

Significance
Virtual chemical space, i.e., the set of all conceivable drug-sized molecules, offers an abundance of unexplored quadrants which might contain novel bioactive compounds. Exploring these quadrants has to balance novelty of compounds with their synthetic accessibility. Forward-synthetic databases create novel chemical matter by combining existing and readily available building blocks, thereby biasing the resulting products toward synthesizability. To date, no one has tested whether molecules from such databases can be made in large numbers with an acceptable probability, and we investigated this question here. Moreover, we analyze whether the emerging molecules are binding to a G protein-coupled receptor (GPCR) target and whether ligands can be considered novel in the sense that they represent chemotypes which have not been described for this target before.
Keywords: de novo design, parallel synthesis, highly designed libraries, docking, forward synthetic libraries
Abstract
Forward-synthetic databases are an efficient way to enumerate chemical space. We explored here whether these databases are good sources of novel protein ligands and how many molecules are obtainable and in which time frame. Based on docking calculations, series of molecules were selected to gain insights into the ligand structure–activity relationship. To evaluate the novelty of compounds in a challenging way, we chose the β2-adrenergic receptor, for which a large number of ligands is already known. Finding dissimilar ligands is thus the exception rather than the rule. Here we report on the results, the successful synthesis of 127/240 molecules in just 2 weeks, the discovery of previously unreported dissimilar ligands of the β2-adrenergic receptor, and the optimization of one series to a KD of 519 nM in only one round. Moreover, the finding that only 3 of 240 molecules had ever been synthesized before indicates that large parts of chemical space are unexplored.
Chemical space, the set of all theoretically possible drug-sized molecules, is a vast playground when one is searching for modulators of protein targets with pharmacological relevance. It is estimated to contain at least substances (1). Many quadrants of this space have been well explored, while others are underinvestigated or have not yet delivered biologically active chemical matter.
Forward-synthetic databases, such as our previously developed database concept Screenable Chemical Universe Based on Intuitive Data Organization (SCUBIDOO) (2) and others (3, 4), are efficient ways to move in the direction of more complete coverage of chemical space. These databases are powerful tools because each compound is the product of two (or more) readily available building blocks. It is one of the unique features of SCUBIDOO that the relationships between individual compounds are explicitly stored in the database. This means that queries such as “show me all products coming from reductive amination” or “which products involve building block X and reaction Y” can be answered with a single mouse click. This facilitates extraction of tailored subsets and medicinal chemistry reasoning. However, the question of whether these databases will be good sources for novel and potent protein ligands was largely unanswered (5).
There are two main ways to screen such databases, either exhaustively or using strategies that limit the computational effort necessary while keeping the exploration scope intact. An example of the first, exhaustive, strategy appeared during the revision of this article (6). As a case exemplifying the second line of thought, we report on the successful application of SCUBIDOO to identify previously unreported or synthesized ligands for one of the best-investigated (and thus, in this context, challenging) G protein-coupled receptors (GPCRs), the β2-adrenergic receptor (β2AR), here. We started the campaign from SCUBIDOO’s “small” (S) subset of ∼10,000 products, to determine whether these would be enough to actually capture diverse molecules in the end. To select compounds for synthesis, we used docking, which we have shown earlier to be able to identify novel scaffolds for GPCR targets (7–9). To demonstrate that ligand selection can be tailored, we wished to explore the binding cavity of the β2AR in more detail. To that end, this site was partitioned into three subcavities (Fig. 1 A and B): the orthosteric pocket (OP, green), the secondary binding pocket (SBP, blue) and the tertiary binding pocket (TBP, orange). We aimed to find ligands that would lie along the OP→SBP or OP→TBP axis, respectively. Hence, compound selection happened in two independent processes. This partitioning of pockets and exploration directions fits with the SCUBIDOO philosophy of bipartite products (i.e., each product consisting of a building block [bb] A and B), each pocket being filled by one bb.
Fig. 1.

(A) Sliced surface view of the binding site of the in an inactive conformation (PDB code 2RH1) with the predicted binding mode of compound red_A05B51. The OP, SBP, and TBP are colored green, blue, and orange, respectively. (B) Binding-site representation illustrating the product design strategy. (C) Dose–response curve of red_A05B51 against -Nb80 (active state, green curve) and -Nb69 (basal state, red curve). The difference in affinity between the two conformations denotes red_A05B51 as the agonist candidate.
