

Abstract
Aims/hypothesis
The aim of this study was to elucidate the impact of 3′-phosphoinositide-dependent protein kinase-1 (PDPK1) in vascular endothelial cells on the maintenance of pancreatic beta cell mass and function.
Methods
Male vascular endothelial cell-specific Pdpk1-knockout mice (Tie2+/−/Pdpk1flox/flox mice) and their wild-type littermates (Tie2−/−/Pdpk1flox/flox mice; control) were used for this study. At 12 weeks of age, an IPGTT and OGTT were conducted. Pancreatic blood flow was measured under anaesthesia. Thereafter, islet blood flow was measured by the microsphere method. Mice were killed for islet isolation and further functional study and mRNA was extracted from islets. Pancreases were sampled for immunohistochemical analyses.
Results
During the IPGTT, the blood glucose level was comparable between knockout mice and control flox mice, although serum insulin level was significantly lower in knockout mice. During the OGTT, glucose tolerance deteriorated slightly in knockout mice, accompanied by a decreased serum insulin level. During an IPGTT after pre-treatment with exendin-4 (Ex-4), glucose tolerance was significantly impaired in knockout mice. In fact, glucose-stimulated insulin secretion of isolated islets from knockout mice was significantly reduced compared with control flox mice, and addition of Ex-4 revealed impaired sensitivity to incretin hormones in islets of knockout mice. In immunohistochemical analyses, both alpha and beta cell masses were significantly reduced in knockout mice. In addition, the CD31-positive area was significantly decreased in islets of knockout mice. The proportion of pimonidazole-positive islets was significantly increased in knockout mice. mRNA expression levels related to insulin biosynthesis (Ins1, Ins2, Mafa, Pdx1 and Neurod [also known as Neurod1]) and beta cell function (such as Gck and Slc2a2) were significantly decreased in islets of knockout mice. Microsphere experiments revealed remarkably reduced islet blood flow. In addition, mRNA expression levels of Hif1α (also known as Hif1a) and its downstream factors such as Adm, Eno1, Tpi1 (also known as Ets1), Hmox1 and Vegfa, were significantly increased in islets of knockout mice, indicating that islets of knockout mice were in a more hypoxic state than those of control flox mice. As a result, mRNA expression levels related to adaptive unfolded protein response and endoplasmic reticulum stress-related apoptotic genes were significantly elevated in islets of knockout mice. In addition, inflammatory cytokine levels were increased in islets of knockout mice. Electron microscopy revealed reduced endothelial fenestration and thickening of basal membrane of vascular endothelium in islets of knockout mice.
Conclusions/interpretation
Vascular endothelial PDPK1 plays an important role in the maintenance of pancreatic beta cell mass and function by maintaining vascularity of pancreas and islets and protecting them from hypoxia, hypoxia-related endoplasmic reticulum stress, inflammation and distortion of capillary structure.
Electronic supplementary material
The online version of this article (10.1007/s00125-019-4878-1) contains peer-reviewed but unedited supplementary material, which is available to authorised users.
Keywords: 3-Phosphoinositide-dependent protein kinase-1 (PDPK1), Capillary structure, Endothelial cells, Hypoxia, Pancreatic beta cells, Vascularity

Introduction
Diabetes is the most prevalent and serious metabolic disease and the number of diabetic individuals has been increasing markedly all over the world. The hallmark of the disease is insulin resistance and pancreatic beta cell dysfunction. Under diabetic conditions, insulin target tissues, such as liver, muscle and fat, become resistant to insulin and pancreatic beta cell function gradually deteriorates [1–3]. Beta cell failure and/or dysfunction is strongly associated with type 2 diabetes and may lead to the onset of hyperglycaemia. Therefore, amelioration of impaired insulin secretion might be a prospective therapeutic goal.
Several studies have reported that pancreatic islets are densely vascularised and many mediators, such as insulin, regulate the islet blood flow, which is highly involved in the regulation of insulin secretion [4, 5].
Kondo et al reported that vascular endothelial cell-specific insulin receptor knockout (VENIRKO) or insulin like growth factor-1 receptor knockout (VENIFARKO) resulted in reduced angiogenesis of the retina [6]. However, these models did not present insulin resistance. Mukai et al reported that endothelial cell-specific constitutive activation of Akt suppresses vascular lesion formation via increased NO production, preservation of functional endothelial layer and suppression of inflammatory and proliferative changes in the vascular wall [7]. Indeed, these studies elucidated the role played by insulin–phosphoinositide 3-kinase (PI3K) signalling in endothelial cells. However, neither of the studies conducted assessment on pancreatic beta cells.
Recently, it was reported that endothelial cell-specific insulin receptor knockout (EndoIRKO) mice presented glucose intolerance and insulin resistance, which was a different phenotype from those used by Kondo et al, probably for the following reasons. First, the mice were backcrossed to C57BL/6, and VE-Cadherin Cre mice were used instead of Tie-2 Cre mice. Second, the mice presented as insulin resistant because of delayed insulin delivery to systemic organs, except for the liver and olfactory bulb in which the fenestrated endothelium of the capillaries freely permits paracellular passage of macromolecules [8]. In this model, no morphological and functional changes in the islets were observed. On the contrary, endothelial cell-specific Irs2-knockout (ETIRS2KO) mice also showed insulin resistance mainly in skeletal muscle due to reduced capillary recruitment [9]. Intriguingly, reduced glucose-stimulated insulin secretion (GSIS) in vivo due to decreased islet blood flow was also observed in this model without any morphological changes in islets [10]. As seen in these previous reports, many things still remain to be unravelled about the influence of insulin–PI3K signalling of endothelial cells on pancreatic beta cells.
Therefore, in the present study, we focused on 3′-phosphoinositide-dependent protein kinase-1 (PDPK1), a serine-threonine kinase that mediates downstream signalling of PI3K and regulates the activity of Akt, to further elucidate the impact of insulin–PI3K signalling in endothelial cells on pancreatic beta cells. We investigated the role played by endothelial PDPK1 in the maintenance of pancreatic beta cell mass and function using vascular endothelial cell-specific Pdpk1-knockout mice.
