

Abstract
Purpose of Review:
Immune dysregulation is a defining feature of myelodysplastic syndromes (MDS). Recently, several studies have further defined the complex role of immune alterations within MDS. Herein we will summarize some of these findings and discuss the therapeutic strategies currently in development.
Recent Findings:
Immune alterations in MDS are complex, heterogeneous and intertwined with clonal hematopoiesis and stromal cell dysfunction. Inflammation in MDS proceeds as a vicious cycle, mediated in large part by secreted factors, which induce cell death and activate innate immune signaling. Therapeutic targeting of this variable immune dysregulation has led to modest responses thus far, but incorporation of the growing repertoire of immunotherapy brings new potential for improved outcomes.
Summary:
The immune milieu is variable across the spectrum of MDS subtypes, with a changing balance of inflammatory and suppressive cellular forces from low to high risk disease.
Keywords: myelodysplastic syndromes, inflammation, immune dysregulation, bone marrow microenvironment, immunotherapy
Introduction
Myelodysplastic syndromes (MDS) constitute a heterogeneous group of clonal bone marrow neoplasms defined by hematopoietic dysplasia, cytopenias, and a variable risk of transformation into secondary acute myeloid leukemia (AML). An diagnosis of MDS is based primarily on morphological dysplasia in the bone marrow and clinically documented cytopenias that can range from indolent disease with mild anemia to aggressive disease on the verge of AML.[1] Underlying these disparate clinical presentations are the phenomena of clonal hematopoiesis secondary to recurrent genetic alterations and a varying degree of immune and microenvironment dysregulation. Pre-clinical and clinical studies have shown that chronic or unresolved inflammation disrupts immune function and alters the bone marrow microenvironment, thus contributing to disease initiation and progression (Figure 1).[2–6] Though this environment is complex, an improved understanding of the altered immune mechanisms in MDS pathogenesis may identify new targets and lead to novel therapeutics. This article will review the recent advances in characterization of the role of the immune system in MDS pathogenesis and outline the progress of related therapeutic strategies.
Figure 1.
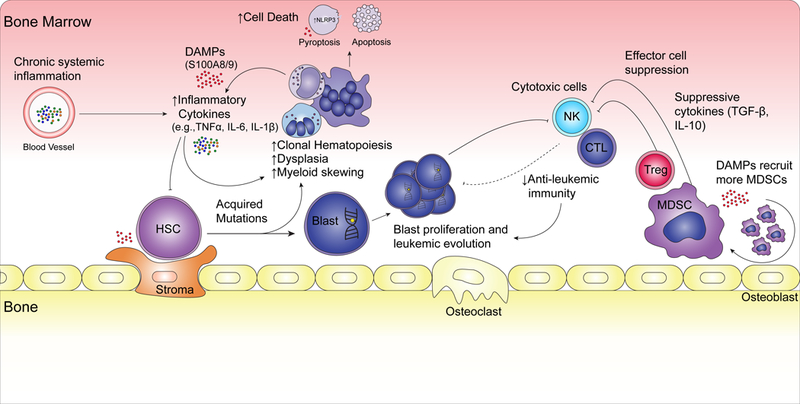
Overview of Immune Dysregulation in MDS.
Part I: New and Established Mechanisms of Immune Dysregulation in MDS
Inflammation and cell death in MDS
Abnormal cytokine secretion patterns play a major role in immune dysregulation in MDS.[7, 8] While many cytokines and growth factors have abnormal levels in MDS, among those with the greatest impact include tumor necrosis factor alpha (TNFα), interleukin (IL)-6, and IL- 1β. Increased levels of TNFα, in particular, have been shown in the bone marrow and serum of MDS patients and are associated with several effects, including increased apoptosis, suppression of hematopoiesis, higher bone marrow cellularity, and activation of downstream signaling pathways and transcription factors.[9–13] High levels of intramedullary apoptosis have been noted across the disease spectrum and are initiated in part secondary to TNFα exposure, which increases first apoptosis signal (Fas) receptor signaling, a canonical apoptotic pathway.[14, 15, 11, 7, 16–19] Similarly, IL-6 has also been implicated in aspects of the MDS phenotype, including induction of increased cell proliferation and hypoferremia due to increased hepcidin, which contributes to anemia.[20–22] Clinically, aberrant cytokine secretion patterns, specifically elevated levels of TNFα and IL-6, are also associated with reduced quality of life and inferior leukemia-free and overall survival.[23, 24, 12] Recently, pyroptosis, a novel mode of inflammatory cell death, has been elucidated in MDS.[25, 26] Pyroptosis proceeds from the assembly of the nucleotide-binding domain and leucine-rich repeat pyrin (NLRP) domain pattern recognition receptors, specifically NLRP3. This in turn induces cell death via a caspase-1 dependent mechanism and stimulates production of IL-iβ and IL-18. Moreover, pyroptosis and resultant ineffective hematopoiesis can be reversed with blockade of S100A9 via a high affinity decoy receptor (CD33-IgG) in vitro.[25]
Somatic mutations and immune disruption
Less clear in MDS is the role of commonly associated somatic mutations in creating an inflammatory milieu within the bone marrow, though evidence for mutational effects on immune disruption is growing. It was recently reported that haploinsufficiency for the gene encoding the ribosomal protein Rps14, which occurs in the 5q minus syndrome, a distinct subtype of MDS, leads to upregulation of the p53 pathway and subsequent erythroid differentiation blockade.[27] Moreover, this model showed this blockade was mediated by increased expression of S100A8 and S100A9, which are danger associated molecular pattern (DAMP) proteins, leading to a reversible disruption of erythropoiesis. Another group also modeled 5q minus syndrome by knocking out mDia1 and miR-146a, found on the deleted 5q.[28] This resulted in upregulation of TNFα and IL-6 as well as expansion of myeloid derived suppressor cells (MDSCs). Overall, these studies convey the impact of mutations within the myeloid clone and the consequences for immune regulation through both cytokine upregulation and a disrupted balance of immune regulatory components. Other commonly mutated genes in MDS, including ten-eleven translocation 2 (TET2) and DNA methyltransferase 3a (DNMT3a), also disrupt immune regulation. Recent work demonstrated that macrophages bearing a TET2 mutation display impaired resolution of inflammation and increased production of IL-6 and IL-1β as compared to wild type TET2 macrophages.[29] In mice with selective knockout of myeloid Tet2, transcripts of IL-6 and IL-1β, as well as several C-X-C motif chemokines (e.g., Cxcl1, Cxcl2, Cxcl3) were found to be elevated in macrophages compared to those with wild type Tet2.[30] This finding was reported with the groundbreaking observation that clonal hematopoiesis of indeterminate potential (CHIP), previously thought to predispose patients mainly to hematologic malignancies such as MDS and AML, also significantly increased the risk of cardiac disease, which was thought, in part, to increased inflammatory activation of macrophages in the vasculature [30]. Finally, mutations in either TET2 or DNMT3A lead to increased expression of arginase-1, an enzyme found in various myeloid cells including macrophages and MDSCs that catalyzes the breakdown of arginine, a required amino acid in T cell proliferation.[31, 32] Upregulation of arginase-1 within the bone marrow appears to mimic the bone marrow microenvironment found in low risk MDS with increased inflammatory response and proliferation.