With the workflow described in this article, we selected 240 molecules by creating an array of building-block combinations. Thus, instead of cherry-picking compounds, we selected miniseries, thereby optimizing building-block use and the possibility for structure–activity relationship (SAR) reasoning. We attempted synthesis of all 240 molecules, obtained 127 in the first round, and assayed each molecule in a recently described “reverse pharmacology” assay (10), leading to a rich SAR and insights into ligand efficacy. A second round of synthesis was aimed at affinity and reaction optimization. Based on the intercorrelated nature of the molecule arrays combined with the assay data, we were able to improve one ligand by 40-fold for a final affinity of 519 nM in this single round of optimization. Moreover, minimal adjustments to the reductive amination synthesis protocol in the second round yielded another 14 of the original 240 molecules. Of note, the total effective time to design, dock, produce, and test the first batch was only 6 weeks. Even more important, none of the compounds had ever been described as a ligand of the β2AR before, and none of the ligands had ever been synthesized.
Results
Although the choice is arbitrary from a chemistry point of view, we will consistently call the building block placed in the OP “bb.A” (Fig. 1B). Every compound is named after the identification code of its bb.A and bb.B parts, preceded by an identifier for the reaction: “red_” for reductive amination and “amd_” for amide coupling. The 2D depictions of all building blocks and the corresponding generated products are available in SI Appendix, Schemes S1–S3.
Reaction and Product Selection.
Product selection was building-block and docking based, as described in detail in Methods and illustrated in the flowchart (Fig. 2). We first determined products from the docking of the SCUBIDOO S subset of 9,994 compounds that would fit well in the OP, regardless of the quality of the interactions in the SBP or TBP. For each direction (OP→SBP or OP→TBP), analysis of all docked products and their derivatives led to the choice of a single reaction which was compatible with binding poses along the desired axis. To keep the number of products to be synthesized within feasible limits, we chose 5 bb.As for each direction. The number of 24 bb.Bs stemmed from the fact that the reaction blocks used during parallel synthesis had wells.
Fig. 2.

Schematic flowchart illustrating the complete process used to select the molecules to be docked and synthesized. Boxes 1 (red background) and 2 (green background) are detailed in SI Appendix, Figs. S2 and S3, respectively. Solid lines signify steps that were taken in this study. Dashed lines are for steps that we would have taken in the event of unsatisfactory outcomes of the respective decision points.
As an example illustrating how the selection process worked in the OPSBP direction, compound 1 (SI Appendix, Fig. S1A) is discussed. It is a product from the S subset whose docking pose predicts that its bb.A (appearing as A04 in SI Appendix, Scheme S2) optimally interacts with the OP, while its bb.B (B53 in SI Appendix, Fig. S1B) leaves several interaction possibilities in the SBP unsatisfied. Despite this, it appeared at rank 131 in the original docking. The bb.A of compound 1 contains a protonated primary amine interacting with the highly conserved Asp and a triazole moiety which engages Asn and Ser. The subpar bb.B of compound 1 is positioned in the SBP and contains a carboxylic acid moiety which is not involved in any interactions. Compound 1 is predicted to emerge from a reductive amination reaction by combining A04 and B53. Most importantly, this reaction is compatible with an extension of A04 toward the SBP. We therefore extracted the sublibrary of 1,195 products containing A04 (obtained via reductive amination with one of the 1,195 bb.Bs from SCUBIDOO) and docked it. Visual inspection of the top-ranked poses revealed a consistent binding mode for the A04 portion in the OP for most of the products and yielded the first candidates for bb.Bs that would engage in favorable interactions in the SBP. These results therefore provided additional evidence that reductive amination is well suited to explore the OPSBP direction. In a completely analogous manner, the other four bb.As for the OPSBP direction were determined by looking first at the close analogs of A04 and afterward at the remaining virtual hits coming from reductive amination in the original docking ranked list of 9,994 products. In summary, the five bb.As were selected because of their predicted interaction with Asp, while offering distinct interactions with the bottom of the OP. While also different products obtainable by reactions other than reductive amination were found in the top ranks of the docking of the S subset, these were rare and consequently, reductive amination was chosen as the common reaction. The joint analysis of all bb.Bs that were ranked favorably in the five individual sublibrary dockings (for each bb.A) led to the identification of 24 bb.Bs that would lead to high-ranking products with all 5 bb.As. They appear as red_A01B01–red_A05B24 in SI Appendix, Scheme S3.
