

Abstract
FAK is a tyrosine kinase overexpressed in cancer cells and plays an important role in the progression of tumors to a malignant phenotype. Except for its typical role as a cytoplasmic kinase downstream of integrin and growth factor receptor signaling, related studies have shown new aspects of the roles of FAK in the nucleus. FAK can promote p53 degradation through ubiquitination, leading to cancer cell growth and proliferation. FAK can also regulate GATA4 and IL-33 expression, resulting in reduced inflammatory responses and immune escape. These findings establish a new model of FAK from the cytoplasm to the nucleus. Activated FAK binds to transcription factors and regulates gene expression. Inactive FAK synergizes with different E3 ligases to promote the turnover of transcription factors by enhancing ubiquitination. In the tumor microenvironment, nuclear FAK can regulate the formation of new blood vessels, affecting the tumor blood supply. This article reviews the roles of nuclear FAK in regulating gene expression. In addition, the use of FAK inhibitors to target nuclear FAK functions will also be emphasized.
Keywords: Nuclear FAK, Cancer, Transcription factors, Gene expression, Inhibitors
Background
Numerous studies on the potential link between FAK and different kinds of cancer have gradually revealed the biological mechanisms by which FAK promotes the development and progression of cancer [1]. FAK is a tyrosine kinase with a molecular weight of 125kD, playing a vital role in cellular communication, especially in cell signaling systems [2]. Wang et al. [3] revealed that increased mRNA levels, protein levels, and the activation of FAK were positively associated with cancer metastasis and invasion and frequently inversely correlated with better clinical cancer sample results in the detection of human cancer samples. Relevant studies have found that FAK was overexpressed and/or over-phosphorylated in multiple cancer cells, responsible for cell migration [4], survival [5], proliferation [6], and adhesion [7]. In addition, FAK is strongly associated with the occurrence and development of tumors [2, 8] and regarded as a functional protein in the cytoplasm, typically functioning in a kinase-dependent manner [9]. Firstly, FAK receives different extracellular signals coming from cell-surface transmembrane receptors including integrins, cytokines, growth factors, and G protein-coupled receptors. After that, FAK activates and triggers subsequent signaling cascades in a variety of cellular activities [10, 11]. FAK can also participate in the signal transduction process in tumor vessel, mediating the vessel permeability [12–14]. The FERM domain of FAK can combine with the cytoplasmic region of vascular endothelial calcium mucin. It is important for cell-cell adhesive junctional structures, an integral part of keeping vascular integrity [15]. Furthermore, FAK is essential for maintaining vascular functions in tumor angiogenesis. Lees et al. [16] found that FAK recovered the vascular leakage defect through the activation of kinase domain. And it is a fact that cytokines induce vascular growth factor expression by the FAK signaling pathway. For example, via Src-FAK-STAT3 signaling, IL-6 induces VEGF-C expressions [17]. As a result, FAK kinase activity is required for tumor growth [18], angiogenesis [17], and vascular permeability [19]. These show that FAK is a typical multifunctional protein which integrates and transduces signals into cancer cells via integrin or growth factor receptors. Tumor stem cells are few tumor cells which are present in malignant cells and believed to be the source of cancer cells. They have the ability to proliferate, self-renew and generate heterogeneous tumor cells, maintaining the vitality of the tumor cell population [20, 21]. Yoon et al. [22] found that FAK promoted cancer stem cells (CSCs) renewal and drug resistance by functioning in survival signaling. For example, FAK and the extracellular signal-regulated kinase (ERK1/2) pathway are involved in the regulation of growth and metastasis of liver cancer stem cells (LCSCs) [23]. The use of the anticancer drug salinomycin inhibited the activity of FAK and ERK1/2, resulting in the increased stiffness of LCSCs [24]. Another study has shown that changes in the stiffness of living cells might affect numerous cellular physiological activities [25]. FAK can affect the growth of LCSCs through this mechanism of the regulation of cell stiffness. Cheng et al. [26] targeted HIC1 and RassF1A methylation, induced the transformation of mesenchymal stem cells (MSCs) and the cell stiffness was lost. It is suggested that Tumor cells are softer than normal cells, mainly due to loss of cytoskeletal support [27, 28]. And the loss of stiffness can represent a phenotype of tumor development which facilitates cancer cell migration and adapts to other tissues [29, 30]. Taken together, these results indicate that FAK is closely related to biological behaviors such as survival, migration, invasion, and proliferation of CSCs. Based on those findings, FAK can be regarded as a target for cancer therapy.
Actually, investigators have found that FAK was also functional in the nucleus [31]. FAK can enter the nucleus and regulates gene expression to influence tumorigenesis [32]. In the nucleus, activated FAK binds to transcription factors to regulate gene expression. Inactive FAK synergizes with different E3 ligases to promote the turnover of transcription factors [33]. FAK affects tumor survival and growth by altering the transcription [34]. In this review, some regulation modes of nuclear FAK are discussed. We focus on nuclear FAK regulating gene expression in different cancer cells. FAK regulates gene expression by affecting the expression of transcription factors. Furthermore, we emphasize that nuclear FAK also has an important role in the study of cancer, which is positively related to the occurrence and development of tumors.
FAK can shuttle between cytoplasm and nucleus
The structure of FAK
In humans, FAK is composed of the N-terminal containing the FERM domain, the central kinase domain, and the C-terminal with the FAT domain (Fig. 1) [35, 36]. The FERM domain consists of approximately 300 amino acid residues, binding directly to the intracellular portion of the transmembrane protein receptors [37]. The kinase domain refers to the 390–650 amino acid region which is highly conserved. It has at least 6 tyrosine phosphorylation sites [38, 39], which is the key to FAK signaling. The FAT domain is responsible for interacting with primary adhesion plaque components such as Paxillin [40], Talin [41], Grab2 [42], Rgnef/p190RhoGef [43], and vascular endothelial growth factor receptor 3 (VEGFR3) [44]. Notably, FAK contains binding sides for more than 50 proteins, permitting FAK to function as a kinase and molecular scaffolds [45].
Fig. 1.
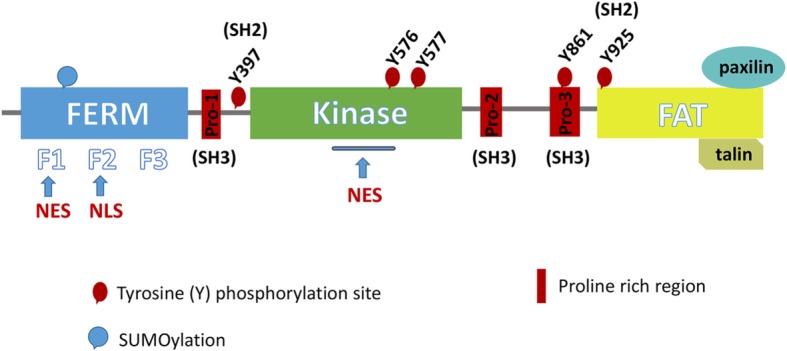
Schematic structure of FAK. The N-terminal FERM is shown in blue, containing three subdomains of F1, F2, and F3. The SUMOylation is located in the FERM domain. NES1 is located in the F1 leaf and NLS is located in the F2 leaf. The kinase domain is shown in green containing NES2 and the FAT domain is shown in yellow responsible for interacting with primary adhesion plaque components such as Paxillin and Talin. There are proline-rich regions between the domains and they are binding sites of Src homology 3 (SH3). Significantly, there are five important tyrosine phosphorylation sites. Among them, Y397 and Y925 are binding sites of Src homology 2 (SH2)
It was reported that FAK had nuclear export signals (NES) in the kinase domain and nuclear localization signals (NLS) in the F2 lobe of the FERM domain [31, 46], which led to the shuttle of FAK between the focal adhesions (FAs) and the nucleus. Further analysis of NLS and NES showed that bare alkaline residue clusters in the surface of NLS were K190, K191, K216, K218, R221, and K222 and the NES was composed of a leucine-rich amino acid sequence [34]. And it was showed that FAK had two NES sequences actually [36, 1, 47]. One was NES1 located in the F1 lobe and the other was NES2 located in the kinase domain. Although both of them are conservative in the evolutionary process, only NES2 has complete biological nuclear export activities [46, 1]. In addition, apart from facilitating phosphatidylinositol 4,5-bisphosphate (PIP2) lipid binding [48], the residues of F2 lobe (KAKTLR) can also function as a nuclear localization sequence [49]. Post-translational modifications of FAK include binding protein inhibitors of activated STAT1 (PIAS1) to the FERM domain of FAK and the addition of a small ubiquitin-related modifier (SUMO) to the Lys152 residue [50]. SUMOylation is always in connection with nuclear import signals and the SUMO of nuclear FAK is related to FAK activation [51]. Under the action of stimulation or biomolecules, such as chemical and/or genetic stress, FAK can occur nuclear recruitment [52, 53]. The nuclear export sequences and nuclear localization sequences are crucial for the entry of FAK into the nucleus and the process of FAK entering the nucleus requires physiological or chemical induction.
