

Abstract
Triglyceride deposit cardiomyovasculopathy (TGCV) is a phenotype primarily reported in patients carrying genetic mutations in PNPLA2 encoding adipose triglyceride lipase (ATGL) which releases long chain fatty acid (LCFA) as a major energy source by the intracellular TG hydrolysis. These patients suffered from intractable heart failure requiring cardiac transplantation. Moreover, we identified TGCV patients without PNPLA2 mutations based on pathological and clinical studies. We provided the diagnostic criteria, in which TGCV with and without PNPLA2 mutations were designated as primary TGCV (P-TGCV) and idiopathic TGCV (I-TGCV), respectively. We hereby report clinical profiles of TGCV patients. Between 2014 and 2018, 7 P-TGCV and 18 I-TGCV Japanese patients have been registered in the International Registry. Patients with I-TGCV, of which etiologies and causes are not known yet, suffered from adult-onset severe heart disease, including heart failure and coronary artery disease, associated with a marked reduction in ATGL activity and myocardial washout rate of LCFA tracer, as similar to those with P-TGCV. The present first registry-based study showed that TGCV is an intractable, at least at the moment, and heterogeneous cardiovascular disorder.
Keywords: Adipose triglyceride lipase, Atherosclerosis, Rare disease, Triglyceride-deposit cardiomyovasculopathy, Triglyceride metabolism
Triglyceride (TG) and orphan diseases
TG is a major energy source for mammals. In normal condition, TG is either received via the diet, or synthesized endogenously and stored in adipose tissues. When required, TG is hydrolyzed by various enzymes called lipases and releases long-chain fatty acid (LCFA), which is delivered to non-adipose tissues for the production of ATP. It has been known that the ectopic TG deposition in non-adipose tissues causes some orphan diseases. In 1953, Jordans reported two brothers with phenotype of skeletal myopathy and vacuolar formation of peripheral leukocytes, called Jordans’ anomaly [1]. Fifty years later, Fischer et al. found that this phenotype is associated with mutations in PNPLA2 [2] encoding adipose TG lipase (ATGL) [3, 4], an essential molecule located in cytoplasmic lipid droplets for the intracellular TG hydrolysis [5, 6], and designated this phenotype as neutral lipid storage disease with myopathy (NLSD-M). Clinical manifestations of NLSD-M appeared variable from mild to severe symptoms [7–13], which could be at least partially explained by function of mutated ATGL proteins [14]. Another phenotype of NLSD involving the skin was reported as NLSD with ichthyosis (NLSD-I) by Chanarin and Dorfman in the 1970s [15–17]. The genetic cause of NLSD-I was found to be mutations in ABHD5 encoding CGI-58, a co-enzyme of ATGL [18]. Using skin fibroblasts and iPS cells from patients with NLSDs, unique intracellular metabolism of TG has been extensively analyzed. These cell-biological experiments showed that cytoplasmic lipid droplets are dynamic cellular organelles interacting with ATGL, CGI-58, and other proteins, and could be a therapeutic target [19–23].
Discovery of TG-deposit cardiomyovasculopathy (TGCV) with PNPLA2 (ATGL) mutation
Since the early 1980s, patients with Jordans’ anomaly and severe heart failure (HF), though very rare, had been reported in Japan [24]. In the early 2000s, our institution started to take care of two patients with severe HF and vacuolar formation in peripheral leukocytes. HF was progressive and intractable, and a couple of years later, they became candidates for cardiac transplantation (CTx). Preoperative examination of their hearts exhibited dilated cardiomyopathy-like morphology in chest X-ray and ultrasonography; however, endomyocardial biopsy specimens showed neutral lipid deposition in cardiomyocytes [25]. When they underwent CTxs, pathological and biochemical analyses of their explanted hearts were performed, demonstrating that their coronary arteries showed unusual coronary atherosclerosis with TG deposition in endothelial and smooth muscle cells (SMCs). We named this novel phenotype as TGCV [26–28]. These patients were identified as homozygous for genetic mutations in PNPLA2 encoding ATGL, which is also known to be responsible for NLSD-M as described above [2].
Postmortem analyses revealed undiagnosed individuals with TGCV
Retrospective postmortem analyses of autopsied cases identified individuals with TGCV phenotype who had TG deposit in both myocardium and coronary arteries, as presented in Fig. 1. A 38-year-old man suddenly died irrespective of intensive treatment for coronary artery disease (CAD) and HF. His heart was heavy in weight and hypertrophied with multiple myocardial fibrous scars. Coronary arteries showed diffuse and concentric stenosis in multi-vessels. Biochemical analyses and imaging mass spectrometry showed TG deposition in both myocardium and coronary arteries [29, 30]. TG-deposit SMCs were observed in his renal and mesenteric arteries as well (data not shown). These data mimic genetic ATGL deficiency; however, the immunoreactive mass of ATGL was detected, and the genetic test using genomic DNA extracted from stored specimens showed no mutation in all exons and exon/intron boundaries of PNPLA2 gene (data not shown). In addition, pathological records showed that he did not have skeletal myopathy.
Fig. 1.

Pathological analysis of the autopsied heart of a 38-year-old man with TGCV phenotype without PNPLA2 mutation. Panel a The transverse section of the autopsied heart stained with Masson’s trichrome showed circumferential patchy fibrosis of the left ventricular wall. The letters A, L, R, and P denotes anterior, left, right, and posterior, respectively. Panel b Lipid droplets (LDs) stained with oil red O in the cytoplasm of cardiomyocytes. Panel c Immunostaining for ATGL (Cell Signaling, Danvers, MA). Cardiomyocytes showed positive reactivity for ATGL. Panel d Coronary arteries with diffuse concentric-type stenosis. Panel e The transverse section of coronary artery was stained with Masson’s trichrome. Coronary artery revealed intimal thickening and fibroatheromatous lesions. Panel f Double-staining of Sudan black B and α-smooth muscle actin (Dako, Tokyo, Japan). Smooth muscle cells (brown color) with lipid droplet (blue color) distributed diffusely in the media and intima (arrows in Panel f). Asterisk represents the vascular lumen. Panel g TG (m/z 879.7) was identified as green and blue colors depending on the intensity. TG signals were diffusely detected in the arterial wall by imaging mass spectrometry. Green color denotes relatively higher intensity of TG than blue color. Myocardial and coronary TG contents (3.64 and 19.44 mg/g of tissue, respectively) were higher in this patient, compared with each of control group (1.4 ± 1.0, and 6.2 ± 4.8 mg/g of tissue, respectively). The detailed clinical profile of this patient is reported as Case 10 in the reference [29].