This procedure was repeated for the OPTBP direction, leading to 5 more bb.As and 24 bb.Bs (SI Appendix, Scheme S1). The reaction that emerged for this direction was amide formation. The corresponding products are amd_A06B25–amd_A10B48 (SI Appendix, Scheme S3).
In total, 5,975 and 61,150 products were docked for the OPSBP and OPTBP directions, respectively. The difference comes from the number of available products for each of the two reactions in SCUBIDOO.
Finally, we were left with 120 () ligands based on reductive amination, designed to explore the OPSBP direction, and another 120 ligands based on amide formation for the OPTBP direction.
Chemical Space Coverage.
The OPSBP direction was explored by docking 5,975 of the 2,704,307 products (0.22% only) resulting from reductive amination in SCUBIDOO. These numbers are similar at the building-block level: There are 1,042 primary amines in SCUBIDOO, 613 of which match the pharmacophore of an aromatic ring connected to a protonable amine via one to four carbons. Selecting 5 amines therefore corresponds to a 0.8% “exploration rate.” We also docked all protonable amines in SCUBIDOO (more than 3 million products) and compared the top 500 to the top 500 of the S subset to gauge the influence of the stratified balanced sampling (2) used to derive the subsets. The S subset docking featured 43% more reactions (20 vs. 14) and 49% more building blocks (673 vs. 452) and had more diversity, as the most frequent bb appeared only 26 times instead of 54. Thus, despite docking fewer molecules, we covered more chemical space.
For the OPTBP direction, 61,150 products of 6,630,786 (0.91%) available for amide coupling were docked. There are 1,332 building blocks with a carboxylic acid in SCUBIDOO and only 64 which feature an additional protonable amine (with 5 selected for synthesis, this corresponds to 0.4% and 7.8%, respectively).
Synthesis of Products Based on Amide Coupling.
Synthesis of the 120 amide coupling products proceeded smoothly. To avoid self-reaction of the five bb.As, their amino acidic nitrogen was protected with a Boc (tert-butyloxycarbonyl) group. The reaction was performed under standard conditions (SI Appendix, Scheme S4) and afforded the desired products with moderate to excellent yields (i.e., 38–92%; Dataset S1). The Boc-protected amino acids were then reacted in parallel with 24 different bb.Bs (SI Appendix, Scheme S1) and purified by preparative HPLC. The acidic removal of the Boc protection followed by analytical purification yielded 102 of the 120 planned compounds (83% synthesis success; SI Appendix, Fig. S4A).
Synthesis of Products Based on Reductive Amination.
In the case of the reductive amination products, the 5 bb.As were first reacted with acetic acid and then with the 24 different bb.Bs (SI Appendix, Scheme S2) to form the corresponding imines. After 20 min, an excess of NaBH(OAc)3 (1.5 eq) was added to reduce the imines to secondary amines (SI Appendix, Scheme S5). The low success rate of this second pool of products (25 compounds delivered of 120 planned, 21% synthesis success; SI Appendix, Fig. S4B and Dataset S1) can be explained by (i) the instability of several of the resulting products, (ii) their highly polar nature, and (iii) the high reactivity of the bb.As. This observation is in agreement with studies showing that more polar products in parallel synthesis are more likely to fail (11–13). In our case, due to the Boc protection, the amide pool is more lipophilic than the amination pool. The distribution of the logP for successfully synthesized products vs. the failed ones is illustrated in SI Appendix, Fig. S5. Despite the use of a deficit amount (0.9 eq) of the 24 bb.Bs, the ratio tertiary amine/secondary amine (i.e., double addition/single addition) was mostly higher than 1, resulting in low amounts of desired product to be potentially isolated. The unstable and polar nature of the products also complicated the purification process and some compounds had to be discarded because of low UHPLC purity (<75%) or insufficient amount (<0.5 mg).
Radioligand Displacement Assays and Structure–Activity Relationship.