The nuclear localization of FAK
FAK can shuttle between the cytoplasm and the nucleus. Membrane anchors such as FAs may immobilize FAK at the adhesion sites, keeping FAK in the cytoplasm. But FAs such as Paxillin and Zyxin families can also migrate to the nucleus [40, 54, 55]. Although neither protein links DNA directly, they can serve as coreceptors of transcription factors. Thus the interaction of FAK with those proteins may jointly regulate nuclear translocation of integrin adherent structures [56]. For example, the ectopic expression of PIAS1 promotes FAK c-terminal protein cleavage, focal adhesion maturation, and FAK nuclear localization in non-small cell lung cancer (NSCLC) cells. Moreover, nuclear p125 FAK was found to be significantly phosphorylated at the Y861 site by specifically expressing the FAK fragments and the amino-terminal domain FAK fragments could regulate the nuclear localization and apoptotic in endothelial cells [57]. Constanzo et al. [58] found that nuclear FAK activity promoted the survival and progression of NSCLC by increasing cellular-extracellular matrix (ECM) interaction and DNA repair regulation. In a related study, neural cell adhesion molecule (NCAM) induces FAK activation and the nuclear import of the C-terminal fragment and N-terminal fragment of FAK [59]. Moreover, it is speculated that the localization of FAK in the nucleus facilitates the formation of FAs complexes by inhibiting autophagy, thereby triggering cell proliferation [47, 58]. It suggests that the nuclear recruitment of FAK requires the participation of other proteins, such as NCAM and the activation of FAK can also affect the nuclear recruitment and regulate DNA repair.
Scientists have found the molecular mechanisms which stimulated FAK from the cytoplasm into the nucleus [31]. Firstly, Lim et al. [34] treated cancer cells with an apoptotic inducer of staurosporine which led to FAK nuclear accumulation. Treatment of myocytes with H2O2 can lead to oxidative stress, promote FAK nuclear localization, and induce myocyte differentiation [60]. These indicate that stress signals promote FAK migration from the cytoplasm to the nucleus. Secondly, the x-linked apoptotic protein inhibitor (XIAP) promotes FAK localization in the FAs, which enhances the phosphorylation of FAK at pY576. Meanwhile, XIAP knockout reduces the phosphorylation of FAK, promoting the shear stress-induced translocation of FAK to the nucleus [61]. The desorption of cells from the matrix may increase the free FAK available in the cytoplasm, leading to FAK nuclear localization by detachment from focal adhesion [31]. Similarly, Aoto et al. [62] mutated the proline-rich region of Pyk2. They mutated proline 859 to alanine, prevented the binding of SH3-containing proteins, and promoted the detachment of Pyk2 from FAs and the nuclear localization of Pyk2. Therefore, the decellularization of cells from the matrix leads to FAK nuclear localization. Thirdly, Lim et al. [63] found that kinase depletion (KD) FAK cells had stronger FAK nuclear localization signals than wild-type (WT) FAK. Studies have shown that FAK inhibitors such as PF-562,271, PF-271, Pfizer, could significantly increase the nuclear localization of FAK [64, 65]. This result is consistent with the previous result of kinase depletion. It is speculated that activated FAK may be localized to the cytoplasm or adhesion preferentially, instead of the nucleus [34]. Inhibitors can modify FAK, inhibit FAK activity and induce it to enter the nucleus. Therefore, FAK inhibition promotes FAK nuclear localization. The three different ways suggest that stress signals, the desorption of cells from the matrix, and the inhibition of FAK may induce FAK into the nucleus.
FAK functions in the nucleus
The function of FAK in regulating p53 pathway
FAK enters the nucleus from the FAs site via FERM NLS. After that, FAK binds to the N-terminal transactivation domain of p53 (Table 1) through its N-terminal FERM domain, decreases the transcriptional activity of p53 and inhibits p53 to activate its downstream gene transcription [67]. A further research found that the binding site of p53 for interaction with FAK was a 7-amino-acid site in the proline-rich region in the N-terminal domain [68]. In this progress, the inactivation of p53 requires F1 leaf interacting with p53, KAKTLR-mediated nuclear localization, F3 leaf connecting murine double minute2 (Mdm2) and proteasome degradation [31]. A Study has found that FAK negatively regulated p53 expression in mesothelioma cell lines and NF2 regulated the interaction of FAK–p53 and Mdm2–p53 [69]. The FERM domain mediates the transfer of FAK into the nucleus and binds to p53, which in turn binds to Mdm2, leading to the degradation of p53 through the ubiquitination pathway and inhibiting apoptosis. FAK promotes p53 transformation by enhancing Mdm2-dependent p53 ubiquitination [31, 82]. This mechanism implies that p53 ubiquitination and Mdm2-mediated proteasomal degradation keep p53 at a low level, promoting cell survival. At this point, this is one of the earliest kinase-independent roles, which requires only FAK’s scaffolding function.
Table 1.
Proteins interacting with nuclear FAK
| Protein | Outcome | Significant Findings |
|---|---|---|
| NCAM | FAK phosphorylation | NCAM induces the nuclear import of the C-terminal fragment and N-terminal fragment of FAK [59] |
| PIAS1 | Post-translational modifications | PIAS1 binds to the FERM domain of FAK, affecting its post-translational modifications [51, 66] |
| MBD2 | Dissociation from HDAC1 | Activate the expression of myogenic proteins and other genes that promote muscle differentiation [60] |
| P53 | Degradation | FAK inhibits p53 to activate its downstream gene transcription [67, 68] |
| Mdm2 | P53 ubiquitination | P53 proteosomal degradation in the nucleus [31, 69] |
| NF2 | Activation | NF2 regulates the interaction of FAK–p53 and Mdm2–p53 [69] |
| PTEN | Upregulation | P53 and PTEN tumor suppressors are co-inactivated in patients and cause cancer metastasis [70] |
| GATA4 | Degradation | FAK inhabits GATA4 expression and reduces the inflammatory responses [58, 65, 71] |
| CHIP | GATA4 ubiquitination | The nuclear FAK interacts with GATA4 and the ubiquitin protein E3 ligase CHIP [58, 65, 72] |
| IL-33 | Upregulation | IL-33 regulates CCL5 expression [73, 74] |
| VEGFR2 | Upregulation | VEGFR2 promotes the formation of tumor neovascularization and tumor growth [75] |
| Sin3A | Activation | FAK participates in the regulation of Runx1 via Sin3A [76] |
| Runx1 | Runx1 complex | The formation of the transcription factor Runx1 complex [77] |
| MEF2 | Forms complex | FAK and MEF2 jointly regulate expression of Jun which is induced by load [78] |
| FIP200 | FAK phosphorylation inhibition | FIP/FAK complex is associated with FAK inactivation after cell detachment [78] |
| EZH2 | EZH2 phosphorylation | FAK affects the transcription and nuclear localization of EZH2 [79] |
| E3 ligase | Ubiquitination | Inactive FAK coordinates with different E3 ligases, promoting transcription factor turnover [65, 71] |
| Sam68 | Activation | Sam68 binds to RNA and signaling molecules to regulate multiple signaling pathways [80] |
| NS | Activation | Active FAK protects the NS from proteasomal degradation [81] |
Nuclear FAK can regulate p53-mediated cell behavior after binding to p53. The earlier studies have shown that almost 50% of tumors had mutations in p53 and p53 inactivation was a key step in tumorigenesis [83, 84]. P53 negatively affects the expression of many genes that promote cell senescence, cell apoptosis and cell death, such as p21, Gadd45, Cyclin G, Bax, Gml, and P2xm. At the same time, p53 inhibits the promoter activity of various cell cycle and anti-apoptotic genes, such as Cyclin, Bax/Bcl2, and IGF-BP3 [85]. For example, apolipoprotein B mRNA editing enzyme catalytic polypeptide-like (APOBEC) is an important source of tumorigenic mutations. Among them, APOBEC3B (A3B) expression is particularly relevant to tumors, such as tumor resistance [86]. Periyasamy et al. demonstrated a negative correlation between A3B expression and p53 expression in different cancer types, as p53 played a direct and critical role in inhibiting A3B expression [87]. The CXCR4 receptor and FAK are believed to regulate the aggressive cancer behavior [70]. Their expression is down-regulated by p53 tumor suppressor and phosphatase and tensin homolog deleted on chromosome ten (PTEN). And p53 and PTEN tumor suppressors are co-inactivated in patients and cause cancer metastasis [88]. A recent study has found the mechanism of stabilizing nuclear p53. The type I phosphatidylinositol phosphate kinase (PIPKI-α) and its product phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5)P) can maintain the stability of nuclear p53. The combination of PIPKI-α and p53 produces PtdIns(4,5)P, which promotes the interaction of small heat shock proteins with p53, thereby stabilizing the nuclear p53 [89]. P53 can also participate in the nucleolar stress pathway [90]. The nuclear mitotic apparatus protein (NuMA) is present in the nucleolus. NuMA can be involved in DNA damage as well as p53-mediated growth arrest and apoptosis [91]. Nuclear FAK binds to p53, reduce p53 levels and regulate the p53 signaling pathway in a kinase-independent manner. And as a multifunctional transcription factor, p53 tumor suppressor proteins regulate cellular processes that affect proliferation, cell cycle checkpoints, and apoptosis.
The function of FAK in regulating inflammation pathway
Inflammation can alter the expression of oncogenes and tumor suppressor genes to promote the transition of cells to malignant tumors. It is estimated that infectious diseases and chronic inflammation account for about 25% of carcinogenic factors [92]. For example, DNA damage associated with inflammation in cancer stem cells can lead to cancer development with invasive clinical features [93]. The ROS/RNS caused by inflammation not only damages DNA, but also damages other biological macromolecules such as proteins and lipids, leading to dysfunction [94]. These indicate that inflammation is also closely related to tumor development. Inflammatory factors such as TNF-α can promote the expression of inflammatory genes through mitogen activated protein kinases (MAPKs) cascade and NF-κB activation [95, 96]. Therefore, inhibiting MAPKs and/or NF-κB pathway may significantly reduce the expression of inflammatory genes [97]. Aulakh et al. [72] found that the inhibition of FAK expression may effectively inhibit vascular cell adhesion factor-1 (VCAM-1) expression. Interestingly, although the inhibition of FAK expression blocks VCAM-1, it does not affect NF-κB activation [10, 65]. In this process, the activation of MAPKs does not affect VCAM-1 expression and FAK inhibition can promote the expression of GATA4 transcription factors [98]. This is mediated by the function of the nuclear FAK scaffold that interacts with GATA4 and the ubiquitin protein E3 ligase chips. Kinase-inhibited FAK has new developments and anti-inflammatory effects in limiting VCAM-1 expression through the nuclear localization and the promotion of GATA4 conversion [58, 65, 71]. This suggests that anti-inflammatory effects can be provided by the nuclear-localized FAK inhibition and it is important that the expressions of FAK and inflammatory cytokine are independent of NF-κB activation.