Methods
Vascular endothelial cell-specific Pdpk1-knockout mice
Mice were kindly provided by K. Kotani (Saso Hospital, Kobe, Japan), W. Ogawa (Division of Diabetes, Metabolism, and Endocrinology, Kobe University Graduate School of Medicine, Kobe, Japan) and M. Kasuga (Research Institute, International Medical Center of Japan, Tokyo, Japan). As described previously [11], vascular endothelial cell-specific Pdpk1-knockout (VE-PDPK1-KO) mice were generated by breeding Pdpk1flox/flox mice, which harbour a modified endogenous Pdpk1 gene in which exons 3 and 4 are flanked by loxP sites, with mice that express the Cre recombinase gene under the control of the Tie2 gene promoter (Tie2-Cre) [12]. The heterozygous offspring of both the loxP-targeted Pdpk1 gene and the Tie2-Cre transgene (Tie2-Cre+/−/Pdpk1flox/+) were then crossed with Pdpk1flox/flox mice to generate VE-PDPK1-KO mice homozygous for the Pdpk1 floxed allele (Tie2-Cre+/−/Pdpk1flox/flox). These mice were all bred from C57BL/6J mice. VE-PDPK1-KO mice were born at the expected Mendelian frequency and their littermates, which did not express Tie2-Cre (Tie2-Cre−/−/Pdpk1flox/flox), were used as control mice. Mice were housed in a 12 h dark–light cycle at a controlled temperature and were allowed free access to water and a standard diet. Male mice were used for all experiments at 12 weeks of age. All aspects of animal care and experiments were performed at the Laboratory Animal Center with the approval of the Animal Research Committee at the Kawasaki Medical School (No. 18-004).
IPGTT and OGTT
After 16 h fasting, d-(+)-glucose (1.5 g/kg) was administered by i.p. injection for the IPGTT or administered orally for the oral glucose tolerance test (OGTT). Blood samples from tail snips were collected at the indicated time point and the blood glucose level was measured using a Free Style Kissei glucose meter (Kissei Pharmaceutical, Tokyo, Japan). For pre-treatment with exendin-4 (Ex-4) in the IPGTT, 100 ng of Ex-4 was given by i.p. injection 15 min before glucose injection. For measurements of active glucagon-like peptide-1 (GLP-1) and glucagon, EDTA and aprotinin were placed in sample collection tubes beforehand. Active GLP-1 level was measured using GLP-1, active form assay (IBL, Gunma, Japan). Glucagon level was determined using glucagon ELISA (Mercodia, Uppsala, Sweden). Serum insulin level was determined using a mouse insulin ELISA kit (Morinaga, Tokyo, Japan).
Pancreatic islet isolation
Isolation of islets from the pancreas of the mice was conducted as previously described [13, 14]. In brief, after ligation of the common bile duct with silk thread at a point close to the duodenal outlet, 2.5 ml of Hanks’ Balanced Salt Solution (HBSS) (Sigma, St Louis, MO, USA) containing 0.6 mg Liberase TL (Roche Diagnostics, Tokyo, Japan) and 25 mmol/l HEPES were injected into the duct. The swollen pancreas was removed and incubated at 37°C for 24 min. The pancreatic tissue was then dispersed by pipetting and washed twice with ice-cold HBSS containing 25 mmol/l HEPES and 10% (wt/vol.) FBS. Thereafter, the islets were manually picked up under a stereoscopic microscope and used immediately for the experiments.
GSIS from isolated pancreatic islets
Size-matched pancreatic islets were prepared (five pancreatic islets/tube) and pre-incubated in KRB–HEPES buffer. The supernatant fraction was replaced with glucose solution (either 3 mmol/l or 16.7 mmol/l) and the mixture was incubated for an additional 60 min at 37°C. The supernatant fraction was recovered and stored at −80°C until use. Thereafter, the solution which contained 16.7 mmol/l glucose and 10 nmol/l Ex-4 was added and incubated for another 60 min at 37°C. The supernatant fraction was collected and stored at −80°C until use.
Islet perifusion
Islet perifusion in vitro was conducted as performed by Hashimoto et al with slight modification [10]. In our study, ten size-matched islets were prepared for analysis.
Measurement of pancreatic and islet blood flow
Mice were anaesthetised with sevoflurane and the body temperature was kept at 37°C. Pancreatic blood flow was measured using MoorFLPI-2 laser-Doppler apparatus (Moor Instruments, Axminster, Devon, UK), in accordance with the manufacturer’s instruction, during the systolic phase. To further elucidate islet blood flow, a microsphere experiment was conducted as performed by Hashimoto et al [10] and Jansson and Hellerström [15], with slight modifications. Polyethylene catheters (SP10: Natsume Seisakusho, Tokyo, Japan) were inserted into the left carotid artery and the left femoral artery. In our study, Dye-Trak microspheres (Triton Technology, Los Angeles, CA, USA) with a mean diameter of 15 mm were injected for 15 s into the ascending aorta. Arterial blood sample was collected from the femoral artery at a constant rate of 0.4 ml/min by using a syringe pump, starting 5 s before the microsphere injection and continued for 60 s. The whole pancreas was removed, blotted, weighed and treated using a freeze–thawing technique to visualise the microspheres and the number of microspheres in islets was counted with a dark field microscope. After counting the number of microspheres, pancreatic tissue was collected again into a tube and the tissue was dissolved with 2% (vol./vol.) Tween80 + 4 mol/l KOH. After sonication, the sample was diluted ten times with PBS and the number of microspheres was counted on the 10 μm pore filter (Triton Technology) with a stereoscopic microscope. Alternatively, the collected blood sample was dissolved with 16 mol/l KOH + 2% (vol./vol.) Tween 80 and the number of microspheres was counted by using a haemocytometer. The blood flow values were calculated according to the following formula: organ blood flow = 0.4 (ml/min) × no. of microspheres in target organ / number of microspheres in blood / weight of target organ. The islet blood flow was expressed per islet weight estimated by multiplying the pancreatic weight with the islet volume fraction of the whole pancreas, which was evaluated by insulin and glucagon double staining.