Scale bars: 1 cm in Panel a, 20 μm in Panel b and c, 5 mm in Panel d, 1 mm in Panel e, 20 μm in Panel f, 200 μm in Panel g
Development of diagnostic methods for TGCV
The above postmortem studies suggested that it is difficult to diagnose TGCV, and many undiagnosed patients should have died, which motivated us to develop diagnostic tools and methods for TGCV. We reported that myocardial scintigraphy with iodine-123-β-methyl iodophenyl-pentadecanoic acid (BMIPP) [31, 32], a radioactive analogue of LCFA, was useful in detecting abnormal LCFA metabolism in patients with TGCV [33, 34]. In addition, we reported the use of automated hematology analyzers to detect Jordans’ anomaly in patients with PNPLA2 mutation [35–37]. Recently, we developed CT-based TG imaging to detect myocardial and coronary TG deposition [34, 38] and selective immunoinactivation assay to measure functional ATGL activities using peripheral leukocytes [39].
Nomenclature, definition, and classification of TGCV
It is well known that disease nomenclature is made not only by their genotypes, but also by their phenotypes in many diseases and by discoverer’s names in some diseases. The nomenclature of TGCV was made by its phenotype that TG accumulated in both myocardium and coronary arteries, resulting from abnormal intracellular metabolism of TG and LCFA (Fig. 2) [26–28]. ATGL is a known enzyme involved in the phenotypic expression of TGCV. The Japan TGCV study group provided the diagnostic criteria for TGCV, in which TGCV with and without PNPLA2 mutations was designated as primary TGCV (P-TGCV) and idiopathic TGCV (I-TGCV), respectively [40–42].
Fig. 2.
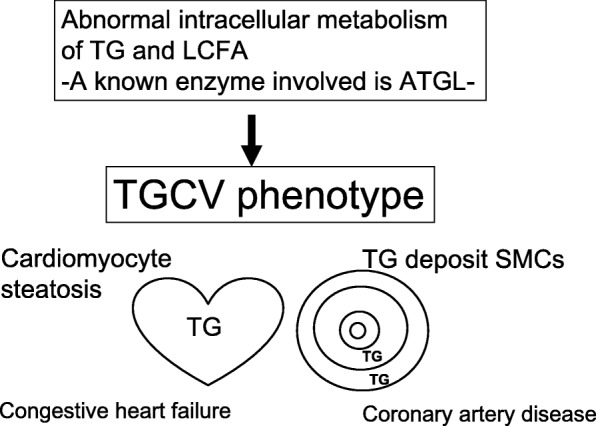
Schematic presentation of the disease concept for TGCV
Pathophysiology of TGCV
The pathophysiological schema of TGCV is shown in Fig. 3. In normal condition (left panel, Fig. 3), LCFAs are taken up through transporters and receptors such as CD36. Some are transported to the mitochondria for β-oxidation, and the remaining LCFAs are utilized as a source of TG and rapidly hydrolyzed by intracellular lipases such as ATGL. In TGCV (right panel, Fig. 3), LCFAs are taken up and used to synthesize TG that cannot be hydrolyzed due to ATGL insufficiency, leading to energy failure and lipotoxicity with massive TG accumulation [28, 43]. It is emphasized that TG-deposit atherosclerosis is an important characteristic of TGCV [44] and distinct from usual cholesterol-deposit atherosclerosis, because the former showed diffuse and concentric narrowing formed by TG-deposit SMCs, whereas the latter showed discrete and eccentric stenosis initiated by the response to injury in the endothelium and accumulation of cholesterol-laden macrophages [45] (Fig. 4). We reported that TG-deposit SMCs and endothelial cells had pro-inflammatory and vulnerable phenotype in vitro [46, 47].
Fig. 3.
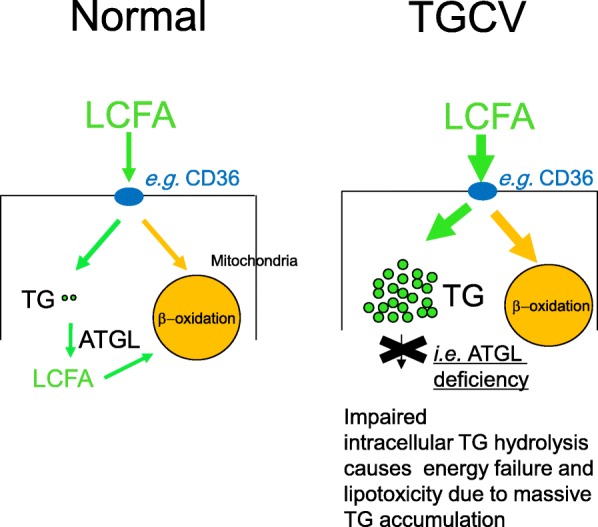
A pathophysiological model for TGCV. Genetic and acquired ATGL deficiency and other causes result in abnormal intracellular metabolism of TG and LCFA, leading to cardiomyocyte steatosis and TG-deposit SMCs. In normal condition (left panel), LCFA is taken up through LCFA transporters and receptors such as CD36 and some of them are transported to mitochondria for β-oxidation and the remaining LCFAs are utilized as a source for TG and rapidly hydrolyzed by intracellular lipases such as ATGL. In TGCV (right panel), LCFAs are taken up and used for the synthesis of TG which can not be hydrolyzed, leading to massive TG accumulation
Fig. 4.