At the same time, a research showed that FAK also induced the expression of inflammatory genes and the products of these genes inhibited the anti-tumor immunity in the microenvironment, leading to the immune escape of tumor [99]. Firstly, researchers found that FAK depletion or inhibition could lead to squamous cell carcinoma regression. Nuclear FAK induces the expression of immunosuppressive molecules cytokines and chemokines, forming an immunosuppressive microenvironment, leading to tumor escape [73]. These factors drive the depletion of CD8+ T cells and the recruitment of regulatory T cells (Tregs) [100], resulting in the depletion of antigen-induced cytotoxic CD8+ T cell activity that allows tumor growth [101]. Tumor invasiveness has a positive correlation with the number and size of nucleoli. In nucleoli, FAK binds a cancer stem cell marker riboflavin and protects it from stress-induced degradation. A further study revealed that nuclear FAK bound to the inflammatory factor IL-33 and regulated the expression of chemokine ligand 5 (CCL5) and growth stimulation expressed gene 2 (ST2) [74]. IL-33 binds to CD8 T cells, leading to tumor cells to escape the recognition of CD8 T cells [102]. Deletion of CCL5 reduces tumor-infiltrating Treg cells, resulting in regression of FAK-WT tumors [103]. ST2 is secreted into the tumor environment as a decoy receptor, resulting in competitive inhibition of IL-33/ST2 autocrine and paracrine signals [104]. Those studies demonstrate the roles of FAK in the nucleoli. FAK protects nucleocapsid proteins from proteasomal degradation that is essential for breast cancer growth. In general, FAK-IL33 regulation is similar to FAK-GATA4 regulation, indicating that nuclear FAK is actually a scaffold promoting transcription factor turnover and regulating of inflammatory factor expression.
The potential roles of nuclear FAK
The formation of blood vessels plays an important role in the occurrence and development of tumors [105]. A study has confirmed that anti-tumor effects could be achieved by inhibiting neovascularization [106]. Targeting tumor vascular endothelial cells to inhibit tumor angiogenesis and block tumor blood supply has become a research hotspot for current anti-tumor. FAK is an indispensable protein in embryonic angiogenesis and regulates angiogenesis in kinase-independent and kinase-dependent manners. In endothelial cell (EC), FAK acts in a kinase-independent manner, regulating cell survival and barrier function. FAK deletion or inhibition of activity reduces EC proliferation and migration [107]. This suggests that FAK acts primarily as a kinase that regulates EC-mediated angiogenesis. Further mechanistic analysis revealed that FAK could regulate the expression of vascular endothelial growth factor receptor 2 (VEGFR2). Nuclear FAK is directly involved in the transcriptional regulation of VEGFR2 via the VEGFR2 promoter-associated RNA polymerase II complex [75, 65]. VEGFR2 is the central substance of angiogenesis. It can bind to VEGF-C and VEGF-D, regulating vascular endothelial cells and lymphatic endothelial cells, promoting lymphangiogenesis and blood vessel formation, and regulating lymphocyte migration. This shows that FAK is also important to maintain the tumor microenvironment. In addition, nuclear FAK can promote the formation of tumor neovascularization and tumor growth.
The SIN3 transcriptional regulatory protein family member A (Sin3A) is a core component of a multiprotein transcriptional repressor complex [108, 109]. Nuclear FAK is involved in the regulation of the formation of the transcription factor Runx1 complex by interacting with Sin3A [76]. Runx1 regulates the expression of insulin-like growth factor binding protein 3 (IGFBP3). IGFBP3 is an extracellular secretory protein that binds to IGF and regulates IGF signaling. IGFBP3 also exhibits ligand-independent function in cultured mammalian cells. In cancer, IGFBP3 regulates cell cycle progression, affecting cell proliferation and tumor growth [110]. The roles of FAK in the nucleus are largely mediated by the FERM domain, and the FAT domain is also involved. It forms a complex with myocyte enhancer factor 2 (MEF2) transcription factors and upregulates transcriptional activity under mechanical stress [111]. Early evidence showed that FIP200 colocalized with nuclear FAK in focal adhesions. Furthermore, the FIP/FAK complex is associated with FAK inactivation after cell detachment [78]. Serrels et al. [73] found that FAK activity increases in squamous carcinoma cells compared to normal keratinocytes and thus FAK nuclear localization is related to cell transformation. And then a recent study has found that FAK affected the transcription and nuclear localization of zeste homolog 2 (EZH2) by regulating the transcriptional activities of p53 and E2F2/3 [79]. In high blood pressure, FAK and FAK-related non-kinase (FRNK) enter the nucleus. Protein kinase C (PKC) mediates the nuclear translocation of FAK and FRNK [112]. In the nucleus, FAK and FRNK can bind to different nuclear proteins, such as Src mitosis-associated protein 68 (Sam68) and fibrin, then targeting different nuclear regions [80]. Nucleolus is a non-membrane nuclear structure that regulates ribosome biogenesis and cell proliferation [113]. Proteins associated with nucleoli, such as nucleolar phosphoprotein B23 and nuclear stabilizing protein (NS), play an important role in genomic protection, ribosome synthesis, and stem cell proliferation [114, 115]. Tancioni et al. inhibited FAK activity and led to a decrease in proteasome-mediated NS levels. They found the mechanism of FAK in nucleoli by which active FAK protected the NS from proteasomal degradation and Akt-mTOR pathway regulated the stability of NS in breast cancer cells [81]. In general, activated FAK in the nucleus controls a variety of transcription factors leading to changes in gene regulation. Meanwhile, inactive FAK coordinates with different ubiquitin protein ligase E3 that promotes transcription factor turnover by enhancing ubiquitination (Fig. 2).
Fig. 2.

FAK functions in the nucleus. The activated FAK phosphorylates Rgnef and paxillin to promote focal adhesions assembly. In the cytoplasm, FAK regulates the development of cancer. Via the FERM structure, cell de-adhesion and/or kinase inhibition can promote FAK shuttle from cytoplasm to the nucleus. After entering the nucleus, active FAK binding to transcription factors (TFs) regulates cancer-related gene expression. Inactive FAK coordinates with different E3 ligases promoting turnover of TFs by enhancing ubiquitination. By regulating gene expression, nuclear FAK can be involved in the regulation of tumor angiogenesis, cell cycle, apoptosis, cell proliferation, immune escape, and tumor growth
FAK inhibitors
There are two important topics in the field of cancer research: one is tumor molecular imaging and the other is targeted molecular therapy. According to the consensus of many publications, FAK can be used as a promising target for anticancer therapies [116]. Currently, targeting FAK as a method of treating tumors mainly focuses on the use of drugs to inhibit its kinase activity and scaffold function. And reported FAK inhibitors are mainly small molecule inhibitors [1]. Small chemical molecules with good drug-forming properties can inhibit the phosphorylation of FAK and block the signal transduction through the cell membrane, thereby inhibiting the proliferation and spread of cancer cells [117]. Therefore, the research on FAK inhibitors is very promising. The pharmacodynamic activities of FAK inhibitors that have entered preclinical or clinical studies can be divided into two categories according to their mechanisms: ATP-dependent and ATP-independent [118]. The ATP-dependent FAK inhibitors can affect the binding of ATP to FAK and block FAK phosphorylation. The ATP-independent FAK inhibitors do not pass through the ATP binding site, but directly targets the FAK site, such as the FAK Y397 phosphorylation site [119]. Experimental results also showed that those small molecule FAK inhibitors could inhabit cell migration [3], survival [120], proliferation [121], and adhesion [122]. FAK inhibitors also can inhibit nuclear active FAK phosphorylation and regulate its related signaling pathways, such as the p53 signaling pathway, the inflammatory signaling pathway, the tumor angiogenesis-related pathway, and the immune escape signaling pathway. These pathways are closely related to tumor survival, migration, invasion, growth. For example, Dao et al. [123] found that 1,3,5-triazinic inhibitors of FAK could resist angiogenesis in HUVEC cells and have anticancer effects on various cancer cells. On this basis, they designed and synthesized a new compound containing a 1,2,4-triazine core as an inhibitor of FAK. And the compound can effectively inhibit the proliferation of U-87MG and HCT-116 cancer cells and exhibit a good anti-tumor effect [124]. In addition, Qu et al. [125] synthesized a class of FAK inhibitors, named Sul-DPPYs and it could effectively inhibit the activity of FAK and treat pancreatic cancer as a potent FAK inhibitor. Experiments have shown that treatment of NSCLC cells with CXCR4 and FAK inhibitors such as WZ811 and PF-573228 can inhibit their ability to migrate and invade [70, 88]. Inducing expression of p53 and p21 in ECs by down-regulating FAK may result in damage to angiogenesis and tumor growth [31]. What’s more, Roslin2 or 1-benzyl-15,3,5,7-tetraazetidine[3.3.1.1~3,7~] decane (R2) compounds disrupt FAK and p53 proteins that subsequently suppress tumor growth [85, 126]. In addition, small molecule inhibitors can inhibit FAK-mediated immune escape [73]. VS-4718 can inhibit the expression of immunosuppressive molecules such as IL-33 and CCL5, and reduce Tregs in the tumor environment [74]. Although inhibitors inhibit FAK phosphorylation, it is also possible to selectively induce nuclear localization. For example, PF-562,271 can block the phosphorylation of FAK at Y397 site and significant increase the nuclear localization of inactive FAK [64, 65]. The inactive FAK enters the nucleus and binds to E3 ligase to regulate the expression of transcription factors. And the mechanism of FAK kinase inhibitor targeting immunosuppressive may represent an effective immunomodulatory therapy. The development of FAK inhibitors is currently underway, and many inhibitors have shown therapeutic effects on cancer. Therefore, research on FAK inhibitors is also one of the research hotspots and it is also one of the directions for the development of anti-tumor drugs.
