

Abstract
A series of seventeen hydroxyl-containing sphingosine 1-phosphate receptor 1 (S1PR1) ligands were designed and synthesized. Their in vitro binding potencies were determined using [32P]S1P competitive binding assays. Compounds 10a, 17a, 17b, and 24 exhibited high S1PR1 binding potencies with IC50 values ranging from 3.9 to 15.4 nM and also displayed high selectivity for S1PR1 over other S1P receptor subtypes (IC50 > 1000 nM for S1PR2–5). The most potent compounds 10a, 17a, 17b, and 24 were subsequently radiolabeled with F-18 in high yields and purities. MicroPET studies in cynomolgus macaque showed that [18F]10a, [18F]17a, and [18F]17b but not [18F]24 crossed the blood brain barrier and had high initial brain uptake. Further validation of [18F]10a, [18F]17a, and [18F]17b in preclinical models of neuroinflammation is warranted to identify a suitable PET radioligand to quantify S1PR1 expression in vivo as a metric of an inflammatory response.
Introduction
Sphingosine 1-phosphate (S1P) is a membrane-derived lysophospholipid that plays a critical regulatory role in inflammatory diseases.1–3 S1P acts by modulating five highly specific G protein-coupled receptors (GPCRs, S1PR1–5) and has high binding affinities (nanomolar) with these five receptors.4–8 The unique patterns of cellular and temporal expression determine the specific roles of S1PR1–5 in each organ system. Modulation of these receptors is particularly important in the central nervous, cardiovascular, and immune systems. Among these five-receptor subtypes, S1PR1 is one of the most abundant receptors in the entire GPCR superfamily. S1PR1 was originally called the endothelial differentiation growth factor receptor 1 (EDG-1). It is particularly enriched in endothelial cells and vascular smooth muscle cells, and also expressed on immune cells such as lymphocytes.9 The relevance of S1PR1 in clinical disease has become readily apparent with the U.S. Food and Drug Administration (FDA) approval of the S1PR1 modulator fingolimod (FTY720) for treating relapsing-remitting multiple sclerosis (RR-MS). MS is a chronic autoimmune, inflammatory disease caused by lymphocytic infiltration that leads to a demyelinating neurodegenerative disease with no cure. RR-MS is the most common form, characterized by intermittent relapses that lead to increasing functional disability that in large part may be mediated by neuroinflammatory responses in the brain.10 Fingolimod, structurally similar to S1P, functionally modulates S1PR1 on lymphocytes, leading to S1PR1 downregulation and subsequent lymphocyte trapping in lymph nodes. This reduces the peripheral blood lymphocyte count and presumably lymphocyte infiltration in the brain. The effects of fingolimod on S1PR1 expression in the central nervous system (CNS) itself and its contribution to treatment efficacy are not well understood. S1PR1 is expressed in neurons and glia in the CNS, including astrocytes, which modulate inflammatory responses throughout the gray and white matter; microglia, the resident macrophages in the CNS, and oligodendrocytes, which produce the myelin needed for nerve conduction. In the in vitro models of demyelination induced by lysophosphatidylcholine (LPS), fingolimod reduced demyelination and neuronal process extension in the CNS, presumably through modulation of S1PR1.11–13 It also inhibited microglial activation and reduced oligodendrocyte apoptosis.13 Specific deletion of S1PR1 in astrocytes resulted in decreased experimental autoimmune encephalomyelitis (EAE) pathology in vivo and a loss of fingolimod efficacy, suggesting that fingolimod also acts on S1PR1 in astrocytes.14,15 Fingolimod-treated MS patients exhibited reduced brain volume loss, lesion numbers, and severity,14,16–18 confirming the in vitro and animal model findings that S1PR1 pathways play important roles in anti-inflammation and neuroprotection. A S1PR1-targeted positron emission tomography (PET) radiotracer would help us understand the pathophysiology of S1PR1 in the CNS. Imaging S1PR1 in vivo could be used to develop therapies that improve upon the initial treatment successes with fingolimod. Furthermore, S1PR1 is a therapeutic target of great interest for a number of other inflammatory diseases, including inflammatory bowel disease19 and atherosclerotic disease,20,21 as well as cancer, where S1PR1 expression promotes treatment resistance22,23 and enhances metastatic potential.23–26 S1PR1 imaging would enable mechanistic studies and S1PR1-specific readouts of targeted therapy in a wide range of inflammatory diseases. Using PET with the promising C-11 radiotracer [11C] TZ3321, we demonstrated increased S1PR1 expression at inflammatory response sites in three animal models: the rat EAE model of MS, the ApoE−/− mouse femoral artery wire-injury model of neointimal hyperplasia, and the rat carotid injury model of vascular inflammation.27–29 The longer half-life of F-18 (T1/2 = 109.8 min) reduces time constraints on tracer production, distribution, and permits longer imaging sessions for higher target-to-reference ratios than is possible with C-11. Therefore, the PET radiochemistry community has focused on the development of potent and selective [18F]-labeled S1PR1 radiotracers to quantitatively measure the changes of S1PR1 expression in response to inflammation. Several [18F]-labeled small molecules have been characterized for their suitability to be [18F]-labeled PET radiotracers for imaging S1PR1 in vitro and in vivo (Fig. 1). However, either they had inadequate in vivo metabolic stability or did not penetrate the blood brain barrier (BBB). For instance, three [18F]-labeled fingolimod derivatives, [18F]3, [18F]4a, [18F]4b reported by Haufe’s group showed fast kinetics, low selectivity, or defluorination in vivo, which prevented their transfer into clinical investigation.30,31 Our group explored oxadiazole-containing analogues and reported two [18F]fluoroethoxy radiotracers, one having an azetidine-3-carboxylic acid ([18F]5), and another tracer having a 1H-1,2,3-triazole-4-methanol ([18F]6).32,33 Although the in vivo study suggested that the mouse liver uptake of [18F]5 had a strong positive correlation with acute liver inflammation induced by LPS injection, this radiotracer has limited penetration of the BBB for imaging neuroinflammation. Our preliminary data revealed that [18F]6 exhibited a high rat brain uptake with 0.71%ID per gram at 60 min post injection and no in vivo defluorination. A specific S1PR1 ligand reduced the uptake of [18F]6 binding to S1PR1 in tissue sections of the inflamed brain from an LPS-induced neuroinflammation mouse model. But follow-up microPET imaging studies of [18F]6 in cynomolgus macaque suggested this radiotracer did not penetrate the nonhuman primate (NHP) brain (seen in ESI†). Species differences in the BBB penetration ability of PET radiotracers have been previously reported for other PET neuroimaging radiotracers.34 Consequently, neither [18F]5 nor [18F]6 is a suitable PET S1PR1 radiotracer to assess the neuroinflammatory responses in humans. To overcome the above-mentioned challenges in the identification of a suitable [18F]-labeled S1PR1 radiotracer for neuroinflammation response in CNS disorders, we focused on further optimization of the oxadiazole analogues by replacing the carboxylic acid moiety with hydroxyl group(s) in the terminal of substituted side chain. Our previous rat biodistribution of [18F]6 showed good brain uptake compared to carboxylic acid containing analogues. Herein, we reported the syntheses of new serial of terminal hydroxyl-containing S1PR1 compounds, in vitro binding assays to determine their binding potencies and selectivity for S1PR1, radiosyntheses of four F-18 radiotracers, and microPET studies of these four radiotracers in the cynomolgus macaque brain to investigate their in vivo kinetics and ability to penetrate the BBB.
Fig. 1.

Structures of fingolimod and S1PR1 radiotracers.
Results and discussion
To identify an [18F]-labeled radiotracer with high potency, high selectivity, and the capability to penetrate the BBB. Initially, the (1,2,3-triazol-4-yl)methanol moiety in compound 6 was replaced by hydroxyl methyl functional group to generate compound 10a, we then replaced the trifluoromethyl group with a cyano or hydrogen group (Scheme 1). Next, we turned our attention to retaining the trifluoromethyl group and replaced the hydroxyl methyl group of 10a with alkyl alcohols, amino alcohols, and ether alcohols. In total, seventeen new analogues were designed, synthesized and screened for in vitro and in vivo properties. The syntheses of the target compounds are depicted in Schemes 1–5 following reported procedures with necessary modifications.32,33 For compounds 10a–c, syntheses were initiated with 4-(hydroxymethyl)-benzonitrile (7) treated with hydroxylamine hydrochloride in presence of NaHCO3 to yield amidoxime 8. Condensation and cyclization of the amidoxime 8 with aromatic carboxylic acids 9a–c in DMF in presence of 2-(1H-benzotriazole-1-yl) 1,1,3,3-tetra-methylaminium tetrafluoroborate (TBTU) and a catalytic amount of 1-hydroxybenzo-triazole hydrate (HOBt) to generate compounds 10a–c (Scheme 1).
Scheme 1.

Syntheses of 10a–c. Reagents and conditions: (a) Hydroxylamine hydrochloride, NaHCO3, methanol, reflux; (b)TBTU, HOBt, DIPEA, DMF, RT–120 °C.
Scheme 5.

Syntheses of (±)-24. Reagents and conditions: (a) Hydroxylamine hydrochloride, NaHCO3, methanol, reflux; (b) 9a, TBTU, HOBt, DIPEA, DMF, RT–120 °C; (c) 4-methylmorpholine N-oxide, OsO4, THF, H2O, RT.
Compounds 13a–c were synthesized by employing a similar procedure described for compounds 10a–c. Treatment of compounds 11a–c with hydroxylamine hydrochloride yielded the intermediates 12a–c, which were subjected to condensation and then cyclization with compound 9a to yield compounds 13a–c. Compound 13d was prepared by deprotection of tert-butyloxy-carbonyl (Boc) group using 4.0 M HCl in dioxane (Scheme 2).
Scheme 2.

Syntheses of 13a–d. Reagents and conditions: (a) Hydroxylamine hydrochloride, NaHCO3, methanol, reflux; (b) 9a, TBTU, HOBt, DIPEA, DMF, RT–120 °C; (c) 4.0 M HCl in dioxane, RT.
Compound 10a was utilized as a key intermediate for the syntheses of analogues 15a–b and 17a–g. Swern oxidation of the primary alcohol in compound 10a with oxalyl chloride in the presence of triethylamine at −78 °C to yield the intermediate aldehyde 14,32 followed by reductive amination using corresponding amines generated the amino alcohol compounds 15a–b (Scheme 3).
Scheme 3.

Syntheses of 15a–b and 17a–g. Reagents and conditions: (a) Oxalyl chloride, DMSO, CH2Cl2, triethylamine, −78 °C–RT; (b) NaBH3CN amines, acetic acid, methanol, RT; (c) cyanuric chloride, DMF, CH2Cl2, RT; (d) NaH, alcohols, THF, reflux; (e) 4.0 M HCl in dioxane, RT.