Histological and immunohistochemical analyses of islets
Isolated pancreases were fixed overnight with 4% (vol./vol.) paraformaldehyde at 4°C. Tissues were routinely processed for paraffin embedding and 4 μm sections were cut and mounted on silanised slides. The sections were incubated with a mixture of primary antibodies (sc-130624 glucagon antibody (79/bB10) mouse monoclonal IgG; diluted 1:100 by PBS supplemented with 1% (wt/vol.) BSA (1% BSA) and sc-9168 insulin antibody (H-86) rabbit polyclonal IgG; diluted 1:200 by 1% BSA; Santa Cruz Biotechnology, Santa Cruz, CA, USA). For secondary antibody, donkey anti-rabbit IgG (H+L) secondary antibody Alexa Fluor 488 conjugate (A-21206, Life Technologies, Carlsbad, CA, USA) diluted 1:1000 by 1% BSA and donkey anti-mouse IgG (H+L) secondary antibody Alexa Fluor 594 conjugate (A-21203, Life Technologies, Carlsbad, CA, USA) diluted 1:1000 by 1% BSA were applied. Pancreatic islet proliferation was identified by staining sections with rabbit anti-Ki-67 polyclonal antibody (Abcam, Cambridge, MA, USA). For primary antibodies, guinea pig polyclonal IgG anti-insulin antibody (ab7842; diluted 1:100 by 1% BSA) and rabbit polyclonal IgG anti-Ki67 antibody (ab15580; diluted 1:500 by 1% BSA) were used. For secondary antibody, goat anti-guinea pig IgG (H+L) secondary antibody Alexa Fluor 488 conjugate (A-11073, Life Technologies, Carlsbad, CA, USA) diluted 1:1000 by 1% BSA and donkey anti-rabbit IgG (H+L) secondary antibody Alexa Fluor 594 conjugate (A-21207, Life Technologies, Carlsbad, CA, USA) were applied. To investigate cell apoptosis in pancreatic islets, a TUNEL assay was performed using a DeadEnd Fluorometric TUNEL System (DeadEnd; cat. no. G3250, Promega, Madison, WI, USA) as previously reported [16]. For CD31 staining, isolated pancreases were embedded in Tissue-Tek Optimal Cutting Temperature compound (Funakoshi, Tokyo, Japan) and frozen by liquid nitrogen. Sections (10 μm) were cut and mounted on slides in a cryostat. The sections were incubated with primary antibody (purified rat anti-mouse CD31, cat. no. 550274; BD Pharmingen) and CD31 was detected by using Anti-Ig HRP Detection Kits (cat. no. 551013, BD Pharmingen, San Diego, CA, USA) in accordance with the manufacturer’s instruction followed by H&E counterstaining. Immunohistochemical staining for pimonidazole was conducted as performed by Hashimoto et al [10] and Takikawa et al [17], with slight modifications. Hypoxyprobe -1 Plus Kit (HP2-100, Hypoxyprobe, Burlington, MA, USA) was used in accordance with the manufacturer’s instruction. In our study, pancreases were collected 1 h after pimonidazole injection.
Morphometric analysis
The image analysis software NIH Image (version 1.61; http://rsbweb.nih.gov/ij/) was used to calculate the pancreas area and islet area. Using a total of nine sections (three sections from three different areas of the pancreas) for each group of mice, beta cell mass was estimated via the following formula: cell mass (mg) = average of islet area per section / average of pancreas area per section × weight of pancreas × beta cell ratio (average of beta cell number / cell number in islet). Observations were made using a minimum of 50 islets and, when quantified, were expressed as a percentage of the total number of islet cells. CD31-positive area and vascular structure numbers/mm2 in islets were calculated using NIH Image (version 1.61; http://rsbweb.nih.gov/ij/).
Electron microscopy
The islets were fixed in a solution of 2.5% (vol./vol.) glutaraldehyde soon after isolation. The samples were fixed for 2 h in 2.5% (vol./vol.) glutaraldehyde buffered to pH 7.4 with phosphate buffer and treated with osmium tetroxide for 1 h at 4°C. The tissues were dehydrated with graded concentration ethanol and then embedded in Epon 812 (Nakarai, Kyoto, Japan). Thin sections were cut with a Leica Ultracut S (Leica Microsystems, Tokyo, Japan) with a diamond knife and stained with uranyl acetate followed by lead citrate [18]. Electron micrographs were taken with a JEOL JEM-1400 electron microscope (JEOL, Tokyo, Japan) operated at 80 kV.
RNA preparation and quantitative PCR
Total RNA was extracted using an RNeasy lipid tissue mini kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. cDNA was produced from mRNA using TaqMan reverse transcription reagents (Applied Biosystems, Foster City, CA, USA). Quantitative PCR (qPCR) was performed using a 7500 Real-Time PCR system (Applied Biosystems). The relative expression levels were compared by normalisation to the expression levels of β-actin. The primers were purchased from Takara Bio Co. (Shiga, Japan); sequences are shown in ESM Table 1.
Statistics
Results are expressed as mean ± SEM. Differences between two groups were tested for statistical significance using Student’s t test. Differences among more than two groups were tested using the Tukey–Kramer method. p values less than 0.05 were considered to denote statistical significance.
Results
Glucose stimulated insulin secretion was impaired in islets of VE-PDPK1-KO mice compared with control flox mice both in vivo and in vitro
IPGTT conducted when mice were 12 weeks old revealed lower insulin levels in VE-PDPK1-KO mice compared with control flox mice, although there was no significant difference in blood glucose levels (Fig. 1a,b). Next, the islet perifusion experiment in vitro was conducted to investigate GSIS. Both first and second phases of GSIS were impaired in islets of VE-PDPK1-KO mice after exposure to high-glucose KRB buffer (Fig. 1c).
Fig. 1.

IPGTT and OGTT revealed lower insulin levels in VE-PDPK1-KO mice, and an isolated islet perifusion study revealed a deterioration in GSIS in knockout mice. (a, b) Blood glucose levels (a) and serum insulin levels (b) during an IPGTT (n=7 mice). (c) Islet perifusion experiment; n=5 (control flox mice) or n=4 (VE-PDPK1-KO mice). (d–g) Blood glucose, insulin, active GLP-1 and glucagon levels during an OGTT; n=7 (control flox mice) or n=8 (VE-PDPK1-KO mice) (h, i) IPGTT conducted after 100 ng Ex-4 pre-treatment; n=7 (control flox mice) or n=8 (VE-PDPK1-KO mice). Black circles, VE-PDPK1-KO mice; white circles, control flox mice. Values are means ± SEM. *p<0.05 vs control flox mice
In the OGTT, glucose tolerance was slightly but significantly deteriorated in VE-PDPK1-KO mice, accompanied by reduced serum insulin levels (Fig. 1d,e). Unexpectedly, during the OGTT, active GLP-1 levels were significantly increased in the knockout mice, although there was no difference in serum glucagon levels (Fig. 1f,g). These results suggested that sensitivity to incretin hormone was impaired in knockout mice. Therefore, an IPGTT was conducted after pre-treatment with Ex-4. This showed impaired glucose tolerance and reduced insulin levels in the knockout mice (Fig. 1h,i). These results suggested that sensitivity to incretin hormone was definitely impaired in VE-PDPK1-KO mice.