The 127 products from both pools were tested in a single-dose radioligand displacement assay against the stabilized in an active and its basal conformation, respectively, thereby providing indications toward the efficacy of each compound (10). For the 40 products showing the highest displacement of dihydroalprenolol at any of the two receptor fusions, the assay was repeated twice, yielding triplicate values. Finally, the top 15 compounds were selected for full dose–response curve measurements to obtain values (Table 1).
Table 1.
Affinity of the compounds for the AR-Nb80 and the AR-Nb69
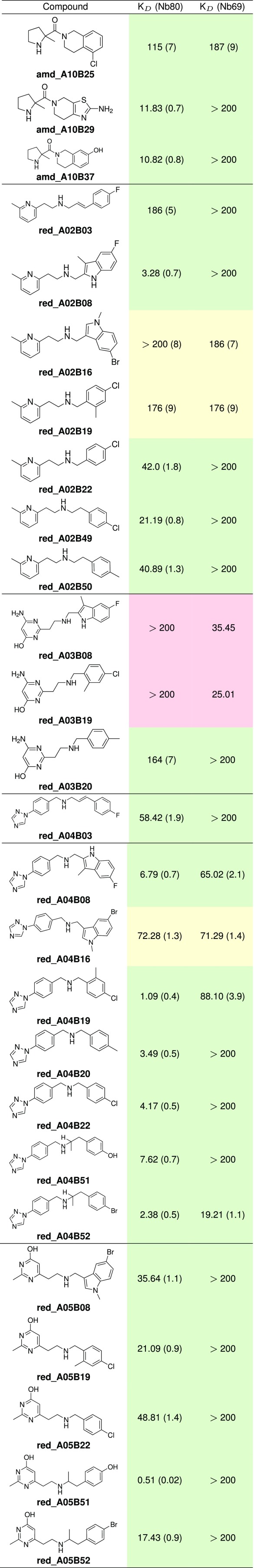 |
All reported KD values are in μM. Values in parentheses are SEs of the mean. Green cells denote agonist candidate (AC) products, red cells inverse agonist candidate (IAC) products, and orange cells antagonist candidate (AntC) products.
Only 3 products in the top 15 belong to the amide pool. All were derivatives of A10 and constitute unprecedented scaffolds compared with known ligands (ECFP4 Tanimoto similarity = 0.21, 0.21, 0.20 to the closest match in ChEMBL for amd_A10B25, amd_A10B37, and amd_A10B29, respectively). A deeper discussion of the SAR is in SI Appendix.
The amination pool yielded a higher hit rate (40%), with 9 of 25 compounds showing reasonable binding affinities (i.e., below 200 M in SI Appendix, Fig. S6). Four of the five bb.As (80%) were involved in at least one bioactive compound. Remarkably, one of the bb.As (A03) was part of two products with opposite efficacy: one inverse agonist candidate (IAC), red_A03B19, and one agonist candidate (AC) (see Methods for definition), red_A03B20. Moreover, one bb.B (B19) was involved in three products with different efficacy profiles (red_A02B19, red_A03B19, and red_A04B19). This illustrates the complexity of fragment-based procedures, where even small changes in one of the building blocks can have a drastic impact on the product binding mode and thus efficacy.
Reaction and Compound Optimization.
Subsequent to the first-round measurements described in the preceding paragraph, we attempted to resynthesize products which initially had failed to complete the matrix (SI Appendix, Fig. S6). Moreover, we suggested additional bb.Bs aiming at improved affinity and gaining additional understanding of activation. To that end, four close analogs of the active bb.Bs of the first round were selected. They contained one additional carbon atom between the benzene and the carbonyl groups (B49 is an analog of B20, for example). This should increase bb.B flexibility, thus facilitating steric complementarity with the SBP. Furthermore, two of the close analogs contained a ketone instead of the original aldehyde (B51 and B52), aimed at increasing hydrophobic contacts in the SBP. Finally, three of the close analogs were targeting the sidechain of Asp192ECL2 via a hydrogen (B51) or a halogen bond (B49 and B52). Asp192ECL2 has been shown to interact with known agonists (ref. 14 and PDB 4LDL) and we wanted to further investigate its possible role in activation. Reactions in the second round were performed on a larger scale (SI Appendix) and preparative HPLC purification afforded 30 of 44 compounds planned (68% synthesis success; SI Appendix, Fig. S4C, Bottom).
The SAR of the additional compounds correlated strongly with our design hypotheses. First, the additional rotatable bond improves the affinities of products based on the four additional bb.Bs over their respective first-round analogs. For instance, red_A02B50’s affinity was improved 42-fold over that of the original red_A02B20. This suggests that slightly more flexible extensions indeed allow higher shape complementarity in the SBP. Focusing on Asp192ECL2 resulted only in AC products with high selectivity for -Nb80. All products (except red_A04B52) targeting this residue had more than 100-fold selectivity for the active over the inactive conformation. This corroborates a possible role of Asp192ECL2 in activation (14). Finally, the products based on ketone bb.Bs (B51 and B52) yielded four products with medium to high affinities against -Nb80, confirming the importance of additional hydrophobic contacts. Of note, our highest-affinity compound (red_A05B51) with a of only 519 nM was obtained by fulfilling all three design hypotheses simultaneously: adding an additional rotatable bond, increasing hydrophobic contacts by using a ketone (which provides an additional methyl group), and interacting with Asp192ECL2 via a hydrogen bond.
Discussion
In this first application of SCUBIDOO, we have investigated whether forward-synthetic libraries based on existing building blocks can yield previously undescribed ligands even for a target for which vast numbers of ligands are already known. It could be argued that such databases contain little novelty as the building blocks they are based on are often derived from molecules with known biological affinity. However, we have found here that none of the ligands synthesized was found in SciFinder, i.e., has not, to the best of our knowledge, ever been made before (only the syntheses of three nonbinders, amd_A04B02, amd_A04B18, and amd_A05B02, have been described earlier). Moreover, we identified three ligands in the amide pool (amd_A10B25, amd_A10B37, and amd_A10B29) that have very low Tanimoto similarities compared with the closest known ligands and can thus be considered chemotypes without precedence. This sheds a bright light on the easily accessible novel chemical space that is right around the corner.
We have capitalized on the strengths of the database to efficiently extract ligands for the . The strategy is based on combinatorial building-block selection, using the fact that products in SCUBIDOO share reactions and building blocks. We coupled this in silico discovery procedure with parallel synthesis and a screening platform. In the first round, all steps took only about 6 weeks full-time equivalent and allowed us to create 127 previously nonexisting data points for the .
The highly designed combinatorial building-block selection procedure was enabled by docking. bb.As and bb.Bs were selected for binding-site complementarity and chemical diversity, leading to distinct binding modes. We made use of the fact that docking is often quite tolerant toward unsatisfied interaction opportunities. Normally, such molecules would be regarded as false positives, but we still accepted them if one half was forming favorable interactions. In doing so, we turned one of docking’s weaknesses into a strength. It is important to note that we never docked individual building blocks, only products (i.e., molecules consisting of a bb.A and a bb.B). This has two advantages: First, we avoid potential issues arising from the fact that many present-day docking scoring functions are optimized for molecules that are larger than fragments; second, we avoid artifacts caused by interactions of atom groups in a building block that would disappear during a reaction. The iterative process of subset extraction and docking described here eventually yielded diverse molecules, despite starting from only ∼10,000 compounds. As the process runs through more than one cycle, it can also be adapted: In our case, we noticed that several building blocks available in house might interact favorably in the SBP and included them, thus exploring this particular area of chemical space in more depth. Crucially, the selection was also driven by constraints imposed by synthetic chemistry: The number of 24 bb.Bs corresponded to the number of wells in the reaction blocks used, ensuring efficient use of resources. Parallel synthesis proved to be successful for amide coupling (83% success rate), but more challenging for reductive amination (21%). A second round of synthesis focusing on optimization of the reductive amination reaction conditions showed an improved synthesis rate of 68%, however, demonstrating that optimization of yields is well within reach. For the first round, a dedicated chemist would probably have obtained even higher synthesis success rates, had she tried to synthesize each product individually. This would have required several iterations, probably necessitating different reaction conditions, and would have likely taken several months. In this study, synthesis and purification were achieved within a few days and still yielded 127 of 240 molecules. Importantly, the initial SAR obtained with these compounds let us prioritize which compounds would be worthwhile to attempt again and/or optimize.
