

A likely contributor to the increased incidence of non-albicans candidemias involving Candida glabrata is the ease with which this yeast acquires azole resistance, in large part due to induction of the ATP-binding cassette transporter-encoding gene CDR1. Azole drugs lead to induction of Pdr1 transactivation, with a central model being that this factor binds these drugs directly. Here we provide evidence that Pdr1 is activated without azole drugs by the use of genetic means to inhibit expression of azole drug target-encoding gene ERG11. These acute reductions in Erg11 levels lead to elevated Pdr1 activity even though no drug is present. A key transcriptional regulator of the ERG pathway, Upc2A, is shown to directly bind to the PDR1 and CDR1 promoters. We interpret these data as support for the view that Pdr1 function is responsive to ergosterol biosynthesis and suggest that this connection reveals the normal physiological circuitry in which Pdr1 participates.
KEYWORDS: Candida glabrata, Pdr1, Upc2A, azole resistance, ergosterol, gene regulation, transcription factors
ABSTRACT
A crucial limitation in antifungal chemotherapy is the limited number of antifungal drugs currently available. Azole drugs represent the most commonly used chemotherapeutic, and loss of efficacy of these drugs is a major risk factor in successful treatment of a variety of fungal diseases. Candida glabrata is a pathogenic yeast that is increasingly found associated with bloodstream infections, a finding likely contributed to by its proclivity to develop azole drug resistance. C. glabrata often acquires azole resistance via gain-of-function (GOF) mutations in the transcription factor Pdr1. These GOF forms of Pdr1 drive elevated expression of target genes, including the ATP-binding cassette transporter-encoding CDR1 locus. GOF alleles of PDR1 have been extensively studied, but little is known of how Pdr1 is normally regulated. Here we test the idea that reduction of ergosterol biosynthesis (as occurs in the presence of azole drugs) might trigger activation of Pdr1 function. Using two different means of genetically inhibiting ergosterol biosynthesis, we demonstrated that Pdr1 activity and target gene expression are elevated in the absence of azole drug. Blocks at different points in the ergosterol pathway lead to Pdr1 activation as well as to induction of other genes in this pathway. Delivery of the signal from the ergosterol pathway to Pdr1 involves the transcription factor Upc2A, an ERG gene regulator. We show that Upc2A binds directly to the PDR1 and CDR1 promoters. Our studies argue for a physiological link between ergosterol biosynthesis and Pdr1-dependent gene regulation that is not restricted to efflux of azole drugs.
INTRODUCTION
Invasive candidiasis is caused primarily by Candida albicans, but a recent trend is the disturbing increase in infections caused by non-albicans species (1–3). Candida glabrata is the second most common species associated with candidiasis, and infections by these species are associated with increasingly common reduced antifungal susceptibility. The limited number of distinct antifungal drug classes makes resistance a serious threat to continued effective chemotherapy (reviewed in reference 4).
The most commonly used antifungal drug class is represented by azole compounds. Anti-Candida chemotherapy routinely utilizes fluconazole, a drug that can be administered orally and that has good selectivity for the target enzyme of the pathogen, lanosterol α-14 demethylase (recently discussed in reference 5). This enzyme is encoded by the ERG11 gene in the Candida genera. The ERG11 gene is essential for production of the fungal sterol ergosterol, a critical component of the fungal plasma membrane. Loss of ERG11 is a lethal event or causes a profound growth defect in most Candida species (6–8).
Resistance to fluconazole is most commonly associated with single amino acid substitution mutations in the gene encoding a Zn2Cys6 zinc cluster-containing transcription factor called Pdr1 (recently reviewed in reference 9). These mutations yield a gain-of-function (GOF) phenotype and lead to the elevated transcription of downstream target genes. The ATP-binding cassette (ABC) transporter-encoding CDR1 gene is one of the principal targets of Pdr1 and is required for the elevated fluconazole resistance seen in such PDR1 GOF mutant strains (10, 11). The GOF alleles of PDR1 cause chronically increased transcription of downstream target genes through the enhanced ability to activate gene expression (12).
Experiments reported from several groups demonstrated that wild-type Pdr1 activity is responsive to challenge with fluconazole, leading to strong autoregulatory induction of PDR1 itself as well as to activation of CDR1 gene transcription (10–12). Both biochemical and genetic approaches were used to argue that azole drugs bind directly to Pdr1 and that this binding leads to activation of Pdr1 function (13).
An intrinsic complication of the use of fluconazole to induce Pdr1 function is its concomitant inhibition of ergosterol biosynthesis. We wanted to test if it were possible to separate the presence of fluconazole from a block in ergosterol production at the level of ERG11. To do this, we utilized two different repressible promoter systems that could be transcriptionally repressed via completely different means. Irrespective of how ERG11 transcription was halted, this was followed by activation of Pdr1 and increased transcription of its target genes. We found that induction of Pdr1 target genes also required the ergosterol-regulated Upc2A transcription. Upc2A is required for normal expression of ergosterol biosynthetic genes. Strikingly, chromatin immunoprecipitation (ChIP) indicated that Upc2A was able to bind to a site in the PDR1 and CDR1 promoters, providing a direct link between ergosterol biosynthesis and a key determinant of azole resistance. Our data provide the first evidence tying control of Pdr1-dependent gene expression that impacts azole resistance to the activity of the biosynthetic pathway inhibited by azole drugs. We propose that coordinated control of ergosterol biosynthesis and of integral membrane proteins such as Cdr1 is required for regulation of permeability through the plasma membrane.
RESULTS
Fluconazole activates expression of Erg11, Cdr1, and Pdr1 at both the protein and mRNA levels.
Previous work has established that fluconazole challenge leads to a rapid and robust increase in transcription of a range of different genes involved in ergosterol biosynthesis, including ERG11 genes as well as PDR1 and its target genes such as CDR1 (14, 15). To directly examine the link between mRNA levels and steady-state protein levels, we generated immunological probes against Cdr1 and Erg11 by raising a polyclonal antiserum or attaching a 3× hemagglutinin (HA) epitope, respectively, to these proteins. We have already described a rabbit antiserum that can detect Pdr1 (16). We inserted a 3× HA tag at the C-terminus of the ERG11 gene and ensured that this epitope-tagged allele supported normal growth and azole susceptibility (data not shown). This strain was designated BVGC3. We grew BVGC3 to mid-log phase and then challenged this strain with fluconazole for various times. Whole-cell protein extracts were prepared and analyzed by Western blotting with the indicated antibodies.
Cdr1, Pdr1, and Erg11 were all induced by fluconazole at the level of each protein, with induction easily seen at 1 h after drug exposure (Fig. 1A). Quantitation of these blots demonstrated that the fold induction level was somewhat greater for Cdr1 and Pdr1 but was more than 2-fold above the control level for Erg11 (Fig. 1B). Blotting for tubulin confirmed that the levels of loading were equal for these samples. These data confirm that fluconazole treatment induces these genes at both the transcriptional and translational levels.
FIG 1.
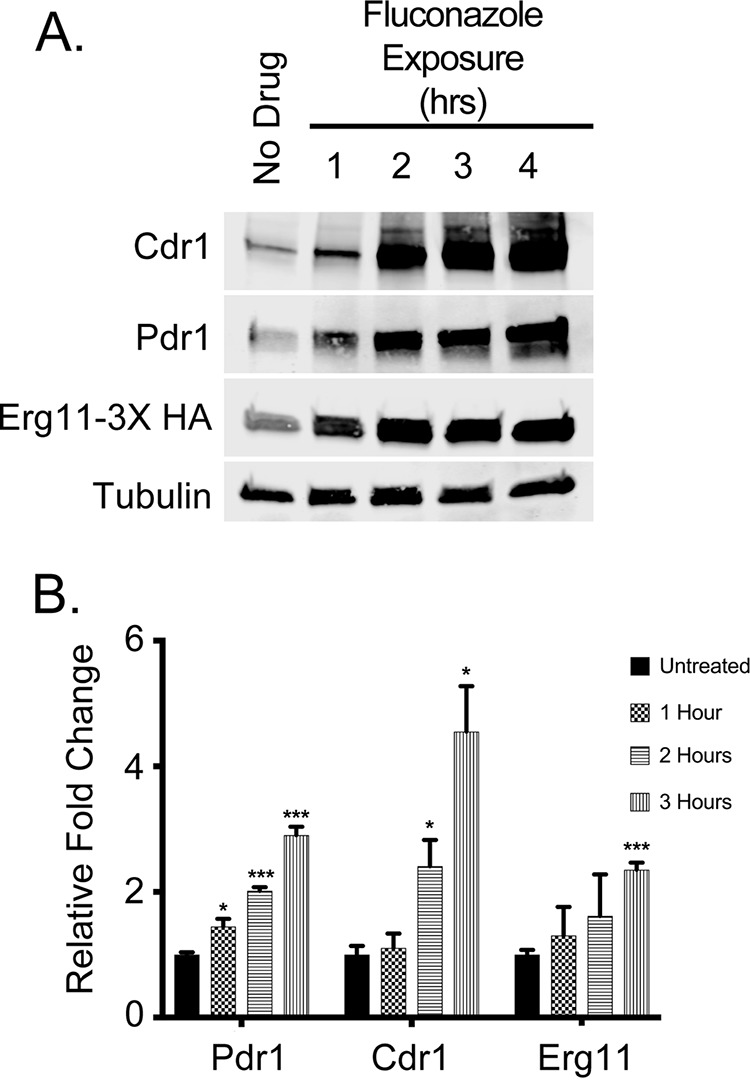
Western blot analysis of fluconazole induction of Cdr1, Pdr1, and Erg11. A C. glabrata strain containing an epitope-tagged version of ERG11 (ERG11-3× HA) was grown to early log phase and then treated with 20 μg/ml fluconazole for the times indicated. An aliquot was withdrawn prior to drug challenge (No Drug) to provide a control for non-drug-stimulated levels. Whole-cell protein extracts were prepared and analyzed by Western blotting using rabbit antibodies against Cdr1 or Pdr1 and mouse monoclonal antibodies against the HA epitope (Erg11-3× HA) or β-tubulin. (B) The fold changes of Cdr1, Pdr1, and Erg11 levels were calculated by normalizing later time points to the initial protein level determined as described for panel A.