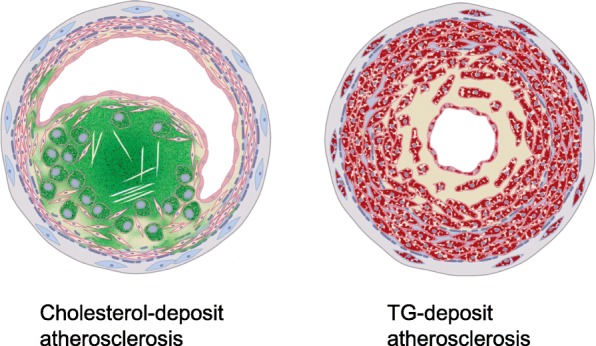
Schemes for cholesterol- (Left) and TG-deposit atherosclerosis (Right). In cholesterol-deposit atherosclerosis, cholesterol (green) accumulates in macrophages, leading to eccentric stenosis. In TG-deposit atherosclerosis, TG (red) accumulates in SMCs, leading to concentric stenosis, which is a major feature of TGCV
A clinical case presentation of I-TGCV
A 58-year-old woman was referred to our hospital due to sudden chest tightness with ST-segment elevation in the electrocardiogram, followed by cardiopulmonary arrest. Under the diagnosis of acute myocardial infarction, she underwent coronary artery bypass grafting (CABG). Past history included type 2 diabetes mellitus requiring insulin treatment and hemodialysis. Cytoplasmic vacuoles in her peripheral polymorphonuclear leukocytes were observed less frequently (< 10% of neutrophils), compared with that in genetic ATGL deficiency (panel A in Fig. 5). ATGL activity in peripheral leukocytes was very low, comparable to that of genetic ATGL deficiency, as shown in Table 1. Myocardial washout rate (WOR) of BMIPP was defective in scintigraphy (panel B in Fig. 5). Pathological analyses of endomyocardial biopsy specimens demonstrated numerous vacuoles filled with stained lipid but positive reactivity for ATGL in cardiomyocytes and adipocytes (right, panel C in Fig. 5). Coronary CT angiogram showed diffuse narrowing coronary arteries, and in TG imaging [25], outside-in involvement of diffuse and abundant lipid components expressed as low CT numbers was seen within the wall in a peninsular pattern (arrows in panel D in Fig. 5). Her laboratory data and imaging tests were similar to those observed in TGCV with genetic ATGL deficiency, except for the conserved expression of ATGL protein in the myocardium. However, it is noted that the case was clinically distinct from genetic ATGL deficiency because there was no skeletal myopathy and no elevation of MM type creatine kinase. Genetic tests showed no mutations or substitutions in any of the exons or intron/exon boundaries of genes encoding ATGL, 1-acylglycerol-3-phosphate O-acyltransferase, hormone-sensitive lipase, or GOS2 (data not shown).
Fig. 5.

Laboratory and imaging examinations for TGCV. a Representative images of May-Giemsa staining of blood smears were shown from patients with P-TGCV and I-TGCV. b Bull’s eye images for BMIPP scintigrams from patients with P-TGCV and I-TGCV. The first scan was performed 20 min post-injection to determine early BMIPP uptake, and the second scan was performed 200 min later to study delayed uptake using myocardial SPECT after patients were injected with 123I-BMIPP. WOR was calculated with the Hear Risk View-S (HRSV) software as the difference between early and delayed images (reference value, 19.4 ± 3.2%). c A patient with P-TGCV showed numerous vacuoles (Panel a, H&E) in cardiomyocytes that stained positively for oil red O (ORO) (inset in Panel b). Furthermore, no positive reactivity for ATGL observed in any of cell types (Panel b, ATGL). Cardiomyocytes of patients with I-TGCV showed numerous vacuoles (Panel c, HE) filled with stained lipid (inset in Panel d, ORO), whereas positive reactivity for ATGL observed not only in adipocytes but also in cardiomyocytes (arrows in Panel d, ATGL). Scale bars: Panels a-d, 30 μm. d Coronary CT angiograms (CTA) from patients with P-TGCV and I-TGCV are shown. Bars in CTA correspond to Panels a-d, which are short axial sections of the left anterior descending coronary artery. The segmentation of the coronary artery lumen and wall was done using a workstation (Ziostation 2, Ziosoft, Japan). Constitutive components were classified into 4 colors with the original analysis software as follows. Colors indicate the CT number (yellow, − 25–0; orange, 0–40; green, 40–215; red, 215–700 Hounsfield unit [HU] (M@XNET, Tokyo, Japan) in Panels a-d. Yellow or orange areas indicate lipid components, red shows blood, and green shows the arterial wall without calcification or lipids. Black arrows in Panels a, b, and c indicate outside-in protrusion, which is the characteristics for TGCV
Table 1.
Patients’ characteristics of Primary and Idiopathic TGCV
| Primary | Idiopathic | |
|---|---|---|
| n = 7 | n = 18 | |
| General Status | ||
| Age (years) | 55.7 ± 12.7 | 64.6 ± 14.7 |
| Sex (female, male) (n) | (2, 5) | (9, 9) |
| BMI (kg/m2) | 19.4 ± 3.4 | 25.4 ± 5.0 |
| Family history for CVD | 7 | 13 |
| ATGL expression | ||
| PNPLA2 mutation | Yes | No** |
| ATGL activities in leukocytes (nmol/h/mg)* | 5.3 ± 8.3 | 12 ± 9 |
| (reference value 52 ± 13 nmol/h/mg) | ||
| Vacuole formation in polymorphonuclear leukocytes (%) | ~ 100% | < 10% |
| Heart disease | ||
| Mean age of symptom onset (years) | 37.7 ± 9.2 | 55.9 ± 12.5 |
| Angina at rest (n) | 3 | 10 |
| Dyspnea or palpitation (n) | 4 | 8 |
| Clinical diagnosis at registration | ||
| Angina pectoris | 1 | 13 |
| (rest, effort) (n) | (1,0) | (11, 2) |
| Heart failure (n) | 5 | 8 |
| Critical arrhythmia (n) | 4 | 1 |
| History of myocardial infarction (n) | 0 | 4 |
| NYHA classification (I, II, III, IV) (n) | (1, 1, 2, 3) | (2, 5, 11, 0) |
| Coronary angiography or CT angiogram | ||
| Affected branch (single vessel, multivessels) (n) | (0, 5) | (4,14) |
| Diffuse narrowing (n) | 5 | 18 |
| Washout rate in BMIPP scintigram (%) | 一3.2 ± 4.8 | 1.4 ± 8 |
| (reference value 19.4 ± 3.2%) | ||
| Treatment history | ||
| Percutaneous coronary intervention (n) | 0 | 7 |
| Coronary artery bypass grafting (n) | 0 | 5 |
| Cardiac transplantation (n) | 2 | 0 |
| Comorbidity | ||
| Skin lesions (n) | 0 | 0 |
| Skeletal myopathy (n) | 7 | 0 |
| Diabetes mellitus (n) | 2 | 15 |
| Outcome | ||
| Death (n) | 5 | 3 |
| (before, after registration) | (3, 2) | (2, 1) |
*Three patients with P-TGCV and fourteen with I-TGCV were enrolled
We did not have opportunity for the measurement in the remaing four patients with P-TGCV and four with I-TGCV
**Two patients were dismissed before the genetic analysis
The Japan TGCV study group certified I-TGCV according to the diagnostic guideline
Abbreviations: CT Computed tomography, CVD Cardiovascular disease, TGCV Triglyceride deposit cardiomyovasculopathy
Clinical characteristics of P- and I-TGCV
Table 1 shows the clinical characteristics of 7 and 18 patients with P- and I-TGCV, respectively, registered to the international registry for NLSD and TGCV between February 2014 and March 2018 in Japan. Both TGCV types were adult onset with chest pain at rest or dyspnea and palpitation. Most patients with either types of TGCV developed severe HF or CAD with diffuse narrowing multivessel lesions or both. Myocardial metabolism of LCFA, detected by WOR of BMIPP and ATGL activities in peripheral leukocytes, was reduced in both TGCV types. Most patients with P-TGCV developed intractable and critical HF, as reported recently [26, 48, 49]. Two of them underwent CTx [26, 48]. Many patients with I-TGCV required percutaneous coronary intervention and CABG. As comorbidity, neither type of TGCV had skin lesions, which suggests that TGCV is not associated with NLSD-I. All patients with P-TGCV had skeletal myopathy, whereas none of those with I-TGCV did. Five of 7 and 3 of 18 registered patients with P- and I-TGCV, respectively, died.
