered almost four decades ago as natural products produced by the soil bacteria Bacillus circulans and were given the company compound codes Bu-2470, EM49, 333–25 and Bu-1880 by the then Squibb Institute (Princeton), Bristol-Banyo Research Institute (Tokyo) and Shionogi Research Laboratory (Osaka) (Kato and Shoji, 1980; Konishi, et al., 1983; Meyers, et al., 1973; Meyers, et al., 1973; Meyers, et al., 1976; Meyers, et al., 1974; Parker and Rathnum, 1975; Parker and Rathnum, 1973; Shoji, et al., 1976; Shoji, et al., 1980; Sugawara, et al., 1983). Eventually, a consensus on the nomenclature was reached and these octapeptins were alphabetically designated into four sub-groups, A, B, C, and D, based on minor structural differences in their core scaffold (Meyers, et al., 1976). Research on this class had been limited to a handful of publications before the 1980s, though very recently a new member of this class was discovered (Qian, et al., 2012).
The octapeptins display good activity against Gram-negative bacteria (Konishi, et al., 1983; Meyers, et al., 1973; Meyers, et al., 1973; Meyers, et al., 1974; Shoji, et al., 1976; Shoji, et al., 1980; Storm, et al., 1977). Despite their resemblance to the polymyxins on a structural level (Table 2), the octapeptins exhibit marked differences in their biological profile: (i) unlike the polymyxins, the octapeptins display activity against Gram-positive bacteria, yeast, fungi and protozoa (Konishi, et al., 1983; Meyers, et al., 1973; Meyers, et al., 1973; Meyers, et al., 1974; Shoji, et al., 1976; Shoji, et al., 1980; Storm, et al., 1977), and (ii) most importantly, the octapeptins retain activity against polymyxin-resistant Gram-negative bacteria (Storm, et al., 1977).
Following their discovery, the octapeptin class was essentially neglected by researchers for the next four decades, presumably due to preferential development of β-lactam and fluoroquinolone antibiotics. Hence, there is no structure-activity relationship (SAR) information available for octapeptins. The chemistry and biology of the octapeptins need to be revisited and their therapeutic potential evaluated within the scope of modern drug discovery procedures. In the present study, we identify biosynthetic pathway of the octapeptins and report the synthesis of a representative natural octapeptin (octapeptin C4) and a number of novel analogues. Furthermore, we characterise their three-dimensional structure, mode of action, and in vitro and in vivo antimicrobial activity and toxicity.
Results and Discussion
Identification and annotation of the octapeptin biosynthetic.
In the present study, we obtained a draft whole genome sequence for B. circulans ATCC 31805, from which we identified and annotated the complete octapeptin biosynthetic gene cluster (BGC).
Analysis of the assembled genome sequence for B. circulans ATCC 31805 using AntiSmash 4 identified a NRPS containing BGC that had characteristics consistent with production of the octapeptins. The octapeptin BGC (Accession LN999013) contains eight NRPS modules that are over distributed three open reading frames (Figure 1). The NRPS genes in the octapeptin cluster obey the colinearity principle, and the amino acid specificity predictions modules are an closely match for the known octapeptin structures (Figure 1, Table 1). Modules one and four of the octapeptin BGC contain epimerisation domains, in agreement with the presence of D-amino acids in the corresponding positions of the octapeptins. The presence of predicted DCL-specific condensation domains downstream of each of these epimerisation domains is also consistent with of incorporation of a D-amino acids.
Table 1.
Substrate specificity predictions for adénylation domains in the octapeptin biosynthetic gene clusters.
| Prediction* | Pocket** | Observed*** | |
|---|---|---|---|
| A1 | Dab | DVGEISAIDK | Dab |
| A2 | Dab | DVGEISAIDK | Dab |
| A3 | Dab | DVGEISAIDK | Dab |
| A4 | Leu | DAWIIGAIVK | Phe |
| A5 | Phe | DAFTIGAICK | Leu |
| A6 | Dab | DVGEISAIDK | Dab |
| A7 | Dab | DVGEISAIDK | Dab |
| A8 | Leu | DAWIIGAIVK | Leu |
Substrate specificity prediction furnished by SANDPUMA algorithm (Chevrette et al. 2017),
10 aa Statchelhaus code from NRPS-predictor 2 output,
Residue observed in Octapeptin A1.
Consistent with the N-acyl group found in the octapeptins, the first C-domain in the octapeptin BGC was annotated as a starter C-domain. This motif is common in the first domain of lipopeptides and indicates condensation of and incoming fatty acyl-CoA initiates the biosynthesis. There is no obvious acyl-CoA ligase candidate in the octapeptin cluster, this may indicate that the gene responsible for this step in the biosynthesis is located elsewhere in the chromosome, as was previously observed for the polymyxins.
In addition to the core NRPS enzymes responsible for biosynthesis of the peptide backbone, the octapeptin cluster contains an enzyme closely related (98% aa identity) to the transaminase PlpA found in the pelgipeptin BGC. Enzymes of this type are known to convert aspartate β-semialdehyde to Dab (which is the substrate for A domains 1,2,3,6 and 7 in the octapeptin biosynthetic pathway (Figure 1). The cluster also contains a Sfp-type 4′-phosphopantiethienyl transferase as well as regulatory proteins and transporters.
In toto synthesis and in vitro antibacterial activity.
Owing to their nonribosomal biosynthetic origins, octapeptins are produced as a mixture of closely related components (Konishi, et al., 1983; Meyers, et al., 1973; Parker and Rathnum, 1975; Parker and Rathnum, 1973; Shoji, et al., 1976; Shoji, et al., 1980; Sugawara, et al., 1983). In the present study, we synthesised for the first time a pure major component, octapeptin C4, via a highly efficient and robust Fmoc-based solid-phase synthesis procedure. Antimicrobial activity was subsequently screened against a panel of ATCC and clinical isolates of polymyxin-susceptible and -resistant Gram-negative strains of P. aeruginosa, A. baumannii, K. pneumoniae and Enterobacter cloacae, and Gram-positive E. faecium and S. aureus (Table 2).
The octapeptins displayed good antibacterial activity against the polymyxin-susceptible strains of P. aeruginosa and K. pneumoniae (Table 2). Octapeptin C4 (MICs 4–16 μg/mL) was less potent against the polymyxin-susceptible strains compared to both polymyxin B and colistin (0.25–1 μg/mL). Notably, the octapeptins displayed poor activity against the polymyxin-susceptible strains of A. baumannii tested, presumably due to differences in the outer membrane lipid A of this species compared to other Gram-negatives. Compared to polymyxin B and colistin (MICs 4–128 μg/mL), the octapeptins showed improved activity (MICs 0.5–32 μg/mL) against polymyxin-resistant clinical isolates of P. aeruginosa, A. baumannii and K. pneumoniae (Table 2). However, octapeptins MCC5387, FADDI-116, −117, and −118 were generally less active against the polymyxin-resistant strains (MIC ≥32 μg/mL). Furthermore, we screened the activity of the octapeptins against a panel of mcr isolates of K. pneumoniae and Escherichia coli (Table S4). Interestingly, FADDI-114 – 118 showed comparable MICs to colistin and polymyxin B (MICs 4–16 μg/mL) against the mcr isolates, whereas FADDI-118 was essentially inactive (MICs ≥16 μg/mL). We speculate that the comparable MICs between the octapeptins and the polymyxins is due to the relatively low levels of polymyxin-resistance conferring modified lipid A in the outer membrane of these isolates; which is reflected by their MICs which show a relatively low level of polymyxin resistance. Static time-kill studies were performed to evaluate the antimicrobial kinetics of polymyxin B (Figure 4a), colistin, and octapeptin C4 (Figure 4b) against a polymyxin-resistant P. aeruginosa strain FADDI-PA070. Polymyxin B and colistin showed no antimicrobial effect at 1 or 4 μg/mL, following the same activity profile as the control. Polymyxin B at 32 μg/mL showed six log10 CFU/mL kill from 1–4 h, followed by regrowth, whereas colistin at 32 μg/mL showed poor killing activity. In comparison, octapeptin C4 displayed nearly a six log10 CFU/mL kill with no bacterial growth being detected between 0.5–6 h at 32 μg/mL, and nearly 4 log10 CFU/mL reduction compared to the control from 2–6 h at 4 μg/mL. However, regrowth of resistant bacteria was evident at 24 h, resulting in a final three and five log10 CFU/mL reduction for polymyxin B and octapeptin C4, respectively.
Figure 4.
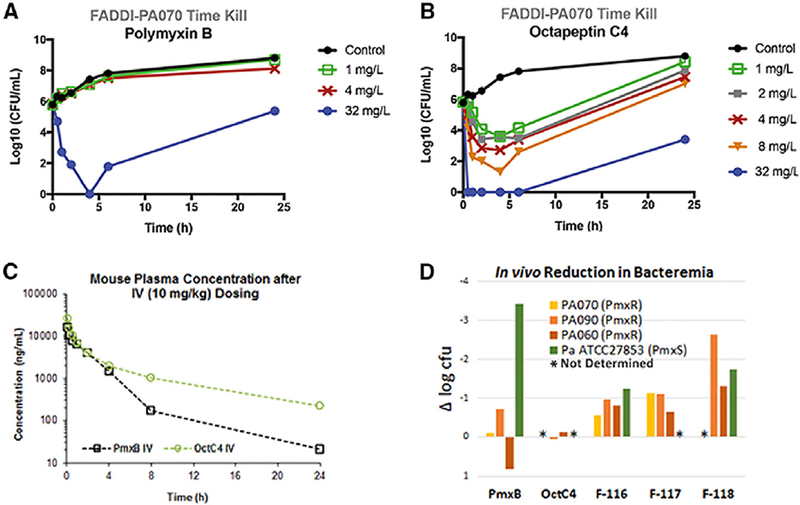
Time-kill profiles of (a) polymyixn B, and (b) octapeptin C4 against P. aeruginosa FADDI-PA070 (MIC of polymyxin B ≥ 128 mg/L; MIC of octapeptin C4 = 1 mg/L). (c) Pharmacokinetic profiles of polymyxin B and octapeptin C4 after an intravenous (10 mg/kg) dose in mice. (d) In vivo efficacy against polymyxin-resistant (FADDI-PA070, FADDI-PA090 and FADDI-PA060) and -susceptible strain of P. aeruginosa (ATCC 27853) in a mouse bacteremia model. Polymyxin B, octapeptin C4 and analogs were dosed at 4 mg/kg intravenously.