To further investigate islet function, GSIS was conducted in vitro. GSIS was significantly impaired at high glucose concentrations (Fig. 2a). Moreover, addition of Ex-4 to high-glucose solution revealed a reduced reaction to Ex-4 in islets of VE-PDPK1-KO mice (Fig. 2b). Islet insulin content was significantly decreased in knockout mice (Fig. 2c). mRNA expression levels related to insulin biosynthesis, such as Ins1, Ins2, Mafa, Pdx1 and Neurod, were all significantly reduced in VE-PDPK1-KO mice (Fig. 2d–h). In addition, mRNA expression levels related to GSIS, such as Gck and Slc2a2, were significantly decreased in the knockout mice (Fig. 2i,j). Furthermore, Glp1r expression levels were also significantly decreased in VE-PDPK1-KO mice, suggesting that there is reduced sensitivity to incretin hormone in knockout mice (Fig. 2k). As Pdx1 and Mafa are essential for differentiation of pancreatic beta cells, we evaluated the mRNA expression level of Ngn3, a marker of endocrine precursor cells, to confirm whether differentiation of pancreatic beta cells was affected by endothelial cell-specific Pdpk1 knockout. As shown in ESM Fig. 1, Ngn3 mRNA expression level was comparable between control flox and knockout mice. Therefore, we think that beta cell differentiation was not affected in these mice but rather that beta cell de-differentiation was not facilitated by endothelial cell-specific Pdpk1 knockout.
Fig. 2.

GSIS of isolated islets and mRNA expression levels related to insulin, its biosynthesis and beta cell function. (a) GSIS of isolated islets from control flox and VE-PDPK1-KO mice (n=8 mice), in 3.3 mmol/l glucose (low glucose, LG) and 16.7 mmol/l glucose (high glucose, HG), without and with addition of 10 nmol/l Ex-4. (b) Change in GSIS after addition of Ex-4 in HG (n=8 mice). (c) Islet insulin content (n=8 mice). (d–l) Expression levels of mRNA (reported as fold vs β-actin) related to insulin biosynthesis (d–h) and beta cell function (i–l) (n=7 or 8 mice, as indicated). Values are the means ± SEM. *p<0.05, KO vs f/f; †p<0.05 vs LG f/f; ‡p<0.05 vs LG KO. f/f, control flox mice; HG, high glucose (16.7 mmol/l); KO, VE-PDPK1-KO mice; LG, low glucose (3.3 mmol/l)
Both pancreatic beta cell mass and alpha cell mass were reduced in VE-PDPK1-KO mice compared with control flox mice
Next, the morphology of islets was investigated by insulin and glucagon double staining. This revealed significantly reduced beta cell mass and alpha cell mass in VE-PDPK1-KO mouse islets (Fig. 3a–c). In TUNEL assay, the proportion of TUNEL-positive nuclei was significantly higher in islets from knockout mice than in islets from control flox mice (Fig. 3d,e). In addition, the proportion of Ki-67-positive nuclei was significantly lower in knockout mouse islets (Fig. 3f,g). These results suggested that proliferation of beta cells was impaired and apoptosis of beta cells was increased in VE-PDPK1-KO mice. Corresponding to these immunohistochemical analyses, mRNA expression levels related to apoptosis such as Bax, Casp8 and Casp3 were significantly increased in VE-PDPK1-KO mice while the mRNA expression level of level of Bcl2, an anti-apoptotic gene, was significantly reduced (Fig. 3h–k). In addition, mRNA expression levels related to cell replication and proliferation, such as Ccnd1 and Irs2, were significantly decreased in VE-PDPK1-KO mice (Fig. 3l–p). These results supported the notion that beta cell mass reduction was induced by increased apoptosis and decreased proliferation of beta cell in the knockout mice.
Fig. 3.

Evaluation of morphology of islets and mRNA expression levels related to beta cell proliferation, apoptosis and beta cell mass. (a) Representative images of insulin and glucagon double staining of pancreatic sections in control flox and VE-PDPK1-KO mice; scale bars, 50 μm; green, insulin; red, glucagon; blue, DAPI. (b, c) Quantification of beta (b) and alpha (c) cell mass (n=4 mice). (d) Representative images of TUNEL assay. White arrow indicates TUNEL-positive beta cell; scale bars, 50 μm; red, insulin; blue, DAPI. (e) TUNEL-positive beta cells were counted and divided by total beta cell number (n=4 mice). (f) Representative images of Ki67 staining. White arrow indicates Ki67-positive beta cell; scale bars, 50 μm; green, insulin; blue, DAPI. (g) Quantification of Ki67-positive beta cells (n=4 mice). (h–p) Expression levels of mRNA (reported as fold vs β-actin) related to apoptosis (h–k) and beta cell mass (l–p) (n=6 or 8 mice, as indicated). Values are the means ± SEM. *p<0.05, **p<0.01, KO vs f/f. f/f, control flox mice; KO, VE-PDPK1-KO mice
Both pancreatic and islet blood flows were significantly reduced in VE-PDPK1-KO mice compared with control flox mice
Next, pancreatic blood flow was investigated. Intriguingly, pancreatic blood flow was significantly decreased in VE-PDPK1-KO mice, although blood pressure and pancreas weight were comparable between the knockout mice and their control flox mice (Fig. 4a–c, and ESM Fig. 2). Next, a microsphere experiment was conducted to precisely evaluate islet blood flow. The number of microspheres in islets was significantly decreased in VE-PDPK1-KO mice (Fig. 4d,e). Whole pancreatic blood flow, which was calculated by microsphere experiment, was comparable with that measured by laser-Doppler apparatus (Fig. 4f). Intriguingly, islet blood flow was drastically reduced in knockout mice (Fig. 4g). In addition, CD31-positive area and capillary density were significantly decreased in VE-PDPK1-KO mice (Fig. 4h–j). As a result, the percentage of pimonidazole-positive islets was significantly increased in VE-PDPK1-KO mice (Fig. 4k), suggesting that islets of the knockout mice were in a more hypoxic state compared with control flox mice. In fact, mRNA levels of Hif1α (also known as Hif1a) and its downstream genes Adm, Eno1, Tpi1 (also known as Ets1), Hmox1 and Vegfa were significantly increased in the knockout mice (Fig. 4l–q).