Chlorination of compound 10a was achieved by reaction with cyanuric chloride in DMF to afford the intermediate chloride 16, followed by nucleophilic substitution with various alcohols to yield compounds 17a–e and 17g. Compound 17f was generated by deprotection of Boc group of 17e (Scheme 3).
Compounds 20a–b were synthesized using the corresponding 4-(2-hydroxy-ethoxy)benzonitrile (18a) or 4-((2,3-dihydroxy-propoxy)methyl)benzonitrile (18b) to generate amidoximes 19a or 19b, followed by condensation and cyclization with 9a to yield compounds 20a or (±)-20b (Scheme 4).
Scheme 4.

Syntheses of 20a–b. Reagents and conditions: (a) Hydroxylamine hydrochloride, NaHCO3, methanol, reflux; (b) 9a, TBTU, HOBt, DIPEA, DMF, RT–120 °C.
Finally, compound (±)-24 was prepared from 4-vinylbenzonitrile (21) reacted with hydroxylamine hydrochloride to yield the intermediate compound 22, which was subjected to condensation followed by cyclization with compound 9a to yield compound 23. The terminal olefin in compound 23 was oxidized with 4-methylmorpholine N-oxide and catalytic OsO4 in THF resulting in target compound (±)-24 (Scheme 5).
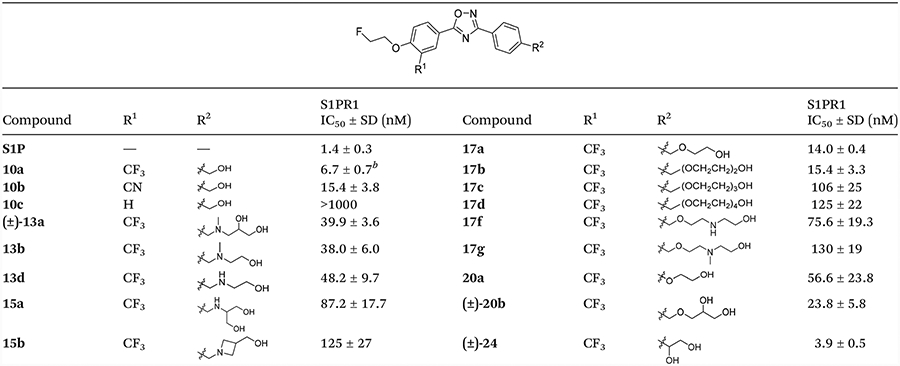
We determined the in vitro binding potency of these compounds to S1PR1 as a measure of biological activity by following our published radioligand assay using freshly synthesized [32P]S1P.32,33,35 The S1PR1 binding data of these new analogues are presented in Table 1. Here we present our observations regarding structure activity relationships. First, among compounds 10a–c, compound 10a with an electron withdrawing trifluoromethyl at the meta-position of the oxadiazole moiety displayed a high binding potency with an IC50 value of 6.7 nM for S1PR1 receptor.32 Replacement of the trifluoro-methyl with a cyano, also an electron withdrawing group, resulted in a 2.3-fold decreased potency (10b, IC50 = 15.4 ± 3.8 nM). Removal of the trifluoromethyl group significantly reduced the binding potency of compound 10c (IC50 > 1000 nM). Because the CF3 group is a key functional group for retaining high S1PR1 potency, we retained the trifluoromethyl group in our further exploration of new analogues. In the second series, different amino alcohols were introduced for compounds (±)-13a, 13b, 13d, 15a, and 15b. We wanted to determine if different amino alcohol-containing groups caused significant changes of S1PR1 binding potency. Our in vitro data showed that all the amino alcohol derivatives (±)-13a, 13b, 13d, 15a, and 15b were less potent than compound 10a with IC50 values of 39.9, 38.0, 48.2, 87.2, and 125 nM, respectively. The above results suggest that the amino alcohols are not favorable to improve the S1PR1 binding potency.
Table 1.
Structures and binding potencies (mean ± SD) of S1P and newly synthesized compounds against S1PR1a
|
IC50 values were determined with at least 3 independent experiments, each run was performed in duplicate.
Ref. 32.
In the third series, we PEGylated compound 10a using different PEG units. PEGylation has been reported as an effective strategy for improving the solubility, stability, pharmacodynamics, and pharmacokinetics of drug candidates interacting with biologically interesting proteins or peptides.36–42 The addition of a small number of PEGylaton units (n = 2, 3, 4) is frequently used to improve a compound’s hydrophilicity which may facilitate crossing the BBB. As one example, [18F]AV45, the well-known radiotracer used for imaging β-amyloid (Aβ) aggregates in the AD brain contains three PEGylation units.41 Therefore, we next synthesized compounds 17a–d with 1–4 PEGs and tested their binding potency. Our in vitro binding data showed that compound 17a and 17b with one and two PEG units had similar binding potencies with IC50 values of 14.0 and 15.4 nM, respectively. Compound 17c (IC50 = 106 nM) with three PEGs, had approximately 16-fold lower potency compared to 10a. Compound 17d (IC50 = 125 nM) with four PEGs also showed further decreased binding potency.
The in vitro S1P binding data of these new PEG-containing compounds suggested that the addition of one or two PEG units retained S1PR1 binding potency, but the extension with more PEG chains (>2 units) decreased S1PR1 binding potency. In addition, for compounds 17f and 17g, in which a secondary or tertiary amine was introduced in the PEG chains, S1PR1 binding potencies decreased with IC50 values of 75.6 and 130 nM, respectively. Finally, we modified compound 20a with a 2-hydroxyethoxyl group directly linked to a phenyl group, this approach also reduced binding potency (IC50 = 56.6 nM) when compared to compound 17a with a 2-hydroxy ethoxyl linked with a benzyl group. Further optimization of 17a was conducted by introducing a terminal hydroxyl group using glycerin to yield compound (±)-20b, which showed an IC50 value of 23.8 nM, which is less potent than the glycol tail compound 17a. The alkyl di-alcohol compound (±)-24, which incorporated an ethylene glycol directly linked with phenyl group was found to be the most potent S1PR1 ligand of this study with an IC50 value of 3.9 nM. Because compounds 10a, 17a, 17b, and (±)-24 each had high binding potency for S1PR1 with IC50 < 20 nM, these four compounds were screened to determine their binding potency to other S1P receptor subtypes S1PR2, 3, 4 and 5 to check their binding selectivity. Our in vitro data shown in Table 2 revealed that all four compounds had no significant binding toward S1PR2–5 (IC50 > 1000 nM), indicating that they are selective for S1PR1 over other S1PR subtypes S1PR2, 3, 4, and 5.
Table 2.
Binding potencies (mean ± SD) of 10a, 17a–b, and (±)-24 toward S1PR1–5 and Clog P values
| IC50a (nM) | ||||||
|---|---|---|---|---|---|---|
| Compd. | S1PR1 | S1PR2 | S1PR3 | S1PR4 | S1PR5 | Clog Pb |
| S1P | 1.4 ± 0.3 | 3.6 ± 0.5 | 0.4 ± 0.2 | 151 ± 82 | 3.1 ± 1.1 | |
| 10a | 6.7 ± 0.7 | >1000 | >1000 | >1000 | >1000 | 4.00 |
| 17a | 14.0 ± 0.4 | >1000 | >1000 | >1000 | >1000 | 4.02 |
| 17b | 15.4 ± 3.3 | >1000 | >1000 | >1000 | >1000 | 3.85 |
| (±)−24 | 3.9 ± 0.5 | >1000 | >1000 | >1000 | >1000 | 3.09 |
IC50 values were determined with at least 3 independent experiments, each run was performed in duplicate; assays for compounds which showed no activity (IC50 > 1000 nM) were repeated twice.
Clog P values were calculated by PerkinElmer ChemDraw® 16.0.1.4 (77).
Consequently, these four compounds were radiolabeled with F-18 to check their potential to be neuroimaging PET radiotracers for quantifying S1PR1 expression in the brain. Prior to radiolabeling, the corresponding precursors 27a–d were synthesized by following Scheme 6. Methoxymethyl (MOM) protected hydroxybenzimidamide 25a–d were prepared by following the general procedure described for 12a–c with MOM protected benzonitriles. The condensation and cyclization of 25a–d and 4-hydroxy-3-(trifluoro-methyl)benzoic acid afforded 26a–d, which were subjected to O-alkylation with ethylene ditosylate to afford MOM-protected precursors 27a–d. The radiosyntheses of [18F]10a, [18F]17a, [18F]17b, and (±)-[18F] 24 were successfully achieved by using nucleophilic reaction between each tosylate precursor and [18F]KF in acetonitrile with Kryptofix 222, followed by deprotection of MOM group. The radioactive products were purified using a semi-preparative high performance liquid chromatography (HPLC) and then formulated using 10% of ethanol saline solution. Each dose of [18F]10a, [18F]17a, [18F]17b, and (±)-[18F]24 was authenticated by co-injecting with corresponding cold standard compound 10a, 17a, 17b, and (±)-24, respectively. These four radiotracers were achieved with radiochemical yields of 35–40%, radiochemical purities >98%, and molar activities >74 GBq μmol−1 (decay corrected to end of synthesis, EOS).
Scheme 6.

General syntheses of precursors 27a–d and radiotracers [18F]10a, [18F]17a, [18F]17b, and (±)-[18F]24. Reagents and conditions: (a) 4-Hydroxy-3-(trifluoromethyl)benzoic acid, TBTU, HOBt, DIPEA, DMF, RT–120 °C; (b) ethylene ditosylate, K2CO3, CH3CN, 90 °C; (c) [18F]KF, Kryptofix 222, K2CO3, CH3CN, 110 °C, 15 min, 6 N HCl, 5 min.
To check if each radiotracer had the capability to penetrate the BBB with sufficient accumulation in the animal brain, microPET studies of the brain of a cynomolgus macaque were performed for each F-18 labeled radiotracer on different days. Macaque did not have studies more frequently than every 2 weeks to allow the subject to recover. Time activity curves (TACs) showing brain uptake and washout from our microPET studies of these four F-18 radiotracers are shown in Fig. 2, brain TACs indicated that three radiotracers [18F]10a, [18F]17a, and [18F]17b were able to cross the BBB and had high initial brain uptake in the nonhuman primates.
Fig. 2.

Representative time-activity curves of microPET studies for [18F]10a, [18F]17a, [18F]17b and (±)-[18F]24 in brains of nonhuman primates. The curves indicated that three radiotracers [18F]10a, [18F]17a and[18F]17b achieved the max SUV value fast, at 4–6 min post injection. [18F]10a had the highest initial brain uptake while [18F]17b exhibited relatively slow washout kinetics. (±)-[18F]24 displayed the lowest initial uptake in the brain, indicating it has no capability in penetrating the BBB of nonhuman primate.