Conclusion
Current researches on FAK focus on the roles of FAK in FAs. FAK is a cytoplasmic non-receptor protein tyrosine kinase that phosphorylates different targets in cells. FAK also has a very important position in cell signal transduction. It is the center of intracellular and extracellular signal transduction and mediates multiple signaling pathways. FAK can be used as a platform to participate in the assembly of protein complexes and a bridge to participate in the signal transduction between proteins. Similarly, FAK also plays an important role in tumor cell signal transduction, mediating the tumor progression to a malignant invasion phenotype. Through these kinase-dependent mechanisms, FAK can regulate biological behaviors of tumor cells such as adhesion, migration, invasion, proliferation and survival.
Since FAK has a nuclear export signal, a nuclear localization signal, and the SUMOylation in the FERM domain related to nuclear import signals. FAK can also enter the nucleus via biological mechanisms. Nuclear FAK controls various transcriptional networks such as the p53 signaling pathway, the inflammatory signaling pathway, the immune escape, and angiogenesis, influencing multiple cancer cell functions. The inhibition of nuclear FAK expression can affect the biological behavior of tumor cells such as aging, apoptosis and immune escape. However, the regulation mechanism of FAK in the nucleus remains to be further studied. For example, FAK regulates p21 cyclin-dependent kinase inhibitor gene expression in a kinase-dependent or kinase-independent manner, but how FAK regulates the expression of the p21 gene in two ways remains unclear. The molecular mechanism is still unclear. In tumor angiogenesis, whether FAK regulates the expression of related molecules through other pathways still needs to be studied. Furthermore, both FAK and p53 can participate in the regulation of nucleolar associated proteins expression in the nucleolus. But it is not clear whether they interact. Therefore, there are still many problems in this field that have not yet been solved. In the future, it is necessary to explore its molecular mechanisms, which is crucial for studying the occurrence and development of tumors. Furthermore, further study of the roles of nuclear FAK may uncover new mechanisms that promote tumor development.
Acknowledgements
Not applicable
Abbreviations
- A3B
APOBEC3B
- APOBEC
Apolipoprotein B mRNA editing enzyme catalytic polypeptide-like
- CCL5
Chemokine ligand 5
- CSCs
Cancer stem cell
- EC
Endothelial cell
- ECM
Extracellular matrix
- ERK1/2
Extracellular signal-regulated kinase
- EZH2
Zeste homolog 2
- FAK
Focal adhesion kinase
- FAs
Focal adhesions
- FAT
C-terminal
- FRNK
FAK-related non-kinase
- IGFBP3
Insulin-like growth factor binding protein 3
- KAKTLR
FERM F2 lobe
- KD
Kinase depletion
- LCSCs
Liver cancer stem cells
- MAPKs
Mitogen activated protein kinases
- Mdm2
Murine double minute2
- MEF2
Myocyte enhancer factor 2
- MSCs
Mesenchymal stem cells
- NCAM
Neural cell adhesion molecule
- NES
Nuclear export signals
- NLS
Nuclear localization signals
- NS
Nuclear stabilizing protein
- NSCLC
Non-small cell lung cancer
- NuMA
Nuclear mitotic apparatus protein
- PIAS1
Protein inhibitor of activated STAT1
- PIP2
Phosphatidylinositol 4,5-biphosphate
- PIPKI-α
Type I phosphatidylinositol phosphate kinase
- PKC
Protein kinase C
- PtdIns(4,5)P
Phosphatidylinositol 4,5-bisphosphate
- PTEN
Tensin homolog deleted on chromosome ten
- R2
1-benzyl-15,3,5,7-tetraazetidine[3.3.1.1~3,7~] decane
- Sam68
Src mitosis-associated protein 68
- SH2
Src homology 2
- SH3
Src homology 3
- Sin3A
SIN3 transcriptional regulatory protein family member A
- ST2
Growth stimulation expressed gene 2
- SUMO
Small ubiquitin-related modifier
- TF
Binding transcription factor
- Tregs
Regulatory T cells
- VCAM-1
Vascular cell adhesion factor-1
- VEGFR2
Vascular endothelial growth factor receptor 2
- VEGFR3
Vascular endothelial growth factor receptor 3
- WT
Wild-type
- XIAP
X-linked apoptotic protein inhibitor
Authors’ contributions
All the authors contributed for the preparation of this manuscript. JZ and LT were responsible for confirming the topic. JZ were responsible for writing the first draft of this article. LT and QY contributed to furtherly editing and polishing the manuscript. All authors read and approved the final manuscript.
Funding
This research was supported by the Fundamental Research Funds for the Central Universities (No.2018CDYJSY0055; 2018CQDYSG0021); the Natural Science Foundation of China (No.31670952).
Availability of data and materials
Not applicable
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Jin Zhou, Email: zhoujin0801@163.com.
Qian Yi, Email: yiqian2010@yeah.net.
Liling Tang, Email: tangliling@cqu.edu.cn.
References
- 1.Lee BY, Timpson P, Horvath LG, Daly RJ. FAK signaling in human cancer as a target for therapeutics. Pharmacol Ther. 2015;146:132–149. doi: 10.1016/j.pharmthera.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 2.Wang B, Qi X, Li D, Feng M, Meng X, Fu S. Expression of pY397 FAK promotes the development of non-small cell lung cancer. Oncol Lett. 2016;11(2):979–983. doi: 10.3892/ol.2015.3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crompton B, Wang S, Hwang E, Guha R, Boxer M, McKnight C, Shen M, Melong N, Veinotte C, Conway A, Berman J, Hall M, Davis M, Stegmaier K. High-throughput chemical screening identifies focal adhesion kinase and Aurora kinase B inhibition as a synergistic treatment combination in Ewing sarcoma. Cancer Res. 2017:77. [DOI] [PMC free article] [PubMed]
- 4.Fan T, Chen J, Zhang L, Gao P, Hui Y, Xu P, Zhang X, Liu H. Bit1 knockdown contributes to growth suppression as well as the decreases of migration and invasion abilities in esophageal squamous cell carcinoma via suppressing FAK-paxillin pathway. Mol Cancer. 2016;15. [DOI] [PMC free article] [PubMed]
- 5.Skinner HD, Giri U, Yang L, Woo SH, Story MD, Pickering CR, Byers LA, Williams MD, El-Naggar A, Wang J, Diao L, Shen L, Fan YH, Molkentine DP, Beadle BM, Meyn RE, Myers JN, Heymach JV. Proteomic profiling identifies PTK2/FAK as a driver of Radioresistance in HPV-negative head and neck Cancer. Clin Cancer Res. 2016;22(18):4643–4650. doi: 10.1158/1078-0432.CCR-15-2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balsas P, Palomero J, Eguileor A, Leonor Rodriguez M, Carmela Vegliante M, Planas-Rigol E, Sureda-Gomez M, Cid MC, Campo E, Amador V. SOX11 promotes tumor protective microenvironment interactions through CXCR4 and FAK regulation in mantle cell lymphoma. Blood. 2017;130(4):501–513. doi: 10.1182/blood-2017-04-776740. [DOI] [PubMed] [Google Scholar]
- 7.Nader GP, Ezratty EJ, Gundersen GG. FAK, Talin and PIPKIgamma regulate endocytosed integrin activation to polarize focal adhesion assembly. Nat Cell Biol. 2016;18(5):491–503. doi: 10.1038/ncb3333. [DOI] [PubMed] [Google Scholar]
- 8.Walker S, Foster F, Wood A, Owens T, Brennan K, Streuli CH, Gilmore AP. Oncogenic activation of FAK drives apoptosis suppression in a 3D-culture model of breast cancer initiation. Oncotarget. 2016;7(43):70336–70352. doi: 10.18632/oncotarget.11856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brami-Cherrier K, Gervasi N, Arsenieva D, Walkiewicz K, Boutterin MC, Ortega A, Leonard PG, Seantier B, Gasmi L, Bouceba T, Kadare G, Girault JA, Arold ST. FAK dimerization controls its kinase-dependent functions at focal adhesions. EMBO J. 2014;33(4):356–370. doi: 10.1002/embj.201386399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yurdagul A, Jr, Sulzmaier FJ, Chen XL, Pattillo CB, Schlaepfer DD, Orr AW. Oxidized LDL induces FAK-dependent RSK signaling to drive NF-kappa B activation and VCAM-1 expression. J Cell Sci. 2016;129(8):1580–1591. doi: 10.1242/jcs.182097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schoenherr C, Serrels B, Proby C, Cunningham DL, Findlay JE, Baillie GS, Heath JK, Frame MC. Eps8 controls Src- and FAK-dependent phenotypes in squamous carcinoma cells. J Cell Sci. 2014;127(24):5303–5316. doi: 10.1242/jcs.157560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen Q, Yi B, Ma JB, Ning JL, Wu LZ, Ma DQ, Lu KZ, Gu JT. Alpha(2)-adrenoreceptor modulated FAK pathway induced by dexmedetomidine attenuates pulmonary microvascular hyper-permeability following kidney injury. Oncotarget. 2016;7(35):55990–56001. doi: 10.18632/oncotarget.10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jean C, Chen XL, Nam JO, Tancioni I, Uryu S, Lawson C, Ward KK, Walsh CT, Miller NLG, Ghassemian M, Turowski P, Dejana E, Weis S, Cheresh DA, Schlaepfer DD. Inhibition of endothelial FAK activity prevents tumor metastasis by enhancing barrier function. J Cell Biol. 2014;204(2):247–263. doi: 10.1083/jcb.201307067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu D, Liu T, Lin LM, Li S, Hang XM, Sun YQ. Exposure to endosulfan increases endothelial permeability by transcellular and paracellular pathways in relation to cardiovascular diseases. Environ Pollut. 2017;223:111–119. doi: 10.1016/j.envpol.2016.12.051. [DOI] [PubMed] [Google Scholar]