Differential diagnosis of TGCV
Myocardial disorders such as dilated cardiomyopathy, hypertrophic cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy, mitochondrial cardiomyopathy, alcoholic heart disease, and metabolic myocardial disorders (e.g., Fabry disease, Pompe disease, cholesteryl ester storage disease) need to be differentiated from TGCV [41, 42].
Furthermore, known diabetic and metabolic heart diseases need to be differentiated from TGCV. One is diabetic cardiomyopathy, which was originally defined as cardiomyopathy without significant stenosis in epicardial coronary arteries [50]. Another concept is epicardial fat accumulation, which is the oeverdeposition of TG in physiological tissues. TGCV is distinct from these two entities because TGCV is characterized by the ectopic deposition of TG in the cardiomyocytes and SMCs with apparent involvement of epicardial coronary arteries, as shown in Figs. 1 and 5.
Academia-initiated development of specific treatment for TGCV
We found that the chow with tricaprin, TG form of capric acid, improved LCFA metabolism, lipid deposition, cardiac function, and life span in ATGL-targeted mice [4], raising a therapeutic hypothesis that capric acid may be an alternative energy source and reduce TG deposition and lipotoxicity in TGCV [51]. Based upon these data, the Osaka University Hospital manufactured GMP-graded capsules containing the active gradients called CNT-01. We developed the assay to measure plasma capric acid levels [52, 53]. After finishing toxicity tests using rats and dogs required, we are finally conducting investigator-initiated clinical trials.
Comparison between NLSD-I, NLSD-M, and TGCV
As mentioned above, the nomenclature of TGCV was made by its phenotype that TG accumulated in both myocardium and coronary arteries, resulting from abnormal intracellular metabolism of TG and LCFA (Figs. 2 and 3). As described in the first paragraph in this letter, there have been known related disorders; NLSD-M and NLSD-I. Figure 6 shows the comparison of phenotype and genotype between TGCV and NLSDs. NLSD-M and NLSD-I are caused by mutations in PNPLA2 and ABHD5, mainly involved in the skeletal muscle and skin, respectively. Genotype of P-TGCV is known to be PNPLA2 mutation which is responsible for NLSD-M as well.
Fig. 6.
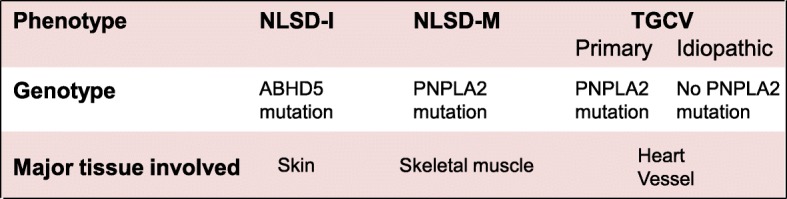
Relationship between TGCV and NLSDs. Comparison of phenotype and genotype between NLSD-I, NLSD-M and TGCV
Issues to be resolved
The following points are important focus for future researches:
-
Possible clinical continuum between P-TGCV and NLSD-M
As mentioned above, both P-TGCV and NLSD-M is caused by genetic ATGL deficiency. It would be of interest to know whether patients with NLSD-M have TG-deposit atherosclerosis, which is the important feature for P-TGCV.
-
Etiologies of I-TGCV and its prevalence in countries other than Japan
As shown in Table 1, 13 out of 18 patients with I-TGCV had family history of cardiovascular disease, suggesting that any genetic factors might be involved in the pathogenesis of I-TGCV. The mechanism underlying downregulation of ATGL activities of I-TGCV and possible involvement of other lipases and related enzymes is of significance to elucidate. In order to elucidate these issues, the development of screening methods for the diagnosis of I-TGCV is under way in our laboratory.
Conclusions
TGCV is a severe cardiovascular disorder named by its phenotype of cardiomyovascular TG deposition, of which etiologies seem heterogeneous.
Methods
-
Pathological, laboratory, and clinical imaging
Standard procedures were performed as described (please see legends of Figs. 1 and 5).
-
International registry for NLSD/TGCV
On the World Rare Disease Day 2014, we launched the international registry for neutral lipid storage diseases, TG-deposit cardiomyovasculopathy, and related disorders (Clinical Trial gov. NCT02830763).The present patients with TGCV were registered according to the study protocol after obtaining written consent. The protocol was approved by the Osaka University Hospital Ethical Committee (approval no. 13204).
Acknowledgments
The authors thank Dr. Kosuke Tsukamoto for playing a part in constructing the international registry. The authors would like to thank Ms. Mao Ishikawa and Yoshie Yabe for their excellent secretarial work.
Funding
This study was partially supported by research grants from the Ministry of Health, Labour and Welfare and the Japan Agency of Medical Research and Development (A-MED; grant no. 17ek0109092h0003), Nihon Medi-Physics Co., Ltd., and the Tochino Foundation to KH.
Availability of data and materials
The datasets generated and/or analyzed in the current study are available from the corresponding author on reasonable request.