The brain standard uptake value (SUV) for [18F]10a, [18F]17a, and [18F]17b reached a maximum at 4–6 min after tracer injection. [18F]10a had the highest initial brain uptake and quick washout kinetics from the brain while the brain washout kinetics of [18F]17b was relatively slow. For the most potent compound (±)-24, with an IC50 value of 3.9 nM, our microPET studies of (±)-[18F]24 indicated that it did not penetrate the BBB and its brain accumulation was very low. Lipophilicity of small molecule drug is a key parameter affecting penetration of the BBB. log P value is an index of small molecular lipophilicity. Most drugs that are active in the central nervous system have log P values that range from 1.5 to 2.7,43 though some have values beyond this range. The calculated log P (Clog P) values (Table 2) of these four tracers were 4.00, 4.02, 3.85, and 3.09 for [18F]10a, [18F]17a, [18F]17b, and (±)-[18F]24, respectively. The most potent radiotracer (±)-[18F]24 (IC50 value of 3.9 nM for S1PR1) had a log P value of 3.09 which would predict better BBB penetration. The unanticipated low nonhuman primate brain uptake of (±)-[18F]24 despite its lipophilicity may have resulted from the two hydroxyl groups causing different in vivo pharmacological properties, such as binding to P-glycoprotein (Pgp).44 Further investigation would be needed to understand the inability of (±)-[18F]24 in penetrating BBB.
Together, our microPET studies in nonhuman primates suggested that radiotracers [18F]10a, [18F]17a, and [18F]17b have potential to be promising S1PR1 PET radiotracers for imaging the neuroinflammatory response in vivo. Further investigations in animal models of neuroinflammatory diseases are warranted to characterize their radiopharmaceutical properties.
Conclusions
In conclusion, we developed a series of new S1PR1 ligands and screened in vitro binding potencies for all the newly synthesized compounds. Compounds 10a, 17a, 17b, and (±)-24 exhibited high potency (IC50 < 20 nM) and high selectivity for S1PR1 (IC50 > 1000 nM over S1PR2, 3, 4, and 5). The radiosyntheses of [18F]10a, [18F]17a, [18F]17b, and (±)-[18F]24 were accomplished with good yields (35–40%), high radiochemical and chemical purities (>98%), and reasonable molar activities (>74 GBq μmol−1). MicroPET studies in cynomolgus macaque indicated that [18F]10a, [18F]17a, and [18F]17b can cross the BBB of NHPs and have good brain uptake while (±)-[18F]24 was not able to penetrate the BBB. Further in vivo evaluation of [18F]10a, [18F]17a, and [18F]17b in animal models of inflammatory disease mechanisms is needed to check the suitability of these radiotracers for assessing the neuroinflammatory response by measuring the S1PR1 expression prior to translation of a lead radiotracer to clinical investigations.
Experimental section
General information
All reagents and chemicals were obtained from standard commercial sources and used without further purification unless otherwise stated. Organic reactions were carried out under inert nitrogen and moisture-free with dry solvent. Thin layer chromatography (TLC) was used to monitor the reaction. Final organic products were purified by flash column chromatography using 230–400 mesh silica gel purchased from Silicycle. Melting points were determined on a MEL-TEMP 3.0 apparatus without correction. All deuterated solvents were purchased from Cambridge Isotope Laboratories. 1H and 13C NMR spectra were recorded on a 400 MHz Varian instrument. Chemical shifts were reported in parts per million (ppm) and were calibrated using a residual undeuterated solvent as an internal reference (CDCl3: δ 7.26 ppm; CD3OD: δ 3.31 ppm; DMSO-d6: δ 2.50 ppm; acetone-d6: δ 2.05 ppm). Multiplicities are indicated by s (singlet), d (doublet), t (triplet), q (quartet), p (pentet), h (hextet), m (multiplet) and br (broad). High-resolution positive ion mass was acquired by a Bruker MaXis 4G Q-TOF mass spectrometer with electrospray ionization source. The welfare of the animals conformed to the requirements of National Institutes of Health (NIH). This work was conducted at the NHP Facility of Washington University in St Louis with approval from the IACUC.
(E)-N′-Hydroxy-4-(hydroxymethyl)benzimidamide (8).
Synthesis of compound 8 followed the published procedure.33
General procedure for the synthesis of 10a–c
To a round-bottom flask equipped with a stir bar was added acid 9a–c (1.0 eq.), HOBt (0.2 eq.), TBTU (1.0 eq.), DIPEA (3.0 eq.), and DMF (5.0 mL mmol−1). The reaction mixture was stirred for 0.5 h followed by adding amidoxime 8 (1.0 eq.). The reaction mixture was stirred for 1 h at room temperature (RT), then refluxed in a pre-heated 120 °C oil-bath for 4 h and monitored by TLC. After cooling, the reaction mixture was diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with 1 M HCl, saturated brine, and dried over anhydrous MgSO4. After filtration and concentration, the residue was purified on a silica gel column to give the product 10a–c.
(4-(5-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)phenyl)methanol (10a).
The characterization is found in our previous publication.32
2-(2-Fluoroethoxy)-5-(3-(4-(hydroxymethyl)phenyl)-1,2,4-oxadiazol-5-yl)benzonitrile (10b).
Compound 10b was purified by flash chromatography, eluted with hexane/ethyl acetate (1/1, v/v) to afford white solid product. Yield: 30%, MP: 150–151 °C. 1H NMR (400 MHz, acetone-d6) δ 8.52–8.34 (m, 2H), 8.14–7.95 (m, 2H), 7.64–7.43 (m, 3H), 5.04–4.85 (m, 2H), 4.78 (d, J = 5.0 Hz, 2H), 4.73–4.60 (m, 2H), 4.49 (br, 1H). 13C NMR (101 MHz, acetone-d6) δ 173.8, 168.6, 163.1, 146.3, 134.2, 133.5, 127.1, 126.8, 125.1, 117.4, 114.6, 113.7, 102.9, 81.5 (d, JC–F = 170 Hz), 69.1 (d, JC–F = 18.1 Hz), 63.3. HRMS (ESI) calcd for C18H14F1N3O3 [M + H]+ 340.1092, found 340.1087.
(4-(5-(4-(2-Fluoroethoxy)phenyl)-1,2,4-oxadiazol-3-yl)-phenyl)-methanol (10c).
Compound 10c was purified by flash chromatography, eluted with hexane/ethyl acetate (1/1, v/v) to afford white solid product. Yield: 45%, MP: 140–142 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.21–8.12 (m, 2H), 8.05 (d, J = 8.2 Hz, 2H), 7.54 (d, J = 8.0 Hz, 2H), 7.24 (d, J = 8.9 Hz, 2H), 5.38 (s, 1H), 4.94–4.74 (m, 2H), 4.61 (s, 2H), 4.53–4.34 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 175.5, 168.5, 162.4, 146.8, 130.4, 127.4, 127.3, 125.0, 116.5, 115.9, 83.2, 81.6, 68.0, 67.8, 62.9. HRMS (ESI) calcd for C17H15FN2O3 [M + H+] 315.1139, found 315.1139.
4-(((2,3-Dihydroxypropyl)(methyl)amino)methyl)benzonitrile (11a).
To a round-bottom flask equipped with a stir bar was added 4-(bromomethyl)benzonitrile (3.9 g, 20.0 mmol), 3-(methylamino)propane-1,2-diol (2.5 g, 23.0 mmol), K2CO3 (6.9 g, 50.0 mmol), and acetonitrile (50 mL). The mixture was stirred overnight at RT. Then, the mixture was concentrated under reduced pressure, the residue was purified by flash chromatography, eluted with hexane/ethyl acetate (1/1, v/v) to afford oil product. (3.0 g, 68%) 1H NMR (400 MHz, CDCl3) δ 7.64–7.56 (m, 2H), 7.44–7.36 (m, 2H), 3.88–3.79 (m, 1H),3.77–3.66 (m, 2H), 3.60–3.44 (m, 2H), 2.70–2.60 (m, 1H),2.43–2.34 (m, 1H), 2.25 (s, 3H).
4-(((2-Hydroxyethyl)(methyl)amino)methyl)benzonitrile (11b).
To a round-bottom flask equipped with a stir bar was added 4-(bromomethyl)benzonitrile (3.0 g, 15.3 mmol), 2-(methylamino)ethan-1-ol (1.0 g, 13.3 mmol), and acetonitrile (20 mL). After cooling to 0 °C, triethylamine (2.8 g, 27.7 mmol) was added to the mixture dropwise. The mixture was warmed to RT and stirred overnight. Then, the mixture was concentrated in vacuum, and the crude residue was purified by flash chromatography, eluted with hexane/ethyl acetate (3/2, v/v) to afford the semi-solid product. (2.0 g, 79%) 1H NMR (400 MHz, CDCl3) δ 7.63–7.58 (m, 2H), 7.42 (d, J = 7.9 Hz, 2H), 3.68–3.59 (m, 5H),2.63–2.57 (m, 2H), 2.23 (s, 3H).
Tert-Butyl (4-cyanobenzyl)(2-hydroxyethyl)carbamate (11c).
To a round-bottom flask equipped with a stir bar was added 4-formylbenzonitrile (6.0 g, 45.8 mmol), ethanolamine (2.8 g, 45.8 mmol), and methanol (80 mL). The mixture was stirred in a pre-heated 60 °C oil-bath for 6 h, then cooled to 0 °C followed by adding sodium borohydride (8.7 g, 229 mmol) portionwise. The reaction was warmed to room temperature (RT) slowly and stirred for an additional 5 h. Then, the reaction mixture was concentrated under reduced pressure, and the residue was dissolved in ethyl acetate (200 mL), washed with water (100 mL) and saturated brine (100 mL), and dried over anhydrous MgSO4. After filtering and concentration, the residual solid was re-dissolved in dichloromethane (100 mL). To the above solution was added di-tert-butyl dicarbonate (6.7 g, 30.6 mmol) and 1 N NaOH (17.0 mL). The mixture was stirred at RT overnight, then the mixture was washed with water and saturated brine, dried over anhydrous MgSO4. After filtering and concentration, the crude semi-solid product 11c was obtained. (8.6 g, 68%) 1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 7.8 Hz, 2H), 7.38 (d, J = 8.0 Hz, 2H), 4.58 (s, 2H), 3.74 (s, 2H), 3.52–3.29 (m, 3H), 1.41 (s, 9H).