Fig. 4.

Pancreatic and islet blood flow, CD31 and pimonidazole staining and mRNA expression levels related to hypoxia, ER stress and inflammation. (a) Representative images of the laser-Doppler experiment for evaluation of pancreatic blood flow. The pancreas is indicated by a red circle. The intensity of the red colour was measured using MoorFLPI-2 apparatus. (b) Pancreatic blood flow, shown as arbitrary units (AU) (n=8 mice). (c) Blood pressure (n=8 mice). (d–g) Representative images of darkfield microscopy (d). Islets are shown in white and the arrows point to microspheres in the islet, which appear as small black dots; the microspheres were counted to evaluate islet blood flow (e). Pancreatic (f) and islet (g) blood flow evaluated by microspheres (n=8 mice). (h–j) Representative images of CD31 staining (h). The black dotted line indicates an islet and brown staining indicates capillaries. The brown area was measured and divided by total islet area to give the CD31-positive area in islet area (%) (i), and the brown structures were counted and divided by total islet area to give capillary density (j) (n=4 mice); scale bars, 50 μm. (k) Percentage of pimonidazole-positive islets (n=6 mice). (l–q) Expression levels of Hif1α mRNA and its downstream factors (reported as fold vs β-actin) (n=7 mice). (r–y) Expression levels of mRNA (reported as fold vs β-actin except for t, in which expression level of sXbp1 [fold vs β-actin] was divided by that of tXbp1 [fold vs β-actin]) related to adaptive UPR and ER stress-related apoptotic genes (r–w) and inflammation (x, y) (n=7 or 8 mice, as indicated). Values are the means ± SEM. *p<0.05 and **p<0.01, KO vs f/f; in (x), p=0.075 for KO vs f/f. f/f, control flox mice; KO, VE-PDPK1-KO mice
As islets of VE-PDPK1-KO mice were in a relatively more hypoxic state than those of control flox mice, we next investigated the endoplasmic reticulum (ER) stress-related adaptive unfolded protein response (UPR) and apoptotic gene expression levels. mRNA expression levels related to adaptive UPR, such as Bip, Fkbp11 and sXbp1/tXbp1, were significantly increased in VE-PDPK1-KO mice (Fig. 4r–t). In addition, the expression levels of ER stress-related apoptotic genes, such as Atf3, Ddit3 and Trb3 (also known as Trib3), were all significantly elevated in knockout mice (Fig. 4u–w). Furthermore, expression levels of inflammatory cytokine genes, Il1-β (also known as Il1b) and Tnf-α (also known as Tnf), were increased in islets of knockout mice (Fig. 4x,y). These results suggest that hypoxia-induced ER stress and inflammation led to the decreased pancreatic beta cell mass and impairment of beta cell function.
Furthermore, electron microscope-elucidated vascular structure in islets was markedly altered in VE-PDPK1-KO mice (Fig. 5a,b). In control flox mice, endothelial cells were aligned along the basal membrane and they exhibited structured fenestrations (Fig. 5a). On the other hand, in VE-PDPK1-KO mice, there were less endothelial cells along basal membrane, so-called ‘empty sleeves’, and thickening of the basal membrane was observed (Fig. 5b). We also evaluated mRNA expression levels of angiocrine factors such as Hgf and Fgf2, and found no difference between control flox and VE-PDPK1-KO mice (ESM Fig. 3).
Fig. 5.
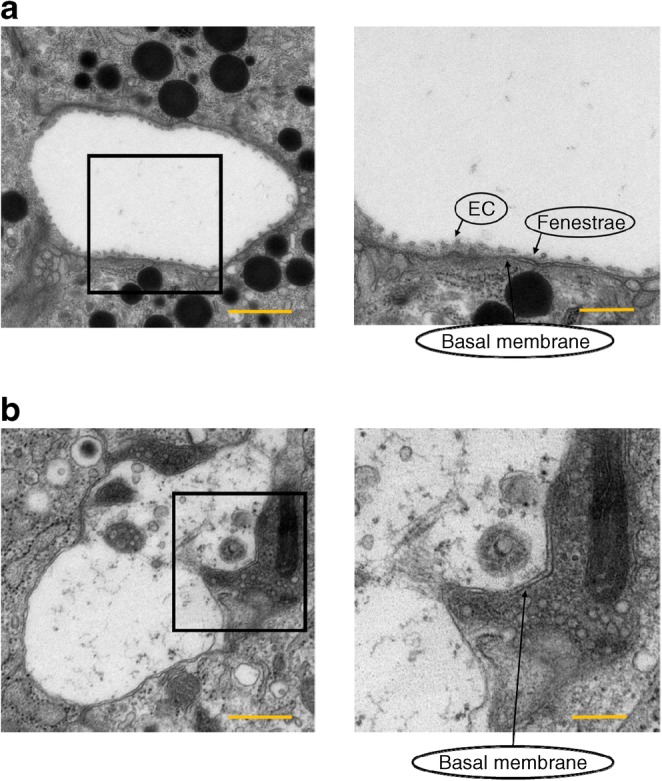
Vascular structure of islets from VE-PDPK1-KO mice and control flox mice. Electron microscopic images showing capillaries surrounded by pancreatic beta cells in (a) control flox mice and (b) VE-PDPK1-KO mice. EC, endothelial cells. Images are representative of four mice. Magnification ×5000 (left), scale bars, 1 μm; or ×10,000 (right), scale bars, 500 nm
Discussion
In the present study, we elucidated endothelial-specific ablation of Pdpk1 impaired GSIS by reducing pancreatic and/or islet blood flow. We also demonstrate that these decreases in blood flow induce hypoxia in islets of VE-PDPK1-KO mice and that hypoxia-induced ER stress and inflammation lead to beta cell mass reduction and impairment of beta cell function accompanied by morphological changes of capillaries in islets (Fig. 6).
Fig. 6.