Toll-like and IL-1 Receptor Signaling in MDS
Toll-like receptor (TLR) signaling is normally involved in the immune response to foreign pathogens, however, in MDS, TLRs and their downstream effectors are aberrantly activated.[33–35] Mouse studies have demonstrated that activation of TLR signaling with low dose lipopolysaccharide (LPS) alters hematopoiesis by increasing numbers of hematopoietic stem cells (HSCs) and myeloid skewing.[36] Overexpression of a TLR4 ligand, S100A9, in a transgenic model also induced phenotypic characteristics of MDS, including cytopenias and dysplastic hematopoiesis.[37] A study of TNFα receptor-associated factor 6 (TRAF6), a downstream effector of TLR signaling, demonstrated that overexpression impairs hematopoiesis via ubiquitination of RNA splicing factors in MDS. Dysregulation of splicing led to Cdc42 activation and subsequent hematopoietic defects.[38] Moreover, ex vivo inhibition of TLR4 signaling in primary MDS monocytes resulted in decreased production of IL-1β, IL-6 and TNFα, indicating the importance and therapeutic potential of this pathway.[35]
IL-1 receptor (IL-1R) activation also plays a critical role in innate immune dysfunction in MDS. Stimulation of TLR and IL-1R resulted in downstream activation of IL-1 receptor associated kinase 1 (IRAK1), which bound to TRAF6 and led to activation of NFκB (nuclear factor kappa-light-chain-enhancer of activated B cells), a critical transcription factor for pro-survival, pro-inflammatory and anti-apoptotic target genes.[39–41] Small molecule inhibition of IRAK1 resulted in impaired cell growth and increased apoptosis of MDS-like cells, while having a minimal effect on healthy CD34+ cells.[41] In another study, transcriptional analysis of high risk MDS and AML samples identified increased transcription of IL-1 receptor associated protein (IL1RAP), a necessary part of the IL-1R signaling complex, when compared to normal controls. In another context, inactivation of TNF and IL-1β signaling sensitized leukemia stem cells to NFκB inhibition both in vivo and in wtro.[42] Oncogenic signaling and leukemic proliferation downstream of IL-1R has also been demonstrated as a key regulator of leukemic cell expansion and progression in AML.[43] This was further shown to be attenuated by downstream inhibition of p38 mitogen associated protein kinase (p38MAPK, or p38). This pathway likely has implications for high risk MDS and leukemic progression. Indeed, p38 has been long proposed as a potential target in MDS and current trials are underway (Table 1 and discussed below). TLR and IL-1R hyperactivation ultimately result in downstream activation of signals, such as NFκB, that increase secretion of inflammatory cytokines, disrupt hematopoiesis, and cause leukemic expansion. Further elucidation of the signaling cross talk within these pathways and therapeutic potential should continue to be developed.
Table 1:
Therapies evaluating various immune targets in MDS.
| Class | Therapy | Target | Mechanism | Clinical Trial | Status |
|---|---|---|---|---|---|
| TLR signaling | OPN-305 | TLR2 | Humanized anti-TLR2 mAB | NCT02363491: second-line treatment for lower risk MDS | Phase I/II, recruiting |
| CX-01 | TLR4 | Heparin-derived polysaccharide disrupts interaction between TLR4 and HMGB1 | NCT02995655: combination therapy with azacitidine in R/R MDS and AML | Phase I, recruiting | |
| Cytokine inhibitors | Etanercept | TNFα | Soluble TNFa receptor | NCT00118287: combination azacitidine and Etanercept in MDS | Phase I/II, completed |
| Siltuximab | IL-6 | Chimeric monoclonal antibody to IL-6 | NCT01513317: siltuximab vs. placebo with best supportive care in anemic patients with IPSS low or intermediate-1 risk MDS | Phase II, terminated | |
| Luspatercept | TGFβ | ||||
| p38 inhibitor | Talmapimod (SCIO-469) | p38 | Inhibitor of p38α | NCT00113893: open label study for any patient with MDS | Phase II, completed |
| ARRY-614 | p38/Tie2 | Inhibitor of p38 and Tie2 | NCT00916227: ARRY-614 in patients with IPSS low or intermediate-1 risk MDS | Phase I, completed | |
| Checkpoint inhibitor | Pembrolizumab (MK-3475) | PD-1 | Monoclonal antibody to PD-1 | NCT01953692: Pembrolizumab in patients with blood cancers | Phase Ib, active not recruiting |
| Nivolumab | PD-1 | Monoclonal antibody to PD-1 | NCT02464657: safety and efficacy of nivolumab in combination with idarubicin and cytarabine in MDS and AML | Phase I/II, recruiting | |
| Nivolumab | PD-1 | Monoclonal antibody to PD-1 | NCT 03417154: nivolumab and cyclophosphamide in R/R AML and high risk MDS | Phase II, not yet recruiting | |
| Ipiliumab | CTLA-4 | Monoclonal antibody to CTLA-4 | NCT02530463: nivolumab and/or ipililumab, with or without azacitidine in MDS | Phase II, recruiting | |
| Trispecific Killer Cell Engager | CD16/IL-15/CD33 TRiKE | CD16/CD33 | anti-CD16 single chain variable fragment (scFv) to engage NK cells and anti-CD33 scFv with a modified IL-15 linker | NCT03214666: CD16/IL-15/CD33 TRiKE for CD33 hematological malignancies | Phase I/II, not yet recruiting |
| Dual Affinity Retargeting molecule | Flotetuzumab (MGD006) | CD123/CD3 | DART recognizing CD123 and CD3 | NCT02152956: safety of MGD006 in R/R AML or IPSS intermediate-2 or high risk MDS | Phase I, recruiting |
| Monoclonal Antibody | BI 836858 | CD33 | Monoclonal antibody to CD33 | NCT02240706: BI836858 vs. best supportive care in patients with IPSS low or intermediate-1 risk MDS | Phase I/II, recruiting |
| Vaccine therapies | PR1 Leukemia Peptide Vaccine | Stimulates host’s immune system to mount a cytotoxic T lymphocyte response to tumor cells | NCT00004918: Vaccine Immune Adjuvant in Chronic Myeloid Leukemia (CML), AML or MDS | Phase I/II, completed | |
| DEC-205/NY-ESO-1 Fusion Protein CDX-1401 | Stimulates host’s immune system to mount a cytotoxic T lymphocyte response to tumor cells | NCT03358719: DEC-205/NY-ESO-1 Fusion Protein CDX-1401, Poly ICLC, Decitabine, and Nivolumab in Treating Patients With MDS or AML | Phase I, not yet recruiting | ||
| NK cellular therapy | Lirilumab | KIR2DL½L3 | Monoclonal antibody to KIR2DL½L3 | NCT 02599649: nivolumab and/or lirilumab, with or without azacitidine in MDS | Phase II, recruiting |
| Activated NK cells | - | Expansion of NK cells ex vivo in presence of irradiated K562 cell line | NCT02123836: NK cell therapy in AML and MDS | Phase I, recruiting | |
| Activated NK cells | - | Donor NK cell transfusion | NCT01898793: Cytokine-induced Memory-like NK Cells in Patients With AML or MDS | Phase I/II, recruiting | |
| Activated NK cells | Donor NK cell transfusion | NCT02890758: Universal Donor NK Cell Therapy in Combination with ALT803 | Phase I, recruiting | ||
| CAR T cell targeting NKG2D-ligand | CART T cells targeting NKG2D-ligands on myeloid cells | NCT02203825: Safety Study of Chimeric Antigen Receptor Modified T-cells Targeting NKG2D-Ligands in MDS/AML/MM | Phase I, not yet recruiting |
Immune Landscape from Low Risk to High Risk MDS
The immune disruption present in MDS also affects cell types outside of the myeloid compartment and these effects vary from low to high risk disease. Patients with low risk MDS harbored increased numbers of Th17 cells, cells which have also been strongly associated with autoimmunity.[44] Elevation in the numbers of Th17 in low risk MDS was also associated with increased levels of cytotoxic T cells and pro-inflammatory cytokines; however, in high risk MDS, Th17 cells declined while levels of inhibitory cytokines, such as IL-10 and soluble IL-2R, increased.
The role and abundance of immune suppressor cells differs greatly across the disease spectrum. Several reports have demonstrated that higher risk disease is accompanied by an increase in MDSCs (MDSCs).[2, 45, 37, 13] MDSCs and their effects were functionally important in the pathogenesis of MDS in a transgenic model.[37] Overexpression of the DAMP, S100A9, in this model resulted in accumulation of bone marrow MDSCs and CD33-dependent IL-10 and TGFβ secretion. Furthermore, disruption of MDSC function via either blockade of the CD33:S100A9 interaction or forced maturation of MDSCs with all-trans retinoic acid (ATRA) resulted in rescue of hematopoiesis.[37] MDSCs were shown to be distinct from the MDS clone via sorting and sequencing, suggesting that some MDSCs arise as a response to the mutated clone and are not mutated themselves in MDS.[37] MDSCs in MDS possessed increased in expression of CXCR4, or CD184, a critical chemokine receptor in homing to the bone marrow, along with CX3CR1.[2] Authors speculated that elevated CXCR4 increased MDSC homing and retention within the bone marrow, thus increasing marrow-specific suppressive effects of these cells. Regulatory T cells (Tregs) were also increased in high risk disease, along with MDSCs, while in low risk MDS, Tregs are less abundant.[2, 44, 3] One potential mechanism of this reduction is through increased expression of interferon regulatory factor-1(IRF-1), a suppressor of Foxp3 expression.[46] Taken together, these data indicate a dynamic role of inflammatory and suppressive cells subsets over the spectrum of MDS.