Structure-activity relationships.
At present, we have made significant progress in understanding polymyxin SAR (Gallardo-Godoy, et al., 2016; Storm, et al., 1977; Vaara, 2013; Velkov, et al., 2010), however, there is virtually no information on the SAR of the superior antimicrobial activity of the octapeptins against polymyxin-resistant strains. To optimise derivatives with variations in hydrophobicity to explore potential differences in protein binding and in vivo efficacy (vide infra), we made a series of novel octapeptin derivatives with substitutions that probed the variations between the octapeptins and polymyxins in the β-hydroxy vs. alkyl fatty acyl chain component and the Thr8 vs. Leu8 alterations (Table 2).
The octapeptins are N-terminally acylated with longer nonanoyl (C9) or decanoyl (C10) β-hydroxy-fatty acyl chains, as opposed to the polymyxins, which are acylated with shorter heptanoyl (C7) or octanoyl (C8) fatty acyl chains (Meyers, et al., 1973; Meyers, et al., 1973; Meyers, et al., 1974; Parker and Rathnum, 1975; Parker and Rathnum, 1973; Puar, 1980; Rosenthal, et al., 1977; Rosenthal, et al., 1976; Shoji, et al., 1980; Sugawara, et al., 1983). It may be significant that the β-hydroxyl group in the octapeptin fatty acyl chain is separated from the cyclic peptide component by the same number of bonds as the Thr2 hydroxyl substituent in the polymyxin structure. Previous literature reports have shown that the N-terminal fatty acyl substituent is essential for antimicrobial activity as the activities of deacylated octapeptins against P. aeruginosa were 20- to 100-fold lower than the polymyxins (Storm, et al., 1977). This is not unlike the deacylated nonapeptide forms of colistin or polymyxin B (Viljanen and Vaara, 1984). Specifically, the naturally occurring octapeptins possess either the straight chain 3(R)-hydroxy-decanoic acid or the branched chain 8-methyl-3(R)-hydroxy-nonanoic acid or 8(S)-methyl-3(R)-hydroxy-decanoic acids (Meyers, et al., 1973; Shoji, et al., 1980; Sugawara, et al., 1983). Our studies indicate that the hydroxyl group appears to be dispensable, as substitution of the N-terminus with non-branched decanoyl (C10) fatty acyl chains had no major effect on the antimicrobial activity of octapeptins (FADDI-115 vs. octapeptin C4, Table 2), notably retaining activity against polymyxin-resistant strains. Decreasing the length of the N-terminal fatty acyl chain from decanoyl (C10) to octanoyl (C8) decreased the antimicrobial activity against polymyxin-susceptible and -resistant strains, particularly against A. baumannii and K. pneumoniae, but not P. aeruginosa (e.g. FADDI-115 vs. FADDI-117, Table 2).
A significant difference between the polymyxins and octapeptins is the absence of Thr at position 8 in the heptapeptide ring of the octapeptins, where it is replaced with Leu. Notably, reverting this residue to the polymyxin substituent (Leu→Thr position 8 substitution; FADDI-116, −118 and MCC5387) did not dramatically impact the antibacterial activity compared to octapeptin C4.
NMR structure determination.
1H and 13C assignments (Data S1) were derived from analysis of 2D NOESY, ROESY and 2D TOCSY spectra, as well as 2D 13C-HSQC and 2D 13C-H2BC spectra (Figures S2, S3 & Data S1). The 2D NOESY spectra recorded at 20–25°C of either peptide displayed only intra and sequential NOE cross peaks. At 3–5°C, a medium range NOE between D-Phe4-Hα/Dab6-HN and another very weak NOE between D-Dab1-Hα/Dab3-HN were observed (Data S1) and are indicative of a pair of a turn-like motifs spanning residues 3 to 6 and from the lipid to Dab3. These motifs are in low abundance as evidence by strong, sequential Hα (i)-HN(i+1) NOEs combined with 3JHNHα and amide temperature coefficients that were not significantly reduced for these residues (Data S1). While a reduced amide temperature coefficient for Dab7 (OctC4 = −0.9 ppb/K, FADDI-115 = −2.0 ppb/K) indicates a significantly protected amide, there was no supporting structural data to infer a hydrogen bond acceptor as part of a regular turn motif. The amide chemical shifts of the two peptides in water were very similar (Figure S2a), as expected for a dynamically structured lipopeptide that differs only by a hydroxy substitution. Notably, no NOEs were observed to indicate an interaction between the decanoyl lipid tail and the main body of the cyclic peptide (Figure S3b).
To investigate the structure of the lipopeptides in a more functional environment (e.g., phospholipids) we recorded NMR data in the presence of an excess of deuterated n-dodecylphosphocholine (d38-DPC) micelles as a membrane mimetic. Titrating increasing amounts of d38-DPC micelle into ~2.0 mM octapeptin C4 led to widespread chemical shift changes in the 1D 1H NMR spectrum of the lipopeptide under conditions of fast exchange on the chemical shift timescale, reaching an end-point at approximately 100–120 mM DPC. General broadening of the signals was consistent with a bound lipopeptide/micelle complex with a combined molecular mass as expected to be in the order of 20–30 kDa. Assignments of the 1H and 13C signals of the lipopeptides (Data S1) were similarly carried out from analysis of 2D TOCSY (50 ms mixing), NOESY (50–200 ms mixing) and 13C HSQC spectra and by comparison with those spectra recorded in acetate-buffered water (vide supra).
Concordant with the formation of a slowly tumbling micelle/lipopeptide complex was a substantial increase in the size and number of the cross peaks in the 2D NOESY spectra (Figure S2). Notably, NOEs were assigned that identified spatial proximity between the decanoyl protons and the aromatic protons of D-Phe4 (Figure S3). Moreover, the high quality of the 2D NOESY spectra facilitated the calculation of the structure of each lipopeptide using a total of 222 (octapeptin C4) and 198 (FADDI-115) experimentally determined distance constraints, within the program CYANA 2.1. Final statistics are documented in Data S1.
The structures showed highly converged ensembles with an overall backbone r.m.s.d. of 0.05 Å (octapeptin C4) and 0.06 Å (FADDI-115) (Figure 2c) with both lipopeptides showing very similar overall structures. The striking feature is a three pronged fork motif constructed in part by the decanoyl lipid tail, which drops down past the side chain of Dab3 and packs against the aromatic side chain of the D-Phe4-Leu5 diad, completing the hydrophobic fork. Scrutiny of the NOEs revealed 6 additional, weak NOE contacts for octapeptin C4 between the lipid tail and the side chain of Dab7, which pull the lipid tail inward and orient the Dab7 side chain downward compared to that observed in FADDI-115. A plausible driving interaction is likely between the β-hydroxyl group in octapeptin C4 and the charged sidechain of Dab7, or perhaps the hydroxyl interacts with the hydrophilic region of the micelle. While these and other subtle structural variations are likely reflected in the observed differences in the amide chemical shifts (Figure S2), the high-resolution NMR structures (Figure 2a) clearly show an overall structural similarity of octapeptin C4 and FADDI-115 as anticipated. Collectively, our NMR data show that the structures of octapeptin C4 and FADDI-115 are substantially more compact compared to polymyxin B. Notably, both octapeptin lipopeptides display a very similar three pronged hydrophobic triad, consisting of the N-terminal hydroxyl-fatty acyl chain, and the D-Phe-Leu segment. It is likely that this shared hydrophobic motif is instrumental in their mode of action against polymyxin-resistant strains as discussed below.
Figure 2.
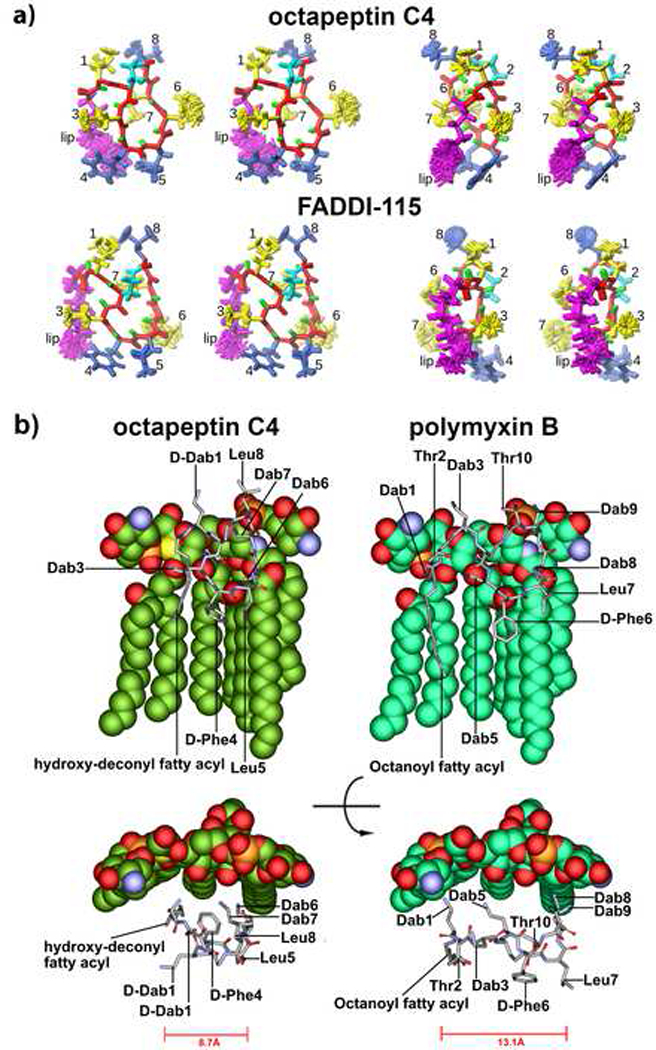
(a) Stereo view of the superposition of the final 20 NMR structures of octapeptin C4 (top) and FADDI-115 (bottom) viewed from the front (left) or side (right) as determined by NMR. The backbone is shown in red and the cyclized side-chain of Dab2 in cyan. Hydrophobic residues are shown in blue and positively charged Dab residues in yellow. (b) SAR model for the interaction of octapeptin C4 with 4-amino-4-deoxy-L-arabinose (L-Ara4N) modified lipid A which confers polymyxin-resistance (top and side views are shown). For comparison the polymyxin B-lipid A complex is shown in identical views.