Development of an acutely repressible form of ERG11.
In order to quickly block ERG11 expression, we adapted a camphor-regulated expression system developed in Saccharomyces cerevisiae for use in C. glabrata. This system is based on a fusion protein between the bacterial camphor repressor protein (CamR) and the strong viral transactivator protein VP16. CamR binds strongly to DNA until exposed to its substrate, camphor, a natural product (17). Thus, the CamR-VP16 activates expression of genes with the CamR binding site (camO) placed as an upstream activation sequence. Our goal was to develop a genetic means of inhibiting ergosterol biosynthesis that would allow us to evaluate if direct binding of fluconazole to Pdr1 was required for induction or if a reduction in Erg11 activity was sufficient.
We generated a C. glabrata strain that produced CamR-VP16 from the S. cerevisiae TDH1 (ScTDH1) promoter. This strain was designated BVGC7. To produce the camphor-repressible allele of ERG11, we replaced the ERG11 promoter in BVGC7 with a multimerized camO element upstream of the ScCYC1 TATA region (camPr) to obtain BVGC9. This strain produced Erg11 in a camphor-sensitive manner (Fig. 2A).
FIG 2.

Characterization of camphor-regulated ERG11 allele. (A) A diagram showing the control of the camR-regulated camPr-ERG11 fusion gene. In the absence of camphor, camR-VP16 can bind to and induce camPr-ERG11 expression. The presence of camphor causes camR-VP16 to be released from this promoter, and ERG11 expression is strongly inhibited. (B) The presence of the camPr-ERG11 allele confers a camphor-sensitive growth phenotype. Strains containing both ScTDH1-driven camR-VP16 and camPr-ERG11 (BVGC9) or only ScTDH1-driven camR-VP16 (BVGC7) were grown overnight and then diluted into media containing or lacking camphor as indicated. Cultures were allowed to grow at 30μC, and optical density was measured using a spectrophotometer. (C) Mid-log phase cultures of BVGC9 grown in the absence (Untreated) or the presence (Treated) of 50 μM camphor for 2 h were harvested and ergosterol levels measured as described previously (41). (D) Cultures of the indicated strains were grown to the mid-log phase and then diluted for plating on rich media containing the listed additions. “Vehicle” represents the solution used to solubilize ergosterol.
We compared the levels of sensitivity of the isogenic BVGC7 and BVGC9 strains with respect to growth in the presence of camphor. Since ERG11 is essential for normal growth in C. glabrata, we anticipated that inappropriate transcriptional repression would cause a growth defect when the only source of ERG11 was the camphor-repressible promoter. This was found to be the case, as BVGC9 growth was quite sensitive to the presence of camphor (Fig. 2B). We also assayed ergosterol levels in BVGC9 cells grown for 2 h in the presence or absence of camphor. This assay showed that ergosterol levels were strongly reduced when cells were grown under camphor-repressing conditions (Fig. 2C).
Finally, we tested the camphor sensitivity of BVGC9 growth on solid yeast extract-peptone-dextrose (YPD) media. Serial dilutions of BVGC7 (transactivator only) and BVGC9 (both transactivator-driven and camO-driven ERG11) were plated on media with the vehicle used to solubilize ergosterol, with ergosterol, with camphor plus vehicle, or with camphor plus ergosterol (Fig. 2D). Growth of BVGC9 was sensitive to camphor, while BVGC7 grew normally. Addition of ergosterol along with camphor rescued growth of BVGC9 in the presence of camphor. These data are consistent with the view that the camO-ScCYC1-ERG11 fusion gene makes sufficient Erg11 enzyme to support growth of cells but does so only in the absence of camphor. To determine if expression changes of PDR1 and CDR1 would respond to diminished levels of Erg11 and to confirm that regulation of ERG11 was consistent with the growth data, we measured both the mRNA and protein levels of these loci.
BVGC9 was grown to the mid-log phase and then treated with 50 μM camphor for 1 to 4 h. Total RNA and whole-cell protein extracts were prepared at each time point for analysis by reverse transcription-quantitative PCR (qRT-PCR) and Western blotting, respectively. The presence of camphor led to a prompt and sustained suppression of ERG11 mRNA in BVGC9 (Fig. 3D) but not in the strain containing only the camphor-dependent transactivator (BVGC7) (Fig. 3A). Transcription of both PDR1 and CDR1 was elevated similar time courses during camphor-mediated ERG11 repression in BVGC9. These changes in mRNA levels were reflected in protein accumulation (Fig. 3E), as anticipated. Importantly, Erg11-3× HA levels were reduced by approximately 50% after the addition of camphor and continued to decline over the course of this treatment. Abundances of both Pdr1 and Cdr1 were elevated within an hour and remained at that level during the camphor treatment. Control experiments (Fig. 3A to C) performed using the transactivator-only strain (BVGC7) showed no significant changes in expression of any of these genes or their corresponding proteins in response to camphor. Minor changes in CDR1 transcription were seen at later time points with camphor treatment but were not seen via Western blotting.
FIG 3.

Both PDR1 and CDR1 are induced upon transcriptional inactivation of the camPr-ERG11 fusion. (A) The strain containing only the camR-VP16 transactivator (BVGC7) was grown to the mid-log phase and then treated with 50 μM camphor for the indicated lengths of time. Total RNA was prepared from each culture at each time point. Levels of the indicated mRNAs were determined by qRT-PCR. (B) Whole-cell protein extracts were prepared from the indicated BVGC7 cultures, and then the levels of Pdr1, Cdr1, Erg11-3× HA, and tubulin were determined by Western blotting with appropriate antisera. (C) Quantitation of Western blots prepared as described in the panel B legend. (D) BVGC9 (which contains both transactivator-responsive and camphor-responsive ERG11 alleles) was grown to the mid-log phase and treated with camphor to inactivate ERG11 expression for various times. RNA levels of ERG11, PDR1, and CDR1 were measured using qRT-PCR. (E) Whole-cell protein extracts were prepared from BVGC9 cultures treated with camphor for various times or left untreated. Western blot analyses were used to measure expression of the indicated proteins. (F) Quantitation of Western blots prepared as described for panel E.
Response of the ERG pathway to defects in ERG11 transcription.
While the data presented above demonstrate that loss of normal ERG11 mRNA levels triggered PDR1 and CDR1 transcriptional increases, we wanted to determine if other genes in the ERG pathway were sensitive to ERG11 mRNA levels. We selected 9 ERG genes (ERG1, ERG2, ERG3, ERG4, ERG5, ERG6, ERG7, ERG9, and ERG10) to represent the behavior of the rest of the pathway (see Fig. 4A). BVGC9 was grown to the mid-log phase and then treated with camphor for 2 h or left untreated. We isolated mRNA from these cultures and analyzed levels of the pathway genes by qRT-PCR. The differences in the gene expression levels seen under camphor-treated versus untreated conditions are presented as ratios (Fig. 4B).
FIG 4.

Effects of camphor-mediated transcriptional repression on the ergosterol pathway. (A) Relevant steps in the ergosterol biosynthetic pathway are indicated. The addition of azole drugs blocks Erg11 function and causes production of a toxic sterol. Ac-CoA, acetate coenzyme A. (B) BVGC9 cells were left untreated or grown in the presence of camphor for 3 h. Total RNA was prepared and levels of the indicated ERG pathway genes determined by qRT-PCR. Relative expression values were determined by normalizing the expression levels seen after camphor treatment to the level of each transcript produced in untreated cells (Baseline). (C) Caspofungin treatment at a subinhibitory level caused a growth defect similar to that seen after addition of camphor to BVGC9. (D) Caspofungin challenge induces FKS2 transcription but not PDR1 or CDR1 transcription. Cells were grown with the echinocandin drug or left untreated for 2 or 4 h. Total RNA was prepared and levels of transcripts of interest assessed by qRT-PCR. (E) BVGC9 cells were grown as described for panel C and whole-cell protein extracts prepared at the indicated time points. Cdr1, Erg11-3× HA, and tubulin levels were determined by Western blot analysis. (F) Quantitation of the Western blot data in panel E.