General procedure for the synthesis of 12a–c
To a round-bottom flask equipped with a stir bar was added 11a–c (1.0 eq.), hydroxylamine hydrochloride (2.0 eq.), NaHCO3 (4.0 eq.), and methanol (5.0 mL mmol−1). The reaction was refluxed and stirred in a pre-heated 75 °C oil-bath for 6 h. The reaction mixture was cooled to room temperature, and the precipitate was filtered off and washed with methanol. The filtrate was concentrated under reduced pressure to give the product 12a–c and used directly without further purification.
(E)-4-(((2,3-Dihydroxypropyl)(methyl)amino)methyl)-N′-hydroxybenzimidamide (12a).
Yield: 51%, MP: 160–164 °C. 1H NMR (400 MHz, MeOD-d4) δ 7.60 (d, J = 7.8 Hz, 2H), 7.37 (d, J = 7.9 Hz, 2H), 3.85–3.78 (m, 1H), 3.66–3.58 (m, 3H), 3.57–3.43 (m, 3H), 2.52–2.45 (m, 2H), 2.25 (s, 3H).
(E)-N′-Hydroxy-4-(((2-hydroxyethyl)(methyl)amino)methyl)-benzimidamide (12b).
Yield: 60%, MP: 162–165 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.57 (s, 1H), 7.61 (d, J = 7.1 Hz, 2H), 7.30 (d, J = 7.4 Hz, 2H), 5.76 (s, 2H), 4.39 (s, 1H), 3.50 (s, 4H), 2.42 (t, J = 6.2 Hz, 2H), 2.15 (s, 3H).
Tert-Butyl (E)-(4-(N′-hydroxycarbamimidoyl)benzyl)(2-hydroxyethyl)carbamate (12c).
Yield: 71%, MP: 138–141 °C. 1H NMR (400 MHz, CDCl3) δ 7.66–7.53 (m, 2H), 7.35–7.26 (m, 2H), 5.30 (s, 2H), 4.86 (s, 2H), 4.52 (s, 2H), 3.71 (s, 2H), 3.47–3.31 (m, 2H), 1.46 (s, 9H).
(±)-3-((4-(5-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)benzyl)(methyl)amino)propane-1,2-diol (13a).
Compound 13a was synthesized according to the general procedure as described for compound 10a–c. Yield: 40%, white solid, MP: 101–103 °C. 1H NMR (400 MHz, MeOD-d4) δ 8.29 (d, J = 7.1 Hz, 2H), 8.01 (d, J = 8.0 Hz, 2H), 7.47 (d, J = 8.1 Hz, 2H),7.35 (d, J = 9.4 Hz, 1H), 4.83–4.69 (m, 3H), 4.48–4.37 (m, 3H), 3.87–3.80 (m, 1H), 3.63 (s, 2H), 3.57–3.46 (m, 2H), 2.53–2.49 (m, 2H), 2.28 (s, 3H). 13C NMR (101 MHz, MeOD-d4) δ 174.3, 168.5, 159.8, 142.1, 133.3, 129.5, 126.9, 126.6 (q, JC–F = 5.4 Hz), 125.4, 122.9 (q, JC–F = 274 Hz), 119.3 (q, JC–F = 31 Hz), 116.4, 113.8, 81.2 (d, JC–F = 171 Hz), 68.9, 68.6 (q, JC–F = 20 Hz), 64.9, 62.0, 60.1, 41.7. HRMS (ESI) calcd for C22H23F4N3O4 [M + H+] 470.1697, found 470.1694.
2-((4-(5-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)benzyl)(methyl)amino)ethan-1-ol (13b).
Compound 13b was prepared according to the general procedure as described for compound 10a–c. Yield: 34%, white solid, MP: 108–111 °C. 1H NMR (400 MHz, CDCl3) δ 8.45 (s, 1H), 8.34 (d, J = 8.7 Hz, 1H), 8.11 (d, J = 8.1 Hz, 2H), 7.45 (d, J = 8.1 Hz, 2H),7.17 (d, J = 8.8 Hz, 1H), 4.90–4.74 (m, 2H), 4.47–4.37 (m, 2H), 3.68–3.62 (m, 4H), 2.64 (t, J = 5.3 Hz, 2H), 2.26 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 174.2, 168.8, 159.9, 142.1, 133.3, 129.4, 127.8 (q, JC–F = 5.25 Hz), 127.6, 125.6, 122.7 (q, JC–F = 274 Hz), 120.2 (q, JC–F = 32 Hz), 117.1, 113.4, 81.2 (d, JC–F = 172 Hz), 68.4 (d, JC–F = 21.2 Hz), 62.0, 58.5, 58.4, 41.6. HRMS (ESI) calcd for C21H21F4N3O3 [M + H+] 440.1592, found 440.1588.
Tert-Butyl (4-(5-(4-(2-fluoroethoxy)-3-(trifluoromethyl)-phenyl)-1,2,4-oxadiazol-3-yl)benzyl)(2-hydroxyethyl)-carbamate (13c).
Compound 13c was prepared according to the general procedure as described for compound 10a–c. Yield: 42%, white solid, MP: 120–122 °C. 1H NMR (400 MHz, CDCl3) δ 8.47 (s, 1H), 8.35 (d, J = 8.8 Hz, 1H), 8.13 (d, J = 8.0 Hz, 2H), 7.38 (d, J =7.7 Hz, 2H), 7.18 (d, J = 8.8 Hz, 1H), 4.92–4.75 (m, 2H), 4.56 (s, 2H), 4.50–4.36 (m, 2H), 3.75 (s, 2H), 3.46 (s, 2H), 2.90 (br, 1H),1.47 (s, 9H).
2-((4-(5-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)benzyl)amino)ethan-1-ol (13d).
To a solution of 13c (250 mg, 0.48 mmol) in methanol (5.0 mL) was added 4 M HCl in dioxane (5.0 mL). The reaction was stirred at RT for 6 h, then the solvent was removed under reduced pressure and the residue was dissolved in ethyl acetate. The solution was washed with saturated NaHCO3, water, and dried over MgSO4. After filtering and concentration, the final product was obtained. (193 mg, 95%) MP: 126–130 °C. 1H NMR (400 MHz, MeOD-d4) δ 8.35 (d, J = 4.9 Hz, 2H), 8.06 (d, J = 8.1 Hz, 2H), 7.50 (d, J = 8.1 Hz, 2H), 7.39 (d, J = 9.3 Hz, 1H), 4.84–4.68 (m, 2H), 4.51–4.39 (m, 2H), 3.85 (s, 2H), 3.71–3.64 (m, 2H), 2.73 (t, J = 5.6 Hz, 2H). 13C NMR (101 MHz, MeOD-d4) δ 174.4, 168.4, 159.8, 142.8, 133.4, 128.7, 127.1, 126.7 (q, JC–F = 5.1 Hz), 125.4, 124.3, 121.6, 116.4, 113.9, 81.2 (d, JC–F = 171 Hz), 68.7 (d, JC–F = 20 Hz), 60.1, 52.5, 50.3. HRMS (ESI) calcd for C20H19F4N3O3 [M + H+] 426.1435, found 426.1432.
General procedure for the synthesis of 15a–b
To a round-bottom flask equipped with a stir bar was added aldehyde 1432,33 (1.0 eq.), amine (1.5 eq.), methanol (14 mL mmol−1), and acetic acid (0.5 mL mmol−1). The reaction mixture was stirred for 1 h at which time sodium cyanoborohydride (1.0 eq.) was added. The reaction mixture was stirred overnight and diluted with water. The precipitate was filtered off and washed with water to give off-white solid product 15a–b.
2-((4-(5-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)benzyl)amino)propane-1,3-diol (15a).
Yield: 50%, off-white solid, MP: 135–137 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.47–8.41 (m, 1H), 8.34 (s, 1H), 8.04 (d, J = 8.2 Hz, 2H), 7.57 (d, J = 8.0 Hz, 3H), 4.89–4.70 (m, 2H), 4.63–4.50 (m, 2H), 4.45 (s, 2H), 3.88 (s, 2H), 3.49–3.35 (m, 4H), 2.61–2.53 (m, 1H). 13C NMR (101 MHz, DMSO-d6) δ 174.0, 168.2, 159.5, 145.6, 134.1, 128.6, 127.0, 126.6 (q, JC–F = 5.1 Hz), 124.2, 122.4 (q, JC–F = 274 Hz) 117.9, 116.0, 115.1, 109.6, 81.7 (d, JC–F = 162 Hz), 68.8 (q, JC–F = 20 Hz), 61.1, 60.5, 50.5. HRMS (ESI) calcd for C21H21F4N3O4 [M + H+] 456.1541, found 456.1537.
(1-(4-(5-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)benzyl)azetidin-3-yl)methanol (15b).
Yield: 23%, white solid, MP: 114–116 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.45 (d, J = 8.8 Hz, 1H), 8.36–8.32 (m, 1H), 8.12 (d, J = 8.2 Hz, 2H), 7.65–7.55 (m, 3H), 5.49 (s, 1H), 4.88–4.82 (m, 1H), 4.78–4.71 (m, 1H), 4.64–4.57 (m, 1H), 4.57–4.49 (m, 1H), 3.41–3.27 (m, 8H), 2.34–2.21 (m, 1H).
3-(4-(Chloromethyl)phenyl)-5-(4-(2-fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazole (16).
To a round-bottom flask was added cyanuric chloride (253 mg, 1.37 mmol) and DMF(2.5 mL). A solution of 10c (500 mg, 1.31 mmol) in dichloromethane (3.5 mL) was added dropwise at RT, and the reaction was continued to stir at RT for overnight. Then, the reaction mixture was diluted with dichloromethane and water. The dichloromethane layer was then separated and concentrated in vacuum to afford crude product (400 mg, 73%). The crude product was used directly for the next step. 1H NMR (400 MHz, CDCl3) δ = 8.47 (d, J = 2.0 Hz, 1H), 8.35 (dd, J = 8.8 Hz, 2.0 Hz, 1H), 8.17 (d, J = 8.0 Hz, 2H), 7.54 (d, J = 8.4 Hz, 2H), 7.18 (d, J =8.8 Hz, 1H), 4.93–4.76 (m, 2H), 4.65 (s, 2H), 4.48–4.36 (m, 2H).
General procedure for the synthesis of 17a–e and 17g
To a round-bottom flask equipped with a stir bar was added alcohols (10 eq.) and THF (2 mL mmol−1). After cooling to 0 °C, sodium hydride (1.8 eq.) was added portionwise, and the reaction vessel was equipped with a reflux condenser and heated to 80 °C and stirred for 30 min. Chloride 16 (1.0 eq.) was added to the flask and the mixture was stirred overnight. The reaction was cooled to room temperature and quenched with water. The crude was extracted with ethyl acetate, washed with saturated brine, and dried over MgSO4. After filtering and concentrated, the residue was purified by flash chromatography on silica gel column to afford the product.