SAR model.
We employed the NMR solution structure of octapeptin C4 to generate a SAR model with the aminoarabinose modified lipid A (Figure 2b), representative of the modified lipid A structure commonly seen in polymyxin-resistant P. aeruginosa strains (Miller, et al., 2011). The SAR model revealed that the four cationic Dab side chains form polar contacts with the Kdo2 (3-Deoxy-D-manno-oct-2-ulosonic acid) sugars, which is unlike the Dab-phosphate contacts in the polymyxin B(Mares, et al., 2009) (Mares et al., 2009). The three-pronged hydrophobic triad of the octapeptin structure forms hydrophobic contacts with the fatty acyl chains of lipid A. Again, this is reminiscent of the polymyxin B-lipid A complex, albeit, the octapeptin triad provides a more focused series of hydrophobic contacts that are all close together. The formation of the triad is a result of the more compact conformation (8.7 Å octapeptin C4 vs. 13.1 Å polymxyin B, Figure 2b) adopted by the octapeptin by virtue of the truncated linear peptide segment and the D-amino residue at position 1, which rotates the N-terminal hydroxyl fatty acyl close to the D-Phe-Leu segments. In comparison, the hydrophobic contacts between the N-terminal fatty acyl and D-Phe-Leu of polymyxin B and the lipid A fatty acyls are further spaced apart, due to the less compact structure of polymyxin B. Polymyxins are heavily reliant on the complementary charge interaction to initiate lipid A complex formation and are easily repelled by the lipid A of polymyxin-resistant strains where the phosphates are modified with cationic sugars such as aminoarabinose (Velkov, et al., 2010). The unique compact conformation and hydrophobic triad of the octapeptins would allow them to effectively bind to lipid A of polymyxin-resistant strains in which the lipid A phosphates are blocked with cationic sugars that normally repel polymyxins.
Interactions with model membranes, lipid A and LPS-binding of lipopeptides to model membranes and lipid A evaluated by SPR.
Polymyxins interact directly with Gram-negative bacterial lipid A (Velkov, et al., 2010). To examine whether octapeptin C4 also possessed affinity for lipid A, its ability to bind to model membranes was compared to that of colistin and polymyxin B using surface plasmon resonance (SPR) with model membrane sensor chips (L1 sensor chip) in a Biacore 3000 system. Affinity for model membranes composed of neutral synthetic lipids 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), or negatively-charged POPC / 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol (POPG) (4:1 molar ratio) more representative of bacterial membranes were compared. An increase in the membrane-binding affinity in the presence of negatively-charged membranes was evident for colistin, polymyxin B and octapeptin C4 when binding to POPC and POPC/POPG membranes was compared (Figure 3a, top & middle). This affinity was further increased with lipid A-containing membranes (Figure 3a, bottom). Interestingly, octapeptin C4 showed an increased ability to bind lipid A compared to colistin and polymyxin B, supporting the SAR model that indicated a stronger hydrophobic binding interaction.
Figure 3.
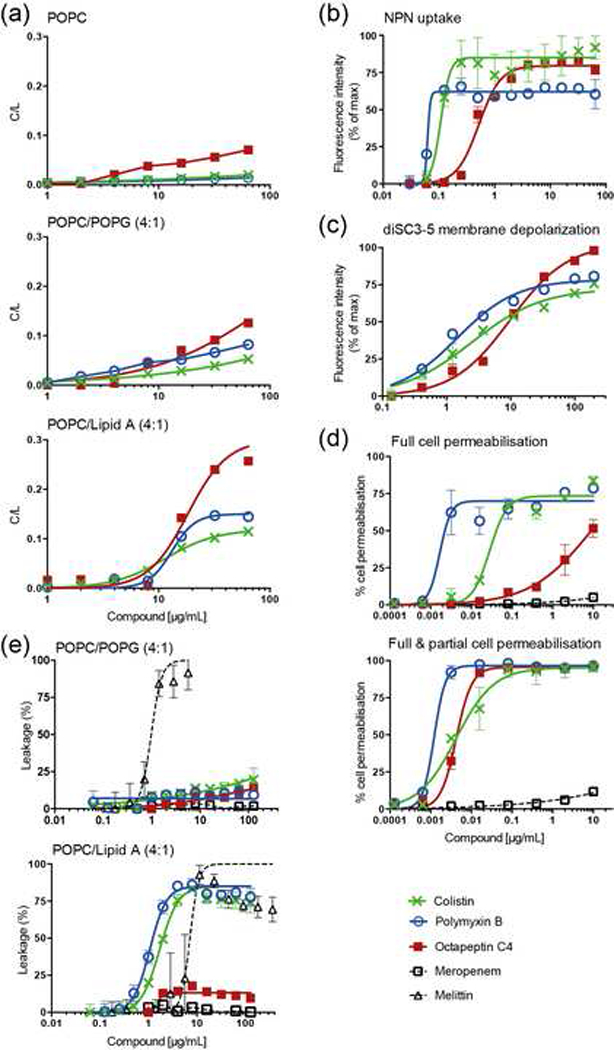
(a) Binding of colistin, polymyxin B and octapeptin C4 to phospholipid bilayers as followed by SPR. Three lipid systems are compared: From top to bottom, POPC, POPC/POPG (4:1 molar ratio), and POPC/E. coli Lipid A (4:1 molar ratio). Data were normalized and are represented as compound-to-lipid ratio (C/L (mol/mol)) using a reporting point at the end of the compound injection. (b) E. coli outer membrane permeabilization as measured with NPN fluorescence emission (excitation at 340 nm and emission at 405 nm) normalized for the maximum. Data points represent mean ± SD (n=3). (c) Membrane depolarization of E. coli cells measured using fluorescence emission (excitation at 620 nm and emission at 670 nm) of diSC3–5 dye following 30 min incubation with lipopeptides. Data points represent mean ± SD (n=3), normalized for the maximum. (d) Cell membrane permeabilisation of E. coli as followed by SYTOX Green fluorescence emission and flow cytometry (laser excitation 488 nm, emission at 530 nm, band pass 30nm). Meropenem, a compound that does not permeabilize E. coli membranes was used as a negative control. Top panel, Percentage of full cell permeabilisation calculated as a measure of the percentage of cells with fluorescence emission signal with intensity in the104 to 105 range. Bottom panel, Percentage of full and partial cell permeabilisation calculated as a measure of the number of cells with fluorescence emission intensity within certain ranges. Data points represent mean ± SD (n=3); 100,000 cells were screened per sample. (e) Leakage of lipid vesicles induced by lipopeptides as followed by carboxyfluorescein (CF) dequenching. Large unilamellar vesicles (LUVs) composed of POPC/POPG (4:1) (top) or POPC/E. coli Lipid A (4:1) (bottom) loaded with 50 mM (CF) were incubated with lipopeptides for 20 min. Melittin and Meropenem were used as a positive and negative controls, respectively. Data points represent mean ± SD (n=3). All data (a-e) are represented by nonlinear regression curve fitting in GraphPad Prism 6.0d.
Impact of lipopeptide treatment on the inner and outer membrane of Gram-negative bacteria. Outer membrane permeability NPN assay.
N-Phenyl-1-naphthylamine (NPN) is an uncharged lipopilic probe with low fluorescence quantum yield in an aqueous environment that becomes fluorescent when partitioned in a hydrophobic environment, such as the one found in lipid membranes (Velkov, et al., 2013; Velkov, et al., 2012). The outer membrane limits the free diffusion of hydrophobic solutes, such as NPN (Nikaido, 2003). However, once permeated, intercalation of NPN into the underlying phospholipid inner leaflet and the cytoplasmic membrane produces an increase in fluorescence emission and can, therefore, be used to examine bacterial outer membrane permeabilisation. Exposure to lipopeptides resulted in permeabilised log-phase E. coli (ATCC 25922) cells, which was seen as an increase in NPN uptake upon incubation with increasing doses of the lipopeptides (Figure 3b). Octapeptin C4 was less effective at permeabilising the membrane, requiring a higher concentration for maximum NPN uptake compared to polymyxin B or colistin, potentially reflecting its reduced potency against polymyxin-sensitive strains.
Membrane depolarisation diSC3–5 assay.
3,3’-Dipropylthiadicarbocyanine iodide (diSC3–5) is a cationic potentiometric carbocyanine probe that can be used to examine changes in the membrane potential of intact bacterial cells (Cheng, et al., 2014). If the bacterial membrane is in its native hyperpolarised state, the cationic dye accumulates in the membrane and its fluorescence emission is self-quenched. However, if membrane potential is disrupted, the dye loses affinity for the membrane and is released into the aqueous environment resulting in an increase in the overall fluorescence emission. Figure 3c shows that octapeptin C4, polymyxin B and colistin induced membrane depolarisation in E. coli cells in a dose-dependent manner. At lower concentrations (< 10 μg/mL) octapeptin C4 was less effective than polymyxin B and colistin, but it was relatively more effective at higher concentrations (> 100 μg/mL).