- 15.Cai J, Wei J, Li S, Suber T, Zhao J. AM966, an antagonist of lysophosphatidic acid receptor 1, increases lung microvascular endothelial permeability through activation of rho signaling pathway and phosphorylation of VE-cadherin. Mediat Inflamm. 2017;2017:6893560. doi: 10.1155/2017/6893560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alexopoulou AN, Lees DM, Bodrug N, Lechertier T, Fernandez I, D'Amico G, Dukinfield M, Batista S, Tavora B, Serrels B, Hodivala-Dilke K. Focal adhesion kinase (FAK) tyrosine 397E mutation restores the vascular leakage defect in endothelium-specific FAK-kinase dead mice. J Pathol. 2017;242(3):358–370. doi: 10.1002/path.4911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang Yu-Han, Yang Hung-Yu, Huang Shiu-Wen, Ou George, Hsu Ya-Fen, Hsu Ming-Jen. Interleukin-6 Induces Vascular Endothelial Growth Factor-C Expression via Src-FAK-STAT3 Signaling in Lymphatic Endothelial Cells. PLOS ONE. 2016;11(7):e0158839. doi: 10.1371/journal.pone.0158839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tan YH, Wood AR, Jia Q, Zhou WW, Luo JY, Yang F, Chen JW, Chen JJ, Sun J, Seong J, Tajik A, Singh R, Wang N. Soft matrices downregulate FAK activity to promote growth of tumor-repopulating cells. Biochem Biophys Res Commun. 2017;483(1):456–462. doi: 10.1016/j.bbrc.2016.12.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hwang JS, Eun SY, Ham SA, Yoo T, Lee WJ, Paek KS, Do JT, Lim DS, Seo HG. PPAR delta modulates oxLDL-induced apoptosis of vascular smooth muscle cells through a TGF-beta/FAK signaling axis. Int J Biochem Cell Biol. 2015;62:54–61. doi: 10.1016/j.biocel.2015.02.014. [DOI] [PubMed] [Google Scholar]
- 20.Goncalves AF, Adlesic M, Brandt S, Hejhal T, Harlander S, Sommer L, Shakhova O, Wild PJ, Frew IJ. Evidence of renal angiomyolipoma neoplastic stem cells arising from renal epithelial cells. Nat Commun. 2017;8. [DOI] [PMC free article] [PubMed]
- 21.Maccalli C, Rasul KI, Elawad M, Ferrone S. The role of cancer stem cells in the modulation of anti-tumor immune responses. Semin Cancer Biol. 2018;53:189–200. doi: 10.1016/j.semcancer.2018.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoon H, Dehart JP, Murphy JM, Lim S-TS. Understanding the roles of FAK in Cancer: inhibitors, genetic models, and new insights. J Histochem Cytochem. 2015;63(2):114–128. doi: 10.1369/0022155414561498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun J, Luo Q, Liu L, Song G. Low-level shear stress promotes migration of liver cancer stem cells via the FAK-ERK1/2 signalling pathway. Cancer Lett. 2018;427:1–8. doi: 10.1016/j.canlet.2018.04.015. [DOI] [PubMed] [Google Scholar]
- 24.Sun J, Luo Q, Liu L, Yang X, Zhu S, Song G. Salinomycin attenuates liver cancer stem cell motility by enhancing cell stiffness and increasing F-actin formation via the FAK-ERK1/2 signalling pathway. Toxicology. 2017;384:1–10. doi: 10.1016/j.tox.2017.04.006. [DOI] [PubMed] [Google Scholar]
- 25.Efremov YM, Dokrunova AA, Efremenko AV, Kirpichnikov MP, Shaitan KV, Sokolova OS. Distinct impact of targeted actin cytoskeleton reorganization on mechanical properties of normal and malignant cells. Biochim Biophys Acta Mol Cell Res. 2015;1853(11):3117–3125. doi: 10.1016/j.bbamcr.2015.05.008. [DOI] [PubMed] [Google Scholar]
- 26.Chen Chih-Cheng, He Bo-Ching, Chen Yao-Li, Lee Kuan-Der, Tung Chun-Hsin, Hsu Chia-Chen, Lin Ping-Yi, Chu Pei-Yi, Leu Yu-Wei, Fu Wei-En, Hsiao Shu-Huei. HIC1 and RassF1A Methylation Attenuates Tubulin Expression and Cell Stiffness in Cancer. International Journal of Molecular Sciences. 2018;19(10):2884. doi: 10.3390/ijms19102884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu Wenwei, Mezencev Roman, Kim Byungkyu, Wang Lijuan, McDonald John, Sulchek Todd. Cell Stiffness Is a Biomarker of the Metastatic Potential of Ovarian Cancer Cells. PLoS ONE. 2012;7(10):e46609. doi: 10.1371/journal.pone.0046609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sharma S, Santiskulvong C, Bentolila LA, Rao J, Dorigo O, Gimzewski JK. Correlative nanomechanical profiling with super-resolution F-actin imaging reveals novel insights into mechanisms of cisplatin resistance in ovarian cancer cells. Nanomed Nanotechnol Biol Med. 2012;8(5):757–766. doi: 10.1016/j.nano.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 29.Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI, Harrington K, Williamson P, Moeendarbary E, Charras G, Sahai E. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol. 2013;15(6):637–646. doi: 10.1038/ncb2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rolli CG, Seufferlein T, Kemkemer R, Spatz JP. Impact of tumor cell cytoskeleton organization on invasiveness and migration: a microchannel-based approach. PLoS One. 2010;5(1). [DOI] [PMC free article] [PubMed]
- 31.Lim S-T, Chen XL, Lim Y, Hanson DA, Vo T-T, Howerton K, Larocque N, Fisher SJ, Schiaepfer DD, Llic D. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol Cell. 2008;29(1):9–22. doi: 10.1016/j.molcel.2007.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kadare G, Gervasi N, Brami-Cherrier K, Blockus H, El Messari S, Arold ST, Girault JA. Conformational dynamics of the focal adhesion targeting domain control specific functions of focal adhesion kinase in cells. J Biol Chem. 2015;290(1):478–491. doi: 10.1074/jbc.M114.593632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kleinschmidt EG, Schlaepfer DD. Focal adhesion kinase signaling in unexpected places. Curr Opin Cell Biol. 2017;45:24–30. doi: 10.1016/j.ceb.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lim ST. Nuclear FAK: a new mode of gene regulation from cellular adhesions. Mol Cells. 2013;36(1):1–6. doi: 10.1007/s10059-013-0139-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lim S-T, Mikolon D, Stupack DG, Schlaepfer DD. FERM control of FAK function - implications for cancer therapy. Cell Cycle. 2008;7(15):2306–2314. doi: 10.4161/cc.6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ceccarelli DFJ, Song HK, Poy F, Schaller MD, Eck MJ. Crystal structure of the FERM domain of focal adhesion kinase. J Biol Chem. 2006;281(1):252–259. doi: 10.1074/jbc.M509188200. [DOI] [PubMed] [Google Scholar]
- 37.Chishti AH, Kim AC, Marfatia SM, Lutchman M, Hanspal M, Jindal H, Liu SC, Low PS, Rouleau GA, Mohandas N, Chasis JA, Conboy JG, Gascard P, Takakuwa Y, Huang SC, Benz EJ, Bretscher A, Fehon RG, Gusella AF, Ramesh V, Solomon F, Marchesi VT, Tsukita S, Tsukita S, Arpin M, Louvard D, Tonks NK, Anderson JM, Fanning AS, Bryant PJ, Woods DF, Hoover KB. The FERM domain: a unique module involved in the linkage of cytoplasmic proteins to the membrane. Trends Biochem Sci. 1998;23(8):281–282. doi: 10.1016/s0968-0004(98)01237-7. [DOI] [PubMed] [Google Scholar]
- 38.Calalb MB, Polte TR, Hanks SK. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase-activity - a role for SRC family kinases. Mol Cell Biol. 1995;15(2):954–963. doi: 10.1128/mcb.15.2.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schlaepfer DD, Hunter T. Evidence for in vivo phosphorylation of the Grb2 SH2-domain binding site on focal adhesion kinase by Src-family protein-tyrosine kinases. Mol Cell Biol. 1996;16(10):5623–5633. doi: 10.1128/mcb.16.10.5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schaller MD, Parsons JT. PP125(FAK)-dependent tyrosine phosphorylation of PAXILLIN creates a high-affinity binding-site for CRK. Mol Cell Biol. 1995;15(5):2635–2645. doi: 10.1128/mcb.15.5.2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen HC, Appeddu PA, Parsons JT, Hildebrand JD, Schaller MD, Guan JL. Interaction of focal adhesion kinase with cytoskeletal protein TALIN. J Biol Chem. 1995;270(28):16995–16999. doi: 10.1074/jbc.270.28.16995. [DOI] [PubMed] [Google Scholar]
- 42.Schlaepfer DD, Hanks SK, Hunter T, Vandergeer P. Integrin-mediated signal-transduction linked to RAS pathway by GRB2 binding to focal adhesion kinase. Nature. 1994;372(6508):786–791. doi: 10.1038/372786a0. [DOI] [PubMed] [Google Scholar]
- 43.Zhai Jinbin, Lin Hong, Nie Zhenying, Wu Junhua, Cañete-Soler Rafaela, Schlaepfer William W., Schlaepfer David D. Direct Interaction of Focal Adhesion Kinase with p190RhoGEF. Journal of Biological Chemistry. 2003;278(27):24865–24873. doi: 10.1074/jbc.M302381200. [DOI] [PubMed] [Google Scholar]
- 44.Garces CA, Kurenova EV, Golubovskaya VM, Cance WG. Vascular endothelial growth factor receptor-3 and focal adhesion kinase bind and suppress apoptosis in breast cancer cells. Cancer Res. 2006;66(3):1446–1454. doi: 10.1158/0008-5472.CAN-05-1661. [DOI] [PubMed] [Google Scholar]