2-((4-(5-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)benzyl)oxy)ethan-1-ol (17a).
Compound 17a was purified by flash chromatography, eluted with hexane/ethyl acetate (2/1, v/v) to afford off-white solid. Yield: 35%, MP: 109–110 °C. 1H NMR (400 MHz, CDCl3) δ 8.42 (s, 1H), 8.30 (dd, J = 8.6, 1.4 Hz, 1H), 8.11 (d, J = 8.1 Hz, 2H), 7.46 (d, J = 8.0 Hz, 2H), 7.13 (d, J = 8.7 Hz, 1H), 4.89–4.71 (m, 2H), 4.62 (s, 2H),4.46–4.32 (m, 2H), 3.82–3.73 (m, 2H), 3.67–3.59 (m, 2H), 2.19 (br, 1H). 13C NMR (101 MHz, CDCl3) δ 174.2, 168.7, 159.6, 141.4, 133.3, 127.9, 127.7 (q, JC–F = 5.2 Hz), 127.6, 126.1, 122.7 (q, JC–F = 271 Hz), 120.2 (q, JC–F = 32 Hz), 117.0, 113.4, 81.3 (d, JC–F = 172 Hz), 72.7, 71.7, 68.4 (d, JC–F = 21 Hz), 61.9. HRMS (ESI) calcd for C20H18F4N2O4 [M + H+] 427.1245, found 427.1240.
2-(2-((4-(5-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)benzyl)oxy)ethoxy)ethan-1-ol (17b).
Compound 17b was purified by flash chromatography, eluted with hexane/ethyl acetate (1/1, v/v) to afford an off-white semi-solid. Yield: 39%. 1H NMR (400 MHz, CDCl3) δ 8.43 (s, 1H), 8.32 (d, J = 8.5 Hz, 1H), 7.94 (d, J = 7.8 Hz, 1H), 7.82 (d, J = 10.3 Hz, 1H),7.71–7.51 (m, 2H), 7.16 (d, J = 8.7 Hz, 1H), 4.91–4.85 (m, 1H),4.79–4.74 (m, 1H), 4.70 (s, 2H), 4.47–4.42 (m, 1H), 4.41–4.35 (m, 1H), 3.79–3.69 (m, 6H), 3.67–3.60 (m, 2H), 2.41 (br, 1H). 13C NMR (101 MHz, CDCl3) δ 174.4, 168.0, 159.5, 141.8, 133.3, 128.0, 127.7 (q, JC–F = 5.4 Hz), 127.2, 126.5, 122.8 (q, JC–F = 274 Hz), 119.0 (q, JC–F = 32 Hz), 116.8, 113.4, 81.2 (d, JC–F = 173 Hz), 72.5, 70.3, 70.0, 68.4 (d, JC–F = 21 Hz), 66.4, 61.8. HRMS (ESI) calcd for C22H22F4N2O5 [M + H+] 471.1583, found 471.1589.
2-(2-(2-((4-(5-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)benzyl)oxy)ethoxy)ethoxy)ethan-1-ol (17c).
Compound 17c was purified by flash chromatography, eluted with hexane/ethyl acetate (1/3, v/v) to afford an off-white semi-solid. Yield: 42%. 1H NMR (400 MHz, CDCl3) δ 8.45 (br, 1H),8.33 (d, J = 8.7 Hz, 1H), 8.13 (d, J = 8.1 Hz, 2H), 7.49 (d, J =8.1 Hz, 2H), 7.16 (d, J = 8.8 Hz, 1H), 4.90–4.75 (m, 2H), 4.64 (s, 2H), 4.49–4.36 (m, 2H), 3.74–3.61 (m, 12H), 2.63 (br, 1H). 13C NMR (101 MHz, CDCl3) δ 174.2, 168.8, 159.6, 141.7, 132.2, 127.9, 127.7 (q, JC–F = 5.2 Hz), 127.6, 125.9, 122.7 (q, JC–F = 271 Hz), 120.2 (q, JC–F = 32 Hz), 117.1, 113.4, 81.3 (d, JC–F = 171 Hz), 72.7, 72.5, 70.7, 70.6, 70.3, 69.7, 68.4 (d, JC–F = 21 Hz), 61.7. HRMS (ESI) calcd for C24H26F4N2O6 [M + H+] 515.1800, found 515.1806.
1-(4-(5-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)phenyl)-2,5,8,11-tetraoxatridecan-13-ol (17d).
Compound 17d was purified by flash chromatography, eluted with hexane/ethyl acetate (1/3, v/v) to afford an off-white semi-solid. Yield: 25%. 1H NMR (400 MHz, CDCl3) δ 8.33 (d, J = 5.7 Hz, 1H), 8.25–8.18 (m, 1H), 8.07–8.00 (m, 2H), 7.40 (d, J =7.9 Hz, 2H), 7.06 (d, J = 8.7 Hz, 1H), 4.80–4.64 (m, 3H),4.59–4.52 (m, 2H), 4.36–4.27 (m, 2H), 3.69–3.54 (m, 14H),3.54–3.48 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 173.9, 168.3, 159.3, 141.8, 132.2, 127.9, 127.7 (q, JC–F = 5.2 Hz), 127.6, 125.9, 122.7 (q, JC–F = 271 Hz), 120.2 (q, JC–F = 32 Hz), 117.1, 113.4, 81.3 (d, J = 172.5 Hz), 72.7, 72.5, 70.6, 70.5, 70.4, 70.3, 70.2, 69.7, 68.4 (d, JC–F = 21 Hz), 61.7. HRMS (ESI) calcd for C26H30F4N2O7 [M + H+] 559.2062, found 559.2066.
Tert-Butyl (2-((4-(5-(4-(2-fluoroethoxy)-3-(trifluoromethyl)-phenyl)-1,2,4-oxadiazol-3-yl)benzyl)oxy)ethyl)(2-hydroxy-ethyl) carbamate (17e).
Compound 17e was synthesized according to the general procedure. The crude product was used directly for the next step reaction without any further purification.
2-((2-((4-(5-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)benzyl)oxy)ethyl)amino)ethan-1-ol (17f).
To a solution of 17e (284 mg, 0.50 mmol) in methanol (5.0 mL) was added 4 M HCl in dioxane (5.0 mL). The reaction was stirred at RT for 6 h, then the solvent was concentrated in vacuum and the residue was dissolved in ethyl acetate. The solution was washed with saturated NaHCO3, water, and dried over MgSO4. After filtering and concentration, the final white solid product was obtained. (199 mg, 85%) MP: 126–130 °C. 1H NMR (400 MHz, CDCl3) δ 8.45 (s, 1H), 8.34 (d, J = 8.7 Hz, 1H), 8.14 (d, J = 8.1 Hz, 2H), 7.46 (d, J = 8.1 Hz, 2H), 7.17 (d, J = 8.8 Hz, 1H), 4.91–4.75 (m, 2H), 4.60 (s, 2H), 4.47–4.38 (m, 2H),4.35–4.29 (m, 2H), 3.74–3.67 (m, 4H), 3.52 (t, J = 5.0 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 174.3, 168.7, 158.6, 141.3, 133.3, 127.8, 127.7 (q, JC–F = 5.25 Hz), 127.6, 126.1, 122.7 (q, JC–F = 275 Hz), 120.3 (q, JC–F = 31 Hz), 117.0, 113.4, 86.3 (d, JC–F = 173 Hz), 72.6, 68.9, 68.4 (d, JC–F = 21.1 Hz), 62.0, 46.0, 44.2. HRMS (ESI) calcd for C22H23F4N3O4 [M + H+] 470.1697, found 470.1694.
2-((2-((4-(5-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)benzyl)oxy)ethyl)(methyl)amino)ethan-1-ol (17g).
Compound 17g was purified by flash chromatography, eluted with ethyl acetate/methanol (50/1, v/v) to afford a pale yellow semi-solid. Yield: 26%. 1H NMR (400 MHz, CDCl3) δ 8.40 (d, J = 1.8 Hz, 1H), 8.29 (dd, J = 8.7, 2.0 Hz, 1H), 8.07 (d, J = 8.1 Hz, 2H), 7.42 (d, J = 8.1 Hz, 2H), 7.11 (d, J = 8.8 Hz, 1H),4.88–4.79 (m, 1H), 4.75–4.65 (m, 1H), 4.54 (s, 2H), 4.44–4.25 (m, 2H), 4.12 (s, 3H), 3.55 (s, 2H), 2.73–2.60 (m, 4H), 2.29 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 174.8, 168.7, 160.2, 159.0, 141.3, 133.3, 128.0, 127.8 (q, JC–F = 5.25 Hz), 127.4, 125.9, 122.8 (q, JC–F = 275 Hz), 120.4 (q, JC–F = 31 Hz), 116.8, 113.4, 72.8, 60.4, 59.9, 57.7, 43.0. HRMS (ESI) calcd for C17H21F4N3O4 [M + H+] 408.1541, found 408.1536.
4-(2-Hydroxyethoxy)benzonitrile (18a).
Compound 18a was synthesized according to the published procedure.45
4-((2,3-Dihydroxypropoxy)methyl)benzonitrile (18b).
Commercially available DL-1,2-isopropylideneglycerol (3.0 g, 22.5 mmol) was added to a suspension of NaH (0.6 g, 25.8 mmol) in THF (30 mL) at 0 °C under nitrogen. The mixture was stirred for 30 min during which time 4-cyanobenzyl bromide (4.0 g, 20.4 mmol) was added dropwise over 5 min followed by warming of the reaction mixture to RT. After 2 h, the reaction mixture was partitioned between ammonium chloride solution and ethyl acetate. The aqueous phase was extracted with ethyl acetate and the combined organics dried over anhydrous MgSO4 and concentrated in vacuum. The crude residue was purified by flash chromatography on silica gel, eluted with hexane/ethyl acetate (1/1, v/v) to afford an off-white solid product. (3.8 g, 90%) MP: 80–83 °C. 1H NMR (400 MHz, CDCl3) δ 7.63 (d, J = 8.1 Hz, 2H), 7.43 (d, J = 7.9 Hz, 2H), 4.61 (s, 2H), 3.97–3.89 (m, 1H), 3.76–3.69 (m, 1H), 3.66–3.56 (m, 3H), 2.75 (s, 1H), 2.29 (s, 1H).
(E)-N′-Hydroxy-4-(2-hydroxyethoxy)benzimidamide (19a).