One effect of increasing suppressive cell subsets is reduction of cytotoxic anti-leukemia immunity. The activation and function of natural killer (NK) cells exert important cytotoxic activity in response to MDS and other myeloid neoplasms, but their function is reduced in high risk MDS.[6, 47] Moreover, NK cells from MDS patients lose in vitro anti-leukemic activity due to a TNFα-induced IL-32 secretion from stromal cells.[48] In this study, IL-32 directly inhibited the activity of NK cells, and it was found that stromal cells from MDS patients produced higher levels of IL-32. The authors concluded that IL-32 impairment of NK cells may contribute to persistence and proliferation of clonal blasts and further progression to leukemia. Leukemic blasts also inhibit NK cell function directly via TGFβ secretion in microvesicles.[49]
Stromal Cell Contributions to MDS
Bone marrow stromal cells arise from mesenchymal stem cells (MSCs) and play a direct role in myeloid skewing and increased inflammatory response in MDS.[50] This was shown in a study that transplanted patient-derived mesenchymal stromal cells into an immunocompromised mouse model and revealed the stromal cells were critical for propagation of the MDS clone.[50] Further studies have shown that MSCs can participate in reciprocal signaling with mutated MDS hematopoietic cells, have altered epigenetic profiles as compared to non-MDS MSCs and drive disease progression starting at initiation.[51] Recent studies have shown MSC epigenetic dysregulation leads to inappropriate activation of the beta-catenin pathway and subsequent disease progression.[52] In addition to their unique epigenetic signature, the transcriptome of MDS-associated stromal cells is also distinct from healthy stromal cells. Transcriptomic analysis of stromal cells in the bone marrow of MDS patients has revealed a transcriptional signature characterized by cellular stress and upregulation of inflammation-associated secreted factors.[53] In addition, alterations of osteolineage progenitors can disrupt hematopoiesis and induce myelodysplasia.[54] The fundamental contributions of non-hematopoietic cells in MDS pathogenesis demonstrates the importance of further investigation into the hematopoietic-stromal interaction.
Part II: Therapeutic Targeting of the Immune System in MDS
Immune Suppression in the Treatment of MDS
Therapies such as anti-thymocyte globulin (ATG) and cyclosporine, both T cell directed therapies, have been found to be effective for some patients with MDS, particularly in the hypoplastic subtype.[55–57] Published studies report response rates to both of these therapies vary greatly (0–100% response with ATG and 33–82% for cyclosporine) and combination therapy is not superior to monotherapy.[58, 59] A single center study of immunosuppressive therapy (IST), including ATG and cyclosporine, demonstrated response rate in low risk disease similar to other standard therapies, however high risk disease patients did not respond as well to IST.[60] A more recent study presented in abstract form was one of the largest studies of immunosuppressive therapy in MDS. Immunosuppressive therapy in this large cohort showed overall response rate of 45%, leading to transfusion independence in 39% of patients.[61] A correlation of response with MDS subtype or other factors was not observed. Over the past decade more has been discovered regarding the role of the immune system in MDS and with these discoveries comes the advent of directed immunotherapies. These therapies fall into distinct groups: targeting of the inflammatory components of MDS, directing cytotoxic cells (including T cells and NK cells) against the MDS clone and targeting the mutant myeloid cells via immune-mediated mechanisms.
Direct Inhibition of Cytokines and Downstream Targets
Early strategies to target abnormal cytokine levels in MDS focused on anti-TNFα therapy.[62] Single agent studies of etanercept, a TNFαreceptor mimetic, and infliximab, a monoclonal antibody to TNFα, showed some early activity, however a phase II trial of infliximab reported low activity and insufficient responses.[62, 63] Combinations with anti-TNF agents have also been underwhelming. Azacitidine, a DNA methyltansferase inhibitor (DNMTi), and etanercept were combined with a reported 72% overall response rate after 3 months of therapy; however, this was a single arm study and response criteria differed from key historical controls with azacitidine alone.[64] Another phase II study of 25 patients combined etanercept with ATG showed the combination was effective in low risk MDS with overall response rate of 56%.[65] Unfortunately, TNFα inhibitors have not been as successful as once hoped and they are not considered standard therapy in MDS at present. IL-6, another cytokine implicated in MDS pathogenesis, has also been used in MDS, with similarly poor results. Siltuximab, a monoclonal antibody to IL-6, was studied in a phase II trial in 2012, but this was terminated early due to lack of efficacy in the primary endpoint (transfusion reduction).[20] Some therapeutic potential, however, exists in newer strategies to target cytokine signaling. Luspatercept, a TGFβ superfamily ligand trap, inhibits downstream SMAD2/3 signaling and improves erythropoiesis in preclinical and early phase studies.[66, 67] This therapy seems to be particularly useful in MDS with a splicing factor 3B subunit 1 (SF3B1) mutation, which is enriched in MDS with ringed sideroblasts. In a phase II study of luspatercept in lower risk MDS showed a 63% rate of hematologic improvement and a 38% of red blood cell transfusion independence. Despite underwhelming evidence in support of cytokine targeting, pre-clinical evaluation of IL-8 inhibition suggests it may be a more effective target than other cytokines, given that high levels of its receptor, CxC chemokine receptor 2 (CXCR2), are associated with adverse prognosis and increased transfusion dependency.[68] Additionally blockade of IL-8 could decrease recruitment of inflammatory cells to the bone marrow and reduce inflammation in MDS.
More recent and ongoing efforts to target innate immune signaling have focused on TLR, IL-1, and DAMP signaling. Overexpression and activation of p38 within the bone marrow is a unifying feature in low risk MDS patients.[69] This molecule, as discussed above, is downstream of the IL-1R and preclinical inhibition has anti-leukemic activity in ex vivo treatment of primary samples.[70, 43] Therefore it is an attractive target as it can be part of many treatment regimens, regardless of mutation subtype. Three small molecule inhibitors of p38 have been used preclinically and clinically: SCIO-469, pexmetinib (ARRY614) and ralimetinib. SCIO-469, inhibits TNFα secretion from myeloid cells and decreases the expression of TNFα and IL-1β from stromal cells in the bone marrow.[71] Data from a phase I/II study, however, indicated rare bone marrow and/or cytogenetic responses, suggesting that clinically this drug may not be as effective as in vitro (NCT00113893).[72] Recent preclinical studies of pexmetinib, a dual inhibitor of p38 and Tie2, the angiopoietin receptor, also demonstrated some hematologic responses.[73] Similar results were seen in the phase I trial of low and intermediate-1 risk MDS, with 5/7 (71%) achieving platelet transfusion independence. However, only three patients (12%) achieved red blood cell transfusion independence (NCT00916227).[74] The newest p38 inhibitor, ramlitinib, has shown some promise as it leads to decreased IL-1 and reduced proliferative effects in an AML model.[43] Targeting p38, thus appears to be of potential utility, especially in low risk MDS, though many studies have only reported preliminary results at this time.