- 45.Cance W. G., Kurenova E., Marlowe T., Golubovskaya V. Disrupting the Scaffold to Improve Focal Adhesion Kinase-Targeted Cancer Therapeutics. Science Signaling. 2013;6(268):pe10–pe10. doi: 10.1126/scisignal.2004021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mousson Antoine, Sick Emilie, Carl Philippe, Dujardin Denis, De Mey Jan, Rondé Philippe. Targeting Focal Adhesion Kinase Using Inhibitors of Protein-Protein Interactions. Cancers. 2018;10(9):278. doi: 10.3390/cancers10090278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ossovskaya V, Lim S-T, Ota N, Schlaepfer DD, Ilic D. FAK nuclear export signal sequences. FEBS Lett. 2008;582(16):2402–2406. doi: 10.1016/j.febslet.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cai X, Lietha D, Ceccarelli DF, Karginov AV, Rajfur Z, Jacobson K, Hahn KM, Eck MJ, Schaller MD. Spatial and temporal regulation of focal adhesion kinase activity in living cells. Mol Cell Biol. 2008;28(1):201–214. doi: 10.1128/MCB.01324-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Frame MC, Patel H, Serrels B, Lietha D, Eck MJ. The FERM domain: organizing the structure and function of FAK. Nat Rev Mol Cell Biol. 2010;11(11):802–814. doi: 10.1038/nrm2996. [DOI] [PubMed] [Google Scholar]
- 50.Kadare G, Toutant M, Formstecher E, Corvol JC, Carnaud M, Boutterin MC, Girault JA. PIAS1-mediated sumoylation of focal adhesion kinase activates its autophosphorylation. J Biol Chem. 2003;278(48):47434–47440. doi: 10.1074/jbc.M308562200. [DOI] [PubMed] [Google Scholar]
- 51.Alanko J, Mai A, Jacquemet G, Schauer K, Kaukonen R, Saari M, Goud B, Ivaska J. Integrin endosomal signalling suppresses anoikis. Nat Cell Biol. 2015;17(11):1412–1421. doi: 10.1038/ncb3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goni GM, Epifano C, Boskovic J, Camacho-Artacho M, Zhou J, Bronowska A, Teresa Martin M, Eck MJ, Kremer L, Graeter F, Gervasio FL, Perez-Moreno M, Lietha D. Phosphatidylinositol 4,5-bisphosphate triggers activation of focal adhesion kinase by inducing clustering and conformational changes. Proc Natl Acad Sci U S A. 2014;111(31):E3177–E3186. doi: 10.1073/pnas.1317022111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Santos AM, Schechtman D, Cardoso AC, Clemente CFMZ, Silva JC, Fioramonte M, Pereira MBM, Marin TM, Oliveira PSL, Figueira ACM, Oliveira SHP, Torriani IL, Gozzo FC, Neto JX, Franchini KG. FERM domain interaction with myosin negatively regulates FAK in cardiomyocyte hypertrophy. Nat Chem Biol. 2012;8(1):102–110. doi: 10.1038/nchembio.717. [DOI] [PubMed] [Google Scholar]
- 54.Dong JM, Lau LS, Ng YW, Lim L, Manser E. Paxillin nuclear-cytoplasmic localization is regulated by phosphorylation of the LD4 motif: evidence that nuclear paxillin promotes cell proliferation (vol 418, pg 173, 2009) Biochem J. 2009;418:717. doi: 10.1042/BJ20080170. [DOI] [PubMed] [Google Scholar]
- 55.Wang Y, Gilmore TD. Zyxin and paxillin proteins: focal adhesion plaque LIM domain proteins go nuclear. Biochim Biophys Acta Mol Cell Res. 2003;1593(2–3):115–120. doi: 10.1016/s0167-4889(02)00349-x. [DOI] [PubMed] [Google Scholar]
- 56.Matias Sanchez A, Eduardo Shortrede J, Maria Vargas-Roig L, Ines Flamini M. Retinoic acid induces nuclear FAK translocation and reduces breast cancer cell adhesion through Moesin, FAK, and Paxillin. Mol Cell Endocrinol. 2016;430(C):1–11. doi: 10.1016/j.mce.2016.04.021. [DOI] [PubMed] [Google Scholar]
- 57.Lobo M, Zachary I. Nuclear localization and apoptotic regulation of an amino-terminal domain focal adhesion kinase fragment in endothelial cells. Biochem Biophys Res Commun. 2000;276(3):1068–1074. doi: 10.1006/bbrc.2000.3547. [DOI] [PubMed] [Google Scholar]
- 58.Constanzo JD, Tang K-j, Rindhe S, Melegari M, Liu H, Tang X, Rodriguez-Canales J, Wistuba I, Scaglioni PP. PIAS1-FAK interaction promotes the survival and progression of non-small cell lung Cancer. Neoplasia. 2016;18(5):282–293. doi: 10.1016/j.neo.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kleene R, Mzoughi M, Joshi G, Kalus I, Bormann U, Schulze C, Xiao M-F, Dityatev A, Schachner M. NCAM-induced neurite outgrowth depends on binding of calmodulin to NCAM and on nuclear import of NCAM and fak fragments. J Neurosci. 2010;30(32):10784–10798. doi: 10.1523/JNEUROSCI.0297-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Luo S-W, Zhang C, Zhang B, Kim C-H, Qiu Y-Z, Du Q-S, Mei L, Xiong W-C. Regulation of heterochromatin remodelling and myogenin expression during muscle differentiation by FAK interaction with MBD2. EMBO J. 2009;28(17):2568–2582. doi: 10.1038/emboj.2009.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ahn S, Park H. XIAP is essential for shear stress-enhanced Tyr-576 phosphorylation of FAK. Biochem Biophys Res Commun. 2010;399(2):256–261. doi: 10.1016/j.bbrc.2010.07.064. [DOI] [PubMed] [Google Scholar]
- 62.Aoto H, Sasaki H, Ishino M, Sasaki T. Nuclear translocation of cell adhesion kinase beta/proline-rich tyrosine kinase 2. Cell Struct Funct. 2002;27(1):47–61. doi: 10.1247/csf.27.47. [DOI] [PubMed] [Google Scholar]
- 63.Lim S-T, Chen XL, Tomar A, Miller NLG, Yoo J, Schlaepfer DD. Knock-in mutation reveals an essential role for focal adhesion kinase activity in blood vessel morphogenesis and cell motility-polarity but not cell proliferation. J Biol Chem. 2010;285(28):21526–21536. doi: 10.1074/jbc.M110.129999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lietha D, Cai X, Ceccarelli DFJ, Li Y, Schaller MD, Eck MJ. Structural basis for the autoinhibition of focal adhesion kinase. Cell. 2007;129(6):1177–1187. doi: 10.1016/j.cell.2007.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lim S-T, Miller NLG, Chen XL, Tancioni I, Walsh CT, Lawson C, Uryu S, Weis SM, Cheresh DA, Schlaepfer DD. Nuclear-localized focal adhesion kinase regulates inflammatory VCAM-1 expression. J Cell Biol. 2012;197(7):907–919. doi: 10.1083/jcb.201109067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kadare G. PIAS1-mediated sumoylation of focal adhesion kinase activates its autophosphorylation. FASEB J. 2004;18(8):C256. doi: 10.1074/jbc.M308562200. [DOI] [PubMed] [Google Scholar]
- 67.Golubovskaya VM, Finch R, Cance WG. Direct interaction of the N-terminal domain of focal adhesion kinase with the N-terminal transactivation domain of p53. J Biol Chem. 2005;280(26):25008–25021. doi: 10.1074/jbc.M414172200. [DOI] [PubMed] [Google Scholar]
- 68.Golubovskaya VM, Finch R, Zheng M, Kurenova EV, Cance WG. The 7-amino-acid site in the proline-rich region of the N-terminal domain of p53 is involved in the interaction with FAK and is critical for p53 functioning. Biochem J. 2008;411:151–160. doi: 10.1042/BJ20071657. [DOI] [PubMed] [Google Scholar]
- 69.Ou W-B, Lu M, Eilers G, Li H, Ding J, Meng X, Wu Y, He Q, Sheng Q, Zhou H-M, Fletcher JA. Co-targeting of FAK and MDM2 triggers additive anti-proliferative effects in mesothelioma via a coordinated reactivation of p53. Br J Cancer. 2016;115(10):1253–1263. doi: 10.1038/bjc.2016.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dragoj M, Milosevic Z, Bankovic J, Tanic N, Pesic M, Stankovic T. Targeting CXCR4 and FAK reverses doxorubicin resistance and suppresses invasion in non-small cell lung carcinoma. Cell Oncol. 2017;40(1):47–62. doi: 10.1007/s13402-016-0304-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tai H-C, Chang A-C, Yu H-J, Huang C-Y, Tsai Y-C, Lai Y-W, Sun H-L, Tang C-H, Wang S-W. Osteoblast-derived WISP-1 increases VCAM-1 expression and enhances prostate cancer metastasis by down-regulating miR-126. Oncotarget. 2014;5(17):7589–7598. doi: 10.18632/oncotarget.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Aulakh GK, Petri B, Wojcik KM, Colarusso P, Lee JJ, Patel KD. Inhibiting focal adhesion kinase (FAK) blocks IL-4 induced VCAM-1 expression and eosinophil recruitment in vitro and in vivo. J Leukoc Biol. 2018;104(1):147–158. doi: 10.1002/JLB.3MA1117-429R. [DOI] [PubMed] [Google Scholar]
- 73.Serrels A, Lund T, Serrels B, Byron A, McPherson RC, von Kriegsheim A, Gomez-Cuadrado L, Canel M, Muir M, Ring JE, Maniati E, Sims AH, Pachter JA, Brunton VG, Gilbert N, Anderton SM, Nibbs RJB, Frame MC. Nuclear FAK controls chemokine transcription, Tregs, and evasion of anti-tumor immunity. Cell. 2015;163(1):160–173. doi: 10.1016/j.cell.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Serrels Bryan, McGivern Niamh, Canel Marta, Byron Adam, Johnson Sarah C., McSorley Henry J., Quinn Niall, Taggart David, Von Kreigsheim Alex, Anderton Stephen M., Serrels Alan, Frame Margaret C. IL-33 and ST2 mediate FAK-dependent antitumor immune evasion through transcriptional networks. Science Signaling. 2017;10(508):eaan8355. doi: 10.1126/scisignal.aan8355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sun S, Wu H-J, Guan J-L. Nuclear FAK and its kinase activity regulate VEGFR2 transcription in angiogenesis of adult mice. Sci Rep. 2018;8(1). [DOI] [PMC free article] [PubMed]
- 76.Canel M, Byron A, Sims AH, Cartier J, Patel H, Frame MC, Brunton VG, Serrels B, Serrels A. Nuclear FAK and Runx1 cooperate to regulate IGFBP3, cell-cycle progression, and tumor growth. Cancer Res. 2017;77(19):5301–5312. doi: 10.1158/0008-5472.CAN-17-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen Y, Li W, Zhou X. Detecting RUNX1 gene exon 3 mutant site in acute myeloid leukemia patients, involves performing amplification of RUNX1 gene 3 exon mutant site by using primer set that comprises forward primer and reverse primer: China, CN103710437-A[P]. 2014-04-09.