Camphor inhibition of ERG11 transcription led to at least 2-fold induction of all tested genes, with ERG1 showing the greatest increase (up to 4-fold). The coregulation of these pathway genes would be expected from their necessary participation in the multistep biosynthesis of ergosterol, an essential metabolite for the cell.
Blocking ergosterol biosynthesis caused a clear growth inhibition such as has been demonstrated using a doxycycline-repressible allele of ERG11 (7). To ensure that the transcriptional induction of PDR1 and CDR1 represents a response to loss of ergosterol biosynthesis and not to this particular form of growth inhibition, we used the β-glucan synthase inhibitor caspofungin as a different means of preventing normal growth. A sublethal dose (5 pg/ml) of caspofungin was used that led to a level of acute inhibition of BVGC9 similar to that elicited by camphor (Fig. 4C). To confirm that caspofungin caused the expected stress in β-glucan synthesis, mRNA levels for FKS1 and FKS2 were assayed. These genes encode the β-glucan synthase enzyme and represent the direct targets of caspofungin. PDR1 and CDR1 mRNA levels were also measured to compare their responses to this echinocandin. Total RNA was prepared from caspofungin-treated (2 and 4 h) cells or from untreated cells, and mRNAs of interest were quantitated by qRT-PCR (Fig. 4D).
Among these 4 genes, only FKS2 was induced by treatment with caspofungin, as others have documented (18). This gene encodes the echinocandin-inducible form of β-glucan synthase. Notably, neither PDR1 transcription nor CDR1 transcription responded to this block in growth.
We also compared the response of Cdr1 and Erg11-3× HA to caspofungin-induced growth inhibition relative to that caused by camphor addition in BVGC9 (Fig. 4E and F). Caspofungin challenge that caused the observed induction of FKS2 mRNA had no significant effect on the steady-state expression levels of either Cdr1 or Erg11-3× HA. Camphor treatment had the expected effect, leading to induction of Cdr1 and repression of Erg11-3× HA. These data support the interpretation that lowered levels of ERG11 generate a signal leading to an induction of PDR1 and CDR1 transcription that extends beyond the inhibition of growth.
Multiple blocks in the ERG pathway induce PDR1 and CDR1.
Having provided the evidence detailed above showing that camphor-mediated repression of ERG11 transcription led to activation of PDR1 and CDR1 transcription, we constructed three other ERG gene promoter replacements. We selected ERG1, ERG2, and ERG3 for this analysis. In each case, the wild-type promoter was replaced with the camphor-regulated form discussed above in a manner analogous to that used with ERG11. These three new strains were grown to the mid-log phase and then placed on solid rich medium in the presence or absence of camphor (Fig. 5A).
FIG 5.

Camphor-off alleles of other ERG genes also lead to induction of PDR1 and CDR1. (A) Three additional strains were produced that contained both the camR-VP16 transactivator and the camphor-off alleles of ERG1, ERG2, or ERG3. These strains, along with a wild-type control, BVGC9 (camR-VP16, camPr-ERG11) were grown to the mid-log phase and serial dilutions plated on rich medium (YPD) or YPD containing 50 μM camphor. Plates were then incubated at 30μC and photographed. (B) The camphor-repressible ERG2 fusion gene-containing strain was grown and left untreated or challenged with camphor for the indicated times. Total RNA was prepared from each sample and analyzed for expression of ERG2, PDR1, and CDR1 mRNA by qRT-PCR. (C) The transcriptional response of the ERG pathway genes was determined by qRT-PCR from cells grown as described for panel B. (D) The strain with the camphor-repressible ERG3 gene was grown in the absence or presence of camphor as described above followed by the indicated qRT-PCR analyses. (E) The transcriptional responses of the ERG pathway to camphor-mediated ERG3 repression determined across the indicated ERG pathway genes.
The camPr-ERG1 gene was associated with strongly camphor-sensitive growth that was only slightly better than that seen with the BVGC9 strain. The camPr-ERG3 strain was found to grow less well than the camPr-ERG2 strain, which showed no growth reduction in the presence of camphor. Along with the growth phenotypes caused by camphor addition, we also compared the levels of expression of both the PDR1/CDR1 system and the ERG pathway by qRT-PCR in all these strains.
Replacement of either ERG2 (Fig. 5B) or ERG3 (Fig. 5D) with camPr caused expression of the resulting fusion genes to be strongly repressed upon the addition of camphor, as expected. The modest reduction in growth of the camphor-repressed camPr-ERG3 strain compared to the camPr-ERG2 strain was likely due to accumulation of different ergosterol precursors in these two strains. Similarly, the levels of expression of both PDR1 and CDR1 were elevated upon camphor treatment in both strains. The levels of ERG pathway induction seen in these two strains were also generally similar (compare Fig. 5C and E), although camphor repression of the camPr-ERG2 fusion strain triggered the largest induction of ERG3 mRNA whereas similar treatment of the camPr-ERG3 strain showed no effect on ERG2 mRNA. The levels of expression of the ERG4, ERG7, and ERG10 genes were not altered by repression of either ERG2 or ERG3, while the ERG1, ERG5, ERG6, ERG9, and ERG11 genes were all found to be induced. This suggests the presence of selective regulatory circuitry modulating the different ERG genes rather than a single common response.
Construction of a camPr-ERG1 fusion gene-containing strain (BVGC89) gave rise to a surprising behavior. Even though the addition of camphor to BVGC89 led to a growth defect, expression of the camPr-ERG1 gene was nearly 8-fold higher than that of wild-type ERG1 (Fig. 6A) prior to camphor addition. Similarly, both the PDR1 and CDR1 levels were also elevated prior to camphor addition. These data indicate that this high-level expression of ERG1 mRNA (and presumably of Erg1 protein) caused an issue with the ergosterol biosynthetic pathway that was sufficient to activate PDR1 and CDR1 expression.
FIG 6.
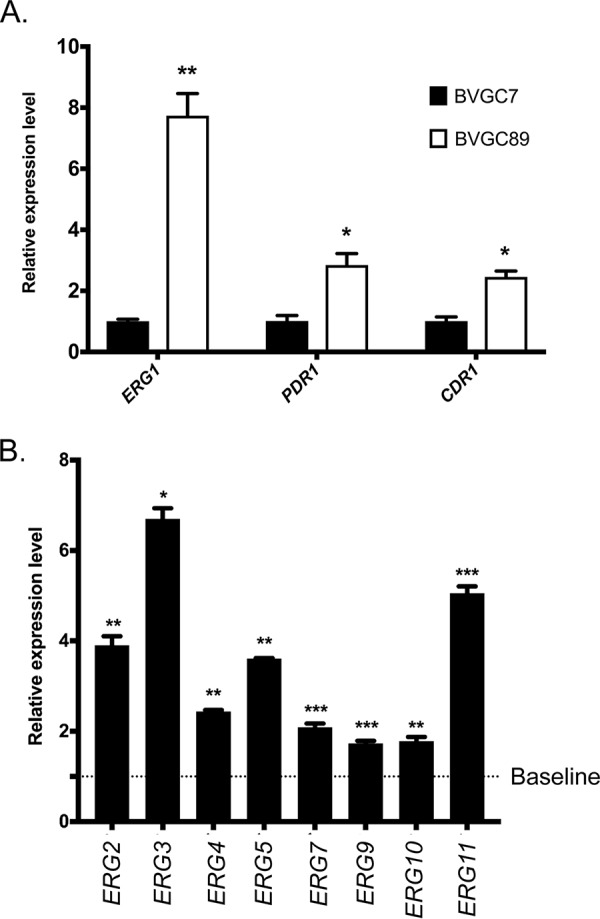
Elevated ERG1 mRNA levels trigger induction of PDR1, CDR1, and ERG pathway genes. Strains containing a camphor-off allele of ERG1 (BVGC89) or the camphor transactivator only (BVGC7) were grown to the mid-log phase, and cells were harvested and used to prepare total RNA. (A) Expression of ERG1, PDR1, and CDR1 was assayed using qRT-PCR. Relative expression level data represent the ratio of the mRNA level detected in BVGC89 to that detected in BVGC7. (B) Expression of ERG pathway genes was determined using qRT-PCR. The relative expression levels were calculated as described for panel A.
Assays of the mRNA levels of ERG pathway genes also demonstrated that these loci were activated by the presence of these high levels of ERG1 mRNA (and presumably of Erg1 protein) (Fig. 6B). In this case, every ERG gene assayed exhibited roughly 2-fold or greater induction.
An alternative signal repressing Erg11 production still led to Pdr1/CDR1 activation.
The camphor-repressible promoter experiments supported the view that a reduction in ergosterol biosynthesis led to the activation of PDR1 and CDR1 transcription. However, as has been demonstrated for doxycycline and fluconazole (19, 20), it is possible that the presence of camphor interacts with and influences azole resistance and possibly PDR1 and CDR1 gene expression.