Activation of TLR signaling within MDS makes the TLR axis a promising therapeutic target and there are several ongoing efforts to test this hypothesis. OPN-305 is a humanized IgG4 monoclonal antibody to TLR2. In a phase I setting for a separate indication it was safe and tolerable, while a phase I/II study is underway in patients with MDS (NCT02363491).[4] [75] Additional mechanisms to target TLR signaling include direct inhibition with CX-01, a heparin-derived polysaccharide. CX-01 disrupts TLR4 interactions with high-mobility group box protein 1 (HMBGP1) and other leukocyte/ adhesion molecules.[76] A phase I trial has begun to evaluate efficacy of CX-01 with azacitidine in relapsed or refractory (R/R) MDS (NCT02995655). Another mechanism to inhibit TLR signaling is through its downstream effectors, the IRAK molecules. Currently only preclinical data has shown IRAK¼ inhibition suppresses mutant myeloid cells through downstream inhibition of NF-kB signaling.[41] Moreover, PF-06650833, an IRAK4 inhibitor, is currently only in phase I clinical trial for rheumatoid arthritis (NCT02996500).[77]
MDSCs are also a targetable source of immune dysregulation; however, only preclinical studies have been done to evaluate these cells as a potential therapeutic target. CD33 is a marker of some MDSCs and therefore a logical target.[78] BI836858 is a Fc-engineered CD33 antibody that binds to the CD33 receptor and 1) prevents the release of immune-suppressive cytokines, 2) reduced reactive oxygen species commonly seen in MDSCs and 3) induce antibody-mediated cytotoxicity to MDSCs.[78] A phase I/II trial of BI836858 is ongoing (NCT02240706).
Vaccine Therapy
Vaccinations against cancer antigens are a novel strategy to resensitize the adaptive immune system to tumor-associated epitopes, thus turning on tumor-directed immune response. Wilms tumor 1 (WT1) is a protein expressed on abnormal cells in MDS and AML and vaccines to the antigen have been in development for some time. Studies using WT1 as a vaccine target have shown mixed results, but some encouraging responses in MDS. [79, 80]. CDX-1401,a vaccine featuring the fusion protein of CD205(DEC-205), a marker on dendritic cells, and cancer-testis antigen 1 (CTAG1 or NY-ESO-1), was shown to effectively induce cytotoxic response from T cells when examined ex vivo and has recently concluded phase I with no clinical outcomes reported to date.[81] Another vaccine target includes the PR1 peptide, an HLA-A2 restricted peptide on myeloid leukemia cells, Initially developed in 2008, the first phase I trial displayed both safety and efficacy of the PR1 vaccine.[82] Last year another phase I/II trial also demonstrated robust immune response (53% patients had at least doubled PR1-specific cytotoxic T cells (CTLs)) and objective clinical response (24% patients had complete, partial or hematological improvement) (NCT00004918).[83] Interestingly, the initial trial has now found that repeated vaccinations lead to the expansion of low-avidity PR1-specific CTLs and loss of high-avidity PR1-specific CTLs.[84] Therefore in the future, vaccine schedules will be carefully considered as more frequent schedules could lead to loss of the high-avidity CTLs.
Repurposing Cytotoxic Cells
Harnessing the cytotoxic immune response to target transformed cells has been a successful approach for many solid tumors and lymphomas, however it has unclear utility in myeloid malignancies. Targeted approaches include both stimulating the endogenous system and reengineering lymphoid-derived cells to target the mutant cells. Checkpoint inhibitors are the most prominent current class of immunotherapy and act through binding of a soluble protein to either the checkpoint receptor (e.g., cytotoxic t-lymphocyte associated protein 4 (CTLA4) and programmed death-1 (PD-1)) or ligand (e.g., PD-L1) and blocking the receptor-ligand interaction which would normally act as an “off” switch, causing a T cell to become anergic or exhausted.[85, 86] Preclinical data suggests that this treatment strategy may be useful in MDS where there is increased expression of PD-1.[87] Additionally, epigenetic modifications of the PD-1 promoter site resulting in increased expression have been implicated patients with DNMTi-resistance.[88] Initial studies using pembrolizumab, a monoclonal antibody against PD-1, were disappointing with the first study showing only 4% overall response rate.[89] However, due to data showing upregulation of PD-1 following hypomethylating agents (HMA), more recent trials have investigated the role of PD-1 therapy in HMA-refractory disease as well as in combination with other therapies.[90] Nivolumab, an IgG4 anti PD-1 antibody, is part of a trial comparing standard of care without and without nivolumab (NCT02464657). Additional trials are enrolling patients following HMA failure (NCT02530463) and in combination with traditional chemotherapies and other immunotherapies (table 1). While currently only theoretical, evidence from melanoma suggests that MDSCs found in the microenvironment could be particularly susceptible to combination PD-1 and c-KIT blockade.[91]
A new class of molecules including bi-specific killer engager (BIKE), bispecific T cell engager (BITE), trispecific killer engager (TriKE) and Dual Affinity Re-targeting (DART) antibodies engage specific epitopes both on malignant cells and cytotoxic cells, bringing these two cell types together in close proximity for target cell killing. The first successful in vitro studies with BIKE therapy in MDS used a CD16xCD33 BIKE to reverse MDSC immunosuppression of NK cells and induce MDSC target cell lysis. This therapy was effective in all patient samples, regardless of disease stage.[92] In AML, addition of a modified IL-15 cross-linker to a CD16xCD33 backbone created a CD16xCD33xIL-15 TRiKE. This therapeutic not only increases NK cell mediated killing but concurrently stimulates NK cell proliferation with IL- 15.[93] In vivo TRiKEs are superior to BIKEs. A phase I/II trial of CD16xCD33xIL-15 TRiKE for AML and high risk MDS is planned (NCT03214666). Unlike BiKEs and TriKES, which are one conjugate polypeptide, DARTs consist of two polypeptide chains linked by a disulfide bond. They have been potent against myeloid malignancies in vitro and in early clinical trials.[94] Results from a phase I study of flotetuzumab (MGD006), a CD123xCD3 DART, in AML and high risk MDS were recently reported to have some anti-leukemic activity with an overall response rate of 43% and enrollment for this study continues (NCT02152956).[95] Future combinations with anti-PD-1 therapy may be advantageous as synergistic toxicity has been observed in an in vitro model with flotetuzumab and check point inhibition.[95]
Engineered NK cell cytotoxicity against dysplastic clones is a new area of research and has shown some effect in AML and high risk MDS.[96, 97] There are multiple potential sources of NK cells for patients, including cord blood, matched donors and autologous harvests.[98] Regardless of type of donation, ex vivo expansion in the presence of K562 leukemia cell line prior patient engraftment to produces immature NK cells with greater cytotoxicity and receptor diversity than unexpanded samples.[99, 100] The ideal method of ex vivo expansion is not yet clear; however, priming with IL-15 did improve NK cell cytotoxicity in a murine myeloma model.[101] A recent trial in Sweden demonstrated NK cell therapy was effective in AML and high risk MDS with 6/16 patients (38%) achieving complete or partial remission following low-intensity lymphodepleting agents and haploidentical NK cell infusion. Additionally, 6 refractory patients became eligible for HSCT, suggesting the NK cell therapy may act as a bridge to transplant in otherwise refractory patients.[97] Trials currently enrolling with NK cells are shown in table 1. Chimeric-antigen receptor (CAR) T cells work similarly to NK cell therapy in that they both are endogenous immune regulators coopted to target malignant clones. The role of CAR T cells in MDS has primarily been explored in vivo with promising results. Early preclinical data showed anti-CD123 CAR T cell therapy eliminates MDS clones in a patient derived xenograft (PDX) model.[102] Additionally, a phase I trial of CAR T cells engineered to recognize NKG2D-ligands, commonly found on MDS clones, is underway (NCT02203825).
Conclusions
Immune dysregulation defines MDS across the broad spectrum of disease (Figure 1). Dysregulated immune signaling stems from elevated levels of inflammatory cytokines and mutations that activate innate immune signaling. The combination of these two events leads to an environment that promotes further mutations, clonal hematopoiesis, increased cell proliferation and abnormal cell death. As the inflammation progresses the immune and stromal compartments become transformed as well and cooperate with further decreased anti-leukemic immunity and increased proliferation of blasts. Regulatory and suppressive cells accumulate, ostensibly to quiet the disrupted immune environment, but also likely contribute to immune suppression and leukemia progression. To alter this disrupted phenotype, several strategies have been attempted. The lack of response to cytokine therapy in MDS has been disappointing, but current strategies and emerging rational clinical trial designs hold some promise. Eventually, given the complexity of the disease, combination of multiple immune focused therapies will likely be needed. Lastly, it must be noted that our current armament of immunotherapies is far more sophisticated than just five years ago, thus, we anticipate an ever-quickening pace of discovery in this burgeoning field.
Footnotes
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Conflict of Interest
Kathryn S. Ivy and P. Brent Ferrell, Jr. declare that they have no relevant conflicts of interest.