SCUBIDOO products facilitate SAR reasoning because of the native building-block– and reaction-based relationships. Every product is the assembly of two building blocks, and the contribution of each building block to each ligand can be evaluated. This post hoc deconstruction enables the straightforward identification of building blocks to be prioritized for optimization. Together with the occurrence of every building block in multiple products, this leads to an unprecedented density of SAR. As an example, the combination of the orthosteric fragment A03 with two different SBP fragments yielded ligands with opposite efficacy. This is remarkable, because it shows that ligand efficacy can be driven from the SBP alone. Such an analysis is less straightforward with ready-made molecules, likely requiring time-consuming retrosynthetic analysis.
In terms of affinity, the present results might be considered lagging behind earlier virtual screens that we conducted (7, 8). However, besides the top compounds (1 in ref. 7 and 3 in ref. 8), which can be considered outliers even in the context of their own campaigns, the remainder of the compounds identified here are in similar ranges. It can also not be ruled out that the protein fusions used in our assay lead to systematic affinity differences, therefore underestimating the affinity against the wild-type receptor. Similarly, while this article was in revision, another study appeared in which the authors explicitly screened 170 million compounds against two targets (6). While the numbers and final optimized affinities of their work are certainly impressive, the strength of our study is the speed and (cost) efficiency with which we obtained our compounds. With only 67,125 molecules docked (which represents only 0.72% of the chemical space covered by reductive amination and amide coupling in SCUBIDOO) after the original 9,994, we still obtained a compound with an affinity of 519 nM. Thus, despite very limited exploration, we identified relevant chemotypes. As, for reasons of combinatorics, chemical space can be grown much faster than computers become more powerful, smart strategies such as ours will be helpful in the midterm future. In the end, the calculation and synthesis capacities necessary to carry out ultralarge screens (6) are not available to most academic groups at present, a fact which has also been commented upon elsewhere (15).
An important difference from the aforementioned studies is also that we did not cherry pick just the top compounds, but were rather aiming for the establishment of a comprehensive SAR. In this context, some low-affinity compounds are to be expected. However, because each compound has multiple similar molecules, even those compounds carry information. Arguably, development of an SAR might not have been necessary for the , but is incredibly helpful for less well-explored targets. Forward-synthetic databases such as SCUBIDOO indeed appear as a valuable source for initial molecule suggestions during efficient ligand discovery projects. This was enabled by combining structure-based in silico ligand screening with parallel synthesis and a semi-high-throughput screening assay platform. Of course, further optimization of compounds will require medicinal chemistry modifications outside of SCUBIDOO, but can be grounded on a solid basis of SAR.
However, we do not consider the medicinal chemistry aspects to be the most important outcomes of this study: The iterative sublibrary-based computational screening approach and the efficient use of each fragment in multiple products put such large arrays of molecules and their biological evaluation within reach even of academic groups and small startups. This will undoubtedly accelerate the speed at which chemical space can be explored.
Methods
Reaction and Product Selection.
Each of the OPSBP and OPTBP directions was addressed separately, but in a similar way, as depicted in Fig. 2. After docking of the S subset of 9,994 products in the basal and active conformations of the receptor, the top 500 poses for each conformation were visually inspected. The products that contained a bb.A with favorable interactions in the OP were retained (step ① in Fig. 2). Importantly, suboptimal interactions of the respective bb.B in the SBP or TBP were not considered sufficient to purge such compounds. For each product, all derivatives featuring the same bb.A and obtainable by the same reaction as the original product (② and ③ in SI Appendix, Fig. S2) were extracted from SCUBIDOO and docked as well (④ in SI Appendix, Fig. S2). The poses of the derivatives were inspected for consistency of the binding mode of the bb.A in the OP (⑤ in Fig. 2), as well as favorable interactions of the bb.B in the SBP or TBP, respectively. Most of the virtual hits showed consistent poses in the basal conformation, and thus we decided to consider only this receptor during further processing, as it also features the larger binding pocket. Optionally, for each bb.A, the prestored, similar, analogs in SCUBIDOO were analyzed and products based on the analog bb.A and the identical reaction extracted from the database and docked (⑥ in SI Appendix, Fig. S3). This iterative process progressed also through the remaining molecules in the original docking ranked list of the S subset (⑦ in SI Appendix, Fig. S3) until sufficiently many bb.A candidates were available (⑧ in SI Appendix, Fig. S3). Besides the selection of a set of bb.As amenable to the same reaction, also a set of bb.Bs that would consistently entertain favorable interactions in the SBP or TBP, respectively, regardless of the bb.As they were coupled to, emerged (⑨ in SI Appendix, Fig. S3). We must note that, during analysis of the docking poses, we realized that some in house bb.Bs might also be valid choices providing favorable SBP or TBP interactions. Thus, we generated the corresponding products, docked them, and supplanted the original pool of BBs from in-house stocks, also to reduce overall costs (B18–B24). These building blocks can thus not be found in the original version of SCUBIDOO.