Inner membrane permeabilisation by FACS analysis of Sytox Green staining.
The uptake of SYTOX® Green was followed by flow cytometry (Figure 3d) and our results suggest that polymyxin B, colistin and octapeptin C4 disrupted both the outer and inner membranes of Gram-negative cells. Interestingly, two distinct cell populations were found when treated with the compounds (see fluorescence histograms in Figure S4), consisting of a population emitting mean fluorescence intensities of 104-105, which matched that of isopropranol-killed dead cells, along with an intermediate fluorescence intensity population of 103-104, much higher than live cells (101 – < 103) but less than control dead cells. Cell populations with distinct fluorescence emission might result either from varying levels of SYTOX® Green uptake, or from cells with different amounts of nucleic acids resulting from different cell division stages. As only a single population was found in the control cells permeabilised with isopropranol, the existence of two populations suggests that polymyxin B, colistin and octapeptin C4 induced different degrees of cell damage. Octapeptin induced a similar ratio of partial cell damage as colistin and polymyxin, but not the same extent of complete cell damage/death at the same concentration (in μg/mL), which correlates with the relative MIC values of the lipopeptides.
Leakage from lipid vesicles induced by lipopeptide.
To examine if bacterial cell permeabilisation by octapeptin C4 and polymyxins is a process dependent on the ability of lipopeptide to compromise lipid bilayer integrity, independent of the presence of lipid A, permeabilisation of lipid vesicles was assessed. Leakage from lipid vesicles was quantified by carboxyfluorescein (CF) fluorescence emission de-quenching. None of the lipopeptides destabilised the membrane of vesicles composed of POPC or POPC/POPG (Figure 3e top). Polymyxin B and colistin destabilised the membrane of vesicles containing lipid A, whereas octapeptin C4 showed minimal leakage (Figure 3e). This suggests that unlike the polymxyins, the ability of octapeptins to bind to the lipid bilayer not entirely lipid A-dependent, but Lipid A is required to disrupt membrane integrity.
Binding of lipopeptides to LPS evaluated by SAXS analysis.
Synchrotron radiation SAXS experiments on the aggregate structure of P. aeruginosa serotype 10 LPS describe a solution of non-lamellar vesicular particles whose morphology changes upon octapeptin titration (Figure S5). In the absence of the octapeptin analog FADDI-117 a solution of large aggregates with an average radius of gyration, Rg ~ 30.2 nm and a maximum particle dimension, Dmax ~ 97 nm was observed. No diffraction peaks, evidence of a partially ordered or lamellar structure, were seen. Upon the addition of FADDI-117 the LPS particles clearly increase in size, observed as a significant increase in forward scattering intensity as the peptide concentration increased. In addition, the average Rg and Dmax increased to ~ 32.4 nm and 106 nm, respectively, at the final titration point. Furthermore, the particles undergo a change in local structure, becoming more ordered, as evidenced by the appearance of low intensity diffraction peaks at high q (~ 0.19 A−1). The same titration of P. aeruginosa serotype 10 LPS using polymyxin B produced similar diffraction peaks at high q (data not shown). A clear change of mesoscopic structure was observed by SAXS in solution for P. aeruginosa serotype 10 LPS upon the addition of octapeptin analog FADDI-117. Recent work has linked such morphological changes including increased LPS aggregate size and/or the formation of more lamellar/ordered particles to decreased endotoxemia (Gutsmann, et al., 2010).
Resistance mechanism − Lipid A isolation and structural elucidation.
To investigate the mechanism of resistance for the octapeptin class, we chose octapeptin C4 as an example. Lipid A was isolated from both the octapeptin-susceptible polymyxin resistant P. aeruginosa FADDI-PA070 strain and a paired octapeptin-resistant strain generated by treating a subculture of FADDI-PA070 with 2 μg/mL or 32 μg/mL concentrations of octapeptin C4. ESI-MS analysis revealed that the mass spectra of lipid A from the octapeptin-susceptible wild-type strain displayed predominant mass peaks at m/z 1732.05, 1748.05, 1863.11, and 1879.11, corresponding to the hexa-acylated lipid A modified by one or two aminoarabinose (L-Ara4N) sugars on its phosphates (Figure S6a) a known polymyxin resistance modification. The peaks at m/z 1651.09 and 1667.08 represent the dephosphorylation (Δm/z = −79.97) of the lipid A species at m/z 1732.05 and 1748.05. The mass peaks at m/z 1560.92, 1576.92, 1692.98, and 1708.98 correspond to the penta-acylated lipid A modified by one or two L-Ara4N moieties on the phosphates. The two main peaks at m/z 1480.95 and 1496.96 represent the removal of one phosphate group from the lipid A species at m/z 1560.92 and 1576.92, respectively. The two peaks at m/z 1930.21 and 1946.20 represent the addition of one palmitate (Δm/z =+237.23) to the lipid A species at m/z 1692.98, and 1708.98; which was confirmed through MS/MS spectrum (Figure S6d).
The mass spectrum profile of the lipid A from the octapeptin-resistant P. aeruginosa FADDI-PA070 exposed to antibiotic at the MIC (2 μg/mL) did not display any noticeable mass peak differences compared to the octapeptin-susceptible wild-type P. aeruginosa FADDI-PA070 strain (Figure S6b), albeit the relative intensities of the lipid A species that lack a hydroxyl group at the secondary C12 fatty acyl chains (m/z 1480.95, 1692.98, and 1863.11) increased dramatically. In comparison, the lipid A profile of the lipid A from the octapeptin-resistant P. aeruginosa FADDI-PA070 isolated at 32 μg/mL displayed significant variations compared to the wild type strain (Figure S6c). Notably, most of the hexa- and penta-acylated lipid A peaks presented in the wild type strain were absent. A noticeable mass peak at m/z 1933.23, unique to the 32 μg/mL resistant strain was seen, which represents the palmitoylation of lipid A to the primary fatty acyl chain (Figures S6f). The MS/MS spectra suggested this was a different compound to the one at m/z 1930.21 (Figure S6 d, e).
Pharmacokinetics.
Following an IV bolus injection of 10 mg/kg in mice, octapeptin C4 had a substantially lower total clearance (4.1 mL/min/kg) and a longer half-life (6.6 h) compared to polymyxin B (7.3 mL/min/kg and 3.6 h, respectively) (Figure 4c & Table S1) (Li, et al., 2003). Polymyxin B was 51% bound to human plasma proteins. In comparison, octapeptin C4 was highly bound to plasma proteins (> 90%), which would account for the extended half-life.
In vivo efficacy in a mouse blood infection model.
A proof-of-concept study was performed using a neutropenic mouse blood infection model to compare the in vivo efficacy of polymyxin B, alongside octapeptin C4, FADDI-116, FADDI-117 and FADDI-118, against a polymyxin-susceptible ATCC27853 strain of P. aeruginosa and three polymyxin-resistant strains, FADDI-PA070, FADDI-PA090 and FADDI-PA060 (Figure 4d & Table S2). Against the polymyxin-susceptible isolate P. aeruginosa ATCC27853, all the octapeptin analogues were ∼ 50% less active than polymyxin B, which was consistent with their less potent in vitro MICs. The novel octapeptin-like lipopeptides (FADDI-116 to −118), but not octapeptin C4, were more effective than polymyxin B at reducing the bacterial burden in the blood against the two polymyxin-resistant P. aeruginosa clinical isolates. We propose that the reduced in vivo efficacy of octapeptin C4 compared to the FADDI lipopeptides is primarily due to its high plasma protein binding (> 90%, human), which in turn is a result of its increased hydrophobicity. To test this hypothesis, we employed the SAR model to selectively reduce the hydrophobicity of the octapeptin scaffold without compromising the antibacterial activity.
We initially selectively reduced the length of the N-terminal fatty acyl group from C10 (octapeptin C4) to C8 (FADDI-117). This had the effect of reducing the plasma protein binding (> 90% vs. 68%, human plasma) and significantly increasing (> 5-fold) the activity against the two polymyxin-resistant isolates. We next selectively replaced position Leu8 with a Thr8 (as per the polymyxin structure), while maintaining the C10 N-terminal fatty acyl length to generate FADDI-116, which again increased the in vivo activity >5-fold against the resistant isolates. Notably, the Leu8→Thr FADDI-116 (85%, human plasma; 92% rat plasma) substitution had a less marked effect on reducing the plasma protein binding than the N-terminal C10→C8 FADDI-117 substitution (68%, human plasma); suggesting that the N-terminal fatty acyl moiety of the octapeptin scaffold is driving the plasma protein binding. Finally, we combined both modifications to generate FADDI-118, which yielded a substantially lower level of plasma protein binding (53%, human plasma; 75% rat plasma) and dramatically improved the in vivo activity > 10-fold against the polymyxin-resistant isolates, compared to octapeptin C4. Interestingly, polymyxin B (69%, human plasma; 83% rat plasma), which incorporates structural characteristics seen in both the octapeptin C4 and FADDI-118 scaffolds, displayed plasma protein binding levels intermediate to that of the octapeptin C4 and FADDI-118 levels. Our in vitro and in vivo efficacy data indicate the significant potential of novel octapeptin-like lipopeptides as new antibiotics against polymyxin-resistant Gram-negative ‘superbugs’.
Safety and tolerability.