To confirm that loss of ERG11 expression was directly responsible for induction of PDR1 and CDR1, we generated a second repressible system for control of ERG11 expression. We replaced the ERG11 promoter with that of the methionine-repressible MET3 gene. This MET3Pr-ERG11 fusion gene was expected to support growth in a methionine-sensitive manner, as addition of methionine would repress the MET3Pr and, subsequently, ERG11 transcription. The native ERG11 promoter present in the ERG11-3× HA allele in BVGC3 was replaced with MET3Pr to create BVGC127. Appropriate transformants were then grown to the mid-log phase, and the ability to support growth on synthetic media with or without methionine was tested (Fig. 7A).
FIG 7.

Methionine-dependent repression of ERG11 transcription induces PDR1 and CDR1. (A) Strains containing the ERG11-3× HA allele under the control of the ERG11 promoter (BVGC3) or regulated by the MET3 promoter (BVGC127) were grown to the mid-log phase and placed on media lacking methionine (CSM) or containing 1 mM methionine (CSM + Methionine). Plates were incubated at 30μC and then photographed. (B) Overnight cultures of the strains described in the panel A legend were diluted into fresh media containing or lacking methionine and grown for 6 h to regulate MET3-ERG11 transcription. Cultures were harvested and used for qRT-PCR analysis for the indicated genes. (C) BVGC3 and BVGC127 were grown as described for panel B, but whole-cell protein extracts were prepared and levels of the proteins of interest assessed by Western blot analysis. (D) Quantitation of Western blotting performed as described for panel C. met, methionine. (E) Results of analysis of ERG pathway genes by qRT-PCR. The dashed line indicates mRNA levels in BVGC127 cultures grown with no methionine, while the bars denote the mRNA levels in this strain grown for 6 h in the presence of methionine.
The presence of methionine prevented growth of the MET3Pr-ERG11-3× HA fusion gene but had no effect on the strain containing a wild-type ERG11Pr-ERG11-3× HA locus. As this result was consistent with a transcriptional limitation of ERG11 expression similar to that which we saw earlier with the camphor-off ERG11 gene, we directly tested whether the induction of ERG11 repression by the MET3Pr-ERG11 gene caused by methionine would trigger a response of the Pdr1-CDR1 circuit. Cells containing the MET3Pr-ERG11-3× HA gene were grown overnight and then freshly inoculated into new media with or without with the addition of methionine. After 6 h of growth, the cells were harvested. Both RNA and whole-cell protein extracts were prepared from these cultures.
Levels of mRNA were assayed for ERG11, PDR1, and CDR1 by qRT-PCR (Fig. 7B). ERG11 mRNA levels were strongly repressed during growth in the presence of methionine. In contrast, both PDR1 and CDR1 transcription levels rose when the fusion gene was repressed by methionine addition. Steady-state protein levels driven by these genes were also measured by Western blotting using appropriate antibodies (Fig. 7C). Both the Pdr1 and Cdr1 protein levels rose as Erg11-3× HA levels dropped, again consistent with the limitation of Erg11 levels and ergosterol biosynthesis leading to an induction of Pdr1 and CDR1. Quantitation of the changes in protein levels revealed that Cdr1 levels rose by approximately 5-fold while Pdr1 levels were elevated 3-fold during methionine repression (Fig. 7D). An identical treatment of an isogenic ERG11-3× HA strain under the control of wild-type ERG11pr showed no response to methionine addition.
The response of other Erg pathway genes was also examined upon methionine-induced repression of the MET3-ERG11 fusion gene (Fig. 7E). The ERG1-ERG7 genes were all found to be induced by 2-fold or greater when ERG11 synthesis was blocked upon methionine addition. These data confirm that, as seen earlier with the camphor-triggered repression of camP-ERG11, methionine addition led to ergosterol limitation and subsequent transcriptional induction of the Erg pathway.
Upc2A provides a transcriptional link between the ERG pathway and the Pdr1/CDR1 regulon.
The data presented above demonstrate an inverse relationship between the levels of transcription of the ERG11 gene and those of PDR1 and CDR1. The Upc2A transcription factor has been demonstrated to be required for normal regulation of expression of several genes in the Erg pathway, including ERG11 (23). In addition, isogenic deletion of Upc2A reduced the levels of PDR1 and CDR1 induced by azole (15). To confirm this, we constructed an isogenic upc2AΔ derivative of the ERG11-3× HA strain and subjected it to fluconazole treatment along with BVGC3. This pair of strains was grown to the mid-log phase. These cultures were then split into two aliquots and allowed to grow further in the presence or absence of fluconazole. Whole-cell protein extracts were prepared, and levels of several proteins of interest were analyzed by Western blotting (see Fig. S2 in the supplemental material).
The absence of Upc2A led to a decrease in the fluconazole induction of both Pdr1 and Cdr1 while eliminating any detectable increase in the levels of Erg11-3× HA (Fig. S2). This suggested that Upc2A might play a role in control of PDR1 and CDR1 transcription that is similar to the role that it plays for ERG11. To further explore the connections between Upc2A and the Pdr regulon, we constructed a upc2AΔ derivative of the strain containing the MET3-ERG11-3× HA fusion gene (BVGC127). As described above, we grew equal aliquots with or without methionine for 6 h. We then analyzed levels of ERG11, PDR1, CDR1, and UPC2A mRNA by qRT-PCR (Fig. 8A).
FIG 8.

UPC2A is required for induction of ERG11, PDR1, and CDR1 when MET3-ERG11 is repressed transcriptionally. (A) Isogenic MET3-ERG11 strains containing (BVGC127) or lacking (BVGC127 upc2AΔ) the UPC2A gene were grown in the absence or presence of 20 μg/ml methionine as described above. Total RNA was prepared and levels of the indicated RNAs determined by qRT-PCR. (B) Whole-cell protein extracts were prepared from the strains grown as described for panel A and levels of expression of proteins of interest assessed by Western blot analysis using antisera described previously and a rabbit anti-Upc2A antibody. (C) Quantitation of the Western blotting results presented in panel B. (D) Strain BVGC127 upc2AΔ was transformed with a low-copy-number plasmid (Vector) or the same plasmid containing a wild-type copy of the UPC2A gene (Vec-UPC2A). Transformants were grown as described for panel A and tested for mRNA levels of the indicated genes by qRT-PCR. (E) Whole-cell protein extracts prepared from the transformants prepared as described in the panel D legend were subjected to Western blot analysis using the listed antibodies. (F) Western blotting data determined as described above were quantitated.
Loss of the UPC2A gene decreased the ability of methionine repression of ERG11 to stimulate expression of both PDR1 and CDR1. We confirmed these data by Western blotting with antibodies against Cdr1, Pdr1, Erg11-3× HA, and Upc2A (Fig. 8B). Note that the addition of methionine lowered levels of Erg11-3× HA but led to elevations of the levels of other four proteins (see Fig. 8C for quantitation).
To confirm that that observed reduction in methionine-induced expression of the Pdr regulon was a consequence of the absence of UPC2A, we used a low-copy-number plasmid vector to complement the upc2AΔ allele with a wild-type version of this gene. We introduced a natMX6-marked vector plasmid containing or not containing a wild-type copy of UPC2A into the MET3-ERG11-3× HA upc2AΔ strain used as described above. Transformants were grown and assayed for methionine-induced expression changes as described above. These data are shown in Fig. 8D to F.
Reintroduction of the wild-type UPC2A gene restored normal induction of both PDR1 and CDR1 upon methionine-induced repression of ERG11 transcription. These data support the view that Upc2A must be present to trigger PDR1 and CDR1 transcriptional induction upon a reduction in ERG11 levels.
Upc2A directly binds to the PDR1 and CDR1 promoters.
Since Upc2A was required for normal transcriptional induction of PDR1 and CDR1 upon Erg11 depletion, we wanted to determine if this effect might be a direct one that is associated with Upc2A binding to these promoters in vivo. To test this idea, we used the rabbit polyclonal anti-Upc2A antibody that we had developed to perform chromatin immunoprecipitation (ChIP) experiments and probed associations of Upc2A with the ERG11, PDR1, and CDR1 promoters (Fig. 9A). ChIP experiments were done in the presence and absence of fluconazole as well as with a upc2AΔ strain as a negative control.
FIG 9.

Upc2A binds directly to ERG11, PDR1, and CDR1 promoter regions. (A) A wild-type (ATCC2001wt) strain was grown in the absence or presence of fluconazole (ATCC2001wt + Fluconazole), along with an isogenic upc2AΔ strain. Total sheared chromatin was prepared from all the strains and immunoprecipitated with anti-Upc2A polyclonal antibody. Immunoprecipitates were analyzed by qPCR using primers specific for the ERG11, PDR1, and CDR1 promoters as well as a region of the ERG11 coding sequence (CDS) as a negative control for enrichment. Percentages of enrichment were calculated by comparing the ratios of each PCR product produced using immunopurified chromatin to that produced in total chromatin. (B) A strain containing the camphor-off allele of ERG11 was grown in the presence or absence of camphor as indicated. Total chromatin was isolated, sheared, and then immunoprecipitated using the anti-Upc2A antibody. Percentages of enrichment of each PCR product were calculated as described for panel A. (C) Model for the coordinate control of ERG11, PDR1, and CDR1 transcription by Upc2A. Based on its striking sequence conservation with S. cerevisiae Upc2, we hypothesize that in the presence of ergosterol, the majority of Upc2A is excluded from the nucleus. Upon limitation of ergosterol, this inhibition is relieved, and Upc2A moves to the nucleus and activates target genes.