Abbreviations
- ATGL
Adipose triglyceride lipase
- BMIPP
Iodine-123-β-methyl iodophenyl-pentadecanoic acid
- CABG
Coronary artery bypass grafting
- CAD
Coronary artery disease
- CTA
CT angiograms
- CTx
Cardiac transplantion
- HF
Heart failure
- Hu
Hounsfield unit
- LCFA
Long-chain fatty acid
- NLSD
Neutral lipid storage disease
- NLSD-I
NLSD with ichthyosis
- NLSD-M
NLSD with myopathy
- SMCs
Smooth muscle cells
- TG
Triglyceride
- TGCV
Triglyceride-deposit cardiomyovasculopathy
- WOR
Washout rate
Authors’ contributions
ML wrote the manuscript and contributed to data analyses and discussion. KH designated the research concept and contributed to the discussion and writing of the manuscript. YoI performed autopsy for the undiagnosed case (Fig. 1), collected data, and contributed to the discussion and writing of the manuscript. KH and JK took care of the clinically identified I-TGCV case (Fig. 5). CH, BZ, JK, KSu, HM, ASu, YH, AT, YaI, KKo, MH, YF, NZ, SYamag, RS, and SYamad collected data and contributed to the discussion. HNak, KaK, E S, S-PH, YNak, TIn, YS, Y Yasui, YNag, ASa, SK, KSh, HH, DN, TId, TA, HNai, and HNag interpreted and discussed the data. KuK designated the registry, collected data, and contributed to the discussion. All the authors have read and revised the manuscript and approved the final manuscript.
Ethics approval and consent to participate
The protocol of the international registry was approved by the Osaka University Hospital Ethical Committee (Approved No. 13204).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Ming Li, Email: liming@cnt-osaka.com.
Ken-ichi Hirano, Phone: +81-6-6872-8215, Email: khirano@cnt-osaka.com.
Yoshihiko Ikeda, Email: yikeda@ncvc.go.jp.
Masahiro Higashi, Email: xhigashi@onh.go.jp.
Chikako Hashimoto, Email: hashimoto@cnt-osaka.com.
Bo Zhang, Email: bozhang@fukuoka-u.ac.jp.
Junji Kozawa, Email: kjunji@endmet.med.osaka-u.ac.jp.
Koichiro Sugimura, Email: ksugimura@cardio.med.tohoku.ac.jp.
Hideyuki Miyauchi, Email: hmiyauchi_circ@yahoo.co.jp.
Akira Suzuki, Email: suzukia@cnt-osaka.com.
Yasuhiro Hara, Email: y-hara@cnt-osaka.com.
Atsuko Takagi, Email: atakagi@cnt-osaka.com.
Yasuyuki Ikeda, Email: yasuikeda@cnt-osaka.com.
Kazuhiro Kobayashi, Email: kazukob@med.kobe-u.ac.jp.
Yoshiaki Futsukaichi, Email: hommefrancophile1950@yahoo.co.jp.
Nobuhiro Zaima, Email: zaimanobuhiro@gmail.com.
Satoshi Yamaguchi, Email: y-satoshi@cnt-osaka.com.
Rojeet Shrestha, Email: cl.biochem@gmail.com.
Hiroshi Nakamura, Email: nakamurah@kure-nh.go.jp.
Katsuhiro Kawaguchi, Email: kawa@komakihp.gr.jp.
Eiryu Sai, Email: eiryu@juntendo.ac.jp.
Shu-Ping Hui, Email: keino@hs.hokudai.ac.jp.
Yusuke Nakano, Email: ysk3ta55@yahoo.co.jp.
Akinori Sawamura, Email: a-sawamu@med.nagoya-u.ac.jp.
Tohru Inaba, Email: inaba178@koto.kpu-m.ac.jp.
Yasuhiko Sakata, Email: sakatayk@cardio.med.tohoku.ac.jp.
Yoko Yasui, Email: yasui@life.osaka-cu.ac.jp.
Yasuyuki Nagasawa, Email: nagasawa@hyo-med.ac.jp.
Shintaro Kinugawa, Email: tuckahoe@med.hokudai.ac.jp.
Kazunori Shimada, Email: shimakaz@juntendo.ac.jp.
Sohsuke Yamada, Email: sohsuke@kanazawa-med.ac.jp.
Hiroyuki Hao, Email: hao.hiroyuki@nihon-u.ac.jp.
Daisaku Nakatani, Email: nakatani@cardiology.med.osaka-u.ac.jp.
Tomomi Ide, Email: tomomi_i@cardiol.med.kyushu-u.ac.jp.
Tetsuya Amano, Email: amanot@aichi-med-u.ac.jp.
Hiroaki Naito, Email: naito.hiroaki@k.nissay-hp.or.jp.
Hironori Nagasaka, Email: nagasaka@cnt-osaka.com.
Kunihisa Kobayashi, Email: nihisak@fukuoka-u.ac.jp.