There was no detectable haemolysis of human red blood cells after exposure to octapeptin C4, polymyxin B or colistin at concentrations up to 100 μg/mL. In vitro assessment of potential nephrotoxicity as measured by monitoring lactate dehydrogenase (LDH) and γ-glutamyltransferase (GGT) release from human tubular (hPT) cells showed that octapeptin C4 exhibited reduced toxicity relative to polymyxin B, colistin and the gentamicin positive control (Figure 5a). A subcutaneous dose of 40 mg/kg of polymyxin B in mice showed acute toxic effects (e.g., decreased respiratory rate, reduced activity). In marked contrast, the same subcutaneous dose of octapeptin C4, FADDI-117 or FADDI-118 showed no adverse effects in mice.
Figure 5.
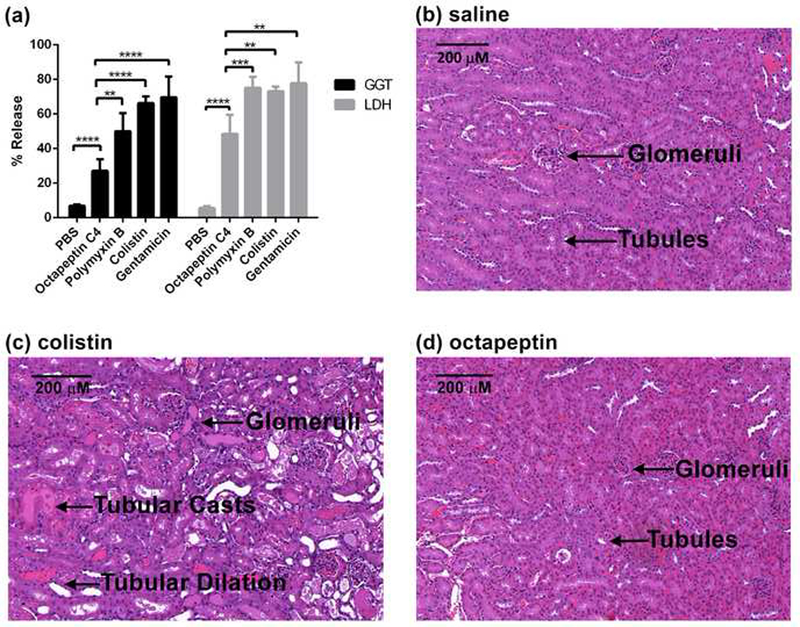
(a) In vitro measurements of drug-induced nephrotoxicity using LDH and GGT release assays in hPT cells. hPT cells were incubated with 128 μg/mL of polymyxin B, colistin and octapeptin C4 for 4 h (n= 6 except for colistin which was n=3). Gentamicin was used as the positive control. Error bars: SD, ns, no statistical significance; **, P<0.01; ****, P<0.0001. (b)-(d) Microscopic images of the cortex section of the kidneys of mice treated with (b) saline control, or an accumulated dose of 72 mg/kg, (c) polymyxin B, or (d) octapeptin C4.
Nephrotoxicity is the major dose-limiting factor for polymyxin B and colistin therapy (Yousef, et al., 2012). The kidneys of three groups of mice (n = 3 per group) treated with octapeptin C4 subcutaneously (12 mg/kg every 2 h, accumulated dose 72 mg/kg in 24 h) were subjected to histopathological examination and compared to the kidneys of mice treated with an identical treatment of polymyxin B, colistin or a saline control (Figure 5b–d & Table S3). Micro- and macro-morphological examination of kidney sections from the octapeptin C4 treated mice revealed no significant lesions in the cortical, medullary or papillary regions. The kidneys of the mice treated with octapeptin C4 essentially resembled the kidneys of mice treated with the saline control and no histological grade was given. In comparison, the kidneys from the colistin and polymyxin B, FADDI-117 and FADDI-118 treated mice displayed damaged tubules, with marked tubular dilation and degeneration. Tubular casts were identified in both the cortex and medulla and the animals were identified to have severe Grade 1–2 lesions. In terms of structure-nephrotoxicity relationships, this finding would suggest that the truncation of the linear tripeptide segment through the deletion of the Dab1-Thr2 segment renders the octapeptin scaffold less nephrotoxic, compared to the polymyxins. Finally, the high protein binding of octapeptin C4 compared to colistin, polymyxin B, FADDI-117 and FADDI-118 may play a protective role in reducing kidney exposure.
Significance
The octapeptin lipopeptides have superior antimicrobial activity against polymyxin-resistant XDR Gram-negative bacteria and potentially octapeptin C4 has reduced nephrotoxicity compared to the last-line antibiotics polymyxin B and colistin. We employed a new SAR model to engineer FADDI-118 by incorporating some of the hydrophilic features of the polymyxins, producing a superior octapeptin analog with reduced plasma protein binding whilst maintaining superior activity against polymyxin-resistant strains. Based on their higher-order activity and lack of cross-resistance, improved octapeptin analogues such as FADDI-118 represent viable clinical candidates if nephrotoxicity can be managed. Overall, this study lays the foundations for the development of octapeptins as a new-generation of novel lipopeptide antibiotics to treat life-threatening XDR infections.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Tony Velkov (Tony.Velkov@unimelb.edu.au).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial strains and growth conditions.
All bacteria were stored at −80ºC in tryptone soya broth (TSB, Oxoid Australia). Prior to experiments, parent strains were sub-cultured onto nutrient agar plates (Medium Preparation Unit, University of Melbourne, VIC, Australia). Overnight broth cultures were subsequently grown in 5 mL of cation-adjusted Mueller-Hinton broth (CaMHB, Oxoid, West Heidelberg, VIC, Australia), from which a 1 in 100 dilution was performed in fresh broth to prepare mid-logarithmic cultures according to the optical density at 500 nm (OD500nm = 0.4 to 0.6). All broth cultures were incubated at 37°C in a shaking water bath (180 rpm).
Animals used, husbandry, housing and ethics permissions
For all animal experiments, Swiss mice (female, 6 weeks, body weight 20 – 25 g) were obtained from Monash Animal Services. Animals were housed in micro-isolated metabolic cages in a temperature (21 ± 3°C) and humidity controlled facility with a 12-h light-dark cycle (06:00–18:00) and acclimatized for 2 days. All animal experiments were approved by the WuXi Institutional Animal Care and Use Committee and the Monash Institute of Pharmaceutical Science Animal Ethics Committee.
METHOD DETAILS
Determination of the nucleotide sequence of the octapeptin biosynthetic locus and prediction of encoded function.
Genomic DNA from Bacillus circulans ATCC 31805 was isolated using a Qiagen genomic DNA extraction kit. Deploying a whole genome shotgun strategy, the genomic DNA was sheared to fragments of approximately 500 base pairs (bp); sequencing libraries were then prepared from the sheared DNA according to Illumina protocols for paired-end sequencing and run on an Illumina GAIIx. The 150 bp, paired-end read set was de novo assembled using Velvet (Zerbino and Birney, 2008). The read set for Bacillus circulans strain ATCC 31805 is available under BioProject PRJEB12658. The sequence for the octapeptin synthetase locus is available under the ENA accession number LN999013.
Materials for peptide sysnthesis.
Trifluoroacetic acid (TFA), and diphenylphosphorylazide (DPPA), triisopropylsilane (TIPS) and hydrazine, were purchased from Sigma-Aldrich (Sydney, NSW, Australia). Diisopropylethylamine (DIPEA) and piperidine were obtained from Auspep (Melbourne, Australia). Fmoc-Dab(ivDde)-OH, Fmoc-D-Dab(Boc)-OH, Fmoc-Dab(Boc)-OH and 1H-benzotriazolium-1-[bis(dimethylamino)methylene]-5-chloro hexafluoro- phosphate-(1), 3-oxide (HCTU) was purchased from Chem-Impex International (USA). Fmoc-D-Phe-OH, Fmoc-Thr(tBu)-OH and Fmoc-Leu-OH were purchased from Mimotopes (Melbourne, Australia). Fmoc-Thr(tBu)-TCP-Resin and Fmoc-Leu-TCP-Resin was obtained from Intavis Bioanalytical Instruments (Germany). Diethyl ether, dichloromethane (DCM), dimethylformamide (DMF), methanol (MeOH) and acetonitrile (MeCN) were obtained from Merck (Melbourne, Australia). For compound MCC5387, Fmoc -amino acids including N-α- Fmoc-N-γ-Boc-L-2,4-diaminobutyric acid (Fmoc-L-Dab(Boc)-OH), N-α-Fmoc-N-γ-Alloc-L-2,4-diaminobutyric acid (Fmoc-L- Dab(Alloc)-OH), O-(1H-6-chlorobenzotriazole-1-yl)-1,1,3,3-tethramethyluronium hexafluorophosphate (HCTU) were purchased from Chem-Impex International Inc. (Wood Dale, IL, USA). 3,4-Dihydro-2H-pyran-2-yl-methoxymethyl polystyrene resin (DHP HM resin 100 – 200 mesh) was purchased from Novabiochem (Merck). triethylsilane (Et3SiH) and diisopropylethylamine (DIPEA), tetrakis(triphenylphosphine)palladium(0) (Pd[PPh3]4), diphenyl phosphoryl azide (DPPA) and phenylsilane (PhSiH3), allyl bromide, hydrazine (H2NNH2), sodium diethyl dithiocarbamate trihydrate (C2H5)2NCSSNa × 3H2O) and anhydrous 1,2-dichloroethane (DCE) were purchased from Sigma-Aldrich. Polymyxin B sulfate (P0972) and vancomycin hydrochloride hydrate (861987) were purchased from Sigma-Aldrich (Sydney, Australia). Cesium carbonate and pyridinium p-toluenesulfonate (PPTS) were purchased from AK Scientific (Union City, CA, USA). All other solvents were HPLC grade and all chemicals were used without further purification.
Synthesis and purification of octapeptin C4 and analogues FADDI-115, FADDI-116, FADDI-177, FADDI-118 and MCC5387.
Octapeptin C4 was synthesised by solid-phase peptide synthesis, with identity confirmed by NMR, HRMS, and (+)-ESI-TOF-MS/MS fragmentation (Becker, et al., 2017). FADDI-115 to 118 peptides were purified by RP-HPLC on a Shimadzu 2010 LC system, employing a Phenomenex Axia column (Luna C8(2), 50 × 21.2 mm ID, 100 Å, 10 micron). Solvent A was 0.1% TFA/water, and Solvent B was 0.1% TFA/acetonitrile. Peptides were eluted with a gradient of 0–60% Solvent B (@ 214 nm) over 60 min at a flow rate of 5 mLmin-1. Fractions collected were analyzed by analytical LC/MS using a Shimadzu 2020 LCMS system incorporating a photodiode array detector (214 nm) coupled to an electrospray ionization source and a single quadrupole mass analyzer. Solvent A was 0.05% TFA/water, and Solvent B was 0.05% TFA/acetonitrile. A Phenomenex column (Luna C8(2), 100 × 2.0 mm ID) was used for RP-HPLC, eluting with a gradient of 0–60% solvent B over 10 min at a flow rate of 0.2 mLmin-1. Mass spectra were acquired in positive ion mode with a scan range of m/z 200 – 2,000. MCC5387 (39 mg) was purified using an Agilent 1260 Infinity Prep HPLC on an Agilent Eclipse XDB-Phenyl 21.2 × 100 mm 5 μm column (flow 20 mL/min, mobile phase A = 0.05% formic acid in water and B = 0.05% formic acid in acetonitrile, gradient 5 → 100% B over 20 min) and then lyophilized. The compound as a formic salt was converted to TFA by re-dissolving the compound with 0.1% TFA in acetonitrile:water (1:1) (concentration of 0.5 mg/mL) and lyophilized. All final compounds were obtained as white powders, as their TFA salts. The synthesis of octapeptin analogues FADDI-115 to FADDI-118 and MCC5387 are outlined in Fig S1 and NMR data are shown in Fig S7.
Determination of minimum inhibitory concentrations (MICs) and static time-kill measurements.
MICs for each strain were determined by the broth microdilution method in 96-well polypropylene microtitre plates (Wiegand, et al., 2008). Bacterial inoculum was prepared at 106 colony-forming units (cfu) per mL with MIC determined following 18 h incubation at 37°C. For P. aeruginosa FADDI-PA070 time kill assay; ~ 20 μL of early log phase bacterial suspensions were inoculated into 50 mL of fresh media with differing concentrations of lipopeptides. The tubes were incubated at 37 oC with shaking and samples were taken from 0.5 to 24 h. The samples were diluted and plated onto nutrient agar with viable cells determined by a ProtoCOL® colony counter and the limit of detection was 20 cfu/mL.
Preparation of lipid vesicles.
SUVs (small unilamellar vesicles, 50 nm diameter) and LUVs (large unilamellar vesicles, 100 nm diameter) were prepared by freeze-thaw fracturing and sorted by size extrusion as previously described (Henriques, et al., 2009). SUVs were used in Surface Plasmon Resonance (SPR) studies, whereas LUVs were used in leakage assays. Synthetic lipids POPC and POPG were obtained from Avanti Polar Lipids (USA), and E. coli F583 diphosphoryl lipid A obtained from Sigma-Aldrich. They were used to prepare model membranes composed of POPC alone, and the mixtures POPC/POPG (4:1 molar ratio) and POPC/lipid A (4:1 molar ratio). All lipid suspensions were prepared in 10 mM HEPES buffer (pH 7.4 containing 150 mM NaCl).
Binding to model membranes evaluated by Surface Plasmon Resonance.
The ability of colistin, polymyxin B and octapeptin C4 to bind to model membranes of various compositions was evaluated by SPR, as previously described (Henriques, et al., 2008). In short, SPR binding affinities were determined on an L1 sensor chip with the Biacore 3000 (GE Lifesciences). SUVs were prepared by freeze-thaw cycles and homogenized by extrusion. The SUVs were then injected into the flow cell of the L1 sensor chip for 2000s at a low flow rate of 2 μL/min to form a lipid bilayer on the chip surface. The binding of the lipopeptides to POPC, POPC/POPG (4:1) and POPC/lipid A (4:1) were compared in a dose-dependent manner with two-fold lipopeptide concentrations ranging from 64 to 1 μg/mL. The compounds were injected for 240s onto lipid bilayers deposited onto L1 surface and the dissociation was followed for 600s. A regeneration step was added before and after every cycle using 20mM CHAPS, which cleaned the chip surface for a new cycle. All assays were repeated at least three times at 25°C and the reference flow cell responses were subtracted from all analysed data. Resulting sensograms were normalised for lipid deposition, compound mass and represented as compound-to-lipid ratio (C/L; mol/mol) as previously described for peptide-to-lipid binding affinities (Torcato, et al., 2013). By selecting a reporting time point towards the end of the compound-to-lipid association phase (t = 190 s) the relative affinities were compared (Henriques, et al., 2010).
Membrane Assays.
For outer membrane permeability assasys, NPN was added to 2 × 106 E. coli ATCC 25922 cells/mL (final NPN concentration of 20 μM) and incubated for 15 min with varying concentrations (64 to 0.015 μg/mL) of lipopeptides with the fluorescence emission intensity recorded (λexc = 340 nm, λem = 405 nm) using a Polarstar-Omega (BMG) spectrophotometer to evaluate NPN uptake by the bacterial cells (Bai, et al., 2012; Loh, et al., 1984). For cytoplasmic membrane depolarisation assasys, mid-log phase (OD ~ 0.6) E. coli ATCC 25922 cells were harvested by centrifugation and resuspended in 5 mM HEPES, 20 mM glucose, pH 7.4. The uptake of diSC3–5 dye into bacterial cells was monitored for 30 min. The fluorescence intensity (λexc = 620 nm, λem = 670 nm) was measured using a Polarstar-Omega (BMG) spectrophotometer. Inner membrane permeability. Suspensions of 1 × 107 E. coli ATCC 25922 cells/mL were incubated for 1 h at 37°C with antibiotics and lipopeptides at varying concentrations (10 to 0.0001 μg/mL) (n = 3). SYTOX® green (Invitrogen/Life Technologies), a DNA binding dye that is fluorescent when bound to nucleic acids and only enters cells with compromised plasma membranes (Roth, et al., 1997), was added at 2 μM to the compound-treated E. coli cells for 5 min at room temperature. Cells were compared for their fluorescence emission profiles by flow cytometry performed with a BD FACSCanto II (BD Biosciences; λexc = 488 nm, λem = 530 nm with 30 nm bandpass (Torcato, et al., 2013).
Leakage from lipid vesicles induced by antimicrobial compounds.
Leakage from lipid vesicles was quantified by carboxyfluorescein (CF) fluorescence emission de-quenching as previously described (Huang, et al., 2009). POPC, POPC/POPG (4:1) or POPC/Lipid A (4:1) lipid vesicle suspensions loaded with 50 mM CF and with a final lipid concentration of 5 μM were incubated with varying lipopeptides concentrations (128 μg/mL to 0.06 μg/mL). The compounds were incubated with lipid vesicles for 20 min and the fluorescence emission intensity (λexc = 485 nm, λem = 520 nm) measured to assess the ability of the antibiotics to disturb the lipid bilayer to a degree that caused CF leakage from the lipid vesicles.
Small angle X-ray scattering (SAXS).
The changes in the morphology of LPS aggregate structure caused by lipopeptides was investigated using SAXS with solutions of P. aeruginosa serotype 10 LPS (Sigma-Aldrich, Melbourne, Australia) aggregates were collected on the SAXS/WAXS beamline of the Australian Synchrotron (Melbourne, Australia) using a Pilatus-1M pixel-array detector (Dectris, Switzerland) (Kirby, et al., 2013). Measurements were performed in IGOR Pro (WaveMetrics) (Ilavsky and Jemian, 2009).
Lipid A isolation and structural elucidation.
P. aeruginosa FADDI-PA070 was subcultured onto nutrient agar plates. Then two resistant strains were isolated by treatment with octapeptin C4 at 2 μg/mL and 32 μg/mL, respectively. The resistant isolates were maintained on agar plates containing octapeptin C4 at 2 μg/mL. Overnight (20 h) cultures were subsequently grown in 5 mL of CaMHB at 37°C, from which a 1:100 dilution was performed into fresh media (octapeptin C4 at 2 μg/mL) to prepare a 100 mL culture which was then grown to an OD600nm = 0.8. Lipid A was isolated by mild acid hydrolysis (Que, et al., 2000). In brief, FADDI-PA070 cells (OD600nm= 0.8) from 100 mL of liquid culture ×via centrifugation at 3,220 g for 20 min, and then washed twice with 5 mL of PBS. The cells were re-suspended in 4 mL of PBS, then chloroform (5 mL) and methanol (10 mL) were then added to the suspension, producing a single-phase Bligh-Dyer mixture (chloroform/methanol/water, 1:2:0.8, v/v) (Bligh and Dyer, 1959). The mixture was centrifuged at 3,220 g for 15 min to remove the supernatant. The LPS pellet was washed once with chloroform/methanol/water (1:2:0.8, v/v) and resuspended in 5.4 mL of hydrolysis buffer (50 mM sodium acetate pH 4.5, 1% sodium dodecyl sulfate (Bligh and Dyer, 1959), and incubated in a boiling water bath for 45 min. To extract the lipids after hydrolysis, the SDS solution was converted into a double-phase Bligh-Dyer mixture by adding 6 mL of chloroform and 6 mL of methanol, forming a chloroform/methanol/water (1:1:0.9, v/v) mixture (Bligh and Dyer, 1959). The lower phase containing lipid A was finally extracted. Samples were dried and stored at 20C for further analysis. Structural analysis of lipid A was performed by electrospray ionization (ESI) tandem mass spectrometry in negative mode performed on a Q-Exactive Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo Fisher).
NMR structural investigations of octapeptin C4 and FADDI-115.
All NMR experiments were conducted on either a Varian Inova or a Bruker AVANCE 600 NMR spectrometer operating at a field strength of 14.1 Tesla equipped with cryoprobes with a Z axis gradient. For experiments recorded in water, 1–2.5 mM of octapeptin C4 or FADDI-115 was dissolved into 330 μL of 50 mM d3-acetate buffer, pH 4.5 containing 90% H2O/10% D2O and placed in a Shigemi microcell. 2D TOCSY (mixing time 30 ms and 80 ms), 2D 13C-HSQC, 2D multiplicity edited 13C -HSQC, 2D 13C-H2BC44 and 2D NOESY (mixing times 150 – 500 ms) were recorded at 25°C and 3–5°C. 2D ROESY experiments (100 – 300 ms) were recorded using a composite pulse for spin locking. Excitation sculpting was used for water suppression in all homonuclear experiments which were recorded with acquision times typically of t1max = 44 ms and t2max = 450 ms. Gradients were used to select coherences in the heteronuclear experiments. For experiments recorded in the presence of DPC micelles, typically 1–2 mM octapeptin C4 or FADDI-115 was dissolved in the same buffer as previous with the addition of 100–150 mM deuterated DPC (d38-DPC) (Novachem). Samples were thorougly mixed on a vortex mixer prior to acquisition. The following experiments were recorded at 25°C: 2D TOCSY (mixing time 50 ms), 2D 13C-HSQC, 2D multiplicity edited 13C-HSQC and 2D NOESY (mixing times 50 – 200 ms). A 2D NOESY experiment (mixing time 150 ms) was also recorded on each lipopeptide dissolved in 50 mM d3-acetate buffer, pH 4.9 (uncorrected), 100% D2O, 100 mM d38-DPC. A 3–9-19 WATERGATE sequence was used for solvent suppression in the 2D NOESY and 2D TOCSY experiments recorded on samples in DPC micelles. Acquisition times were typically t1max = 33 ms and t2max = 340 ms in 2D NOESY spectra. Chemical shifts were referenced to 2,2-dimethyl-2-silapentane-5-sulfonic acid at 1H 0.00 and 13C 0.0 ppm. NMR data was processed using NMRPipe (Delaglio, et al., 1995), and analyzed using SPARKY (http://www.cgl.ucsf.edu/home/sparky/) or XEASY. Structure calculations were performed using restrained simulated annealing in torsion angle space using the program CYANA 2.1 (Guntert, 2004). NOEs in the 2D NOESY data (150 ms mixing time) were assigned using the NOEASSIGN protocol and followed by manual inspection. Non-standard residues were built using MAESTRO (http://www.schröinger.com) and new library entries written for the Dab and the N-terminal fatty acyl residues. The sign of the y coordinate was swapped to invert the chirality for the D-Phe residue. 200 structures were calculated in each round of CYANA and the top 20 selected based on lowest overall target function. No ‘rama’ potential was included in the structure calculations. Structures were visualised and figures prepared using MOLMOL2K (Koradi, et al., 1996).
Binding to model membranes evaluated by Surface Plasmon Resonance.
The ability of colistin, polymyxin B and octapeptin C4 to bind to model membranes of various compositions was evaluated by SPR, as previously described (Henriques, et al., 2008). In brief, SPR binding affinities were determined on an L1 sensor chip with the Biacore 3000 (GE Lifesciences). The SUVs were then injected into the flow cell of the L1 sensor chip for 2000 s at a low flow rate of 2 μL/min to form a lipid bilayer on the chip surface. The binding of the lipopeptides to POPC, POPC/POPG (4:1) and POPC/lipid A (4:1) were compared in a dose-dependent manner with two-fold lipopeptide concentrations ranging from 64 to 1 μg/mL. The compounds were injected for 240 s onto lipid bilayers deposited onto L1 surface and the dissociation was followed for 600 s. A regeneration step was added before and after every cycle using 20mM CHAPS, which cleaned the chip surface for a new cycle. All assays were repeated at least three times at 25°C and the reference flow cell responses were subtracted from all analysed data. Resulting sensograms were normalised for lipid deposition, compound mass and represented as compound-to-lipid ratio (C/L; mol/mol) as previously described for peptide-to-lipid binding affinities. By selecting a reporting time point towards the end of the compound-to-lipid association phase (t = 190 s) the relative affinities were compared.
Mouse pharmacokinetics (PK) studies.
PK of polymyxin B and octapeptin C4 were determined at WuXiAppTec (Shanghai, China). Three mice were used for each compound/dose route combination using longitudinal sampling following a single intravenous (IV) bolus administration at 10 mg/kg or a single subcutaneous (SC) administration at 40 mg/kg. Approximately 30 μL blood was obtained via submandibular or saphenous vein at 0.083, 0.25, 0.5, 1, 2, 4, 8, and 24 h post dosing for the IV group, and with the 0.083 h time-point removed for the SC group. Plasma was separated and samples analysed on an AB Sciex API4000 (octapeptin C4) or AB Sciex Triple Quad 5500 (polymyxin B) LC-MS/MS system using ESI+ SRM detection. Plasma protein binding. Plasma protein binding was determined with pooled healthy human plasma samples from the Australian Red Cross using equilibrium dialysis as previously described (Zavascki, et al., 2008).
Toxicity assessments.
The potential nephrotoxic effect of the lipopeptides was evaluated in a mouse model (Yousef, et al., 2012). Three groups of mice (n = 3 per group) were subcutaneous injected with polymyxin B (sulfate) or octapeptin lipopeptides with a dose of 12 mg/kg every 2 h until an accumulated dose of 72 mg was achieved. At 20 h after the last dose, mice were euthanised and their kidneys removed for fixing and histopathological examination using hematoxylin and rosin staining. A graded scoring matrix was employed to illustrate the degrees of change (Yousef, et al., 2012).
QUANTIFICATION AND STATISTICAL ANALYSIS
All experiments were performed in at least triplicate (n=3, experimental replication). All data are expressed as the mean ± standard deviation (SD) using SPSS V16.0 (SPSS Inc., Chicago, USA) and the differences between groups were compared with one-way ANOVA followed by Dunnett’s multiple comparison procedure. A P value <0.05 were considered as statistically significant.
Supplementary Material
Acknowledgements
J.L. and T.V are supported by research grants from the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (R01A1070896 and R01AI079330). J.L and T.V are also supported by the Australian National Health and Medical Research Council (NHMRC). J.L. is an Australian NHMRC Senior Research Fellow. T.V. is an Australian NHMRC Industry Career Development Level 1 Research Fellow and M.A.C. is an NHMRC Principal Research Fellow (APP1059354). Research conducted by AGG, MSB, AGE, STH, JXH, MATB, and MAC was supported by NHMRC grants APP1005350 and APP1045326, and research by AGG, MSB, AGE, STH, JXH, MATB, MAC and LL by NIH grant R21AI09873¼R33AI098731–03. MSB, JXH and MATB were also supported by a Wellcome Trust Seeding Drug Discovery Award (094977/Z/10/Z). S.T.H. is the recipient of a Discovery Early Career Researcher Award (DE120103152), awarded by the Australian Research Council. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health.
Footnotes
DATA AND SOFTWARE AVAILABILITY
The read set for Bacillus circulans strain ATCC 31805 is available under BioProject PRJEB12658 (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJEB12658). The sequence for the octapeptin synthetase locus is available under the ENA accession number LN999013 (https://www.ebi.ac.uk/ena/data/view/LN999013).
Supplementary information
Supplementary Information includes Figures S1–S6, Tables S1–S4, Data S1 and experimental synthetic procedures and characterisation of FADDI-115, FADDI-116, FADDI-117, FADDI-118/MCC6399 and MCC5387, and can be found with this article online.