Supplementary Material
Acknowledgments
We thank Aline Keils and Eva Beke for technical assistance and Stefania Monteleone and Corey Taylor for critical comments on the manuscript. The purification group of Taros Chemicals is acknowledged for their efforts to make compound isolation possible. P.K. thanks the German Research Foundation (DFG) for Emmy Noether Fellowship KO4095/1-1 and Heisenberg Professorship KO4095/4-1. J.S. and P.K. participated in European Cooperation in Science and Technology (COST) Action CM1207 “GLISTEN” (GPCR-Ligand Interactions, Structures, and Transmembrane Signaling: A European Research Network). We thank Jaykumar Menon and Matthew Todd for initiating the Open Source Pharma movement, at whose Rauischholzhausen 2015 meeting this project was conceived.
Footnotes
Conflict of interest statement: S.S., A.K., and S.H. were employees of Taros Chemicals at the time of the study. D.T. is the founder and CEO of Taros Chemicals.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1818718116/-/DCSupplemental.
References
- 1.Barril X., Computer-aided drug design: Time to play with novel chemical matter. Expert Opin. Drug Discov. 12, 977–980 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Chevillard F., Kolb P., SCUBIDOO: A large yet screenable and easily searchable database of computationally created chemical compounds optimized toward high likelihood of synthetic tractability. J. Chem. Inf. Model. 55, 1824–1835 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Opassi G., Gesù A., Massarotti A., The hitchhiker’s guide to the chemical-biological galaxy. Drug Discov. Today 23, 565–574 (2018). [DOI] [PubMed] [Google Scholar]
- 4.van Hilten N., Chevillard F., Kolb P., Virtual compound libraries in computer-assisted drug discovery. J. Chem. Inf. Model. 59, 644–651 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Klucznik T., et al. , Efficient syntheses of diverse, medicinally relevant targets planned by computer and executed in the laboratory. Chem 4, 522–532 (2018). [Google Scholar]
- 6.Lyu J., et al. , Ultra-large library docking for discovering new chemotypes. Nature 566, 224–229 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kolb P., et al. , Structure-based discovery of -adrenergic receptor ligands. Proc. Natl. Acad. Sci. U.S.A. 106, 6843–6848 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schmidt D., Gunera J., Baker J. G., Kolb P., Similarity- and substructure-based development of -adrenergic receptor ligands based on unusual scaffolds. ACS Med. Chem. Lett. 8, 481–485 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chevillard F., et al. , Binding-site compatible fragment growing applied to the design of -adrenergic receptor ligands. J. Med. Chem. 61, 1118–1129 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Pardon E., et al. , Nanobody-enabled reverse pharmacology on GPCRs. Angew. Chem. Int. Ed. 57, 5292–5295 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Blakemore D. C., et al. , Organic synthesis provides opportunities to transform drug discovery. Nat. Chem. 10, 383–394 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Nadin A., Hattotuwagama C., Churcher I., Lead-oriented synthesis: A new opportunity for synthetic chemistry. Angew. Chem. Int. Ed. 51, 1114–1122 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Cooper T. W., Campbell I. B., Macdonald S. J., Factors determining the selection of organic reactions by medicinal chemists and the use of these reactions in arrays (small focused libraries). Angew. Chem. Int. Ed. 49, 8082–8091 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Ring A. M., et al. , Adrenaline-activated structure of 2-adrenoceptor stabilized by an engineered nanobody. Nature 502, 575–579 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gloriam D. E., Bigger is better in virtual drug screens. Nature 566, 193–194 (2019). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.