References
- 1.Jordans GH. The familial occurrence of fat containing vacuoles in the leukocytes diagnosed in two brothers suffering from dystrophia musculorum progressiva (ERB.) Acta Med Scand. 1953;145(6):419–423. doi: 10.1111/j.0954-6820.1953.tb07038.x. [DOI] [PubMed] [Google Scholar]
- 2.Fischer J, Lefevre C, Morava E, Mussini JM, Laforet P, Negre-Salvayre A, Lathrop M, Salvayre R. The gene encoding adipose triglyceride lipase (PNPLA2) is mutated in neutral lipid storage disease with myopathy. Nat Genet. 2007;39(1):28–30. doi: 10.1038/ng1951. [DOI] [PubMed] [Google Scholar]
- 3.Zimmermann R, Strauss JG, Haemmerle G, Schoiswhol G, Birner-Gruenberger R, Riedere M, Lass A, Neuberger G, Eisenhaber F, Hermetter A, Zechner R. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science. 2004;306(5700):1383–1386. doi: 10.1126/science.1100747. [DOI] [PubMed] [Google Scholar]
- 4.Haemmerle G, Lass A, Zimmermann G, Gorkiewicz G, Meyer C, Rozman J, Heldmaier G, Maier R, Theussl C, Eder S, Kratky D, Wagner EF, Klingenspor M, Hoefler G, Zechner R. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science. 2006;312(5774):734–737. doi: 10.1126/science.1123965. [DOI] [PubMed] [Google Scholar]
- 5.Schweiger M, Schoiswohl G, Lass G, Radner FP, Haemmerle G, Malli R, Graier W, Cornaciu I, Oberer M, Salvayre R, Fischer J, Zechner R, Zimmerman G. The C-terminal region of human adipose triglyceride lipase affects enzyme activity and lipid droplet binding. J Biol Chem. 2008;283(25):17211–17220. doi: 10.1074/jbc.M710566200. [DOI] [PubMed] [Google Scholar]
- 6.Koyabashi K, Inoguchi T, Maeda Y, Nakashima N, Kuwano A, Eto E, Ueno N, Sasaki S, Sawada F, Fujii M, Matoba Y, Sumiyoshi S, Kawate H, Takayanagi R. The lack of the C-terminal domain of adipose triglyceride lipase causes neutral lipid storage disease through impaired interactions with lipid droplets. J Clin Endocrinol Metab. 2008;93(7):2877–2884. doi: 10.1210/jc.2007-2247. [DOI] [PubMed] [Google Scholar]
- 7.Reilich P, Horvath R, Krause S, Schramm N, Turnbull DM, Trenell M, Hollingsworth KG, Gorman GS, Hans VH, Reimann J, MacMillan A, Turner L, Schollen A, Witte G, Czermin B, Holinski-Feder E, Walter MC, Schoser B, Lochmüller H. The phenotypic spectrum of neutral lipid storage myopathy due to mutations in the PNPLA2 gene. J Neurol. 2011;258(11):1987–1997. doi: 10.1007/s00415-011-6055-4. [DOI] [PubMed] [Google Scholar]
- 8.Missaglia S, Tasca E, Angelini C, Moro L, Tavian D. Novel missense mutations in PNPLA2 causing late onset and clinical heterogeneity of neutral lipid storage disease with myopathy in three siblings. Mol Genet Metab. 2015;115(2–3):110–117. doi: 10.1016/j.ymgme.2015.05.001. [DOI] [PubMed] [Google Scholar]
- 9.Pennisi EM, Missaglia S, Dimauro S, Bernardi C, Akman HO, Tavian D. A myopathy with unusual features caused by PNPLA2 gene mutations. Muscle Nerve. 2015;51(4):609–613. doi: 10.1002/mus.24477. [DOI] [PubMed] [Google Scholar]
- 10.Pasanisi MB, Missaglia S, Cassandrini D, Salerno F, Farina S, Andreini D, Agostoni P, Morandi L, Mora M, Tavian D. Severe cardiomyopathy in a young patient with complete deficiency of adipose triglyceride lipase due to a novel mutation in PNPLA2 gene. Int J Cardiol. 2016;207:165–167. doi: 10.1016/j.ijcard.2016.01.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Missaglia S, Maggi L, Mora M, Gibertini S, Blasevich F, Agostoni P, Moro L, Cassandrini D, Santorelli FM, Gerevini S, Tavian D. Late onset of neutral lipid storage disease due to novel PNPLA2 mutations causing total loss of lipase activity in a patient with myopathy and slight cardiac involvement. Neuromuscul Disord. 2017;27(5):481–486. doi: 10.1016/j.nmd.2017.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laforêt P, Stojkovic T, Bassez G, Carlier PG, Clément K, Wahbi K, Petit FM, Eymard B, Carlier RY. Neutral lipid storage disease with myopathy: a whole-body nuclear MRI and metabolic study. Mol Genet Metab. 2013;108(2):125–131. doi: 10.1016/j.ymgme.2012.12.004. [DOI] [PubMed] [Google Scholar]
- 13.Pennisi EM, Arca M, Bertini E, Bruno C, Cassandrini D, D'amico A, Garibaldi M, Gragnani F, Maggi L, Massa R, Missaglia S, Morandi L, Musumeci O, Pegoraro E, Rastelli E, Santorelli FM, Tasca E, Tavian D, Toscano A, Angelini C, Italian NLSD Group Neutral Lipid Storage Diseases: clinical/genetic features and natural history in a large cohort of Italian patients. Orphanet J Rare Dis. 2017;12(1):90. doi: 10.1186/s13023-017-0646-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tavian D, Missaglia S, Redaelli C, Pennisi EM, Invernici G, Wessalowski R, Maiwald R, Arca M, Coleman RA. Contribution of novel ATGL missense mutations to the clinical phenotype of NLSD-M: a strikingly low amount of lipase activity may preserve cardiac function. Hum Mol Genet. 2012;21(24):5318–5328. doi: 10.1093/hmg/dds388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dorfman ML, Hershko C, Eisenberg S, Sagher F. Ichthyosiform dermatosis with systemic lipidosis. Arch Dermatol. 1974;110(2):261–266. doi: 10.1001/archderm.1974.01630080059017. [DOI] [PubMed] [Google Scholar]
- 16.Chanarin I, Patel A, Slavin G, Wills EJ, Andrews TM, Stewart G. Neutral-lipid storage disease: a new disorder of lipid metabolism. Br Med J. 1975;1(5957):553–555. doi: 10.1136/bmj.1.5957.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Igal RA, Rhoads JM, Coleman RA. Neutral lipid storage disease with fatty liver and cholestasis. J Pediatr Gasteroenterol Nutr. 1997;25(5):541–547. doi: 10.1097/00005176-199711000-00011. [DOI] [PubMed] [Google Scholar]