References
- 1.Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–405. doi: 10.1182/blood-2016-03-643544. [DOI] [PubMed] [Google Scholar]
- 2.Kittang AO, Kordasti S, Sand KE, Costantini B, Kramer AM, Perezabellan P et al. Expansion of myeloid derived suppressor cells correlates with number of T regulatory cells and disease progression in myelodysplastic syndrome. Oncoimmunology. 2016;5(2):e1062208. doi: 10.1080/2162402x.2015.1062208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kordasti SY, Ingram W, Hayden J, Darling D, Barber L, Afzali B et al. CD4+CD25high Foxp3+ regulatory T cells in myelodysplastic syndrome (MDS). Blood. 2007;110(3):847–50. doi: 10.1182/blood-2007-01-067546. [DOI] [PubMed] [Google Scholar]
- 4.Ganan-Gomez I, Wei Y, Starczynowski DT, Colla S, Yang H, Cabrero-Calvo M et al. Deregulation of innate immune and inflammatory signaling in myelodysplastic syndromes. Leukemia. 2015;29(7):1458–69. doi: 10.1038/leu.2015.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li AJ, Calvi LM. The microenvironment in myelodysplastic syndromes: Niche-mediated disease initiation and progression. Exp Hematol. 2017;55:3–18. doi: 10.1016/j.exphem.2017.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Epling-Burnette PK, Bai F, Painter JS, Rollison DE, Salih HR, Krusch M et al. Reduced natural killer (NK) function associated with high-risk myelodysplastic syndrome (MDS) and reduced expression of activating NK receptors. Blood. 2007;109(11):4816–24. doi: 10.1182/blood-2006-07-035519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shetty V, Mundle S, Alvi S, Showel M, Broady-Robinson L, Dar S et al. Measurement of apoptosis, proliferation and three cytokines in 46 patients with myelodysplastic syndromes. Leuk Res. 1996;20(11–12):891–900. [DOI] [PubMed] [Google Scholar]
- 8.Mundle SD, Venugopal P, Cartlidge JD, Pandav DV, Broady-Robinson L, Gezer S et al. Indication of an involvement of interleukin-1 beta converting enzyme-like protease in intramedullary apoptotic cell death in the bone marrow of patients with myelodysplastic syndromes. Blood. 1996;88(7):2640–7. [PubMed] [Google Scholar]
- 9.Seipelt G, Ganser A, Duranceyk H, Maurer A, Ottmann OG, Hoelzer D. Induction of TNF-alpha in patients with myelodysplastic syndromes undergoing treatment with interleukin-3. Br J Haematol. 1993;84(4):749–51. [DOI] [PubMed] [Google Scholar]
- 10.Alexandrakis M, Coulocheri S, Xylouri I, Ganotakis E, Eliakis P, Karkavitsas N et al. Elevated serum TNF-alpha concentrations are predictive of shortened survival in patients with high-risk myelodysplastic syndromes. Haematologia (Budap). 1998;29(1):13–24. [PubMed] [Google Scholar]
- 11.Sawanobori M, Yamaguchi S, Hasegawa M, Inoue M, Suzuki K, Kamiyama R et al. Expression of TNF receptors and related signaling molecules in the bone marrow from patients with myelodysplastic syndromes. Leuk Res. 2003;27(7):583–91. [DOI] [PubMed] [Google Scholar]
- 12.Stifter G, Heiss S, Gastl G, Tzankov A, Stauder R. Over-expression of tumor necrosis factor-alpha in bone marrow biopsies from patients with myelodysplastic syndromes: relationship to anemia and prognosis. Eur J Haematol. 2005;75(6):485–91. doi: 10.1111/j.1600-0609.2005.00551.x. [DOI] [PubMed] [Google Scholar]
- 13.Cluzeau T, McGraw KL, Irvine B, Masala E, Ades L, Basiorka AA et al. Pro-inflammatory proteins S100A9 and TNFalpha suppress erythropoietin elaboration in myelodysplastic syndromes. Haematologica. 2017. doi: 10.3324/haematol.2016.158857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Claessens YE, Bouscary D, Dupont JM, Picard F, Melle J, Gisselbrecht S et al. In vitro proliferation and differentiation of erythroid progenitors from patients with myelodysplastic syndromes: evidence for Fas-dependent apoptosis. Blood. 2002;99(5):1594–601. [DOI] [PubMed] [Google Scholar]
- 15.Ribeiro E, Lima CS, Metze K, Lorand-Metze I. Flow cytometric analysis of the expression of Fas/Fasl in bone marrow CD34+ cells in myelodysplastic syndromes: relation to disease progression. Leuk Lymphoma. 2004;45(2):309–13. [DOI] [PubMed] [Google Scholar]
- 16.Gersuk GM, Beckham C, Loken MR, Kiener P, Anderson JE, Farrand A et al. A role for tumour necrosis factor-alpha, Fas and Fas-Ligand in marrow failure associated with myelodysplastic syndrome. British journal of haematology. 1998;103(1):176–88. [DOI] [PubMed] [Google Scholar]
- 17.Bouscary D, De Vos J, Guesnu M, Jondeau K, Viguier F, Melle J et al. Fas/Apo-1 (CD95) expression and apoptosis in patients with myelodysplastic syndromes. Leukemia. 1997;11(6):839–45. [DOI] [PubMed] [Google Scholar]
- 18.Gupta P, Niehans GA, LeRoy SC, Gupta K, Morrison VA, Schultz C et al. Fas ligand expression in the bone marrow in myelodysplastic syndromes correlates with FAB subtype and anemia, and predicts survival. Leukemia. 1999;13(1):44–53. [DOI] [PubMed] [Google Scholar]
- 19.Gyan E, Frisan E, Beyne-Rauzy O, Deschemin JC, Pierre-Eugene C, Randriamampita C et al. Spontaneous and Fas-induced apoptosis of low-grade MDS erythroid precursors involves the endoplasmic reticulum. Leukemia. 2008;22(10):1864–73. doi: 10.1038/leu.2008.172. [DOI] [PubMed] [Google Scholar]
- 20.Garcia-Manero G, Gartenberg G, Steensma DP, Schipperus MR, Breems DA, de Paz R et al. A phase 2, randomized, double-blind, multicenter study comparing siltuximab plus best supportive care (BSC) with placebo plus BSC in anemic patients with International Prognostic Scoring System low- or intermediate-1-risk myelodysplastic syndrome. Am J Hematol. 2014;89(9):E156–62. doi: 10.1002/ajh.23780. [DOI] [PubMed] [Google Scholar]
- 21.Nemeth E, Rivera S, Gabayan V, Keller C, Taudorf S, Pedersen BK et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest. 2004;113(9):1271–6. doi: 10.1172/JCI20945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schipperus MR, Sonneveld P, Lindemans J, van Lom K, Vlastuin M, Abels J. Interleukin-6 and interleukin-1 enhancement of GM-CSF-dependent proliferation of haematopoietic progenitor cells in myelodysplastic syndromes. Br J Haematol. 1991;77(4):515–22. [DOI] [PubMed] [Google Scholar]
- 23.Tsimberidou AM, Estey E, Wen S, Pierce S, Kantarjian H, Albitar M et al. The prognostic significance of cytokine levels in newly diagnosed acute myeloid leukemia and high-risk myelodysplastic syndromes. Cancer. 2008;113(7):1605–13. doi: 10.1002/cncr.23785. [DOI] [PubMed] [Google Scholar]
- 24.Meyers CA, Albitar M, Estey E. Cognitive impairment, fatigue, and cytokine levels in patients with acute myelogenous leukemia or myelodysplastic syndrome. Cancer. 2005;104(4):788–93. doi: 10.1002/cncr.21234. [DOI] [PubMed] [Google Scholar]