Compound 19a was synthesized according to the general procedure as described for compound 12a–c. The crude product was used directly for the next step reaction without further purification. 1H NMR (400 MHz, DMSO-d6) δ 9.41 (s, 1H), 7.56 (d, J = 8.7 Hz, 2H), 6.89 (d, J = 8.7 Hz, 2H), 5.68 (s, 2H), 4.83 (s, 1H), 3.97 (t, J = 4.7 Hz, 2H), 3.68 (d, J = 4.7 Hz, 2H).
(E)-4-((2,3-Dihydroxypropoxy)methyl)-N′-hydroxybenzimidamide (19b).
Compound 19b was synthesized according to the general procedure as described for compound 12a–c. Yield: 45%, MP: 142–144 °C. 1H NMR (400 MHz, MeOD-d4) δ 7.61 (d, J = 8.0 Hz, 2H), 7.39 (d, J = 7.9 Hz, 2H), 4.57 (s, 2H), 3.83–3.75 (m, 1H), 3.64–3.45 (m, 4H).
2-(4-(5-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)phenoxy)ethan-1-ol (20a).
Compound 20a was synthesized according to the general procedure as described for compound 10a–c. Yield: 37%, a white solid. MP: 148–149 °C. 1H NMR (400 MHz, acetone-d6) δ 8.45–8.39 (m, 2H), 8.08 (d, J = 8.9 Hz, 2H), 7.56 (d, J = 8.7 Hz, 1H), 7.13 (d, J =8.9 Hz, 2H), 4.95–4.80 (m, 2H), 4.68–4.56 (m, 2H), 4.18 (t, J =4.9 Hz, 2H), 4.05 (br, 1H), 3.92 (d, J = 4.3 Hz, 2H). 13C NMR (101 MHz, acetone-d6) δ 174.1, 168.4, 161.8, 159.9, 133.7, 128.9, 126.8 (q, JC–F = 5.1 Hz), 123.2 (q, JC–F = 273 Hz), 119.3, 119.0, 116.8, 114.9, 114.4, 81.6 (d, JC–F = 170 Hz), 69.9, 68.9 (d, JC–F = 20 Hz), 60.3. HRMS (ESI) calcd for C19H16F4N2O4 [M + H+] 413.1119, found 413.1113.
(±)-3-((4-(5-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)benzyl)oxy)propane-1,2-diol (20b).
Compound 20b was synthesized according to the general procedure as described for compound 10a–c. Yield: 38%, white solid, MP: 102–105 °C. 1H NMR (400 MHz, MeOD-d4) δ 8.34–8.28 (m, 2H), 8.04 (d, J = 8.1 Hz, 2H), 7.50 (d, J = 8.0 Hz, 2H), 7.35 (d, J =8.5 Hz, 1H), 4.85–4.69 (m, 2H), 4.61 (s, 2H), 4.48–4.38 (m, 2H),3.88–3.81 (m, 1H), 3.67–3.51 (m, 4H). 13C NMR (101 MHz, MeOD-d4) δ 174.3, 168.4, 159.8, 142.1, 133.3, 127.6, 126.9, 126.6 (q, JC–F = 5.35 Hz), 125.6, 122.9 (q, JC–F = 273 Hz), 119.3 (q, JC–F = 31 Hz), 116.3, 113.8, 81.2 (d, JC–F = 170 Hz), 72.3,71.6, 70.9, 68.6 (q, JC–F = 20.1 Hz), 63.1. HRMS (ESI) calcd for C21H20F4N2O5 [M + H+] 457.1381, found 457.1385.
(E)-N′-Hydroxy-4-vinylbenzimidamide (22).
Compound 22 was synthesized according to the general procedure as described for compound 12a–c. Yield: 95%. The detailed procedure was described in the published literature.46
5-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-3-(4-vinylphenyl)-1,2,4-oxadiazole (23).
Compound 23 was synthesized according to the general procedure as described for compound 10a–c. The crude product was used directly for the next step reaction without further purification.
(±)-1-(4-(5-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)phenyl)ethane-1,2-diol (24).
To a 50 mL of round bottle flask was added 18 (327 mg, 0.86 mmol), 4-methylmorpholine N-oxide (122 mg, 1.03 mmol) and THF/H2O (3/1, 12.0 mL). The mixture was stirred for 5 min before adding OsO4 (2.5 wt% in tert-butanol, 545 μL). The reaction was stirred continuously at RT overnight. Then, the reaction mixture was diluted with ethyl acetate and water. The ethyl acetate layer was separated and concentrated in vacuum to afford crude product. Crude product was purified by flash chromatography, eluted with hexane/ethyl acetate (1/2, v/v) to furnish the final product as a white semi-solid. (120 mg, 34%) 1H NMR (400 MHz, MeOD-d4) δ 8.39–8.31 (m, 2H), 8.08 (d, J =8.2 Hz, 2H), 7.55 (d, J = 8.2 Hz, 2H), 7.38 (d, J = 8.8 Hz, 1H), 4.85–4.70 (m, 3H), 4.50–4.39 (m, 2H), 3.72–3.60 (m, 2H). 13C NMR (101 MHz, MeOD-d4) δ 174.4, 168.5, 159.9, 145.8, 133.3, 126.9, 126.7 (q, JC–F = 5.2 Hz), 126.6, 125.6, 122.9 (q, JC–F = 273 Hz), 119.4 (q, JC–F = 32 Hz), 116.4, 113.8, 81.2 (d, JC–F = 171 Hz), 74.1, 68.6(d, JC–F = 20.2 Hz), 67.1. HRMS (ESI) calcd for C19H16F4N2O4 [M + H+] 413.1119, found 413.1115.
(E)-N′-Hydroxy-4-((methoxymethoxy)methyl)benzimidamide (25a).
Compound 25a was synthesized according to the general procedure as described for compound 12a–c. Yield: 79%, a pale white solid. MP: 158–159 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.64 (s, 1H), 7.70–7.62 (m, 2H), 7.37–7.26 (m, 2H),5.80 (s, 3H), 4.69–4.61 (m, 2H), 4.53 (s, 2H), 3.30 (s, 2H).
(E)-N′-Hydroxy-4-((2-(methoxymethoxy)ethoxy)methyl)benzimidamide (25b).
Compound 25b was synthesized according to the general procedure as described for compound 12a–c. Yield: 60%, a white solid. MP: 150–151 °C. 1H NMR (400 MHz, MeOD-d4) δ 7.62 (d, J = 8.1 Hz, 2H), 7.38 (d, J = 8.1 Hz, 2H), 4.63 (s, 2H), 4.57 (s, 2H), 3.74–3.63 (m, 4H), 3.34 (s, 3H).
(E)-N′-Hydroxy-4-(2,4,7,10-tetraoxaundecan-11-yl)benzimidamide (25c).
Compound 25c was synthesized according to the general procedure as described for compound 12a–c. Yield: 50%, a white solid. MP: 140–142 °C. 1H NMR (400 MHz, MeOD-d4) δ 7.65 (d, J = 8.1 Hz, 2H), 7.41 (d, J = 8.1 Hz, 2H), 4.66 (s, 2H), 4.60 (s, 2H), 3.73–3.65 (m, 8H), 3.37 (s, 3H).
(E)-4-(2,4,7,9-Tetraoxadecan-5-yl)-N′-hydroxybenzimidamide (25d).
Compound 25d was synthesized according to the general procedure as described for compound 12a–c. Yield: 62%, a yellow solid. MP: 170–172 °C. 1H NMR (400 MHz, CDCl3) δ 8.40 (s, 1H), 8.19 (d, J = 8.8 Hz, 1H), 8.11 (d, J = 8.1 Hz, 2H), 7.50 (d, J = 8.1 Hz, 2H), 7.04 (d, J = 8.6 Hz, 1H),4.91–4.87 (m, 1H), 4.75–4.71 (m, 1H), 4.69–4.64 (m, 3H),3.84–3.72 (m, 2H), 3.40 (s, 3H), 3.31 (s, 3H).
General procedure for the synthesis of 26a–d
To a round-bottom flask equipped with a stir bar was added 4-hydroxy-3-(trifluoromethyl)benzoic acid (1.0 eq.), HOBt (0.2 eq.), TBTU (1.0 eq.), DIPEA (3.0 eq.), and DMF (5.0 mL mmol−1). The reaction mixture was stirred for 0.5 h followed by adding methoxymethyl (MOM) protected amidoxime 25a–d (1.0 eq.). The reaction mixture was stirred for 1 h at room temperature, then refluxed in a pre-heated 120 °C oil-bath for 4 h and monitored by TLC. After cooling, the reaction mixture was diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with 1 M HCl, saturated brine, and dried over anhydrous MgSO4. After filtration and concentration under reduced pressure, the crude residue was purified on a silica gel column to give 26a–d.
4-(3-(4-((Methoxymethoxy)methyl)phenyl)-1,2,4-oxadiazol-5-yl)-2-(trifluoromethyl)phenol (26a).
Compound 26a was purified by flash chromatography, eluted with hexane/ethyl acetate (2/1, v/v) to afford a white solid. Yield: 50%, white solid, MP: 130–135 °C. 1H NMR (400 MHz, DMSO-d6) δ = 11.83 (br, 1H), 8.29–8.24 (m, 2H), 8.07 (d, J = 8.0 Hz, 2H), 7.55 (d, J = 8.0 Hz, 2H), 7.27 (d, J = 8.8 Hz, 1H), 4.70 (s, 2H), 4.63 (s, 2H), 3.33 (s, 3H).
4-(3-(4-((2-(Methoxymethoxy)ethoxy)methyl)phenyl)-1,2,4-oxadiazol-5-yl)-2-(trifluoromethyl)phenol (26b).
Compound 26b was purified by flash chromatography, eluted with hexane/ethyl acetate (3/2, v/v) to afford a white solid. Yield: 45%, white solid, MP: 120–122 °C. 1H NMR (400 MHz, CDCl3) δ 8.34 (s, 1H), 8.04–7.93 (m, 3H), 7.53–7.45 (m, 1H), 7.41 (d, J = 8.1 Hz, 2H), 6.99 (d, J = 8.6 Hz, 1H), 4.73 (s, 2H), 4.62 (s, 2H),3.86–3.76 (m, 4H), 3.41 (s, 3H).
4-(3-(4-(2,4,7,10-Tetraoxaundecan-11-yl)phenyl)-1,2,4-oxadiazol-5-yl)-2-(trifluoromethyl)phenol (26c).
Compound 26c was purified by flash chromatography, eluted with hexane/ethyl acetate (3/2, v/v) to afford a white solid. Yield: 60%, MP: 103–105 °C. 1H NMR (400 MHz, CDCl3) δ 8.26 (d, J = 6.1 Hz, 1H), 7.90 (d, J = 7.9 Hz, 2H), 7.80 (d, J = 8.6 Hz, 1H), 7.30 (d, J =7.9 Hz, 2H), 6.83 (d, J = 8.6 Hz, 1H), 4.61 (s, 2H), 4.54 (s, 2H), 3.85–3.70 (m, 8H), 3.34 (s, 3H), 2.86 (s, 2H).