- 18.Lefevre C, Jobard F, Caux F, Bouadjar B, Karaduman A, Heilig R, Lakhdar H, Wollenberg A, Verret JL, Weissenbach J, Ozguc M, Lathrop M, Prud'homme JF, Fischer J. Mutations in CGI-58, the gene encoding a new protein of the esterase/lipase/thioesterase subfamily, in Chanarin-Dorfman syndrome. Am J Hum Genet. 2001;69(5):1002–1012. doi: 10.1086/324121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salvayre R, Negre A, Radom J, Douste-Blazy L. Independence of triacylglycerol-containing compatments in cultured firbroblasts from Wolman disease and multisystemic lipid storage myopathy. FEBS Lett. 1989;250(1):35–39. doi: 10.1016/0014-5793(89)80679-9. [DOI] [PubMed] [Google Scholar]
- 20.Hilaire N, Salvayre R, Their JC, Bonnafe MJ, Negre-Salvayre A. The turnover of cytoplasmic triacylglycerols in human fibroblasts involves two separate acyl chain length-dependent degradation pathway. J Biol Chem. 1995;270(10):27027–27034. doi: 10.1074/jbc.270.45.27027. [DOI] [PubMed] [Google Scholar]
- 21.Nakamura K, Hirano K, Wu SM. iPS cell modeling of cardiometabolic diseases. J Cardiovasc Transl Res. 2013;6(1):46–53. doi: 10.1007/s12265-012-9413-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tavian D, Missaglia S, Castagnetta M, Degiorgio D, Pennisi M, Coleman RA, Dell’Era P, Mora C, Angelini C, Coviello DA. Generation of induced pluripotent stem cells as disease modelling of NLSDM. Mol Genet Metab. 2017;121(1):28–34. doi: 10.1016/j.ymgme.2017.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Missaglia Sara, Coleman Rosalind, Mordente Alvaro, Tavian Daniela. Neutral Lipid Storage Diseases as Cellular Model to Study Lipid Droplet Function. Cells. 2019;8(2):187. doi: 10.3390/cells8020187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hasebe M. A case of Jordans' anomaly. Rinsho Ketsueki. 1983;24(5):553–558. [PubMed] [Google Scholar]
- 25.Oshima Y, Hirota H, Nagai H, Izumi M, Nakaoka Y, Osugi T, Fujio Y, Tateyama H, Kikui M, Yamauchi-Takihara K, Kawase I. Specific cardiomyopathy caused by multisystemic lipid storage in Jordans' anomaly. Circulation. 2002;106(2):280–281. doi: 10.1161/01.CIR.0000018080.37316.D6. [DOI] [PubMed] [Google Scholar]
- 26.Hirano K, Ikeda Y, Zaima N, Sakata Y, Matsumiya G. Triglyceride deposit cardiomyovasculopathy. N Engl J Med. 2008;359(22):2396–2398. doi: 10.1056/NEJMc0805305. [DOI] [PubMed] [Google Scholar]
- 27.Hirano K. A novel clinical entity: triglyceride deposit cardiomyovasculopathy. J Atheroscler Thromb. 2009;16(5):702–705. doi: 10.5551/jat.1669. [DOI] [PubMed] [Google Scholar]
- 28.Hirano K, Tanaka T, Ikeda Y, Yamaguchi S, Zaima N, Kobayashi K, Suzuki A, Sakata Y, Sakata Y, Kobayashi K, Toda T, Fukushima N, Ishibashi-Ueda H, Tavian D, Nagasaka H, Hui SP, Chiba H, Sawa Y, Hori M. Genetic mutations in adipose triglyceride lipase and myocardial up-regulation of peroxisome proliferated activated receptor-gamma in patients with triglyceride deposit cardiomyovasculopathy. Biochem Biophys Res Commun. 2014;443:574–579. doi: 10.1016/j.bbrc.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 29.Ikeda Y, Zaima N, Hirano K, Mano M, Kobayashi K, Yamada S, Yamaguchi S, Suzuki A, Kanzaki H, Hamasaki T, Kotani J, Kato S, Nagasaka H, Setou M, Ishibashi-Ueda H. Coronary triglyceride deposition in contemporary advanced diabetics. Pathol Int. 2014;64(7):325–335. doi: 10.1111/pin.12177. [DOI] [PubMed] [Google Scholar]
- 30.Nakanishi T, Kato S. Impact of diabetes mellitus on myocardial lipid deposition: an autopsy study. Pathol Res Pract. 2014;210(12):1018–1025. doi: 10.1016/j.prp.2014.04.008. [DOI] [PubMed] [Google Scholar]
- 31.Goodman MM, Kirsch G, Knapp FF., Jr Synthesis and evaluation of radioiodinated terminal p-iodophenyl-substituted alpha- and beta-methyl-branched fatty acids. J Med Chem. 1984;27:390–397. doi: 10.1021/jm00369a027. [DOI] [PubMed] [Google Scholar]
- 32.Kurata C, Tawarahara K, Taguchi T, Aoshima S, Kobayashi A, Yamazaki N, Kawai H, Kaneko M. Myocardial emission computed tomography with iodine-123-labeled beta-methyl-branched fatty acid in patients with hypertrophic cardiomyopathy. J Nucl Med. 1992;33(1):6–13. [PubMed] [Google Scholar]
- 33.Hirano K, Ikeda Y, Sugimura K, Sakata Y. Cardiomyocyte steatosis and defective washout of iodine-123-beta-methyl iodophenyl-pentadecanoic acid in genetic deficiency of adipose triglyceride lipase. Eur Heart J. 2015;36(9):580. doi: 10.1093/eurheartj/ehu417. [DOI] [PubMed] [Google Scholar]
- 34.Higashi M, Ikeda Y, Miyauchi H, Zaima N, Suzuki A, Li M, Kobayashi K, Naito H, Hirano K. Imaging modalities for triglyceride deposit Cardiomyovasculopathy. Ann Nucl Cardiol. 2017;3(1):94–102. doi: 10.17996/anc.17-00028. [DOI] [Google Scholar]
- 35.Suzuki A, Nagasaka H, Ochi Y, Kobayashi K, Nakamura H, Nakatani D, Yamaguchi S, Yamaki S, Wada A, Shirata Y, Hui SP, Toda T, Kuroda H, Chiba H, Hirano K. Peripheral leucocyte anomaly detected with routine automated hematology analyzer sensitive to adipose triglyceride lipase deficiency manifesting neutral lipid storage disease with myopathy/triglyceride deposit cardiomyovasculopathy. Mol Genet Metab Rep. 2014;1:249–253. doi: 10.1016/j.ymgmr.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Inaba T, Ishizuka K, Suzuki A, Yuasa S, Saito K, Kodama M, Hamada M, Hongo F, Fujita N, Hirano K. Basic utility of Pentra series automated hematology analyzer for screening Jordans' anomaly. Int J Lab Hematol. 2017;39:e1–e3. doi: 10.1111/ijlh.12570. [DOI] [PubMed] [Google Scholar]