- 25.Basiorka AA, McGraw KL, Eksioglu EA, Chen X, Johnson J, Zhang L et al. The NLRP3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood. 2016;128(25):2960–75. doi: 10.1182/blood-2016-07-730556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sallman DA, Cluzeau T, Basiorka AA, List A. Unraveling the Pathogenesis of MDS: The NLRP3 Inflammasome and Pyroptosis Drive the MDS Phenotype. Front Oncol. 2016;6:151. doi: 10.3389/fonc.2016.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schneider RK, Schenone M, Ferreira MV, Kramann R, Joyce CE, Hartigan C et al. Rps14 haploinsufficiency causes a block in erythroid differentiation mediated by S100A8 and S100A9. Nat Med. 2016;22(3):288–97. doi: 10.1038/nm.4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mei Y, Zhao B, Basiorka AA, Yang J, Cao L, Zhang J et al. Age-related inflammatory bone marrow microenvironment induces ineffective erythropoiesis mimicking del(5q) MDS. Leukemia. 2017. doi: 10.1038/leu.2017.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cull AH, Snetsinger B, Buckstein R, Wells RA, Rauh MJ. Tet2 restrains inflammatory gene expression in macrophages. Exp Hematol. 2017;55:56–70 e13. doi: 10.1016/j.exphem.2017.08.001. [DOI] [PubMed] [Google Scholar]
- 30.Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med. 2017;377(2):111–21. doi: 10.1056/NEJMoa1701719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cull AH, Mahendru D, Snetsinger B, Good D, Tyryshkin K, Chesney A et al. Overexpression of Arginase 1 is linked to DNMT3A and TET2 mutations in lower-grade myelodysplastic syndromes and chronic myelomonocytic leukemia. Leuk Res. 2017;65:5–13. doi: 10.1016/j.leukres.2017.12.003. [DOI] [PubMed] [Google Scholar]
- 32.Pellagatti A, Esoof N, Watkins F, Langford CF, Vetrie D, Campbell LJ et al. Gene expression profiling in the myelodysplastic syndromes using cDNA microarray technology. British journal of haematology. 2004;125(5):576–83. doi: 10.1111/j.1365-2141.2004.04958.x. [DOI] [PubMed] [Google Scholar]
- 33.Maratheftis CI, Andreakos E, Moutsopoulos HM, Voulgarelis M. Toll-like receptor-4 is up-regulated in hematopoietic progenitor cells and contributes to increased apoptosis in myelodysplastic syndromes. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007;13(4):1154–60. doi: 10.1158/1078-0432.ccr-06-2108. [DOI] [PubMed] [Google Scholar]
- 34.Wei Y, Dimicoli S, Bueso-Ramos C, Chen R, Yang H, Neuberg D et al. Toll-like receptor alterations in myelodysplastic syndrome. Leukemia. 2013;27(9):1832–40. doi: 10.1038/leu.2013.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Velegraki M, Papakonstanti E, Mavroudi I, Psyllaki M, Tsatsanis C, Oulas A et al. Impaired clearance of apoptotic cells leads to HMGB1 release in the bone marrow of patients with myelodysplastic syndromes and induces TLR4-mediated cytokine production. Haematologica. 2013;98(8):1206–15. doi: 10.3324/haematol.2012.064642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Varney ME, Melgar K, Niederkorn M, Smith M, Barreyro L, Starczynowski DT. Deconstructing innate immune signaling in myelodysplastic syndromes. Exp Hematol. 2015;43(8):587–98. doi: 10.1016/j.exphem.2015.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen X, Eksioglu EA, Zhou J, Zhang L, Djeu J, Fortenbery N et al. Induction of myelodysplasia by myeloid-derived suppressor cells. J Clin Invest. 2013;123(11):4595–611. doi: 10.1172/JCI67580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fang J, Bolanos LC, Choi K, Liu X, Christie S, Akunuru S et al. Ubiquitination of hnRNPA1 by TRAF6 links chronic innate immune signaling with myelodysplasia. Nat Immunol. 2017; 18(2):236–45. doi: 10.1038/ni.3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Breccia M, Alimena G. NF-kappaB as a potential therapeutic target in myelodysplastic syndromes and acute myeloid leukemia. Expert Opin Ther Targets. 2010;14(11):1157–76. doi: 10.1517/14728222.2010.522570. [DOI] [PubMed] [Google Scholar]
- 40.Culver-Cochran AE, Starczynowski DT. Chronic innate immune signaling results in ubiquitination of splicing machinery. Cell Cycle. 2018:1–4. doi: 10.1080/15384101.2018.1429082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rhyasen GW, Bolanos L, Fang J, Jerez A, Wunderlich M, Rigolino C et al. Targeting IRAK1 as a therapeutic approach for myelodysplastic syndrome. Cancer cell. 2013;24(1):90–104. doi: 10.1016/j.ccr.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li J, Volk A, Zhang J, Cannova J, Dai S, Hao C et al. Sensitizing leukemia stem cells to NF- kappaB inhibitor treatment in vivo by inactivation of both TNF and IL-1 signaling. Oncotarget. 2017;8(5):8420–35. doi: 10.18632/oncotarget.14220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carey A, Edwards DK, Eide CA, Newell L, Traer E, Medeiros BC et al. Identification of Interleukin-1 by Functional Screening as a Key Mediator of Cellular Expansion and Disease Progression in Acute Myeloid Leukemia. Cell Rep. 2017;18(13):3204–18. doi: 10.1016/j.celrep.2017.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kordasti SY, Afzali B, Lim Z, Ingram W, Hayden J, Barber L et al. IL-17-producing CD4(+) T cells, pro-inflammatory cytokines and apoptosis are increased in low risk myelodysplastic syndrome. British journal of haematology. 2009;145(1):64–72. doi: 10.1111/j.1365-2141.2009.07593.x. [DOI] [PubMed] [Google Scholar]
- 45.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–74. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Perazzio AS, Oliveira JS, Figueiredo VL, Chauffaille ML. Increase of IRF-1 gene expression and impairment of T regulatory cells suppression activity on patients with myelodysplastic syndrome: A longitudinal one-year study. Leuk Res. 2017;55:6–17. doi: 10.1016/j.leukres.2017.01.008. [DOI] [PubMed] [Google Scholar]
- 47.Aggarwal N, Swerdlow SH, TenEyck SP, Boyiadzis M, Felgar RE. Natural killer cell (NK) subsets and NK-like T-cell populations in acute myeloid leukemias and myelodysplastic syndromes. Cytometry B Clin Cytom. 2016;90(4):349–57. doi: 10.1002/cyto.b.21349. [DOI] [PubMed] [Google Scholar]
- 48.Marcondes AM, Mhyre AJ, Stirewalt DL, Kim SH, Dinarello CA, Deeg HJ. Dysregulation of IL-32 in myelodysplastic syndrome and chronic myelomonocytic leukemia modulates apoptosis and impairs NK function. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(8):2865–70. doi: 10.1073/pnas.0712391105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Szczepanski MJ, Szajnik M, Welsh A, Whiteside TL, Boyiadzis M. Blast-derived microvesicles in sera from patients with acute myeloid leukemia suppress natural killer cell function via membrane-associated transforming growth factor-beta1. Haematologica. 2011. ;96(9):1302–9. doi: 10.3324/haematol.2010.039743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Medyouf H, Mossner M, Jann JC, Nolte F, Raffel S, Herrmann C et al. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell. 2014;14(6):824–37. doi: 10.1016/j.stem.2014.02.014. [DOI] [PubMed] [Google Scholar]
- 51.Raaijmakers MH. Disease progression in myelodysplastic syndromes: do mesenchymal cells pave the way? Cell Stem Cell. 2014;14(6):695–7. doi: 10.1016/j.stem.2014.05.010. [DOI] [PubMed] [Google Scholar]