4-(3-(4-(2,4,7,9-Tetraoxadecan-5-yl)phenyl)-1,2,4-oxadiazol-5-yl)-2-(trifluoromethyl)phenol (26d).
Compound 26d was purified by flash chromatography, eluted with hexane/ethyl acetate (1/1, v/v) to afford a white semi-solid. Yield: 50%. 1H NMR (400 MHz, CDCl3) δ 8.38 (s, 1H), 8.14 (d, J = 8.4 Hz, 1H), 8.08 (d, J = 8.1 Hz, 2H), 7.48 (d, J = 8.1 Hz, 2H), 7.02 (d, J = 8.6 Hz, 1H), 4.89–4.84 (m, 1H), 4.73–4.69 (m, 1H), 4.68–4.61 (m, 3H),3.84–3.69 (m, 3H), 3.38 (s, 3H), 3.29 (s, 3H).
General procedure for the synthesis of 27a–d
To a round-bottom flask equipped with a stir bar was added 26a–d (1.0 eq.), ethylene ditosylate (2.0 eq.), K2CO3 (3.0 eq.), and acetonitrile (10 mL mmol−1). The reaction mixture was stirred at RT for 1 h, then refluxed in a pre-heated 90 °C oil-bath overnight and monitored by TLC. The reaction was diluted with ethyl acetate and water, the ethyl acetate layer was washed with saturated brine and dried over anhydrous MgSO4. After filtering and concentrated in vacuum, the crude residue was purified on a silica gel column to give 27a–d.
2-(4-(3-(4-((Methoxymethoxy)methyl)phenyl)-1,2,4-oxa-diazol-5-yl)-2-(trifluoromethyl)phenoxy)ethyl 4-methyl-benzenesulfonate (27a).
Compound 27a was purified by flash chromatography, eluted with hexane/ethyl acetate (3/1, v/v) to afford a white solid. Yield: 30%, MP: 125–127 °C. 1H NMR (400 MHz, CDCl3) δ 8.44 (s, 1H), 8.32 (d, J = 8.8 Hz, 1H), 8.15 (d, J = 8.0 Hz, 2H), 7.81 (d, J = 8.0 Hz, 2H), 7.51 (d, J = 8.0 Hz, 2H),7.35 (d, J = 8.0 Hz, 2H), 7.09 (d, J = 8.8 Hz, 1H), 4.75 (s, 2H), 4.68 (s, 2H), 4.45–4.35 (m, 4H), 3.44 (s, 3H), 2.45 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 174.3, 169.0, 159.2, 145.3, 141.6, 133.5, 132.6, 130.1, 128.2, 128.1, 127.8 (q, JC–F = 5.5 Hz), 127.8, 126.1, 122.8 (q, JC–F = 271 Hz), 120.3 (q, JC–F = 32 Hz), 117.5, 113.5, 96.1, 68.9, 67.3, 66.8, 55.6, 21.8. HRMS (ESI) calcd for C27H25F3N2O7S [M + H+] 579.1407. Found [M + H+] 579.1415.
2-(4-(3-(4-((2-(Methoxymethoxy)ethoxy)methyl)phenyl)-1,2,4-oxadiazol-5-yl)-2-(trifluoromethyl)phenoxy)ethyl 4-methylbenzenesulfonate (27b).
Compound 27b was purified by flash chromatography, eluted with hexane/ethyl acetate (3/1, v/v) to afford a white solid. Yield: 57%, MP: 115–117 °C. 1H NMR (400 MHz, CDCl3) δ 8.37 (d, J = 1.4 Hz, 1H), 8.28–8.23 (m, 1H), 8.10 (d, J = 8.2 Hz, 2H), 7.78 (d, J = 8.2 Hz, 2H), 7.48 (d, J = 8.1 Hz, 2H), 7.32 (d, J = 8.1 Hz, 2H), 7.05 (d, J = 8.8 Hz, 1H), 4.67 (s, 2H), 4.64 (s, 2H), 4.40–4.33 (m, 4H), 3.78–3.67 (m, 4H), 3.37 (s, 3H), 2.41 (s, 3H). 13C NMR (101 MHz, CDCl3): δ 174.1, 168.7, 159.0, 145.2, 141.8, 133.3, 132.3, 129.9, 127.9, 127.8, 127.7, 127.5 (q, JC–F = 5.5 Hz), 125.8, 122.6 (q, JC–F = 271 Hz), 120.0 (q, JC–F = 32 Hz), 117.1, 113.3, 96.5, 72.7, 69.7, 67.3, 66.8, 66.5, 55.2, 21.6. HRMS (ESI) calcd for C29H29F3N2O8S [M + H+] 623.1669, found 623.1660.
2-(4-(3-(4-(2,4,7,10-Tetraoxaundecan-11-yl)phenyl)-1,2,4-oxadiazol-5-yl)-2-(trifluoromethyl)phenoxy)ethyl 4-methyl-benzenesulfonate (27c).
Compound 27c was purified by flash chromatography, eluted with hexane/ethyl acetate (3/1, v/v) to afford a white semi-solid. Yield: 38%. 1H NMR (400 MHz, CDCl3) δ 8.41 (s, 1H), 8.31 (d, J = 8.7 Hz, 1H), 8.13 (d, J = 8.1 Hz, 2H), 7.80 (d, J = 8.2 Hz, 2H), 7.49 (d, J = 8.1 Hz, 2H), 7.34 (d, J =8.1 Hz, 2H), 7.08 (d, J = 8.8 Hz, 1H), 4.67 (s, 2H), 4.65 (s, 2H), 4.44–4.35 (m, 4H), 3.77–3.67 (m, 8H), 3.37 (s, 3H), 2.44 (s, 3H). 13C NMR (101 MHz, CDCl3): δ 174.1, 168.8, 159.0, 145.2, 141.9, 133.3, 132.3, 129.9, 127.9, 127.9, 127.6 (q, JC–F = 5.5 Hz), 127.5, 125.8, 122.6 (q, JC–F = 271 Hz), 120.1 (q, JC–F = 32 Hz), 117.2, 113.3, 96.5, 72.7, 70.6, 69.8, 67.2, 66.8, 66.5, 55.2, 21.6. HRMS (ESI) calcd for C31H33F3N2O9S [M + H+] 667.1932, found 667.1925.
(±)-2-(4-(3-(4-(2,4,7,9-Tetraoxadecan-5-yl)phenyl)-1,2,4-oxadiazol-5-yl)-2-(trifluoromethyl)phenoxy)ethyl 4-methyl-benzenesulfonate (27d).
Compound 27d was purified by flash chromatography, eluted with hexane/ethyl acetate (2/1, v/v) to afford a white semi-solid. Yield: 44%. 1H NMR (400 MHz, CDCl3) δ 8.42 (s, 1H), 8.32 (d, J = 8.7 Hz, 1H), 8.15 (d, J = 8.1 Hz, 2H), 7.81 (d, J = 7.9 Hz, 2H), 7.52 (d, J = 8.2 Hz, 2H), 7.34 (d, J = 8.0 Hz, 2H), 7.09 (d, J = 8.7 Hz, 1H), 4.91–4.86 (m, 1H), 4.75–4.59 (m, 5H), 4.46–4.36 (m, 4H), 3.84–3.70 (m, 2H), 3.38 (s, 3H), 3.29 (s, 3H), 2.44 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 174.6, 168.5, 159.5, 145.5, 143.7, 134.5, 132.4, 130.5, 128.3, 128.0, 127.5, 127.1, 127.0 (q, JC–F = 5.5 Hz), 123.1 (q, JC–F = 271 Hz) 125.8, 118.8, 116.4, 115.4, 96.2, 94.9, 76.9, 71.1, 68.8, 67.3, 55.4, 55.1, 21.5. HRMS (ESI) calcd for C30H31F3N2O9S [M + H+] 653.1775, found 653.1770.
General procedure for the radiosynthesis of [18F]10a, [18F]17a, [18F]17b, and (±)-[18F]24
[18F]KF (~7.4 GBq) aqueous was added to a vial containing Kryptofix 222 (~10 mg), and dried by azeotropic evaporation with acetonitrile (3 × 1 mL) under N2 flow at 110 °C. To the reaction vial was added precursor 27a–d (~2 mg) as a solution in acetonitrile (300 μL). The reaction was placed in a 110 °C oil-bath for 15 min. The reaction was removed from the oil-bath, at which time 6 M HCl (150 μL) was added. The reaction mixture was heated in a 110 °C oil-bath for another 5 min. Then, the reaction was removed from the oil-bath and quenched with 6 M NaOH (150 μL) and diluted with 2.4 mL of the HPLC mobile phase. The mixture was passed through a Sep-Pak® Alumina N Cartridge (Part No. WAT020510) and injected onto the semi-preparation HPLC column (Agilent SB-C18 250 × 9.6 mm, 5 μm, UV = 254 nm, 4.0 mL min−1). The HPLC fraction was collected into a water bottle with 60 mL of water and then trapped on a Sep-Pak® C-18 Cartridge (Part No. WAT020515). The activity was washed out with 0.6 mL of ethanol and 5.4 mL of 0.9% saline. After sterile filtration into a glass vial, the radiolabeled product was ready for quality control (QC) analysis and animal studies. An aliquot of sample was injected onto an analytical HPLC column (Agilent Eclipse XDB-C18 250 × 4.6 mm, 5 μm) to determine the concentration of tracer. Meanwhile, the tracer was authenticated by co-injecting with the non-radiolabeled standard sample solution.
Semi-preparation HPLC conditions.
[18F]10a, [18F]17a, and [18F]17b: Mobile phase, 50% acetonitrile in 0.1 M ammonium formate buffer, pH = 4.2; flow rate, 4.0 mL min−1; UV = 254 nM; tR = 18–21 min. The radiochemical yields were 38%, 40%, and 35%, respectively (decay corrected to EOS).
(±)-[18F]24: Mobile phase, 40% acetonitrile in 0.1 M ammonium formate buffer, pH = 4.2; flow rate, 4.0 mL min−1; UV = 254 nM; tR = 25–27 min. The radiochemical yield was 38% (decay corrected to EOS).
QC HPLC conditions.
[18F]10a: Mobile phase, 75% acetonitrile in 0.1 M ammonium formate buffer, pH = 4.2; flow rate, 1.5 mL min−1; UV = 254 nM; tR = 5.5 min.
[18F]17a: Mobile phase, 70% acetonitrile in 0.1 M ammonium formate buffer, pH = 4.2; flow rate, 1.0 mL min−1; UV = 254 nM; tR = 5.7 min.