- 37.Inaba T, Ishizuka K, Suzuki A, Yuasa S, Saito K, Kodama M, Hongo F, Fujita N, Hirano K. Comparison fo neutrophil distribution patterns in Jordans' anomaly among major automated hematology analyzers. Int J Lab Hematol. 2018;40:e78–e81. doi: 10.1111/ijlh.12842. [DOI] [PubMed] [Google Scholar]
- 38.Kozawa J, Higashi M, Shimomura I, Hirano KI. Intractable Coronary Artery Disease in a Patient with Type 2 Diabetes Presenting With Triglyceride Deposit Cardiomyovasculopathy. Diabetes Care. 2019;42(5):983–986. doi: 10.2337/dc18-2365. [DOI] [PubMed] [Google Scholar]
- 39.Takagi A, Ikeda Y, Kobayashi K, Kobayashi K, Ikeda Y, Kozawa J, Miyauchi H, Li M, Hashimoto C, Hara Y, Yamaguchi S, Suzuki A, Toda T, Nagasaka H, Hirano KI. Newly developed selective immunoinactivation assay revealed reduction in adipose triglyceride lipase activity in peripheral leucocytes from patients with idiopathic triglyceride deposit cardiomyovasculopathy. Biochem Biophys Res Commun. 2018;495(1):646–651. doi: 10.1016/j.bbrc.2017.11.070. [DOI] [PubMed] [Google Scholar]
- 40.Hirano K. Triglyceride deposit cardiomyovasculopathy, TGCV-To overcome this intractable disease one day sooner. Nihon Naika Gakkai Zasshi. 2017;106:2385. doi: 10.2169/naika.106.2385. [DOI] [Google Scholar]
- 41.Miyauchi H, Hashimoto C, Ikeda Y, Li M, Nakano Y, Kozawa J, Sai E, Nagasakwa Y, Sugimura K, Kinugawa S, Kawaguchi K, Shimada K, Ide T, Amano T, Higashi M, Inaba T, Nakamura H, Kobayashi K, Hirano K. Diagnostic criteria and severity score for triglyceride deposit cardiomyovasculopathy. Ann Nucl Cardiol. 2018;4(1):94–100. doi: 10.17996/anc.18-00081. [DOI] [Google Scholar]
- 42.Hirano K, Li M, Ikeda Y. Triglyceride deposit cardiomyovasculopathy. In: Oohashi T, Tsukahawa H, Ramirez F, Barber C, Otsuka F, editors. Human Pathobiochemistry. Luxembourg: Springer, Singapore; 2018. pp. 111–119. [Google Scholar]
- 43.Hara Y, Kawasaki N, Hirano K, Hashimoto Y, Adachi J, Watanabe S, Tomonaga T. Quantitative proteomic analysis of cultured skin fibroblast cells derived from patients with triglyceride deposit cardiomyovasculopathy. Orphanet J Rare Dis. 2013;8:197. doi: 10.1186/1750-1172-8-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ikeda Y, Hirano K, Fukushima N, Sawa Y. A novel type of human spontaneous coronary atherosclerosis with triglyceride deposition. Eur Heart J. 2014;35(13):875. doi: 10.1093/eurheartj/eht522. [DOI] [PubMed] [Google Scholar]
- 45.Ross R. Atherosclerosis-an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 46.Inoue T, Kobayshi K, Inoguchi T, Sonoda N, Fujii M, Maeda Y, Fujimura Y, Miura D, Hirano K, Takayanagi R. Reduce expression of adipose triglyceride lipase enhances tumor-necrosis factor alpha-induced intercellular adhesion molecule-1 expression in human aortic endothelial cells via protein kinase C-dependent activation of nuclear factor-kappa B. J Biol Chem. 2011;286:32045–32053. doi: 10.1074/jbc.M111.285650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin Y, Chiba S, Suzuki A, Yamaguchi S, Nakanishi T, Matsumoto H, Ikeda Y, Ishibashi-Ueda H, Hirano K, Kato S. Vascular smooth muscle cells isolated from adipose triglyceride lipase-deficient mice exhibit distinct phenotype and phenotypic plasticity. Biochem Biophys Res Commun. 2013;434(3):534–540. doi: 10.1016/j.bbrc.2013.03.109. [DOI] [PubMed] [Google Scholar]
- 48.Higashi M, Hirano K, Kobayashi K, Ikeda Y, Issiki A, Otsuka T, Suzuki A, Yamaguchi S, Zaima N, Hamada S, Hanada H, Suzuki C, Nakamura H, Nagasaka H, Miyata T, Miyamoto Y, Kobayashi K, Naito H, Toda T. Distinct cardiac phenotype between two homozygotes born in a village with accumulation of a genetic deficiency of adipose triglyceride lipase. Int J Cardiol. 2015;192:30–32. doi: 10.1016/j.ijcard.2015.05.004. [DOI] [PubMed] [Google Scholar]
- 49.Kaneko K, Kuroda H, Izumi R, Tateyama M, Kato M, Sugimura K, Sakata Y, Ikeda Y, Hirano K, Aoki M. A novel mutation in PNPLA2 causes neutral lipid storage disease with myopathy and triglyceride deposit cardiomyovasculopathy: a case report and literature review. Neuromuscul Disord. 2014;24(7):634–641. doi: 10.1016/j.nmd.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 50.Rubler S, Dlugash J, Yuceoglu YZ, Kumral T, Branwaood AW, Grishman A. A new type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol. 1972;30:595–602. doi: 10.1016/0002-9149(72)90595-4. [DOI] [PubMed] [Google Scholar]
- 51.Suzuki A, Yamaguchi S, Li M, Hara Y, Miyauchi H, Ikeda Y, Zhang B, Higashi M, Ikeda Y, Takagi A, Nagasaka H, Kobayashi K, Magata Y, Aoyama T, Hirano K. Tricaprin rescues myocardial abnormality in a mouse model of triglyceride deposit Cardiomyovasculopathy. J Oleo Sci. 2018;8:983–989. doi: 10.5650/jos.ess18037. [DOI] [PubMed] [Google Scholar]
- 52.Shrestha R, Hui SP, Imai H, Hashimoto S, Uemura N, Takeda S, Fuda H, Suzuki A, Yamaguchi S, Hirano K, Chiba H. Plasma capric acid concentrations in healthy subjects determined by high-performance liquid chromatography. Ann Clin Biochem. 2015;52:588–596. doi: 10.1177/0004563215569081. [DOI] [PubMed] [Google Scholar]
- 53.Shrestha R, Hirano K, Suzuki A, Yamaguchi S, Miura Y, Chen YF, Mizuta M, Chiba H, Hui SP. Change in plasma total, esterified and non-esterified capric acid concentrations during a short-term oral administration of synthetic tricaprin in dogs. Anal Sci. 2017;33:1297–1303. doi: 10.2116/analsci.33.1297. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated and/or analyzed in the current study are available from the corresponding author on reasonable request.