Possible mechanism of how endothelial cell PDPK1 maintains beta cell mass and function. Ablation of endothelial PDPK1 causes a reduction of vascularity in islets, and both pancreatic and islets blood flow is decreased, which leads to hypoxia in islets, which in turn induces ER stress-related apoptosis and inflammation. Furthermore, PDPK1 ablation in endothelial cells leads to distortion of capillary structure in islets. For example, there are fewer endothelial cells along the basal membrane, together with a thickening of the basal membrane. Thus, vascular endothelial PDPK1 plays an important role in the maintenance of pancreatic beta cell mass and function by maintaining the vascularity of the pancreas and islets and protecting them from hypoxia, hypoxia-related ER stress, inflammation and distortion of capillary structure
Our in vitro results suggest that GSIS is impaired (Fig. 1c, Fig. 2a,b). However, the perifusion experiment did not end with a period of low glucose and we did not confirm that the insulin secretion decreased again during low glucose, so we cannot completely exclude the possibility that the preparation was damaged to some extent during the experiment.
In our study, OGTT revealed unexpectedly elevated levels of active GLP-1 in VE-PDPK1-KO mice (Fig. 1f). This might be a compensational elevation due to chronically impaired sensitivity to incretin hormone in islets of the knockout mice. The Glp1r mRNA level was significantly reduced in knockout mice. However, the level of GLP-1 receptor protein was not assessed by western blotting or immunostaining in this study as there was no appropriate antibody commercially available. This could be a limitation of our study. In addition, serum glucagon level was not decreased during OGTT in VE-PDPK1-KO mice although insulin and glucagon double staining revealed decreased alpha cell mass (Fig. 1g, Fig. 3c). This might be explained by the phenomena that blood glucose level was maintained within physiological range, and not hypoglycaemic range. Hyperinsulinaemic–hypoglycaemic clamp might reveal the difference in glucagon levels and needs further investigation to address this question.
Several studies have demonstrated that islets in animal models of diabetes, such as db/db mice, KKay mice and Zucker diabetic fatty rats, are exposed to the hypoxic state in vivo or ex vivo [19–21]. In these models, islets are exposed to high oxygen consumption due to GSIS induced by high blood glucose levels, leading to hypoxia in islets. On the contrary, our study demonstrated that reduced vasculature in pancreatic beta cells led to reduced beta cell mass and function in VE-PDPK1-KO mice.
The integrity of the islet microvasculature is essential for normal islet function as it regulates the transport of nutrients and oxygen in addition to support of adequate paracrine interactions within each islet. However, since previous papers showed that a stronger reduction in vessel density does not always strongly affect islet function in vitro [22, 23], it is not clear whether the density alone is key to the phenotypes of our observation. Therefore, we evaluated capillary morphology by electron microscopy. We elucidated that capillary morphology was markedly altered in our endothelial cell-specific Pdpk1-knockout mice. We think that such alteration in capillary morphology in addition to the reduction of vessel density could, at least in part, explain the deterioration of islet function.
It was reported that beta cell-specific Vegfa knockout results in glucose intolerance and diabetes [24]. Vegfa-knockout mice displayed decreased density of the microvasculature and abnormal morphology of capillaries was observed. The pancreatic endothelial cells of the knockout mice had a thicker cell body and there were few fenestrae and thus the vessel permeability within islets was impaired. Interestingly, a recent report elucidated that overexpression of Vegfa in beta cells actually increased endothelial cell number but impaired islet morphogenesis and both alpha and beta cell proliferation [25]. These studies suggest that the precise control of Vegfa production by developing islet cells is important for normal islet development and vascularisation. In our model, the expression level of Vegfa was significantly increased in VE-PDPK1-KO mice, probably due to increased Hif1α expression. However, PDPK1 knockdown in human umbilical arterial endothelial cells (HUAECs) revealed significantly reduced expression levels of vascular endothelial growth factor receptor 2 (VEGFR2) and this was not observed following IRS2 knockdown (data not shown). It is possible that increased Vegfa could not increase islet vascularity due to reduced VEGFR2 expression in endothelial cells of VE-PDPK1-KO mice.
Electron microscopy observation revealed that cells were aligned along the basal membrane in islets of control flox mice (Fig. 5a). However, we cannot completely rule out that these remaining cells might be pericytes and not endothelial cells. In addition, basement membrane structures with no endothelium, so-called ‘empty sleeves’, were observed in islets of VE-PDPK1-KO mice (Fig. 5b). Nikolova et al reported that pancreatic beta cells do not form a basement membrane by themselves and that they attract endothelial cells and form capillaries with a vascular basement membrane next to pancreatic beta cells by using Vegfa [26]. We assume that endothelial cells were present at an earlier stage of development but left the islets of knockout mice due to lack of PDPK1, as basal membrane structures did exist in islets of both control flox and knockout mice. However, in the context of the report by Nikolova et al, the possibility that endothelial cells were removed during the procedure of digestion for islet isolation cannot be completely discounted.
VE-PDPK1-KO mice presented with reduced alpha and beta cell masses and impaired GSIS and response to Ex-4 in isolated islets. These results were not observed in endothelial cell-specific insulin receptor knockout (EndoIRKO) and endothelial cell-specific Irs2-knockout (ETIRS2KO) mice. There are some hypotheses which might explain the difference in phenotype between our VE-PDPK1-KO mice and these mice. First, VE-PDPK1-KO mice displayed decreased pancreatic blood flow and significantly increased ratio of pimonidazole-positive islets. It might be surprising that the ratio of pimonidazole-positive islets reached approximately 15–20% in control flox mice. However, this result was compatible with the findings in previous reports [27, 28]. Although speculative, we assume that hypoxia is induced to some extent even under normal conditions through some unknown mechanism. In addition, we cannot deny the possibility that pimonidazole shows false positivity under some experimental conditions. In ETIRS2KO mice, vascularity in islets was significantly decreased, while there was no difference in pancreatic blood flow and ratio of pimonidazole-positive islets. This means that islets in VE-PDPK1-KO mice were in a more hypoxic state compared with ETIRS2KO mice. It was reported that moderately hypoxic MIN6 cells presented similar features to VE-PDPK1-KO mice, such as decreased expression of insulin and its transcription factors such as Mafa, Pdx1 and Neurod and Glp1r and impaired GSIS accompanied by activation of ER stress-related apoptotic genes [29]. There might be a threshold of hypoxia level, which determines beta cell survival rate in vivo and which needs further investigation. Second, insulin delivery to pancreatic islets through endothelial cells might be more severely impaired in VE-PDPK1-KO mice than in EndoIRKO and ETIRS2KO mice. It was reported that the PI3K signal plays an important role in transendothelial insulin delivery [30]. PDPK1 is the downstream molecule of PI3K. On the contrary, the insulin receptor could be compensated for by Igf1r and Irs2 could be compensated for by Irs1, as both the insulin receptors and Irs2 function upstream of PI3K. Therefore, it is possible that the mass and function of islets in VE-PDPK1-KO mice were more severely deteriorated than those in EndoIRKO and ETIRS2KO mice. Last, as described above, PDPK1 knockdown in HUAECs caused an apparent decrease in VEGFR2 expression and its phosphorylation, not observed following IRS2 knockdown (data not shown). VEGFR2 plays a very important role in normal microvascular formation including its fenestration. This might partially explain the difference between VE-PDPK1-KO and ETIRS2KO mice.