References
- Bai Y, Liu S, Li J, Lakshminarayanan R, Sarawathi P, Tang C, Ho D, Verma C, Beuerman RW, and Pervushin K (2012). Progressive structuring of a branched antimicrobial peptide on the path to the inner membrane target. J. Biol. Chem. 287, 26606–26617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker B, Butler MS, Hansford KA, Gallardo-Godoy A, Elliott AG, Huang JX, Edwards DJ, Blaskovich MAT, and Cooper MA (2017). Synthesis of octapeptin C4 and biological profiling against NDM-1 and polymyxin-resistant bacteria. Bioorg. Med. Chem. Lett. 27, 2407–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bligh EG, and Dyer WJ (1959). A rapid method of total lipid extraction and purification. Can. J. Biochem Physiol. 37, 911–917. [DOI] [PubMed] [Google Scholar]
- Cheng M, Huang JX, Ramu S, Butler MS, and Cooper MA (2014). Ramoplanin at bactericidal concentrations induces bacterial membrane depolarization in Staphylococcus aureus. Antimicrobial agents and chemotherapy 58, 6819–6827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, and Bax A (1995). NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- Gallardo-Godoy A, Muldoon C, Becker B, Elliott AG, Lash LH, Huang JX, Butler MS, Pelingon R, Kavanagh AM, Ramu S, et al. (2016). Activity and Predicted Nephrotoxicity of Synthetic Antibiotics Based on Polymyxin B. Journal of medicinal chemistry 59, 1068–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould IM (2009). Antibiotic resistance: the perfect storm. Int J Antimicrob Agents 34 Suppl 3, S2–5. [DOI] [PubMed] [Google Scholar]
- Guntert P (2004). Automated NMR structure calculation with CYANA. Methods Mol Biol 278, 353–378. [DOI] [PubMed] [Google Scholar]
- Gutsmann T, Razquin-Olazaran I, Kowalski I, Kaconis Y, Howe J, Bartels R, Hornef M, Schurholz T, Rossle M, Sanchez-Gomez S, et al. (2010). New antiseptic peptides to protect against endotoxin-mediated shock. Antimicrobial agents and chemotherapy 54, 3817–3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques ST, Castanho MA, Pattenden LK, and Aguilar MI (2010). Fast membrane association is a crucial factor in the peptide pep-1 translocation mechanism: a kinetic study followed by surface plasmon resonance. Biopolymers 94, 314–322. [DOI] [PubMed] [Google Scholar]
- Henriques ST, Pattenden LK, Aguilar MI, and Castanho MA (2008). PrP(106–126) does not interact with membranes under physiological conditions. Biophys J 95, 1877–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques ST, Pattenden LK, Aguilar MI, and Castanho MA (2009). The toxicity of prion protein fragment PrP(106–126) is not mediated by membrane permeabilization as shown by a M112W substitution. Biochemistry 48, 4198–4208. [DOI] [PubMed] [Google Scholar]
- Huang YH, Colgrave ML, Daly NL, Keleshian A, Martinac B, and Craik DJ (2009). The biological activity of the prototypic cyclotide kalata b1 is modulated by the formation of multimeric pores. J. Biol. Chem. 284, 20699–20707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilavsky J, and Jemian PR (2009). Irena: tool suite for modeling and analysis of small-angle scattering. J. Appl. Crystallogr. 42, 347–353. [Google Scholar]
- Kato T, and Shoji T (1980). The structure of octapeptin D (studies on antibiotics from the genus Bacillus. XXVIII). J Antibiot (Tokyo) 33, 186–191. [DOI] [PubMed] [Google Scholar]
- Kirby NM, Mudie ST, Hawley AM, Cookson DJ, Mertens HDT, Cowieson N, and Samardzic-Boban V (2013). A low-background-intensity focusing small-angle X-ray scattering undulator beamline. J. Appl. Crystallogr. 46, 1670–1680. [Google Scholar]
- Konishi M, Sugawara K, Tomita K, Matsumoto K, Miyaki T, Fujisawa K, Tsukiura H, and Kawaguchi H (1983). Bu-2470, a new peptide antibiotic complex. I. Production, isolation and properties of Bu-2470 A, B1 and B2. J Antibiot (Tokyo) 36, 625–633. [DOI] [PubMed] [Google Scholar]
- Koradi R, Billeter M, and Wuthrich K (1996). MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph 14, 51–55, 29–32. [DOI] [PubMed] [Google Scholar]
- Li J, Milne RW, Nation RL, Turnidge JD, Smeaton TC, and Coulthard K (2003). Use of high-performance liquid chromatography to study the pharmacokinetics of colistin sulfate in rats following intravenous administration. Antimicrob. Agents Chemother. 47, 1766–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh B, Grant C, and Hancock RE (1984). Use of the fluorescent probe 1-N-phenylnaphthylamine to study the interactions of aminoglycoside antibiotics with the outer membrane of Pseudomonas aeruginosa. Antimicrobial agents and chemotherapy 26, 546–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mares J, Kumaran S, Gobbo M, and Zerbe O (2009). Interactions of lipopolysaccharide and polymyxin studied by NMR spectroscopy. The Journal of biological chemistry 284, 11498–11506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers E, Brown WE, Principe PA, Rathnum ML, and Parker WL (1973). EM49, a new peptide antibiotic. I. Fermentation, isolation, and preliminary characterization. J Antibiot (Tokyo) 26, 444–448. [DOI] [PubMed] [Google Scholar]
- Meyers E, Pansy FE, Basch HI, McRipley RJ, Slusarchyk DS, Graham SF, and Trejo WH (1973). EM49, a new peptide antibiotic. 3. biological characterization in vitro and in vivo. J Antibiot (Tokyo) 26, 457–462. [DOI] [PubMed] [Google Scholar]
- Meyers E, Parker WL, and Brown WE (1976). A nomenclature proposal for the octapeptin antibiotics. J Antibiot (Tokyo) 29, 1241–1242. [DOI] [PubMed] [Google Scholar]
- Meyers E, Parker WL, Brown WE, Linnett P, and Strominger JL (1974). EM49: a new polypeptide antibiotic active against cell membranes. Ann N Y Acad Sci 235, 493–501. [DOI] [PubMed] [Google Scholar]
- Miller AK, Brannon MK, Stevens L, Johansen HK, Selgrade SE, Miller SI, Hoiby N, and Moskowitz SM (2011). PhoQ mutations promote lipid A modification and polymyxin resistance of Pseudomonas aeruginosa found in colistin-treated cystic fibrosis patients. Antimicrobial agents and chemotherapy 55, 5761–5769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido H (2003). Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev 67, 593–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker WL, and Rathnum ML (1975). EM49, a new peptide antibiotic IV. The structure of EM49. J Antibiot (Tokyo) 28, 379–389. [DOI] [PubMed] [Google Scholar]
- Parker WL, and Rathnum ML (1973). EM49, a new peptide antibiotic. II. Chemical characterization. J Antibiot (Tokyo) 26, 449–456. [DOI] [PubMed] [Google Scholar]
- Puar MS (1980). Carbon-13 NMR studies of EM 49 and related octapeptins. J Antibiot (Tokyo) 33, 760–763. [DOI] [PubMed] [Google Scholar]
- Qian CD, Wu XC, Teng Y, Zhao WP, Li O, Fang SG, Huang ZH, and Gao HC (2012). Battacin (Octapeptin B5), a new cyclic lipopeptide antibiotic from Paenibacillus tianmuensis active against multidrug-resistant Gram-negative bacteria. Antimicrobial agents and chemotherapy 56, 1458–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Que NL, Lin S, Cotter RJ, and Raetz CR (2000). Purification and mass spectrometry of six Lipid A species from the bacterial endosymbiont Rhizobium etli. Demonstration of a conserved distal unit and a variable proximal portion. J. Biol. Chem. 275, 28006–28016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal KS, Ferguson RA, and Storm DR (1977). Mechanism of action of EM 49, membrane-active peptide antibiotic. Antimicrobial agents and chemotherapy 12, 665–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal KS, Swanson PE, and Storm DR (1976). Disruption of Escherichia coli outer membranes by EM 49. A new membrane active peptide. Biochemistry 15, 5783–5792. [DOI] [PubMed] [Google Scholar]
- Roth BL, Poot M, Yue ST, and Millard PJ (1997). Bacterial viability and antibiotic susceptibility testing with SYTOX green nucleic acid stain. Appl. Environ. Microbiol. 63, 2421–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoji J, Hinoo H, and Sakazaki R (1976). The constituent amino acids and fatty acid of antibiotic 333–25. (Studies on antibiotics from the genus Bacillus. XII. J Antibiot (Tokyo) 29, 521–525. [DOI] [PubMed] [Google Scholar]
- Shoji J, Sakazaki R, Wakisaka Y, Koizumi K, Matsuura S, Miwa H, and Mayama M (1980). Isolation of octapeptin D (studies on antibiotics from the genus Bacillus. XXVII). J Antibiot (Tokyo) 33, 182–185. [DOI] [PubMed] [Google Scholar]
- Storm DR, Rosenthal KS, and Swanson PE (1977). Polymyxin and related peptide antibiotics. Annu Rev Biochem 46, 723–763. [DOI] [PubMed] [Google Scholar]
- Sugawara K, Yonemoto T, Konishi M, Matsumoto K, Miyaki T, and Kawaguchi H (1983). Bu-2470, a new peptide antibiotic complex. II. Structure determination of Bu-2470 A, B1, B2a and B2b. J Antibiot (Tokyo) 36, 634–638. [DOI] [PubMed] [Google Scholar]
- Torcato IM, Huang YH, Franquelim HG, Gaspar DD, Craik DJ, Castanho MA, and Henriques ST (2013). The antimicrobial activity of Sub3 is dependent on membrane binding and cell-penetrating ability. Chembiochem : a European journal of chemical biology 14, 2013–2022. [DOI] [PubMed] [Google Scholar]
- Vaara M (2013). Novel derivatives of polymyxins. J Antimicrob Chemoth 68, 1213–1219. [DOI] [PubMed] [Google Scholar]
- Velkov T, Deris ZZ, Huang JX, Azad MA, Butler M, Sivanesan S, Kaminskas LM, Dong YD, Boyd B, Baker MA, et al. (2013). Surface changes and polymyxin interactions with a resistant strain of Klebsiella pneumoniae. Innate Immun. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velkov T, Soon RL, Chong PL, Huang JX, Cooper MA, Azad MA, Baker MA, Thompson PE, Roberts K, Nation RL, et al. (2012). Molecular basis for the increased polymyxin susceptibility of Klebsiella pneumoniae strains with under-acylated lipid A. Innate Immun 19, 265–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velkov T, Thompson PE, Nation RL, and Li J (2010). Structure--activity relationships of polymyxin antibiotics. Journal of medicinal chemistry 53, 1898–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viljanen P, and Vaara M (1984). Susceptibility of gram-negative bacteria to polymyxin B nonapeptide. Antimicrobial agents and chemotherapy 25, 701–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegand I, Hilpert K, and Hancock RE (2008). Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 3, 163–175. [DOI] [PubMed] [Google Scholar]
- Yousef JM, Chen G, Hill PA, Nation RL, and Li J (2012). Ascorbic acid protects against the nephrotoxicity and apoptosis caused by colistin and affects its pharmacokinetics. The Journal of antimicrobial chemotherapy 67, 452–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavascki AP, Goldani LZ, Cao GY, Superti SV, Lutz L, Barth AL, Ramos F, Boniatti MM, Nation RL, and Li J (2008). Pharmacokinetics of intravenous polymyxin B in critically-ill patients. Clin Infect Dis 47, 1298–1304. [DOI] [PubMed] [Google Scholar]
- Zerbino DR, and Birney E (2008). Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18, 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.