- 52.Bhagat TD, Chen S, Bartenstein M, Barlowe AT, Von Ahrens D, Choudhary GS et al. Epigenetically Aberrant Stroma in MDS Propagates Disease via Wnt/beta-Catenin Activation. Cancer research. 2017;77(18):4846–57. doi: 10.1158/0008-5472.can-17-0282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen S, Zambetti NA, Bindels EM, Kenswill K, Mylona AM, Adisty NM et al. Massive parallel RNA sequencing of highly purified mesenchymal elements in low-risk MDS reveals tissue- context-dependent activation of inflammatory programs. Leukemia. 2016;30(9):1938–42. doi: 10.1038/leu.2016.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Raaijmakers MH, Mukherjee S, Guo S, Zhang S, Kobayashi T, Schoonmaker JA et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature. 2010;464(7290):852–7. doi: 10.1038/nature08851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lim ZY, Killick S, Germing U, Cavenagh J, Culligan D, Bacigalupo A et al. Low IPSS score and bone marrow hypocellularity in MDS patients predict hematological responses to antithymocyte globulin. Leukemia. 2007;21(7):1436–41. doi: 10.1038/sj.leu.2404747. [DOI] [PubMed] [Google Scholar]
- 56.Haidinger M, Geyeregger R, Poglitsch M, Weichhart T, Zeyda M, Vodenik B et al. Antithymocyte globulin impairs T-cell/antigen-presenting cell interaction: disruption of immunological synapse and conjugate formation. Transplantation. 2007;84(1):117–21. doi: 10.1097/01tp.0000266677.45428.80. [DOI] [PubMed] [Google Scholar]
- 57.Matsuda S, Koyasu S. Mechanisms of action of cyclosporine. Immunopharmacology. 2000;47(2–3):119–25. [DOI] [PubMed] [Google Scholar]
- 58.Sloand EM, Rezvani K. The role of the immune system in myelodysplasia: implications for therapy. Semin Hematol. 2008;45(1):39–48. doi: 10.1053/j.seminhematol.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 59.Parikh AR, Olnes MJ, Barrett AJ. Immunomodulatory treatment of myelodysplastic syndromes: antithymocyte globulin, cyclosporine, and alemtuzumab. Semin Hematol. 2012;49(4):304–11. doi: 10.1053/j.seminhematol.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Haider M, Al Ali N, Padron E, Epling-Burnette P, Lancet J, List A et al. Immunosuppressive Therapy: Exploring an Underutilized Treatment Option for Myelodysplastic Syndrome. Clin Lymphoma Myeloma Leuk. 2016;16 Suppl:S44–8. doi: 10.1016/j.clml.2016.02.017. [DOI] [PubMed] [Google Scholar]
- 61.Stahl M, Deveaux M, de Witte TMM, Neukirchen J, Sekeres MA, Brunner AM et al. The Use of Immunosuppressive Therapy (IST) in Patients with the Myelodysplastic Syndromes (MDS): Clinical Outcomes and Their Predictors in a Large International Patient Cohort. Blood. 2017;130(Suppl 1):422. [Google Scholar]
- 62.Deeg HJ, Gotlib J, Beckham C, Dugan K, Holmberg L, Schubert M et al. Soluble TNF receptor fusion protein (etanercept) for the treatment of myelodysplastic syndrome: a pilot study. Leukemia. 2002;16(2):162–4. doi: 10.1038/sj.leu.2402356. [DOI] [PubMed] [Google Scholar]
- 63.Baron F, Suciu S, Amadori S, Muus P, Zwierzina H, Denzlinger C et al. Value of infliximab (Remicade(R)) in patients with low-risk myelodysplastic syndrome: final results of a randomized phase II trial (EORTC trial 06023) of the EORTC Leukemia Group. Haematologica. 2012;97(4):529–33. doi: 10.3324/haematol.2011.044347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Scott BL, Ramakrishnan A, Storer B, Becker PS, Petersdorf S, Estey EH et al. Prolonged responses in patients with MDS and CMML treated with azacitidine and etanercept. Br J Haematol. 2010; 148(6):944–7. doi: 10.1111/j.1365-2141.2009.08061 .x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Scott BL, Ramakrishnan A, Fosdal M, Storer B, Becker P, Petersdorf S et al. Anti-thymocyte globulin plus etanercept as therapy for myelodysplastic syndromes (MDS): a phase II study. Br J Haematol. 2010;149(5):706–10. doi: 10.1111/j.1365-2141.2010.08145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mies A, Hermine O, Platzbecker U. Activin Receptor II Ligand Traps and Their Therapeutic Potential in Myelodysplastic Syndromes with Ring Sideroblasts. Curr Hematol Malig Rep. 2016;11(6):416–24. doi: 10.1007/s11899-016-0347-9. [DOI] [PubMed] [Google Scholar]
- 67.Platzbecker U, Germing U, Gotze KS, Kiewe P, Mayer K, Chromik J et al. Luspatercept for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes (PACE-MDS): a multicentre, open-label phase 2 dose-finding study with long-term extension study. Lancet Oncol. 2017;18(10):1338–47. doi: 10.1016/S1470-2045(17)30615-0. [DOI] [PubMed] [Google Scholar]
- 68.Schinke C, Giricz O, Li W, Shastri A, Gordon S, Barreyro L et al. IL8-CXCR2 pathway inhibition as a therapeutic strategy against MDS and AML stem cells. Blood. 2015; 125(20):3144–52. doi: 10.1182/blood-2015-01-621631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Navas TA, Mohindru M, Estes M, Ma JY, Sokol L, Pahanish P et al. Inhibition of overactivated p38 MAPK can restore hematopoiesis in myelodysplastic syndrome progenitors. Blood. 2006; 108(13):4170–7. doi: 10.1182/blood-2006-05-023093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shastri A, Will B, Steidl U, Verma A. Stem and progenitor cell alterations in myelodysplastic syndromes. Blood. 2017;129(12):1586–94. doi: 10.1182/blood-2016-10-696062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Navas T, Zhou L, Estes M, Haghnazari E, Nguyen AN, Mo Y et al. Inhibition of p38alpha MAPK disrupts the pathological loop of proinflammatory factor production in the myelodysplastic syndrome bone marrow microenvironment. Leuk Lymphoma. 2008;49(10):1963–75. doi: 10.1080/10428190802322919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sokol L, Cripe L, Kantarjian H, Sekeres MA, Parmar S, Greenberg P et al. Randomized, dose-escalation study of the p38a MAPK inhibitor SCIO-469 in patients with myelodysplastic syndrome. Leukemia. 2013;27(4):977–80. doi: 10.1038/leu.2012.264. [DOI] [PubMed] [Google Scholar]
- 73.Bachegowda L, Morrone K, Winski SL, Mantzaris I, Bartenstein M, Ramachandra N et al. Pexmetinib: A Novel Dual Inhibitor of Tie2 and p38 MAPK with Efficacy in Preclinical Models of Myelodysplastic Syndromes and Acute Myeloid Leukemia. Cancer Res. 2016;76(16):4841–9. doi: 10.1158/0008-5472.CAN-15-3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Garcia-Manero G, Khoury HJ, Jabbour E, Lancet J, Winski SL, Cable L et al. A phase I study of oral ARRY-614, a p38 MAPK/Tie2 dual inhibitor, in patients with low or intermediate-1 risk myelodysplastic syndromes. Clin Cancer Res. 2015;21(5):985–94. doi: 10.1158/1078-0432.CCR-14-1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Reilly M, Miller RM, Thomson MH, Patris V, Ryle P, McLoughlin L et al. Randomized, double-blind, placebo-controlled, dose-escalating phase I, healthy subjects study of intravenous OPN-305, a humanized anti-TLR2 antibody. Clin Pharmacol Ther. 2013;94(5):593–600. doi: 10.1038/clpt.2013.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Park JS, Gamboni-Robertson F, He Q, Svetkauskaite D, Kim JY, Strassheim D et al. High mobility group box 1 protein interacts with multiple Toll-like receptors. American journal of physiology Cell physiology. 2006;290(3):C917–24. doi: 10.1152/ajpcell.00401.2005. [DOI] [PubMed] [Google Scholar]
- 77.Lee KL, Ambler CM, Anderson DR, Boscoe BP, Bree AG, Brodfuehrer JI et al. Discovery of Clinical Candidate 1-{[(2S,3S,4S)-3-Ethyl-4-fluoro-5-oxopyrrolidin-2-yl]methoxy}−7-methoxyisoquinoline-6-carboxamide (PF-06650833), a Potent, Selective Inhibitor of Interleukin- 1 Receptor Associated Kinase 4 (IRAK4), by Fragment-Based Drug Design. J Med Chem. 2017;60(13):5521–42. doi: 10.1021/acs.jmedchem.7b00231. [DOI] [PubMed] [Google Scholar]