In conclusion, vascular endothelial Pdpk1 plays an important role in the maintenance of pancreatic beta cell mass and function. Furthermore, our study demonstrates the different roles in vascular endothelial cells for Pdpk1, Insr and Irs2, all of these genes being important in insulin–PI3K signalling.
Electronic supplementary material
(PDF 386 kb)
Acknowledgements
We would like to express our cordial appreciation to K. Kotani (Saso Hospital, Kobe, Japan), W. Ogawa (Division of Diabetes, Metabolism, and Endocrinology, Kobe University Graduate School of Medicine, Kobe, Japan) and M. Kasuga (Research Institute, International Medical Center of Japan, Tokyo, Japan) for kindly providing Pdpk1flox/flox mice. We also thank Y. Nogami and S. Moriuchi (Research assistants of Kawasaki Medical School, Department of Diabetes, Endocrinology and Metabolism) for help with the animal care. Some of the data were presented as an abstract at the ADA 77th Scientific Sessions in 2017 (San Diego).
Abbreviations
- EndoIRKO
Endothelial cell-specific insulin receptor knockout
- ETIRS2KO
Endothelial cell-specific Irs2-knockout
- Ex-4
Exendin-4
- GLP-1
Glucagon-like peptide-1
- GSIS
Glucose-stimulated insulin secretion
- HBSS
Hanks’ Balanced Salt Solution
- HUAEC
Human umbilical arterial endothelial cells
- PDPK1
3′-Phosphoinositide-dependent protein kinase-1
- PI3K
Phosphoinositide 3-kinase
- UPR
Unfolded protein response
- VEGFR2
Vascular endothelial growth factor receptor 2
- VE-PDPK1-KO
Vascular endothelial cell-specific Pdpk1-knockout
Contribution statement
AO, YO, TM, KKa and HK conceived the study and designed the experiments. AO, TKimu, YO, MS, TKino, KKo, SO, HH, SK and SN conducted the experiments. AO, HH and SN analysed the data. All authors interpreted the data and contributed to discussion. AO drafted the manuscript. All authors critically revised and edited the manuscript and figures. All authors approved the final version of this manuscript. AO is responsible for the integrity of the work as a whole.
Funding
This work was supported by a Grant-in-Aid from the Japan Society for the Promotion of Science (no. 17K16157 to AO and no. 16K09770 to HK) and Research Project Grants from the Kawasaki Medical School (no. 28-101 to AO and no. 27-14 to HK). Additional support was provided by the Japan Foundation for Applied Enzymology (Front Runner of Future Diabetes Research grant to AO).
Data availability
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.
Duality of interest
K.Ka has been an advisor to, received honoraria for lectures from and received scholarship grants from Novo Nordisk Pharma, Sanwa Kagaku Kenkyusho, Takeda, Taisho Pharmaceutical Co., Ltd, MSD, Kowa, Sumitomo Dainippon Pharma, Novartis, Mitsubishi Tanabe Pharma, AstraZeneca, Nippon Boehringer Ingelheim Co., Ltd, Chugai, Daiichi Sankyo and Sanofi. HK has received honoraria for lectures and received scholarship grants from Sanofi, Novo Nordisk, Lilly, Boehringer Ingelheim, MSD, Takeda, Ono Pharma, Daiichi Sankyo, Sumitomo Dainippon Pharma, Mitsubishi Tanabe Pharma, Pfizer, Kissei Pharma, AstraZeneca, Astellas, Novartis, Kowa, Chugai and Taisho Pharma. All other authors declare that there is no duality of interest associated with their contribution to this manuscript.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
10/11/2019
It has been brought to our attention that Fig. 5a showing the vasculature in islets of control flox mice is not in fact an endocrine cell but rather exocrine tissue.
Change history
10/11/2019
It has been brought to our attention that Fig. 5a showing the vasculature in islets of control flox mice is not in fact an endocrine cell but rather exocrine tissue.
References
- 1.Rhodes CJ. Type 2 diabetes-a matter of β-cell life and death? Science. 2005;307(5708):380–384. doi: 10.1126/science.1104345. [DOI] [PubMed] [Google Scholar]
- 2.Alarcon C, Boland BB, Uchizono Y, et al. Pancreatic β-cell adaptive plasticity in obesity increases insulin production but adversely affects secretory function. Diabetes. 2016;65(2):438–450. doi: 10.2337/db15-0792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Halban PA, Polonsky KS, Bowden DW, et al. β-Cell failure in type 2 diabetes: postulated mechanisms and prospects for prevention and treatment. Diabetes Care. 2014;37(6):1751–1758. doi: 10.2337/dc14-0396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Richards OC, Raines SM, Attie AD, et al. The role of blood vessels, endothelial cells, and vascular pericytes in insulin secretion and peripheral insulin action. Endocr Rev. 2010;31(3):343–363. doi: 10.1210/er.2009-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jansson L. The regulation of pancreatic islet blood flow. Diabetes Metab Rev. 1994;10(4):407–416. doi: 10.1002/dmr.5610100405. [DOI] [PubMed] [Google Scholar]
- 6.Kondo T, Vicent D, Suzuma K. Knockout of insulin and IGF-1 receptors on vascular endothelial cells protects against retinal neovascularization. J Clin Invest. 2003;111(12):1835–1842. doi: 10.1172/JCI200317455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mukai Y, Rikitake Y, Shiojima I, et al. Decreased vascular lesion formation in mice with inducible endothelial-specific expression of protein kinase Akt. J Clin Invest. 2006;116(2):334–343. doi: 10.1172/JCI26223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Konishi M, Sakaguchi M, Lockhart SM, et al. Endothelial insulin receptors differentially control insulin signaling kinetics in peripheral tissues and brain of mice. Proc Natl Acad Sci U S A. 2017;114(40):E8478–E8487. doi: 10.1073/pnas.1710625114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kubota T, Kubota N, Kumagai H, et al. Impaired insulin signaling in endothelial cells reduces insulin-induced glucose uptake by skeletal muscle. Cell Metab. 2011;13(3):294–307. doi: 10.1016/j.cmet.2011.01.018. [DOI] [PubMed] [Google Scholar]