Strikingly, Upc2A was found on both the PDR1 and CDR1 promoters. A significant association of Upc2A with CDR1 was seen prior to fluconazole treatment, while Upc2A binding to the PDR1 promoter was detectable only after azole challenge. Fluconazole-inducible Upc2A binding was also detected on the ERG11 promoter, but Upc2A was found on ERG11 even with no azole drug present. These data indicate that Upc2A binds to all three of these promoters and is likely to act as a direct transcriptional regulator of these genes.
Having established the presence of inducible Upc2A binding to promoters of interest after azole challenge, we wanted to determine if binding was also seen when we limited production of ERG11 using our camphor-off ERG11 fusion gene described earlier. The strain containing the camP-ERG11 gene was grown in the presence and absence of camphor as described earlier. ChIP using the anti-Upc2A antibody was carried out on these samples, and the levels of PDR1 and CDR1 promoter DNA recovered in these reactions were quantitated (Fig. 9B).
Repression of the camphor-regulated ERG11 gene led to induction of Upc2A binding to both PDR1 and CDR1. Again, there was a significant Upc2A association with the CDR1 promoter, but not with the PDR1 promoter, in the absence of ERG11 repression. These data support the view that limitation of Erg11 activity, mediated either by the presence of azole drugs or by repression of the camphor-off ERG11 gene, triggers Upc2A binding to target genes (Fig. 9C; discussed below). Upc2A provides a direct connection between ergosterol biosynthesis and expression of genes involved in azole resistance.
DISCUSSION
C. glabrata is an emerging pathogen of increasing importance in infectious disease, certainly due in part to its proclivity to develop azole resistance. Gain-of-function (GOF) mutations in the PDR1 gene have been well established to provide the primary means of developing azole resistance through the overproduction of target genes like CDR1 (21, 22). In C. albicans, an important azole resistance mechanism is provided by substitution mutations with the Erg11 enzyme, the target of azole drugs (reviewed in reference 23), as well as GOF mutations in transcriptional regulators like Tac1 (24) and Mrr1 (25). In both of these Candida species, changes in expression of ABC transporter-encoding genes and ERG pathway genes have been considered to be due to two separate transcriptional circuits. Additionally, direct experiments in C. glabrata have provided evidence that azole drugs can directly bind to and stimulate activity of Pdr1 in this yeast (13).
The data reported here support two additional modifications to our current understanding of azole resistance in C. glabrata. First, by genetically limiting Erg11 activity, we can trigger induction of Pdr1 and its target gene CDR1. This occurs in the absence of any azole drug. Clearly, azole drugs may still act as direct inducers of Pdr1 activity but Pdr1 activation can also occur in response to a reduction in function of the ergosterol biosynthetic pathway. Second, we identify a direct connection between transcriptional control of the ERG pathway and expression of both PDR1 and CDR1 that is manifested by the function of the Upc2A transcription factor. Together, these data argue for the presence of a physiological link between expression of genes producing plasma membrane-localized ABC transporter proteins and the major plasma membrane sterol component ergosterol that extends beyond the presence of the azole drugs (Fig. 9C).
There is precedent for the idea of direct transcriptional cross talk between ERG genes and ABC transporter loci in at least two other organisms. Chromatin immunoprecipitation with microarray technology (ChIP-chip) experiments carried out on C. albicans Upc2 detected binding of this important ERG gene regulator to the CDR1 promoter in this yeast (26). Analysis of the Aspergillus fumigatus transcription factor AtrR indicated that this Zn2Cys6-containing regulator could directly bind to the promoters of both cyp51A (ERG11 homologue) and abcG1 (CDR1 homologue) (27, 28).
The work reported here provides a further understanding of the potential connection between levels of ergosterol pathway function and expression of the Pdr1 regulon in C. glabrata. As mentioned above, this yeast frequently acquires azole resistance and represents an emerging clinical complication. We used two very different signals to induce a limitation of ERG11 production to argue that this is sufficient for induction of Pdr1/CDR1. Previous experiments have demonstrated an unexpected synergy between doxycycline and fluconazole (19). Doxycycline is routinely used as a means to artificially regulate gene expression (29), but the interaction between it and fluconazole complicates interpretations in regard to homeostasis during challenge by this azole drug. We used both a natural product (camphor) and the amino acid methionine as artificial signals to repress ERG11 transcription with the intent of avoiding any complicating interactions with fluconazole or the genes that mediate resistance to this drug. The two compounds had the same effects of repressing ERG11 synthesis and triggering the elevation of Pdr1 activity and attendant target genes.
Previous experiments in Neurospora crassa also showed that transcriptional repression of erg11 in this organism also led to induction of erg genes as well as the ABC transporter-encoding locus cdr4 (30), although no transcription factor was linked to this induction. Repression of erg11 in this system was accomplished by the use of a copper-repressible promoter as well as use of various erg pathway mutants. Copper is both an essential nutrient but also a potent inducer of membrane and oxidative stress (reviewed in reference 31) which could lead to unexpected interactions with regulatory systems in place to control membrane biogenesis and maintenance. However, the range of different signals used to repress ERG11 production (copper, camphor, methionine) makes it less likely that all of these different agents have similar effects that lead to activation of ABC transporter-encoding loci in two very different organisms (filamentous fungus and yeast). The simplest explanation is that limitation of ERG11 production and ergosterol biosynthesis leads to the activation of both genes involved in production of this important plasma membrane lipid and to proteins that are embedded and function in this lipidic environment, namely, ABC transporters.
Detection of Upc2A binding to both the PDR1 and CDR1 promoters provides an attractive model to explain how this physiological circuitry is controlled. Strong evidence that has been provided by work on Upc2 in Saccharomyces cerevisiae indicates that this transcription factor is excluded from the nucleus when ergosterol levels are high (Fig. 9C) (32). Depletion of ergosterol allows Upc2 to move to nuclear target genes and to induce expression of the ERG pathway. Based on experiments using green fluorescent protein (GFP)-labeled Upc2 in S. cerevisiae, roughly 30% of the protein is in the nucleus prior to induction using fluconazole (32).
These data fit well with what is seen by ChIP in C. glabrata with respect to Upc2A DNA binding to the three target promoters examined. Both ERG11 and CDR1 exhibited significant levels of Upc2A association prior to fluconazole treatment (Fig. 9A), while PDR1 showed very little Upc2A prior to drug challenge. Importantly, both PDR1 and CDR1 were still inducible by fluconazole, even in the upc2AΔ strain (Fig. 8A to C). The level of induction was lower than that seen in a strain with intact UPC2A, consistent with this factor playing a role in fluconazole activation of expression of these two genes. It is important that loss of Upc2A has a number of cellular consequences, including a reduction in ergosterol levels, lower baseline expression of ERG genes, and a defective response to anaerobic conditions (15, 33). This spectrum of phenotypes indicates that loss of Upc2A has physiological sequelae that could impact other regulatory circuits such as those represented by Pdr1/CDR1. We are presently mapping the sterol response element (SRE) for Upc2A in both the PDR1 and CDR1 promoters in order to more specifically examine the consequences of loss of Upc2A binding for activation of these two promoters.
Evidence for potential coregulation of Upc2 function and Pdr1 function was also provided by the analysis of SAGA components in S. cerevisiae cells that lacked the ERG3 gene (34). Proteomic analyses of proteins that copurified with Spt7-TAP (SAGA component) led to the detection of both Pdr1 and Upc2 but only in samples derived from erg3 mutant strains. These findings are consistent with our detection of Upc2 recruitment to the PDR1 promoter such as happens only in the presence of an inducing signal like fluconazole or ERG11 transcriptional repression (Fig. 9). These findings in S. cerevisiae and C. glabrata suggest that analyses of factors binding to target promoters that are done in the absence of induction face the risk of missing important interactions. Additionally, while we do not yet know the precise location of the SRE in the PDR1 promoter, two Pdr1 response elements (PDREs) have been mapped to this region (16, 35). This presents the possibility that Pdr1 binding to these PDREs may impact Upc2A recruitment to this region. This regulatory interaction may play a previously undetected role in the control of expression of both PDR1 and, ultimately, its suite of target genes.
MATERIALS AND METHODS
Media, plasmids, and strains.
C. glabrata was grown in rich YPD medium (1% yeast extract, 2% peptone, 2% glucose) or under amino acid-selective conditions in complete supplemental medium (CSM) (Difco yeast nitrogen extract without amino acids, amino acid powder from Sunrise Science Products, 2% glucose). All solid media contained 1.5% agar. Nourseothricin (Jena Bioscience, Jena, Germany) was supplemented to liquid CSM media at 50 μg/ml and to a CSM agar plate at 200 μg/ml to select for C. glabrata strains containing the pBV133 vector (36) and its derivative. The MET3 promoter is repressed by the presence of methionine (Fisher Biotec, Wembley, Australia) in the media as previously shown (37). Liquid CSM lacking methionine was used to induce expression, whereas addition of methionine at 20 μg/ml was used to repress promoter activity. Methionine (1 mM) was used to supplement CSM agar plates. All strains used in this study are listed in Table 1.
TABLE 1.
Strains used in this studya
| Strain | Parent | Genotype or description |
|---|---|---|
| BVGC3 | ATCC 2001 | ERG11-3× HA |
| BVGC7 | BVGC3 | HO::ADH1Pr-camR-VP16 |
| BVGC9 | BVGC7 | HO::ADH1Pr-camR-VP16 camO- CYC1Pr-ERG11-3× HA |
| BVGC85 | BVGC3 | ERG11-3× HA, upc2AΔ |
| BVGC89 | BVGC7 | HO::ADH1Pr-camR-VP16 camO- CYC1Pr-ERG1-3× HA |
| BVGC113 | BVGC7 | HO::ADH1Pr-camR-VP16 camO- CYC1Pr-ERG2-3× HA |
| BVGC125 | BVGC7 | HO::ADH1Pr-camR-VP16 camO-CYC1Pr-ERG3-3× HA |
| BVGC127 | BVGC3 | MET3Pr-ERG11-3× HA |
| BVGC130 | BVGC127 | MET3Pr-ERG11-3× HA, upc2AΔ |
All listed strains are from this study.
C. glabrata transformation.
Cell transformations were performed using a lithium acetate method (38). After being heat shocked, cells were either directly plated onto selective agar plates (for auxotrophic complementation) or grown at 30μC at 200 rpm overnight (for dominant-drug selection). Overnight cultures were then plated on YPD or CSM agar plates supplemented with 50 μg/ml or 200 μg/ml of nourseothricin, respectively. Plates were incubated at 30μC for 24 to 48 h before individual colonies were isolated and screened by PCR for correct insertion of the targeted construct.
Plasmid construction.
All constructs used for homologous recombination into the chromosome were constructed in a pUC19 plasmid vector (New England Biolabs, Ipswich, MA). PCR was used to amplify DNA fragments and Gibson assembly cloning (New England Biolabs, Ipswich, MA) to assemble fragments together. Sequences of the repeated influenza hemagglutinin epitope tag (3× HA) along with S. cerevisiae ADH1 terminator (ScADH1tr) and HIS3MX6 were PCR amplified from the pFA6a-3HA-His3MX6 plasmid (39). This tag element was inserted in place of the stop codon of ERG11. The camphor transactivator (camR-VP16), which was under the control of the ScTDH1 promoter (ScTDH1pr) and ScSTR1 terminator (ScSTR1tr), was amplified from the pSIB619 plasmid (40). It was then combined with the S. cerevisiae codon-optimized form of the nourseothricin resistance gene and fragments from the immediate upstream/downstream regions of the stop codon of the HO gene. The camphor promoter (camP), which included ScADH1 terminator followed by the multiple copies of the camphor repressor operator element (camO) attached to the CYC1 minimal promoter, was amplified from pSIB426 (40). It then was combined with the wild-type version of S. cerevisiae LEU2 (ScLEU2) and with fragments from the upstream/downstream regions of the target gene start codon. The C. glabrata MET3 promoter (MET3pr) was amplified from the pBV133 plasmid (36) and assembled with the upstream/downstream regions of the start codon of the ERG11 gene and wild-type ScLEU2. The UPC2A deletion construct was made by assembling the recyclable cassette from pBV65 (14) and fragments from the immediate upstream/downstream regions of UPC2A. A plasmid-borne copy of wild-type UPC2A was generated by digesting the pBV133 plasmid (36) with SacI and EcoRI (New England Biolabs, Ipswich, MA). The wild-type UPC2A gene was inserted via Gibson assembly with 1 kb of 5′ flanking DNA and 0.25 kb of 3′ flanking DNA along with the open reading frame.
Camphor and methionine sensitivity assay.
Cells were grown in YPD medium to mid-log-phase. Cultures were then serially diluted and spotted onto YPD agar plates containing 50 μM camphor (Sigma-Aldrich, St. Louis, MO). In some experiments, the YPD medium was supplemented with 20 μg/ml ergosterol (Sigma-Aldrich, St. Louis, MO) or Tween 80/ethanol (1:1) as the vehicle control. Ergosterol (2 mg/ml) was made in Tween 80/ethanol (1:1). To test strains containing MET3pr, overnight cultures were serially diluted and freshly inoculated directly onto a CSM agar plate supplemented with 1 mM methionine. All agar plates were incubated at 30μC for 24 to 48 h before imaging was performed.
Total sterol estimation.
Cell total sterol was extracted and measured as previously described (41). In short, cell pellets were lysed in 25% alcoholic potassium hydroxide at 90μC for 2 h. Total sterol was detected by spectrophotometric scanning between the wavelengths of 240 nm and 300nm. The presence of ergosterol in the extracted sample resulted in a four-peak curve with peaks located at approximately 262, 270, 281, and 290 nm.
Quantification of transcript levels by RT-qPCR.
Total RNA was extracted from cells by extraction using TRIzol (Invitrogen, Grand Island, NY) and chloroform (Fisher Scientific, Hampton, NH) followed by purification with RNeasy minicolumns (Qiagen, Redwood City, CA). RNA was reverse transcribed using an iScript cDNA synthesis kit (Bio-Rad, Des Plaines, IL). Assay of RNA via quantitative PCR (qPCR) was performed with iTaq universal SYBR green supermix (Bio-Rad, Des Plaines, IL). Target gene transcript levels were normalized to transcript levels of 18S rRNA.
Antibodies and Western blotting.
Mouse Anti-HA antibody was purchased from Thermo Scientific (Waltham, MA), and IRDye secondary antibodies were purchased from Li-COR (Lincoln, NE). Mouse anti-tubulin antibody was obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology. To produce the anti-Cdr1 polyclonal antibody, the 169 N-terminal amino acids of CDR1 were PCR amplified and cloned in frame as a NcoI/SacI fragment downstream of the 6×-His tag in pET28a+ (EMD Millipore Inc.). Expression was carried out in Escherichia coli BL21(DE3) cells (Thermo Scientific, Waltham, MA). Transformants were grown to the log phase and induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 90 min at 37μC. Cell lysates were prepared using a French press. Protein purification was accomplished using Talon metal affinity resin (TaKaRa Bio USA, Inc.) as described by the manufacturer. Protein fractions were analyzed by staining them with Coomassie blue and by Western blotting using His-specific antibodies. The purified proteins were then dialyzed against phosphate-buffered saline overnight (PBS) and then lyophilized and sent to Pacific Immunology (Ramona, CA) for injection into rabbits to generate polyclonal antibodies. Antiserum generated from these rabbits was received and tested for immunoreactivity against C. glabrata cell lysates. The antiserum was then affinity purified against the purified protein using AminoLink Plus coupling resin (Thermo Scientific, Inc.) according to the manufacturer’s instructions. All antibodies were validated by Western blotting against an appropriate isogenic deletion strain (see Fig. S1 in the supplemental material). In the case of Upc2A, the 200 N-terminal amino acids were amplified and cloned as a NcoI/SacI fragment downstream of the 6×-His tag in pET28a+ (EMD Millipore, Inc.) and transformed into the bacterial expression strain BL21-CodonPlus (Agilent, Santa Clara, CA). Western blotting of C. glabrata whole-cell protein extracts was performed as previously described (42), and extracts were prepared using NaOH/β-mercaptoethanol-based lysis. All Western blot experiments were performed in triplicate, and the results were quantitated using Odyssey software and are presented as averages of these determinations with standard errors.
Characterization of polyclonal antibodies against Cdr1 and Upc2A. (A) Isogenic wt and cdr1Δ cells were grown to the mid-log phase, and whole-cell proteins were extracted and then analyzed by Western blotting using affinity-purified anti-Cdr1 antisera. Molecular mass standards are indicated in kilodaltons on the left; the position of the 170-kDa Cdr1 is shown by the arrow. (B) Whole-cell protein extracts were prepared from isogenic wt and upc2AΔ cells. Extracts were analyzed by Western blotting as described above, but affinity-purified anti-Upc2A antisera were used to develop this blot. Molecular mass standards are shown on the left, and the position of Upc2A is indicated by the arrow. Download FIG S1, TIF file, 0.6 MB (647.4KB, tif) .
Copyright © 2019 Vu et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Fluconazole induction of Pdr1, Cdr1, and Erg11-3× HA requires the presence of the Upc2A transcription factor. (A) Isogenic BVGC3 and BVGC3 upc2AΔ cells were grown to the mid-log phase and then treated with 20 μg/ml fluconazole (+) or allowed to continue to grow (−) for 3 h. Cultures were harvested, whole-cell protein extracts were prepared, and levels of the indicated proteins were assayed by Western blotting. (B) Quantitation of the Western blotting results presented in panel A. The presence of fluconazole is indicated as “+ FLC.” Download FIG S2, TIF file, 0.6 MB (592.2KB, tif) .
Copyright © 2019 Vu et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Chromatin immunoprecipitation and qRT-PCR.
Protein-DNA cross-linking was performed with 1% formaldehyde (Sigma-Aldrich, St. Louis, MO) for 15 min at room temperature with gentle shaking. The reaction was stopped with 250 mM glycine and with 15 min of shaking at room temperature. About 5 × 108 cells were then pelleted, washed with PBS, and subjected to glass bead lysis or stored at −80μC. Cell pellets were resuspended in 600 μl of lysis buffer (50 mM HEPES [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% deoxycholate, 0.1% SDS, 1 mM phenylmethylsulfonyl fluoride [PMSF], 1× fungal proteinase inhibitor cocktail) and lysed with 1 ml of 0.5-mm-diameter glass beads for 10 min (5 intervals of 2 min with 30 s cooling between intervals) at 4μC. Samples were then vortexed and split into 3 AFA Fiber Pre-Slit Snap-Cap (6 × 15 mm) MicroTUBEs (Covaris) (130 μl of sample per tube). Chromatin was sheared with an E220 focused ultrasonicator (Covaris) under the following conditions: peak incident power (W), 175; duty factor, 10%; number of cycles per burst, 200; treatment time, 780 s; temperature, 7μC; sample volume, 130 μl (in the presence of E220 intensifier [pn500141]). The contents of the tubes from each sample were then pooled and centrifuged at 10,000 × g for 5 min at 4μC. The supernatant was transferred into a new tube, and 20 μl was reserved as an input control fraction to verify sonication and as a control for chromatin immunoprecipitation (ChIP) and qPCR. The sheared chromatin was incubated with rabbit polyclonal anti-Upc2A antibody (1:50 dilution) for 2 h before being incubated together with 30 μl of washed protein G Dynabeads (Life Technologies) overnight on a nutator at 4μC. Washing, reversal of cross-links, and purification of DNA processed by the use of ChIP were performed as described in reference 43.
Real-time PCR was performed in triplicate for each separate ChIP experiment using primers designed for regions described below, under the following conditions: 1 cycle of 95μC for 30 s followed by 40 cycles of 95μC for 15 s and 56μC for 30 s on an MyiQ2 Bio-Rad machine. A 0.5-μl volume of the DNA processed by the use of ChIP or of input (diluted 20-fold) DNA was used in a reaction mixture with a 20-μl total volume using SYBR green master mix (Bio-Rad) and a 0.4 μM concentration of each primer. The percent input method was used to calculate the signal of enrichment of the promoter region for each gene. ERG11, CDR1, and PDR1 promoters were analyzed with primers specifically targeting the ERG11 promoter (−561 to −694 relative to the ATG as +1), CDR1 promoter (−476 to −665), and PDR1 promoter (−551 to −651) regions. A region of ERG11, located within the coding sequence (939 to 1042), was used as a negative control.
Statistics.
The Student t test was used to assess the statistical significance of results of comparisons of samples. Paired conditions were used for comparisons of results from the same isolate obtained under different treatment conditions, while unpaired conditions were used for comparisons of results from isolates obtained under the same treatment conditions (*, P < 0.5; **, P < 0.01; ***, P < 0.001).
ACKNOWLEDGMENTS
We thank Dave Rogers and Damian Krysan for helpful discussions and Sanjoy Paul and Lucia Simonicova for a critical reading of the manuscript.
This work was supported by NIH grant GM49825.
Footnotes
Citation Vu BG, Thomas GH, Moye-Rowley WS. 2019. Evidence that ergosterol biosynthesis modulates activity of the PDR1 transcription factor in Candida glabrata. mBio 10:e00934-19. https://doi.org/10.1128/mBio.00934-19.
REFERENCES
- 1.Pfaller MA, Andes DR, Diekema DJ, Horn DL, Reboli AC, Rotstein C, Franks B, Azie NE. 2014. Epidemiology and outcomes of invasive candidiasis due to non-albicans species of Candida in 2,496 patients: data from the Prospective Antifungal Therapy (PATH) registry 2004–2008. PLoS One 9:e101510. doi: 10.1371/journal.pone.0101510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arendrup MC, Patterson TF. 2017. Multidrug-resistant Candida: epidemiology, molecular mechanisms, and treatment. J Infect Dis 216:S445–S451. doi: 10.1093/infdis/jix131. [DOI] [PubMed] [Google Scholar]
- 3.Beardsley J, Halliday CL, Chen SC, Sorrell TC. 2018. Responding to the emergence of antifungal drug resistance: perspectives from the bench and the bedside. Future Microbiol 13:1175–1191. doi: 10.2217/fmb-2018-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Denning DW, Bromley MJ. 2015. Infectious disease. How to bolster the antifungal pipeline. Science 347:1414–1416. doi: 10.1126/science.aaa6097. [DOI] [PubMed] [Google Scholar]
- 5.Berkow EL, Lockhart SR. 2017. Fluconazole resistance in Candida species: a current perspective. Infect Drug Resist 10:237–245. doi: 10.2147/IDR.S118892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Geber A, Hitchcock CA, Swartz JE, Pullen FS, Marsden KE, Kwon-Chung KJ, Bennett JE. 1995. Deletion of the Candida glabrata ERG3 and ERG11 genes: effect on cell viability, cell growth, sterol composition, and antifungal susceptibility. Antimicrob Agents Chemother 39:2708–2717. doi: 10.1128/aac.39.12.2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakayama H, Nakayama N, Arisawa M, Aoki Y. 2001. In vitro and in vivo effects of 14alpha-demethylase (ERG11) depletion in Candida glabrata. Antimicrob Agents Chemother 45:3037–3045. doi: 10.1128/AAC.45.11.3037-3045.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Becker JM, Kauffman SJ, Hauser M, Huang L, Lin M, Sillaots S, Jiang B, Xu D, Roemer T. 2010. Pathway analysis of Candida albicans survival and virulence determinants in a murine infection model. Proc Natl Acad Sci U S A 107:22044–22049. doi: 10.1073/pnas.1009845107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Whaley SG, Rogers PD. 2016. Azole resistance in Candida glabrata. Curr Infect Dis Rep 18:41. doi: 10.1007/s11908-016-0554-5. [DOI] [PubMed] [Google Scholar]
- 10.Vermitsky JP, Earhart KD, Smith WL, Homayouni R, Edlind TD, Rogers PD. 2006. Pdr1 regulates multidrug resistance in Candida glabrata: gene disruption and genome-wide expression studies. Mol Microbiol 61:704–722. doi: 10.1111/j.1365-2958.2006.05235.x. [DOI] [PubMed] [Google Scholar]
- 11.Tsai HF, Krol AA, Sarti KE, Bennett JE. 2006. Candida glabrata PDR1, a transcriptional regulator of a pleiotropic drug resistance network, mediates azole resistance in clinical isolates and petite mutants. Antimicrob Agents Chemother 50:1384–1392. doi: 10.1128/AAC.50.4.1384-1392.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khakhina S, Simonicova L, Moye-Rowley WS. 2018. Positive autoregulation and repression of transactivation are key regulatory features of the Candida glabrata Pdr1 transcription factor. Mol Microbiol 107:747–764. doi: 10.1111/mmi.13913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thakur JK, Arthanari H, Yang F, Pan S-J, Fan X, Breger J, Frueh DP, Gulshan K, Li DK, Mylonakis E, Struhl K, Moye-Rowley WS, Cormack BP, Wagner G, Näär AM. 2008. A nuclear receptor-like pathway regulating multidrug resistance in fungi. Nature 452:604–609. doi: 10.1038/nature06836. [DOI] [PubMed] [Google Scholar]
- 14.Henry KW, Nickels JT, Edlind TD. 2000. Upregulation of ERG genes in Candida species by azoles and other sterol biosynthesis inhibitors. Antimicrob Agents Chemother 44:2693–2700. doi: 10.1128/aac.44.10.2693-2700.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whaley SG, Caudle KE, Vermitsky JP, Chadwick SG, Toner G, Barker KS, Gygax SE, Rogers PD. 2014. UPC2A is required for high-level azole antifungal resistance in Candida glabrata. Antimicrob Agents Chemother 58:4543–4554. doi: 10.1128/AAC.02217-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paul S, Bair TB, Moye-Rowley WS. 2014. Identification of genomic binding sites for Candida glabrata Pdr1 transcription factor in wild-type and rho0 cells. Antimicrob Agents Chemother 58:6904–6912. doi: 10.1128/AAC.03921-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aramaki H, Sagara Y, Hosoi M, Horiuchi T. 1993. Evidence for autoregulation of camR, which encodes a repressor for the cytochrome P-450cam hydroxylase operon on the Pseudomonas putida CAM plasmid. J Bacteriol 175:7828–7833. doi: 10.1128/jb.175.24.7828-7833.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Katiyar SK, Alastruey-Izquierdo A, Healey KR, Johnson ME, Perlin DS, Edlind TD. 2012. Fks1 and Fks2 are functionally redundant but differentially regulated in Candida glabrata: implications for echinocandin resistance. Antimicrob Agents Chemother 56:6304–6309. doi: 10.1128/AAC.00813-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fiori A, Van Dijck P. 2012. Potent synergistic effect of doxycycline with fluconazole against Candida albicans is mediated by interference with iron homeostasis. Antimicrob Agents Chemother 56:3785–3796. doi: 10.1128/AAC.06017-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao Y, Li H, Liu S, Zhang X, Sun S. 2014. Synergistic effect of fluconazole and doxycycline against Candida albicans biofilms resulting from calcium fluctuation and downregulation of fluconazole-inducible efflux pump gene overexpression. J Med Microbiol 63:956–961. doi: 10.1099/jmm.0.072421-0. [DOI] [PubMed] [Google Scholar]
- 21.Sanglard D, Ischer F, Calabrese D, Majcherczyk PA, Bille J. 1999. The ATP binding cassette transporter gene CgCDR1 from Candida glabrata is involved in the resistance of clinical isolates to azole antifungal agents. Antimicrob Agents Chemother 43:2753–2765. doi: 10.1128/AAC.43.11.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vermitsky JP, Edlind TD. 2004. Azole resistance in Candida glabrata: coordinate upregulation of multidrug transporters and evidence for a Pdr1-like transcription factor. Antimicrob Agents Chemother 48:3773–3781. doi: 10.1128/AAC.48.10.3773-3781.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Whaley SG, Berkow EL, Rybak JM, Nishimoto AT, Barker KS, Rogers PD. 2016. Azole antifungal resistance in Candida albicans and emerging non-albicans Candida species. Front Microbiol 7:2173. doi: 10.3389/fmicb.2016.02173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coste AT, Karababa M, Ischer F, Bille J, Sanglard D. 2004. TAC1, transcriptional activator of CDR genes, is a new transcription factor involved in the regulation of Candida albicans ABC transporters CDR1 and CDR2. Eukaryot Cell 3:1639–1652. doi: 10.1128/EC.3.6.1639-1652.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morschhauser J, Barker KS, Liu TT, Blab-Warmuth J, Homayouni R, Rogers PD. 2007. The transcription factor Mrr1p controls expression of the MDR1 efflux pump and mediates multidrug resistance in Candida albicans. PLoS Pathog 3:e164. doi: 10.1371/journal.ppat.0030164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Znaidi S, Weber S, Al-Abdin OZ, Bomme P, Saidane S, Drouin S, Lemieux S, De Deken X, Robert F, Raymond M. 2008. Genomewide location analysis of Candida albicans Upc2p, a regulator of sterol metabolism and azole drug resistance. Eukaryot Cell 7:836–847. doi: 10.1128/EC.00070-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hagiwara D, Miura D, Shimizu K, Paul S, Ohba A, Gonoi T, Watanabe A, Kamei K, Shintani T, Moye-Rowley WS, Kawamoto S, Gomi K. 2017. A novel Zn2-Cys6 transcription factor AtrR plays a key role in an azole resistance mechanism of Aspergillus fumigatus by co-regulating cyp51A and cdr1B expressions. PLoS Pathog 13:e1006096. doi: 10.1371/journal.ppat.1006096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paul S, Stamnes M, Thomas GH, Liu H, Hagiwara D, Gomi K, Filler SG, Moye-Rowley WS. 2019. AtrR is an essential determinant of azole resistance in Aspergillus fumigatus. mBio 10:e02563-18. doi: 10.1128/mBio.02563-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baron U, Bujard H. 2000. Tet repressor-based system for regulated gene expression in eukaryotic cells: principles and advances. Methods Enzymol 327:401–421. doi: 10.1016/S0076-6879(00)27292-3. [DOI] [PubMed] [Google Scholar]
- 30.Hu C, Zhou M, Wang W, Sun X, Yarden O, Li S. 2018. Abnormal ergosterol biosynthesis activates transcriptional responses to antifungal azoles. Front Microbiol 9:9. doi: 10.3389/fmicb.2018.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gaetke LM, Chow CK. 2003. Copper toxicity, oxidative stress, and antioxidant nutrients. Toxicology 189:147–163. doi: 10.1016/S0300-483X(03)00159-8. [DOI] [PubMed] [Google Scholar]
- 32.Yang H, Tong J, Lee CW, Ha S, Eom SH, Im YJ. 2015. Structural mechanism of ergosterol regulation by fungal sterol transcription factor Upc2. Nat Commun 6:6129. doi: 10.1038/ncomms7129. [DOI] [PubMed] [Google Scholar]
- 33.Nagi M, Nakayama H, Tanabe K, Bard M, Aoyama T, Okano M, Higashi S, Ueno K, Chibana H, Niimi M, Yamagoe S, Umeyama T, Kajiwara S, Ohno H, Miyazaki Y. 2011. Transcription factors CgUPC2A and CgUPC2B regulate ergosterol biosynthetic genes in Candida glabrata. Genes Cells 16:80–89. doi: 10.1111/j.1365-2443.2010.01470.x. [DOI] [PubMed] [Google Scholar]
- 34.Dewhurst-Maridor G, Abegg D, David FPA, Rougemont J, Scott CC, Adibekian A, Riezman H. 2017. The SAGA complex, together with transcription factors and the endocytic protein Rvs167p, coordinates the reprofiling of gene expression in response to changes in sterol composition in Saccharomyces cerevisiae. Mol Biol Cell 28:2637–2649. doi: 10.1091/mbc.E17-03-0169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paul S, Schmidt JA, Moye-Rowley WS. 2011. Regulation of the CgPdr1 transcription factor from the pathogen Candida glabrata. Eukaryot Cell 10:187–197. doi: 10.1128/EC.00277-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vu BG, Moye-Rowley WS. 2018. Construction and use of a recyclable marker to examine the role of major facilitator superfamily protein members in Candida glabrata drug resistance phenotypes. mSphere 3:e00099-18. doi: 10.1128/mSphere.00099-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zordan RE, Ren Y, Pan SJ, Rotondo G, De Las Penas A, Iluore J, Cormack BP. 2013. Expression plasmids for use in Candida glabrata. G3 (Bethesda) 3:1675–1686. doi: 10.1534/g3.113.006908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ito H, Fukuda Y, Murata K, Kimura A. 1983. Transformation of intact yeast cells treated with alkali cations. J Bacteriol 153:163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Longtine MS, McKenzie A, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 40.Ikushima S, Zhao Y, Boeke JD. 2015. Development of a tightly controlled off switch for Saccharomyces cerevisiae regulated by camphor, a low-cost natural product. G3 (Bethesda) 5:1983–1990. doi: 10.1534/g3.114.012765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Breivik ON, Owades JL. 1957. Yeast analysis, spectrophotometric semimicrodetermination of ergosterol in yeast. J Agric Food Chem 5:360–363. doi: 10.1021/jf60075a005. [DOI] [Google Scholar]
- 42.Paul S, McDonald WH, Moye-Rowley WS. 2018. Negative regulation of Candida glabrata Pdr1 by the deubiquitinase subunit Bre5 occurs in a ubiquitin independent manner. Mol Microbiol 110:309–323. doi: 10.1111/mmi.14109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chung D, Barker BM, Carey CC, Merriman B, Werner ER, Lechner BE, Dhingra S, Cheng C, Xu W, Blosser SJ, Morohashi K, Mazurie A, Mitchell TK, Haas H, Mitchell AP, Cramer RA. 2014. ChIP-seq and in vivo transcriptome analyses of the Aspergillus fumigatus SREBP SrbA reveals a new regulator of the fungal hypoxia response and virulence. PLoS Pathog 10:e1004487. doi: 10.1371/journal.ppat.1004487. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Characterization of polyclonal antibodies against Cdr1 and Upc2A. (A) Isogenic wt and cdr1Δ cells were grown to the mid-log phase, and whole-cell proteins were extracted and then analyzed by Western blotting using affinity-purified anti-Cdr1 antisera. Molecular mass standards are indicated in kilodaltons on the left; the position of the 170-kDa Cdr1 is shown by the arrow. (B) Whole-cell protein extracts were prepared from isogenic wt and upc2AΔ cells. Extracts were analyzed by Western blotting as described above, but affinity-purified anti-Upc2A antisera were used to develop this blot. Molecular mass standards are shown on the left, and the position of Upc2A is indicated by the arrow. Download FIG S1, TIF file, 0.6 MB (647.4KB, tif) .
Copyright © 2019 Vu et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Fluconazole induction of Pdr1, Cdr1, and Erg11-3× HA requires the presence of the Upc2A transcription factor. (A) Isogenic BVGC3 and BVGC3 upc2AΔ cells were grown to the mid-log phase and then treated with 20 μg/ml fluconazole (+) or allowed to continue to grow (−) for 3 h. Cultures were harvested, whole-cell protein extracts were prepared, and levels of the indicated proteins were assayed by Western blotting. (B) Quantitation of the Western blotting results presented in panel A. The presence of fluconazole is indicated as “+ FLC.” Download FIG S2, TIF file, 0.6 MB (592.2KB, tif) .
Copyright © 2019 Vu et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.