- 78.Eksioglu EA, Chen X, Heider KH, Rueter B, McGraw KL, Basiorka AA et al. Novel therapeutic approach to improve hematopoiesis in low risk MDS by targeting MDSCs with the Fc-engineered CD33 antibody BI 836858. Leukemia. 2017;31(10):2172–80. doi: 10.1038/leu.2017.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Brayer J, Lancet JE, Powers J, List A, Balducci L, Komrokji R et al. WT1 vaccination in AML and MDS: A pilot trial with synthetic analog peptides. Am J Hematol. 2015;90(7):602–7. doi: 10.1002/ajh.24014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Keilholz U, Letsch A, Busse A, Asemissen AM, Bauer S, Blau IW et al. A clinical and immunologic phase 2 trial of Wilms tumor gene product 1 (WT1) peptide vaccination in patients with AML and MDS. Blood. 2009;113(26):6541–8. doi: 10.1182/blood-2009-02-202598. [DOI] [PubMed] [Google Scholar]
- 81.Griffiths EA, Srivastava P, Matsuzaki J, Brumberger Z, Wang ES, Kocent J et al. NY-ESO-1 Vaccination in Combination with Decitabine Induces Antigen-Specific T-Lymphocyte Responses in Patients with Myelodysplastic Syndrome. Clin Cancer Res. 2017. doi: 10.1158/1078-0432.CCR-17-1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rezvani K, Yong AS, Mielke S, Savani BN, Musse L, Superata J et al. Leukemia-associated antigen-specific T-cell responses following combined PR1 and WT1 peptide vaccination in patients with myeloid malignancies. Blood. 2008;111(1):236–42. doi: 10.1182/blood-2007-08-108241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Qazilbash MH, Wieder E, Thall PF, Wang X, Rios R, Lu S et al. PR1 peptide vaccine induces specific immunity with clinical responses in myeloid malignancies. Leukemia. 2017;31(3):697–704. doi: 10.1038/leu.2016.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rezvani K, Yong AS, Mielke S, Jafarpour B, Savani BN, Le RQ et al. Repeated PR1 and WT1 peptide vaccination in Montanide-adjuvant fails to induce sustained high-avidity, epitope-specific CD8+ T cells in myeloid malignancies. Haematologica. 2011;96(3):432–40. doi: 10.3324/haematol.2010.031674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Choi DC, Tremblay D, lancu-Rubin C, Mascarenhas J. Programmed cell death-1 pathway inhibition in myeloid malignancies: implications for myeloproliferative neoplasms. Ann Hematol. 2017;96(6):919–27. doi: 10.1007/s00277-016-2915-4. [DOI] [PubMed] [Google Scholar]
- 86.Boddu P, Kantarjian H, Garcia-Manero G, Allison J, Sharma P, Daver N. The emerging role of immune checkpoint based approaches in AML and MDS. Leuk Lymphoma. 2017:1–13. doi: 10.1080/10428194.2017.1344905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kondo A, Yamashita T, Tamura H, Zhao W, Tsuji T, Shimizu M et al. Interferon-gamma and tumor necrosis factor-alpha induce an immunoinhibitory molecule, B7-H1, via nuclear factor-kappaB activation in blasts in myelodysplastic syndromes. Blood. 2010;116(7):1124–31. doi: 10.1182/blood-2009-12-255125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yang H, Bueso-Ramos C, DiNardo C, Estecio MR, Davanlou M, Geng QR et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia. 2014;28(6):1280–8. doi: 10.1038/leu.2013.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Garcia-Manero G, Tallman MS, Martinelli G, Ribrag V, Yang H, Balakumaran A et al. Pembrolizumab, a PD-1 Inhibitor, in Patients with Myelodysplastic Syndrome (MDS) after Failure of Hypomethylating Agent Treatment. Blood. 2016;128(22):345– [Google Scholar]
- 90.Green C, Yan M, Nalle S, Ma C, Robert A, Zhong J et al. Evidence of Targetable Immune Dysfunction in the Bone Marrow of Patients with Intermediate/High-Risk Myelodysplastic Syndrome Refractory to Hypomethylating Agents. Blood. 2017;130(Suppl 1):4241. [Google Scholar]
- 91.Stahl M, Gedrich R, Peck R, LaVallee T, Eder JP. Targeting KIT on innate immune cells to enhance the antitumor activity of checkpoint inhibitors. Immunotherapy. 2016;8(7):767–74. doi: 10.2217/imt-2016-0040. [DOI] [PubMed] [Google Scholar]
- 92.Gleason MK, Ross JA, Warlick ED, Lund TC, Verneris MR, Wiernik A et al. CD16xCD33 bispecific killer cell engager (BiKE) activates NK cells against primary MDS and MDSC CD33+ targets. Blood. 2014;123(19):3016–26. doi: 10.1182/blood-2013-10-533398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vallera DA, Felices M, McElmurry R, McCullar V, Zhou X, Schmohl JU et al. IL15 Trispecific Killer Engagers (TriKE) Make Natural Killer Cells Specific to CD33+ Targets While Also Inducing Persistence, In Vivo Expansion, and Enhanced Function. Clin Cancer Res. 2016;22(14):3440–50. doi: 10.1158/1078-0432.CCR-15-2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Batlevi CL, Matsuki E, Brentjens RJ, Younes A. Novel immunotherapies in lymphoid malignancies. Nat Rev Clin Oncol. 2016;13(1):25–40. doi: 10.1038/nrclinonc.2015.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rettig MP, Godwin J, Vey N, Fox B, Ballesteros-Merino C, Bifulco CB et al. Preliminary Translational Results from an Ongoing Phase 1 Study of Flotetuzumab, a CD123 x CD3 Dart®, in AML/MDS: Rationale for Combining Flotetuzumab and Anti-PD-1/PD-L1 Immunotherapies. Blood. 2017;130(Suppl 1):1365. [Google Scholar]
- 96.Dolstra H, Roeven MWH, Spanholtz J, Hangalapura BN, Tordoir M, Maas F et al. Successful Transfer of Umbilical Cord Blood CD34+ Hematopoietic Stem and Progenitor- derived NK Cells in Older Acute Myeloid Leukemia Patients. Clin Cancer Res. 2017;23(15):4107–18. doi: 10.1158/1078-0432.CCR-16-2981. [DOI] [PubMed] [Google Scholar]
- 97.Bjorklund AT, Carlsten M, Sohlberg E, Liu LL, Clancy T, Karimi M et al. Complete Remission with Reduction of High-risk Clones following Haploidentical NK Cell Therapy against MDS and AML. Clinical cancer research : an official journal of the American Association for Cancer Research. 2018. doi: 10.1158/1078-0432.Ccr-17-3196. [DOI] [PubMed] [Google Scholar]
- 98.Sarvaria A, Jawdat D, Madrigal JA, Saudemont A. Umbilical Cord Blood Natural Killer Cells, Their Characteristics, and Potential Clinical Applications. Front Immunol. 2017;8:329. doi: 10.3389/fimmu.2017.00329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Domogala A, Blundell M, Thrasher A, Lowdell MW, Madrigal JA, Saudemont A. Natural killer cells differentiated in vitro from cord blood CD34+ cells are more advantageous for use as an immunotherapy than peripheral blood and cord blood natural killer cells. Cytotherapy. 2017;19(6):710–20. doi: 10.1016/j.jcyt.2017.03.068. [DOI] [PubMed] [Google Scholar]
- 100.Lieberman NAP, DeGolier K, Haberthur K, Chinn H, Moyes KW, Bouchlaka MN et al. An Uncoupling of Canonical Phenotypic Markers and Functional Potency of Ex Vivo-Expanded Natural Killer Cells. Front Immunol. 2018;9:150. doi: 10.3389/fimmu.2018.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wagner JA, Rosario M, Romee R, Berrien-Elliott MM, Schneider SE, Leong JW et al. CD56bright NK cells exhibit potent antitumor responses following IL-15 priming. J Clin Invest. 2017; 127(11):4042–58. doi: 10.1172/JCI90387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhang W, Stevens BM, Budde EE, Forman SJ, Jordan CT, Purev E. Anti-CD123 CAR T-Cell Therapy for the Treatment of Myelodysplastic Syndrome. Blood. 2017;130(Suppl 1):1917. [DOI] [PMC free article] [PubMed] [Google Scholar]