- 78.Leidal AM, Debnath J. 'Doubling down' on the autophagy pathway to suppress tumor growth. Genes Dev. 2014;28(11):1137–1139. doi: 10.1101/gad.244681.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gnani Daniela, Romito Ilaria, Artuso Simona, Chierici Marco, De Stefanis Cristiano, Panera Nadia, Crudele Annalisa, Ceccarelli Sara, Carcarino Elena, D’Oria Valentina, Porru Manuela, Giorda Ezio, Ferrari Karin, Miele Luca, Villa Erica, Balsano Clara, Pasini Diego, Furlanello Cesare, Locatelli Franco, Nobili Valerio, Rota Rossella, Leonetti Carlo, Alisi Anna. Focal adhesion kinase depletion reduces human hepatocellular carcinoma growth by repressing enhancer of zeste homolog 2. Cell Death & Differentiation. 2017;24(5):889–902. doi: 10.1038/cdd.2017.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yi XP, Zhou J, Huber L, Qu J, Wang X, Gerdes AM, Li F. Nuclear compartmentalization of FAK and FRNK in cardiac myocytes. Am J Phys Heart Circ Phys. 2006;290(6):H2509–H2515. doi: 10.1152/ajpheart.00659.2005. [DOI] [PubMed] [Google Scholar]
- 81.Tancioni I, Miller NLG, Uryu S, Lawson C, Jean C, Chen XL, Kleinschmidt EG, Schlaepfer DD. FAK activity protects nucleostemin in facilitating breast cancer spheroid and tumor growth. Breast Cancer Res. 2015;17. [DOI] [PMC free article] [PubMed]
- 82.Gillory LA, Stewart JE, Megison ML, Waters AM, Beierle EA. Focal adhesion kinase and p53 synergistically decrease neuroblastoma cell survival. J Surg Res. 2015;196(2):339–349. doi: 10.1016/j.jss.2015.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408(6810):307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 84.Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137(3):413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 85.Stiewe T, Haran TE. How mutations shape p53 interactions with the genome to promote tumorigenesis and drug resistance. Drug Resist Updat. 2018;38:27–43. doi: 10.1016/j.drup.2018.05.001. [DOI] [PubMed] [Google Scholar]
- 86.Law Emily K., Sieuwerts Anieta M., LaPara Kelly, Leonard Brandon, Starrett Gabriel J., Molan Amy M., Temiz Nuri A., Vogel Rachel Isaksson, Meijer-van Gelder Marion E., Sweep Fred C. G. J., Span Paul N., Foekens John A., Martens John W. M., Yee Douglas, Harris Reuben S. The DNA cytosine deaminase APOBEC3B promotes tamoxifen resistance in ER-positive breast cancer. Science Advances. 2016;2(10):e1601737. doi: 10.1126/sciadv.1601737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Periyasamy M, Singh AK, Gemma C, Kranjec C, Farzan R, Leach DA, Navaratnam N, Palinkas HL, Vertessy BG, Fenton TR, Doorbar J, Fuller-Pace F, Meek DW, Coombes RC, Buluwela L, Ali S. p53 controls expression of the DNA deaminase APOBEC3B to limit its potential mutagenic activity in cancer cells. Nucleic Acids Res. 2017;45(19):11056–11069. doi: 10.1093/nar/gkx721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dragoj M, Bankovic J, Sereti E, Stojanov SJ, Dimas K, Pesic M, Stankovic T. Anti-invasive effects of CXCR4 and FAK inhibitors in non-small cell lung carcinomas with mutually inactivated p53 and PTEN tumor suppressors. Investig New Drugs. 2017;35(6):718–732. doi: 10.1007/s10637-017-0494-4. [DOI] [PubMed] [Google Scholar]
- 89.Choi S, Chen M, Cryns VL, Anderson RA. A nuclear phosphoinositide kinase complex regulates p53. Nat Cell Biol. 2019;21(4):462–475. doi: 10.1038/s41556-019-0297-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.James A, Wang Y, Raje H, Rosby R, DiMario P. Nucleolar stress with and without p53. Nucleus. 2014;5(5):402–426. doi: 10.4161/nucl.32235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jayaraman S, Chittiboyina S, Bai Y, Abad PC, Vidi P-A, Stauffacher CV, Lelievre SA. The nuclear mitotic apparatus protein NuMA controls rDNA transcription and mediates the nucleolar stress response in a p53-independent manner. Nucleic Acids Res. 2017;45(20):11725–11742. doi: 10.1093/nar/gkx782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hussain SP, Harris CC. Inflammation and cancer: an ancient link with novel potentials. Int J Cancer. 2007;121(11):2373–2380. doi: 10.1002/ijc.23173. [DOI] [PubMed] [Google Scholar]
- 93.Ohnishi S, Ma N, Thanan R, Pinlaor S, Hammam O, Murata M, Kawanishi S. DNA damage in inflammation-related carcinogenesis and Cancer stem cells. Oxidative Med Cell Longev. 2013. [DOI] [PMC free article] [PubMed]
- 94.Laothong U, Hiraku Y, Oikawa S, Intuyod K, Murata M, Pinlaor S. Melatonin induces apoptosis in cholangiocarcinoma cell lines by activating the reactive oxygen species-mediated mitochondrial pathway. Oncol Rep. 2015;33(3):1443–1449. doi: 10.3892/or.2015.3738. [DOI] [PubMed] [Google Scholar]
- 95.Karin M, Gallagher E. TNFR signaling: ubiquitin-conjugated TRAFfic signals control stop-and-go for MAPK signaling complexes. Immunol Rev. 2009;228:225–240. doi: 10.1111/j.1600-065X.2008.00755.x. [DOI] [PubMed] [Google Scholar]
- 96.Pober JS. Endothelial activation: intracellular signaling pathways. Arthritis Res Ther. 2002;4:S109–S116. doi: 10.1186/ar576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pan X, Wu X, Yan D, Peng C, Rao C, Yan H. Acrylamide-induced oxidative stress and inflammatory response are alleviated by N-acetylcysteine in PC12 cells: involvement of the crosstalk between Nrf2 and NF-kappa B pathways regulated by MAPKs. Toxicol Lett. 2018;288:55–64. doi: 10.1016/j.toxlet.2018.02.002. [DOI] [PubMed] [Google Scholar]
- 98.Wang D, Wang Y, Liu H, Tong C, Ying Q, Sachinidis A, Li L, Peng L. Laminin promotes differentiation of rat embryonic stem cells into cardiomyocytes by activating the integrin/FAK/PI3K p85 pathway. J Cell Mol Med. 2019;23(5):3629–3640. doi: 10.1111/jcmm.14264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rogovskii VS. The linkage between inflammation and immune tolerance: interfering with inflammation in Cancer. Curr Cancer Drug Targets. 2017;17(4):325–332. doi: 10.2174/1568009617666170109110816. [DOI] [PubMed] [Google Scholar]
- 100.Jones KL, Croen LA, Yoshida CK, Heuer L, Hansen R, Zerbo O, DeLorenze GN, Kharrazi M, Yolken R, Ashwood P, Van de Water J. Autism with intellectual disability is associated with increased levels of maternal cytokines and chemokines during gestation. Mol Psychiatry. 2017;22(2):273–279. doi: 10.1038/mp.2016.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Atretkhany KSN, Drutskaya MS, Nedospasov SA, Grivennikov SI, Kuprash DV. Chemokines, cytokines and exosomes help tumors to shape inflammatory microenvironment. Pharmacol Ther. 2016;168:98–112. doi: 10.1016/j.pharmthera.2016.09.011. [DOI] [PubMed] [Google Scholar]
- 102.Nascimento DC, Melo PH, Pineros AR, Ferreira RG, Colon DF, Donate PB, Castanheira FV, Gozzi A, Czaikoski PG, Niedbala W, Borges MC, Zamboni DS, Liew FY, Cunha FQ, Alves JC. IL-33 contributes to sepsis-induced long-term immunosuppression by expanding the regulatory T cell population. Nat Commun. 2017;8. [DOI] [PMC free article] [PubMed]