[18F]17b: Mobile phase, 70% acetonitrile in 0.1 M ammonium formate buffer, pH = 4.2; flow rate, 1.0 mL min−1; UV = 254 nM; tR = 5.6 min.
(±)-[18F]24: Mobile phase, 63% acetonitrile in 0.1 M ammonium formate buffer, pH = 4.2; flow rate, 1.5 mL min−1; UV = 254 nM; tR = 4.6 min.
In vitro S1PR1–5 binding assay
The binding potencies of the newly synthesized compounds were determined by competition against the binding of [32P] S1P to commercial cell membranes expressing recombinant human S1PRs (1, 2, 3, 4, and 5) according to methods reported previously.32,33,35
MicroPET studies in cynomolgus macaque
Male macaques (9–10 kg) were studied with a microPET Focus 220 scanner (Concorde/CTI/Siemens Microsystems, Knoxville, TN). Animals were maintained in facilities with 12-hour dark and light cycles, given access to food, water, and libitum. Animals were also provided a variety of psychologically enriching tasks to prevent inappropriate deprivation. Animals were scanned under anesthesia (induced with ketamine and glycolpyrrolate and maintained with inhalation isoflurane). Core temperature was kept about 37 °C with a heated water blanket. The head was secured in a head holder with the brain in the center of the field of view. Subsequently, a 2 h dynamic emission scan was acquired after administration of ~0.35 GBq of radiotracer via the venous catheter.
PET scans were collected from 0–120 min with the following time frames: 3 × 1 min, 4 × 2 min, 3 × 3 min and 20 × 5 min. Emission data were corrected for dead time, scatter and attenuation and then reconstructed to a final resolution of 2.0 mm full-width half maximum in all 3 dimensions at the center of the field of view. PET and MRI images were co-registered using automated image registration program (AIR). For quantitative analyses, three-dimensional ROI (the global brain) was identified on the MRI and transformed to the reconstructed PET images to obtain time-activity curves. Activity measures were standardized to body weight and the dose of radioactivity injected to yield SUV.
Supplementary Material
Acknowledgements
This work was supported by the USA National Institutes of Health including the National Institute of Neurological Disorders and Stroke, and the National Institute on Aging [NS075527 and NS103988], the National Institute of Mental Health [MH092797], and National Institute of Biomedical Imaging and Bioengineering [EB025815].
This work was also partially supported by USA Department of Energy Training Grant titled “Training in Techniques and Translation: Novel Nuclear Medicine Imaging Agents for Oncology and Neurology” [DE-SC0008432]. American Parkinson Disease Association (APDA), the Greater St Louis Chapter of the APDA and the Barnes Jewish Hospital Foundation.
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ob02609b
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.Proia RL and Hla T, J. Clin. Invest, 2015, 125, 1379–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kono M and Proia RL, Exp. Cell Res, 2015, 333, 178–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blaho VA and Hla T, J. Lipid Res, 2014, 55, 1596–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee MJ, Van Brocklyn JR, Thangada S, Liu CH,Hand AR, Menzeleev R, Spiegel S and Hla T, Science, 1998, 279, 1552–1555. [DOI] [PubMed] [Google Scholar]
- 5.Kohama T, Olivera A, Edsall L, Nagiec MM, Dickson R and Spiegel S, J. Biol. Chem, 1998, 273, 23722–23728. [DOI] [PubMed] [Google Scholar]
- 6.Hla T and Maciag T, J. Biol. Chem, 1990, 265, 9308–9313. [PubMed] [Google Scholar]
- 7.Ishizaka N, Okazaki H, Kurokawa K, Kumada M and Takuwa Y, Biochim. Biophys. Acta, 1994, 1218, 173–180. [DOI] [PubMed] [Google Scholar]
- 8.Wang C, Mao J, Redfield S, Mo Y, Lage JM and Zhou X, Exp. Mol. Pathol, 2014, 97, 259–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maceyka M and Spiegel S, Nature, 2014, 510, 58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lublin FD and Reingold SC, Neurology, 1996, 46, 907–911. [DOI] [PubMed] [Google Scholar]
- 11.Sheridan GK and Dev KK, Glia, 2012, 60, 382–392. [DOI] [PubMed] [Google Scholar]
- 12.Miron VE, Ludwin SK, Darlington PJ, Jarjour AA, Soliven B, Kennedy TE and Antel JP, Am. J. Pathol, 2010, 176, 2682–2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jackson SJ, Giovannoni G and Baker D, J. Neuroinflammation, 2011, 8, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brinkmann V, Br. J. Pharmacol, 2009, 158, 1173–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choi JW, Gardell SE, Herr DR, Rivera R, Lee CW, Noguchi K, Teo ST, Yung YC, Lu M, Kennedy G and Chun J, Proc. Natl. Acad. Sci. U. S. A, 2011, 108, 751–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trkov S, Stenovec M, Kreft M, Potokar M, Parpura V, Davletov B and Zorec R, Glia, 2012, 60, 1406–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Noda H, Takeuchi H, Mizuno T and Suzumura A, J. Neuroimmunol, 2013, 256, 13–18. [DOI] [PubMed] [Google Scholar]
- 18.Radue EW, O’Connor P, Polman CH, Hohlfeld R, Calabresi P, Selmaj K, Mueller-Lenke N, Agoropoulou C, Holdbrook F, de Vera A, Zhang-Auberson L, Francis G, Burtin P and Kappos L, Arch. Neurol, 2012, 69, 1259–1269. [DOI] [PubMed] [Google Scholar]
- 19.Karuppuchamy T, Behrens E. h., González-Cabrera P, Sarkisyan G, Gima L, Boyer JD, Bamias G, Jedlicka P, Veny M, Clark D, Peach R, Scott F, Rosen H and Rivera-Nieves J, Mucosal Immunol., 2016, 10, 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van der Giet M, Tolle M and Kleuser B, Biol. Chem, 2008, 389, 1381–1390. [DOI] [PubMed] [Google Scholar]
- 21.Hughes JE, Srinivasan S, Lynch KR, Proia RL, Ferdek P and Hedrick CC, Circ. Res, 2008, 102, 950–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee H, Deng J, Kujawski M, Yang C, Liu Y, Herrmann A, Kortylewski M, Horne D, Somlo G, Forman S, Jove R and Yu H, Nat. Med, 2010, 16, 1421–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liang J, Nagahashi M, Kim EY, Harikumar KB, Yamada A, Huang WC, Hait NC, Allegood JC, Price MM, Avni D, Takabe K, Kordula T, Milstien S and Spiegel S, Cancer Cell, 2013, 23, 107–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Watson C, Long JS, Orange C, Tannahill CL, Mallon E, McGlynn LM, Pyne S, Pyne NJ and Edwards J, Am. J. Pathol, 2010, 177, 2205–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pyne NJ and Pyne S, Nat. Rev. Cancer, 2010, 10, 489–503. [DOI] [PubMed] [Google Scholar]
- 26.Ponnusamy S, Selvam SP, Mehrotra S, Kawamori T, Snider AJ, Obeid LM, Shao Y, Sabbadini R and Ogretmen B, EMBO Mol. Med, 2012, 4, 761–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu H, Jin H, Yue X, Luo Z, Liu C, Rosenberg AJ and Tu Z, Mol. Imaging Biol, 2016, 18, 724–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jin H, Yang H, Liu H, Zhang Y, Zhang X, Rosenberg AJ, Liu Y, Lapi SE and Tu Z, J. Nucl. Cardiol, 2017, 24, 558–570. [DOI] [PubMed] [Google Scholar]
- 29.Liu H, Jin H, Yue X, Han J, Baum P, Abendschein DR and Tu Z, Mol. Imaging, 2017, 16, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prasad VP, Wagner S, Keul P, Hermann S, Levkau B, Schäfers M and Haufe G, Bioorg. Med. Chem, 2014, 22, 5168–5181. [DOI] [PubMed] [Google Scholar]
- 31.Shaikh RS, Schilson SS, Wagner S, Hermann S, Keul P, Levkau B, Schäfers M and Haufe G, J. Med. Chem, 2015, 58, 3471–3484. [DOI] [PubMed] [Google Scholar]
- 32.Rosenberg AJ, Liu H, Jin H, Yue X, Riley S, Brown SJ and Tu Z, J. Med. Chem, 2016, 59, 6201–6220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo Z, Rosenberg AJ, Liu H, Han J and Tu Z, Eur. J. Med. Chem, 2018, 150, 796–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pike VW, Trends Pharmacol. Sci, 2009, 30, 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rosenberg AJ, Liu H and Tu Z, Appl. Radiat. Isot, 2015, 102, 5–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harris JM and Chess RB, Nat. Rev. Drug Discovery, 2003, 2, 214. [DOI] [PubMed] [Google Scholar]
- 37.Fishburn CS, J. Pharm. Sci, 2008, 97, 4167–4183. [DOI] [PubMed] [Google Scholar]
- 38.Riley T and Riggs-Sauthier J, Pharm. Technol, 2008, 32, 88, 90–92, 94. [Google Scholar]
- 39.Li W, Zhan P, De Clercq E, Lou H and Liu X, Prog. Polym. Sci, 2013, 38, 421–444. [Google Scholar]
- 40.Mattheolabakis G, Wong CC, Sun Y, Amella CA, Richards R, Constantinides PP and Rigas B, J. Pharmacol. Exp. Ther, 2014, 351, 61–66. [DOI] [PubMed] [Google Scholar]
- 41.Kung HF, Choi SR, Qu W, Zhang W and Skovronsky D, J. Med. Chem, 2010, 53, 933–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zha Z, Choi SR, Ploessl K, Lieberman BP, Qu W, Hefti F, Mintun M, Skovronsky D and Kung HF, J. Med. Chem, 2011, 54, 8085–8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pajouhesh H and Lenz GR, NeuroRx, 2005, 2, 541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miller DS, Bauer B and Hartz AMS, Pharmacol. Rev, 2008, 60, 196–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kihara N, Hashimoto M and Takata T, Org. Lett, 2004, 6, 1693–1696. [DOI] [PubMed] [Google Scholar]
- 46.Gilmore JL, Sheppeck JE, Watterson SH, Haque L, Mukhopadhyay P, Tebben AJ, Galella MA, Shen DR, Yarde M, Cvijic ME, Borowski V, Gillooly K, Taylor T, McIntyre KW, Warrack B, Levesque PC, Li JP, Cornelius G, D’Arienzo C, Marino A, Balimane P, Salter-Cid L, Barrish JC, Pitts WJ, Carter PH, Xie J and Dyckman AJ, J. Med. Chem, 2016, 59, 6248–6264. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.