- 10.Hashimoto S, Kubota N, Sato H, et al. Insulin receptor substrate-2 (Irs2) in endothelial cells plays a crucial role in insulin secretion. Diabetes. 2015;64(3):876–886. doi: 10.2337/db14-0432. [DOI] [PubMed] [Google Scholar]
- 11.Tawaramoto K, Kotani K, Hashiramoto M, et al. Ablation of 3-phosphoinositide-dependent protein kinase 1 (Pdk1) in vascular endothelial cells enhances insulin sensitivity by reducing visceral fat and suppressing angiogenesis. Mol Endocrinol. 2012;26(1):95–109. doi: 10.1210/me.2010-0412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol. 2001;230(2):230–242. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- 13.Terauchi Y, Takamoto I, Kubota N, et al. Glucokinase and IrS-2 are required for compensatory beta cell hyperplasia in response to high-fat diet-induced insulin resistance. J Clin Invest. 2007;117(1):246–257. doi: 10.1172/JCI17645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsui J, Terauchi Y, Kubota N, et al. Pioglitazone reduces islet triglyceride content and restores impaired glucose-stimulated insulin secretion in heterozygous peroxisome proliferator-activated receptor-gamma-deficient mice on a high-fat diet. Diabetes. 2004;53(11):2844–2854. doi: 10.2337/diabetes.53.11.2844. [DOI] [PubMed] [Google Scholar]
- 15.Jansson L, Hellerström C. A rapid method of visualizing the pancreatic islets for studies of islet capillary blood flow using non-radioactive microspheres. Acta Physiol Scand. 1981;113(3):371–374. doi: 10.1111/j.1748-1716.1981.tb06909.x. [DOI] [PubMed] [Google Scholar]
- 16.Kimura T, Kaneto H, Shimoda M, et al. Protective effects of pioglitazone and/or liraglutide on pancreatic β-cells in db/db mice: comparison of their effects between in an early and advanced stage of diabetes. Mol Cell Endocrinol. 2015;400:78–89. doi: 10.1016/j.mce.2014.11.018. [DOI] [PubMed] [Google Scholar]
- 17.Takikawa A, Mahmood A, Nawaz A. HIF-1α in myeloid cells promotes adipose tissue remodeling toward insulin resistance. Diabetes. 2016;65(12):3649–3659. doi: 10.2337/db16-0012. [DOI] [PubMed] [Google Scholar]
- 18.Iwashita N, Uchida T, Choi JB, et al. Impaired insulin secretion in vivo but enhanced insulin secretion from isolated islets in pancreatic beta cell-specific vascular endothelial growth factor-A knock-out mice. Diabetologia. 2007;50(2):380–389. doi: 10.1007/s00125-006-0512-0. [DOI] [PubMed] [Google Scholar]
- 19.Li X, Zhang L, Meshinchi S, Dias-Leme C, et al. Islet microvasculature in islet hyperplasia and failure in a model of type 2 diabetes. Diabetes. 2006;55(11):2965–2973. doi: 10.2337/db06-0733. [DOI] [PubMed] [Google Scholar]
- 20.Sato Y, Endo H, Okuyama H, et al. Cellular hypoxia of pancreatic β-cells due to high levels of oxygen consumption for insulin secretion in vitro. J Biol Chem. 2011;286(14):12524–12532. doi: 10.1074/jbc.M110.194738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bensellam M, Maxwell EL, Chan JY, et al. Hypoxia reduces ER-to-Golgi protein trafficking and increases cell death by inhibiting the adaptive unfolded protein response in mouse beta cells. Diabetologia. 2016;59(7):1492–1502. doi: 10.1007/s00125-016-3947-y. [DOI] [PubMed] [Google Scholar]
- 22.Kragl M, Schubert R, Karsjens H et al (2016) The biomechanical properties of an epithelial tissue determine the location of its vasculature. Nat Commun 7:13560 [DOI] [PMC free article] [PubMed]
- 23.Brissova M, Shostak A, Shiota M, et al. Pancreatic islet production of vascular endothelial growth factor-a is essential for islet vascularization, revascularization, and function. Diabetes. 2006;55(11):2974–2985. doi: 10.2337/db06-0690. [DOI] [PubMed] [Google Scholar]
- 24.Lammert E, Gu G, McLaughlin M, et al. Role of VEGF-A in vascularization of pancreatic islets. Curr Biol. 2003;13(12):1070–1074. doi: 10.1016/S0960-9822(03)00378-6. [DOI] [PubMed] [Google Scholar]
- 25.Cai Q, Brissova M, Reinert RB, et al. Enhanced expression of VEGF-A in β cells increases endothelial cell number but impairs islet morphogenesis and β cell proliferation. Dev Biol. 2012;367(1):40–54. doi: 10.1016/j.ydbio.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nikolova G, Jabs N, Konstantinova I, et al. The vascular basement membrane: a niche for insulin gene expression and Beta cell proliferation. Dev Cell. 2006;10(3):397–405. doi: 10.1016/j.devcel.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 27.Olsson R, Carlsson PO. A low-oxygenated subpopulation of pancreatic islets constitutes a functional reserve of endocrine cells. Diabetes. 2011;60(8):2068–2075. doi: 10.2337/db09-0877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lau J, Svensson J, Grapensparr L, Johansson Å, Carlsson PO. Superior beta cell proliferation, function and gene expression in a subpopulation of rat islets identified by high blood perfusion. Diabetologia. 2012;55(5):1390–1399. doi: 10.1007/s00125-012-2476-6. [DOI] [PubMed] [Google Scholar]
- 29.Sato Y, Inoue M, Yoshizawa T, Yamagata K. Moderate hypoxia induces β-cell dysfunction with HIF-1-independent gene expression changes. PLoS One. 2014;9(12):e114868. doi: 10.1371/journal.pone.0114868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang H, Wang AX, Liu Z, Barrett EJ. Insulin signaling stimulates insulin transport by bovine aortic endothelial cells. Diabetes. 2008;57(3):540–547. doi: 10.2337/db07-0967. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 386 kb)
Data Availability Statement
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.