- 103.Collins N, Jiang XD, Zaid A, Macleod BL, Li J, Park CO, Haque A, Bedoui S, Heath WR, Mueller SN, Kupper TS, Gebhardt T, Carbone FR. Skin CD4(+) memory T cells exhibit combined cluster-mediated retention and equilibration with the circulation. Nat Commun. 2016;7. [DOI] [PMC free article] [PubMed]
- 104.Reichenbach DK, Schwarze V, Matta BM, Tkachev V, Lieberknecht E, Liu Q, Koehn BH, Pfeifer D, Taylor PA, Prinz G, Dierbach H, Stickel N, Beck Y, Warncke M, Junt T, Schmitt-Graeff A, Nakae S, Follo M, Wertheimer T, Schwab L, Devlin J, Watkins SC, Duyster J, Ferrara JLM, Turnquist HR, Zeiser R, Blazar BR. The IL-33/ST2 axis augments effector T-cell responses during acute GVHD. Blood. 2015;125(20):3183–3192. doi: 10.1182/blood-2014-10-606830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mathivet T, Bouleti C, Van Woensel M, Stanchi F, Verschuere T, Phng L-K, Dejaegher J, Balcer M, Matsumoto K, Georgieva PB, Belmans J, Sciot R, Stockmann C, Mazzone M, De Vleeschouwer S, Gerhardt H. Dynamic stroma reorganization drives blood vessel dysmorphia during glioma growth. Embo Mol Med. 2017;9(12):1629–1645. doi: 10.15252/emmm.201607445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Demircioglu F, Hodivala-Dilke K. Alpha v beta 3 integrin and tumour blood vessels - learning from the past to shape the future. Curr Opin Cell Biol. 2016;42:121–127. doi: 10.1016/j.ceb.2016.07.008. [DOI] [PubMed] [Google Scholar]
- 107.Cheng H-W, Chen Y-F, Wong J-M, Weng C-W, Chen H-Y, Yu S-L, Chen H-W, Yuan A, Chen JJW. Cancer cells increase endothelial cell tube formation and survival by activating the PI3K/Akt signalling pathway. J Exp Clin Cancer Res. 2017;36. [DOI] [PMC free article] [PubMed]
- 108.Kadosh D, Struhl K. Repression by Ume6 involves recruitment of a complex containing Sin3 corepressor and Rpd3 histone deacetylase to target promoters. Cell. 1997;89(3):365–371. doi: 10.1016/s0092-8674(00)80217-2. [DOI] [PubMed] [Google Scholar]
- 109.Laherty CD, Yang WM, Sun JM, Davie JR, Seto E, Eisenman RN. Histone deacetylases associated with the mSin3 corepressor mediate mad transcriptional repression. Cell. 1997;89(3):349–356. doi: 10.1016/s0092-8674(00)80215-9. [DOI] [PubMed] [Google Scholar]
- 110.Mayo Juan C., Hevia David, Quiros-Gonzalez Isabel, Rodriguez-Garcia Aida, Gonzalez-Menendez Pedro, Cepas Vanesa, Gonzalez-Pola Iván, Sainz Rosa M. IGFBP3 and MAPK/ERK signaling mediates melatonin-induced antitumor activity in prostate cancer. Journal of Pineal Research. 2016;62(1):e12373. doi: 10.1111/jpi.12373. [DOI] [PubMed] [Google Scholar]
- 111.Cardoso AC, Pereira AHM, Ambrosio ALB, Consonni SR, de Oliveira RR, Bajgelman MC, Dias SMG, Franchini KG. FAK forms a complex with MEF2 to couple biomechanical signaling to transcription in cardiomyocytes. Structure. 2016;24(8):1301–1310. doi: 10.1016/j.str.2016.06.003. [DOI] [PubMed] [Google Scholar]
- 112.Koshman YE, Chu M, Engman SJ, Kim T, Iyengar R, Robia SL, Samarel AM. Focal adhesion kinase-related nonkinase inhibits vascular smooth muscle cell invasion by focal adhesion targeting, tyrosine 168 phosphorylation, and competition for p130(Cas) binding. Arterioscler Thromb Vasc Biol. 2011;31(11):2432–U208. doi: 10.1161/ATVBAHA.111.235549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Orsolic I, Jurada D, Pullen N, Oren M, Eliopoulos AG, Volarevic S. The relationship between the nucleolus and cancer: current evidence and emerging paradigms. Semin Cancer Biol. 2016;37-38:36–50. doi: 10.1016/j.semcancer.2015.12.004. [DOI] [PubMed] [Google Scholar]
- 114.Mai W, Huang F, Chen H, Zhou Y, Chen Y. Nervous necrosis virus capsid protein exploits nucleolar phosphoprotein Nucleophosmin (B23) function for viral replication. Virus Res. 2017;230:1–6. doi: 10.1016/j.virusres.2016.12.015. [DOI] [PubMed] [Google Scholar]
- 115.Hein N, Hannan KM, George AJ, Sanij E, Hannan RD. The nucleolus: an emerging target for cancer therapy. Trends Mol Med. 2013;19(11):643–654. doi: 10.1016/j.molmed.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 116.Chatzizacharias NA, Kouraklis GP, Theocharis SE. Focal adhesion kinase: a promising target for anticancer therapy. Expert Opin Ther Targets. 2007;11(10):1315–1328. doi: 10.1517/14728222.11.10.1315. [DOI] [PubMed] [Google Scholar]
- 117.Tiede S, Meyer-Schaller N, Kalathur RKR, Ivanek R, Fagiani E, Schmassmann P, Stillhard P, Hafliger S, Kraut N, Schweifer N, Waizenegger IC, Bill R, Christofori G. The FAK inhibitor BI 853520 exerts anti-tumor effects in breast cancer. Oncogenesis. 2018;7. [DOI] [PMC free article] [PubMed]
- 118.Ma WW. Development of focal adhesion kinase inhibitors in Cancer therapy. Anti Cancer Agents Med Chem. 2011;11(7):638–642. doi: 10.2174/187152011796817628. [DOI] [PubMed] [Google Scholar]
- 119.Golubovskaya VM, Nyberg C, Zheng M, Kweh F, Magis A, Ostrov D, Cance WG. A small molecule inhibitor, 1,2,4,5-Benzenetetraamine Tetrahydrochloride, targeting the Y397 site of focal adhesion kinase decreases tumor growth. J Med Chem. 2008;51(23):7405–7416. doi: 10.1021/jm800483v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhao G, Gong L, Su D, Jin Y, Guo C, Yue M, Yao S, Qin Z, Ye Y, Tang Y, Wu Q, Zhang J, Cui B, Ding Q, Huang H, Hu L, Chen Y, Zhang P, Hu G, Chen L, Wong K-K, Gao D, Ji H. Cullin5 deficiency promotes small-cell lung cancer metastasis by stabilizing integrin beta 1. J Clin Investig. 2019;129(3):972–987. doi: 10.1172/JCI122779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhou B, Wang G-Z, Wen Z-S, Zhou Y-C, Huang Y-C, Chen Y, Zhou G-B. Somatic mutations and splicing variants of focal adhesion kinase in non-small cell lung Cancer, Jnci. J Natl Cancer Inst. 2018;110(2). [DOI] [PubMed]
- 122.Kuenzi BM, Remsing Rix LL, Stewart PA, Fang B, Kinose F, Bryant AT, Boyle TA, Koomen JM, Haura EB, Rix U. Polypharmacology-based ceritinib repurposing using integrated functional proteomics. Nat Chem Biol. 2017;13(12):1222–1231. doi: 10.1038/nchembio.2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dao P, Jarray R, Le Coq J, Lietha D, Loukaci A, Lepelletier Y, Hadj-Slimane R, Garbay C, Raynaud F, Chen H. Synthesis of novel diarylamino-1,3,5-triazine derivatives as FAK inhibitors with anti-angiogenic activity. Bioorg Med Chem Lett. 2013;23(16):4552–4556. doi: 10.1016/j.bmcl.2013.06.038. [DOI] [PubMed] [Google Scholar]
- 124.Dao P, Lietha D, Etheve-Quelquejeu M, Garbay C, Chen H. Synthesis of novel 1,2,4-triazine scaffold as FAK inhibitors with antitumor activity. Bioorg Med Chem Lett. 2017;27(8):1727–1730. doi: 10.1016/j.bmcl.2017.02.072. [DOI] [PubMed] [Google Scholar]
- 125.Qu M, Liu Z, Zhao D, Wang C, Zhang J, Tang Z, Liu K, Shu X, Yuan H, Ma X. Design, synthesis and biological evaluation of sulfonamide-substituted diphenylpyrimidine derivatives (Sul-DPPYs) as potent focal adhesion kinase (FAK) inhibitors with antitumor activity. Bioorg Med Chem. 2017;25(15):3989–3996. doi: 10.1016/j.bmc.2017.05.044. [DOI] [PubMed] [Google Scholar]
- 126.Gu B, Zhu W-G. Surf the post-translational modification network of p53 regulation. Int J Biol Sci. 2012;8(5):672–684. doi: 10.7150/ijbs.4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable