

1. Introduction
Zika virus (ZIKV) belongs to the Flavivirus genus of positive-sense single-stranded RNA viruses, together with dengue virus (DENV), West Nile (WNV), and yellow fever virus (YFV). The ~10.8 kb genome produces a single polyprotein that is co- and post-translationally processed into 10 mature proteins [1, 2]. While ZIKV is transmitted primarily through the bite of an infected Aedes mosquito, there is evidence supporting sexual transmission [3–5]. ZIKV has been detected in sporadic and isolated outbreaks in Africa, Southeast Asia and the Pacific Islands before early 2015 [6, 7], followed by a subsequent outbreak in South and Central America [2, 8]. While most ZIKV infections are associated with mild or asymptomatic viral disease, the magnitude of the recent epidemic has demonstrated that ZIKV can cause neurological syndromes including Guillian-Barre syndrome in adults, and congenital neurological disease such as microcephaly in a developing fetus [9]. The World Health Organization declared the ZIKV epidemic a global Public Health Emergency of International Concern in February 2016 [10], but lifted its designation in November 2016. The emergence and reemergence of ZIKV and similar pathogens around the world requires constant surveillance to facilitate rapid detection of new outbreaks.
Waggoner and Pinsky described methods including viral culture, antibody/antigen detection and RNA detection to identify ZIKV material in human specimens [11]. Although antibody-based assays to detect ZIKV are being developed to enable diagnosis after an acute infection [12, 13], such methods are not only time consuming and laborious, but currently lack regulatory approval in the United States. Similarly, a combination of comparative genomics and protein structure analysis has been utilized to identify regions that could distinguish between antibodies against a panel of flavivirus species and subtypes [14]. Nonetheless, serology-based diagnostics still require optimization and standardization. Genome-based detection methods such as qRT-PCR and/or sequencing can easily distinguish between ZIKV and other Flaviviruses during acute infection [15], with several sets of reagents for nucleic acid tests (NATs) being granted emergency-use authorization for detecting ZIKV in clinical samples. Real-time quantitative PCR (qRT-PCR) is a rapid, sensitive and specific method for virus detection at any stage of acute infection [16–19]. In contrast to conventional assays, qRT-PCR provides high specificity together with easy standardization and a quantitative measurement.
Serum and urine are two of the most commonly collected types of human specimens for testing with qRT-PCR. The composition, manipulation and processing of such samples are critical factors for optimal virus detection. Both RNA extraction efficiency and downstream PCR amplification could be affected by the concentration of virus in such clinical samples, which can impact the ability to detect viral genetic material. In addition, molecules like IgG, hemoglobin and lactoferrin that are present in blood, serum or plasma samples have been described as inhibiting PCR reactions [20]. Anticoagulants such as heparin can also affect the results of PCR [21]. In addition, the presence of these and other inhibitors in RNA extracted from sera is an obstacle for viral RNA amplification by qRT-PCR [22]. Urine is another type of human specimen that is frequently used for viral diagnosis, especially since viral RNA is detectable in urine at a higher load and for a longer period of time than it is in blood or cerebrospinal fluid [23, 24]. A protocol for isolating WNV and ZIKV genetic material from urine has been described [25, 26]; however, the need to optimize ZIKV detection by qRT-PCR and correlate cycle threshold (Ct) values to viral load should be addressed in order to improve the interpretation of data generated by qRT-PCR methods.
This study was consequently designed to establish and optimize a quantitative, sensitive, and specific method for ZIKV RNA detection in a manner that would allow additional downstream experiments. Specifically, our approach would apply the gold standard qRT-PCR method to serum and urine, the two most common human clinical materials used for diagnosis of ZIKV infection. In parallel, we performed additional experiments to determine which type of clinical material, either urine or serum, is ideal for detecting ZIKV-infected individuals. To optimize a qRT-PCR method for ZIKV detection, we spiked different amounts of ZIKV in urine or serum and then investigated the ability of two different reverse transcriptases to efficiently and consistently detect virus genetic material in either type of human fluid.
2. Materials and Methods
2.1. Virus and cells.
Stocks of ZIKV virus strain PRVABC59 were obtained from the Biodefense and Emerging Infections Research Resources Repository (BEI Resources, NR-50240) and used to infect Vero cells with a multiplicity of infection (MOI) of 0.01. Three days following infection, the supernatant was collected and centrifuged at 2,000 rpm for 10 minutes at 4°C, then stored at −80°C. A plaque assay was performed in triplicate to titer the ZIKV stocks by making 10-fold serial dilutions of the viral supernatant prior to using it to infect Vero cells that were 90% confluent. Three days after infection, plaques were counted and the titer of the viral stock was calculated in plaque forming units (PFU)/mL from three biological replicates [27].
2.2. Inoculation of human specimens.
The virus stock was then added to human serum and human urine from 3 healthy donors that were obtained from BioreclamationIVT. The urine was filtered using 0.45 μm filters from Millipore-Sigma before any manipulation. Two hundred microliters of each matrix were spiked with 1 μL of undiluted or 10-fold serial dilutions of the ZIKV strain PRVABC59 stock at 5 × 107 PFU/mL.
2.3. Viral RNA extraction, primers and qRT-PCR.
RNA extraction of spiked samples (200 μL) was performed using QIAamp Viral RNA mini kit (Qiagen). Five microliters of extracted RNA were used for cDNA synthesis using two different reverse-transcriptase enzymes: PrimeScript (Takara) and SuperScript III First-Strand Synthesis SuperMix (Life Technologies). Custom Taqman Primers and Probes were ordered from IDT. Separate primers and probes were designed for the Asian and African lineages, with the African lineage used as a negative control in these sets of experiments. Specifically, a bioinformatics analysis was performed to identify the best sequences for primers and probes. This workflow consisted of: 1) collecting available Asian and African ZIKV complete genomes from the Virus Pathogen Resource (ViPR) database [28], 2) generating a multiple sequence alignment with MAFFT [29], 3) performing a statistical analysis to identify nucleotide positions that significantly differed between Asian and African lineages [30], and 4) confirming the specificity of the reagents using BLAST [31]. The Asian primer set is as follows: (forward primer) ATAACAGCTTTGTCGTGGATG; (reverse primer) TAACCTTGAGCCAGACACTAG; FAM-probe: AGAGCATGGAACAGCTTTCTTGTG. The African primer set is as follows: (forward primer): TGAGAGCATGCTGCTAGC; (reverse primer) TGGCACGGCCATTGCTCG; VIC-probe: TGGATTTGCTTTGGCCTGGTTGG. For the qRT-PCR reaction, 4 μL of diluted cDNA (1:2) were added to the reaction using TaqMan Universal Master Mix II with UNG (Applied Biosystems). An ABI7500 (Applied Biosystem) instrument was used to calculate the Ct values during the qRT-PCR assay and the protocol involved: incubation at 50°C for 2 minutes, polymerase activation at 95°C for 10 minutes; followed by up to 45 cycles of denaturation at 95°C for 15 seconds and annealing/extension at 60°C for 1 minute.
2.4. Standard curves.
Ten-fold serial dilutions of ZIKV genetic material from BEI resources (NR-50244) were used to establish the correlation between Ct and number of molecules/μL of viral RNA. The copy number of RNA (number of molecules/μL) was calculated as: [RNA concentration (g/mL)] / [RNA transcript length (nucleotides) × molecular weight of a nucleotide (330 g/mol)] × Avogadro’s number (6.023 × 1023)]. In the same way, RNA from ZIKV stock (BEI, NR-50240) was serially diluted to establish the correlation between Ct and PFU/mL.
2.5. ZIKV infected individual samples.
Serum and urine samples from three infected individuals were analyzed using the qRT-PCR method described above. Samples from two of these patients were collected during the first clinical visit, while the sample from the third patient was collected 7 days after the first clinical visit.
2.6. Statistical analysis.
A two-tailed paired Student’s t-test was performed to compare the mean values of each data set, with p-values < 0.05 being categorized as significant.
3. Results
3.1. ZIKV detection by qRT-PCR.
In order to determine the best conditions for ZIKV detection, we spiked different concentrations of culture-grown virus (from 2.5×105 to 2.5×10−2 PFU/mL) into either human serum or filtered urine to evaluate the efficiency of detection by qRT-PCR. We purposefully avoided filtration of the serum due to its viscosity. After viral RNA extraction, cDNA was synthesized with two distinct reverse transcriptase enzymes to enable the identification of any differences during downstream virus quantification. The results of this experiment clearly showed differences in the ability to detect ZIKV based on the type of clinical specimen, as well as the reverse transcriptase enzyme used for cDNA synthesis (Figure 1). As expected, both matrix composition and the selected reverse transcriptase contributed to our ability to detect viral genetic material. Based on these results, we decided to perform a more in-depth analysis by comparing these variables side-by-side.
Figure 1. ZIKV detection by qRT-PCR at different concentrations of the virus in human serum and filtered (“F”) urine.
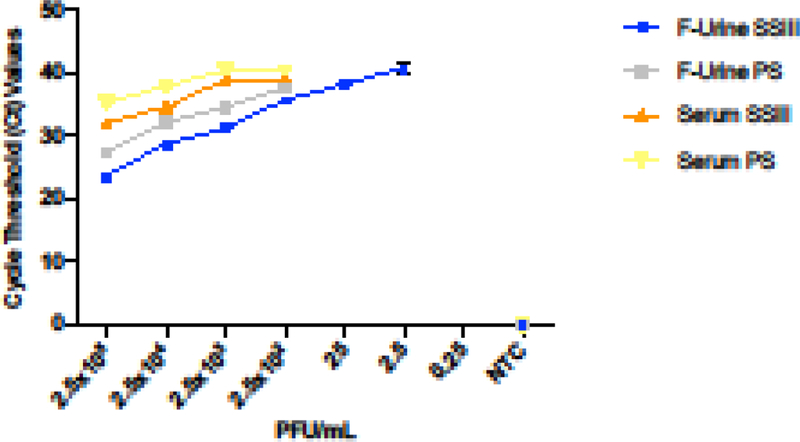
qRT-PCR was carried out using two reverse- transcriptase enzymes: SuperScript III (SSIII) represented in blue for filtered urine (F-Urine) and in orange for serum samples, and PrimeScript (PS) represented in grey and yellow for F-Urine and serum respectively. The experiment was performed in triplicate with the corresponding standard deviation measurement, and the addition of a negative control (NTC).
3.2. Reverse transcriptase sensitivity for ZIKV detection.
We began by studying how the different activities of reverse-transcriptase enzymes could affect ZIKV detection by qRT-PCR. Regardless of the clinical specimen that we used for RNA extraction, the SuperScript III (SSIII) reverse transcriptase enzyme showed significantly lower Ct values in detecting ZIKV by qRT-PCR compared with PrimeScript (PS) (Figure 2). This fact could be interpreted to mean a higher efficiency of SSIII; however, comparing the slopes and R2 values showed minimal difference (data not shown). Notably, the filtered urine samples showed that lower concentrations of the virus were still detectable when the cDNA synthesis was performed with the SSIII enzyme (Figure 2B).
Figure 2: Reverse transcriptase efficiency for ZIKV detection.
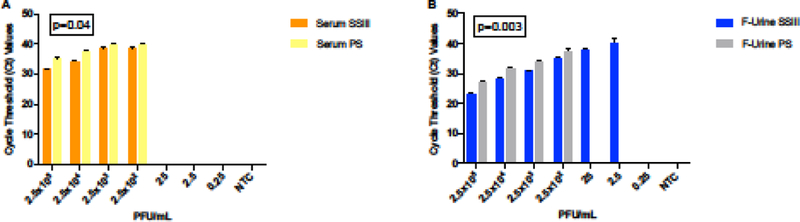
Side-by-side comparison of the effect that reverse-transcriptase SSIII (orange and yellow) or PS (blue and grey) had on ZIKV detection in serum (A) and filtered urine (B) by qRT-PCR. A two-tailed Student’s t-test of the paired means was used to calculate the p- values when comparing the two enzymes in the same matrix.
3.3. ZIKV detection in serum and urine samples spiked with ZIKV.
We then performed a side-by-side comparison to quantify our ability to detect ZIKV in serum and filtered urine by qRT-PCR. This experiment showed that we were better able to detect ZIKV in filtered urine samples when compared against serum (Figures 3A, 3B). Both of the enzymes that we tested showed statistically-significant differences in the Ct values for detecting ZIKV in both types of specimens, showing lower Ct values in the detection of ZIKV in filtered urine when compared with serum. We also found a 100-fold lower limit of detection (LOD) for ZIKV in filtered urine than in serum when we used the SSIII reverse-transcriptase for cDNA synthesis (LOD 2.5 PFU/mL and 250 PFU/mL respectively), in accordance with the previous results (Figure 2B). This observation was determined to be statistically significant (p=0.01) and can be used to better inform future clinical study designs.
Figure 3: ZIKV detection in ZIKV-spiked clinical sample types.
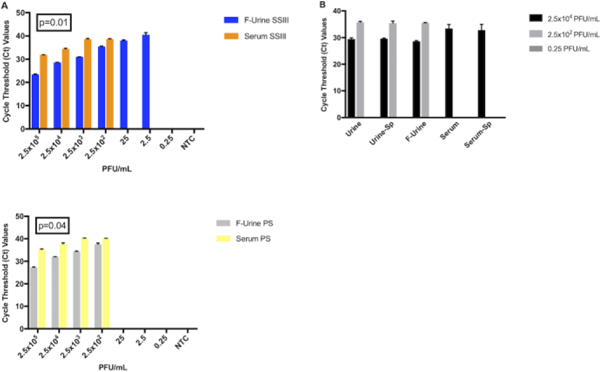
(A) Ct values corresponding to ZIKV detection by qRT-PCR in filtered urine (blue and grey bars) and serum (orange and yellow bars) using either the SSIII (upper panel) or the PS (lower panel) enzyme. A two-tailed Student’s t-test of the paired means was used to calculate the p-values when comparing the each enzyme in both matrices. (B) Effect of filtration (“F”) and spinning (“Sp”) before RNA isolation for ZIKV identification.
It is not always possible to obtain large volumes of samples for processing and analysis. In order to avoid extra and unnecessary steps in the sample preparation process, we repeated the experiment with and without the urine filtration step or the 30 second centrifugation for both matrices prior to RNA extraction. Regardless of whether the samples were filtered or centrifuged, we found no observable differences in our ability to detect ZIKV in spiked urine at the concentration tested (Figure 3B). Similarly, the centrifugation step did not produce any noticeable difference in the detection of virus in serum.
3.4. Quantification and standard curve of ZIKV RNA and infectious particles.
In order to test the sensitivity and specificity of the qRT-PCR assay, we first generated a standard curve using ten-fold serial dilutions of ZIKV genetic material to determine the LOD for the primer-probe set. The detection ranged from 8.6×106 to 0.865 molecules/μL with a R2 value of 0.99, showing a good confidence in correlating Ct values and the number of molecules per microliter (Figure 4A). We then evaluated the lowest detection limit of the qRT-PCR assay using 10-fold dilutions of the ZIKV stocks, which enabled us to quantify the virus concentration (PFU/mL) based on the Ct values generated by the qRT-PCR assay (Figure 4B). The sensitivity of the assay ranged between 1.4 ×107 and 1.4 PFU/mL. The Ct values were linear and correlated extremely well with the concentration of viral ZIKV RNA that we used (R2 value of 0.99). These data allowed us to calculate the concentration of ZIKV (PFU/mL) in infected samples based on the Ct values that were generated by qRT-PCR.
Figure 4: Sensitivity of the qRT-PCR assay.
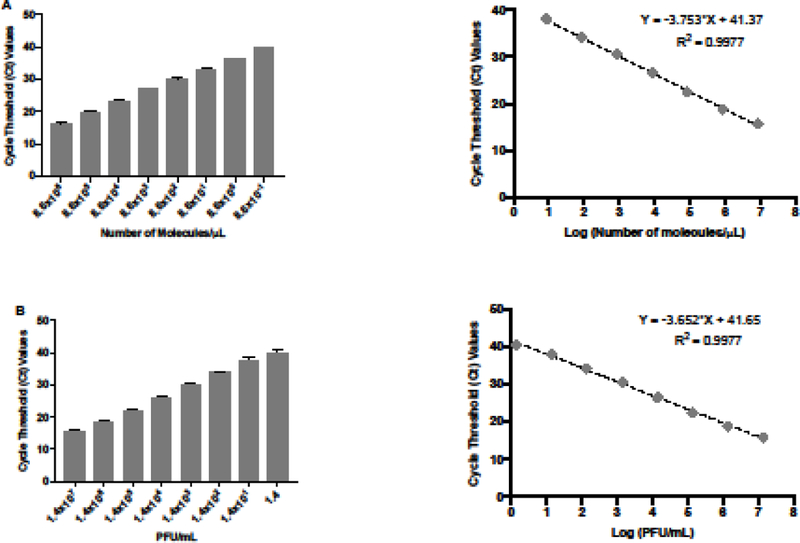
(A) Quantification and standard curve of number of molecules of ZIKV RNA/μL. Ct values were generated by qRT-PCR of extracted 10-fold serial diluted RNA from ZIKV stock. The standard curve was generated with 10-fold serial dilutions of ZIKV RNA. Ct values obtained are represented against the log of the number of molecules of ZIKV RNA. (B) Quantification and standard curve of ZIKV infectious particles. Ct values were generated by qRT-PCR of extracted 10-fold serial diluted RNA from ZIKV stock. The standard curve was generated with 10-fold serial dilutions of ZIKV RNA. Ct values obtained are represented against the log of the quantity of infectious viral particles (PFU/mL).
3.5. ZIKV detection clinical samples from infected individuals.
In order to corroborate our results, we decided to perform these assays in clinical samples collected from ZIKV-infected individuals. To do so, we obtained archived serum and urine samples from three different patients and followed the same experimental protocols as described above. The results from these assays detected ZIKV in one out of the three available urine samples, while ZIKV was not detectable in paired serum samples. Interestingly, the positive urine sample was collected during the first visit to the clinic, which was likely closer to the time of peak viral titer. Similar to our previous results, we observed improved detection of ZIKV in clinical samples when using the SSIII enzyme over the PS enzyme in clinical samples (Figure 5).
Figure 5: ZIKV detection in ZIKV-infected individual clinical samples.
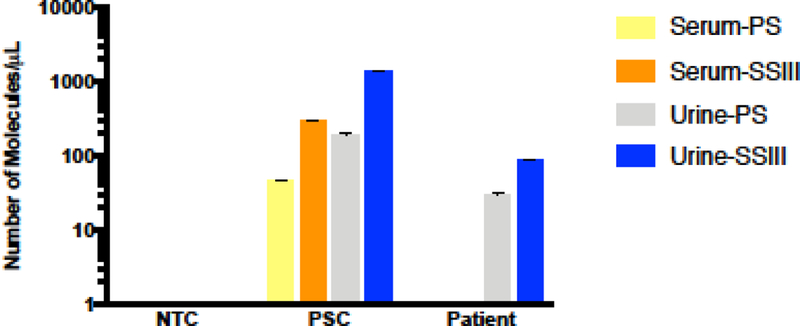
Number of viral molecules/μL were calculated through qRT-PCR after viral RNA isolation and cDNA synthesis with two different RTases (PS, SSIII) in serum and urine samples. ZIKV was detected in one out of three urine samples tested, with sera from all three patients being under the limit of detection for the assay.
4. Discussion
The recent re-emergence and expansion of ZIKV in the western hemisphere shows the need to develop rapid, specific and less-demanding extraction and amplification methods to detect the virus in patients. Selecting the ideal human specimens to detect ZIKV genetic material using qRT-PCR is an important step towards better detection of the virus in a way that is minimally invasive for the patient.
In our study, we have demonstrated that the type of matrix and the reverse transcriptase used in the assay play an important role in the ability to detect ZIKV by qRT-PCR. After performing a more in-depth analysis by comparing these variables side- by-side, we report that the SSIII reverse transcriptase is more suitable under these conditions than the PS enzyme for synthesizing ZIKV cDNA for downstream detection by qRT-PCR in the two matrices tested. These results confirm the need to account for enzyme evaluation when performing such experiments.
Our results demonstrate that the limit of viral RNA detection using these reagents is lower in urine than it is in serum, which concurs with previous reports of detecting WNV RNA at a higher load in urine than in other specimens [23, 24]. However, the underlying reason(s) for this observation are not completely clear. It could possibly be due to factors such as enzyme inhibitors present in the serum or the intrinsic viral burden found in either physiological compartment during ZIKV infection. Of note, we showed that neither the filtration nor the centrifugation steps are critical for sample processing before ZIKV RNA extraction, which may be useful to reduce the time and resources needed to complete similar protocols in clinical laboratories. The use of urine samples for ZIKV infection diagnosis has clinically-relevant implications, since its collection is rapid, noninvasive, and a relatively high volume of material being produced by the patient-especially when compared with specimens derived from blood. It is also interesting that the LOD of ZIKV in urine is lower than it was in serum. Therefore, a negative result for ZIKV by a qRT-PCR assay of patient serum cannot always be interpreted to mean a lack of ZIKV infection and consequently it remains possible for the virus to propagate in and from the patient during acute infection. Previous work has shown that urine samples were found to be positive for ZIKV for a longer period of time than serum samples after the onset of disease [26]. Taking this into consideration, urine is the ideal human clinical sample for ZIKV detection by qRT-PCR, and is very suitable for screening of samples collected in large-scale investigations as well as in epidemiological studies. We were able to validate these results in urine collected from a patient who was diagnosed with ZIKV infection; however, we could not detect the presence of ZIKV in the paired serum from the same patient. Paired samples from two other infected patients gave negative results in both urine and serum, suggesting a longer period of time had elapsed between peak viral titer and reporting at the clinic. Due to the limited number of paired serum and urine samples we are unable to perform more assays on clinical samples from infected individuals. Our results show promise as an alternative to detecting ZIKV in the clinical setting and support our earlier detection experiments performed with artificially “spiked” virus to mimic infected samples.
The standard curve that we generated to quantify the virus concentration (PFU/mL) with these reagents can help identify the clinical stage of the infection (early, acute, late). Given the length of the primers and probes, the specificity, and the sensitivity, we expect that these reagents can be useful in identifying new ZIKV outbreaks and enabling the relevant public health personnel to better control the spread of this pathogen.
5. Conclusions
This study provides a sensitive and specific method to detect ZIKV genomic RNA in two of the most common human specimen types. The qRT-PCR reagents reported here comprise a useful tool for virus detection, quantification and consequently, prevention of viral transmission to other individuals.
Highlights:
Reverse transcriptase selection is critical for improving ZIKV detection by qRT-PCR
The limit of detection of ZIKV by qRT-PCR is lower in urine than in serum
SuperScript III is better than PrimeScript for ZIKV detection in serum and urine
Filtration and centrifugation are not critical protocol steps for qRT-PCR detection
8. Acknowledgments
We gratefully acknowledge the Mexico Emerging Infectious Diseases Clinical Research Network (LaRed) Zik01 study team for providing the serum and urine samples from study participants infected with Zika virus.
6. Funding
This project has been funded in whole or part with federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services under Award Number U19AI110819.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest
The authors declare no conflict of interest.
9. References
- 1.Lindenbach BDRCM, Molecular biology of flaviviruses.Adv Virus Res, 2003. 59: p. 23–61. [DOI] [PubMed] [Google Scholar]
- 2.White MK, W.H.S., David Beckham J, Tyler KL, Khalili K, Zika virus: An emergent neuropathological agent. Ann Neurol, 2016. 80(4): p. 479–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moreira J, Lamas CC, and Siqueira A, Sexual Transmission of Zika Virus: Implications for Clinical Care and Public Health Policy. Clin Infect Dis, 2016. 63(1): p. 141–2. [DOI] [PubMed] [Google Scholar]
- 4.McDonald EM, Duggal NK, and Brault AC, Pathogenesis and sexual transmission of Spondweni and Zika viruses. PLoS Negl Trop Dis, 2017. 11(10): p. e0005990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Musso D, et al. , Detection of Zika virus RNA in semen of asymptomatic blood donors. Clin Microbiol Infect, 2017. 23(12): p. 1001 e1–1001 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duffy MR, et al. , Zika virus outbreak on Yap Island, Federated States of Micronesia. N Engl J Med, 2009. 360(24): p. 2536–43. [DOI] [PubMed] [Google Scholar]
- 7.Galindo-Fraga A, et al. , Zika Virus: A New Epidemic on Our Doorstep. Rev Invest Clin, 2015. 67(6): p. 329–32. [PubMed] [Google Scholar]
- 8.Musso D, Zika Virus Transmission from French Polynesia to Brazil. Emerg Infect Dis, 2015. 21(10): p. 1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Russo FB, Jungmann P, and Beltrao-Braga PCB, Zika infection and the development of neurological defects. Cell Microbiol, 2017. 19(6). [DOI] [PubMed] [Google Scholar]
- 10.WHO, Zika Situation Report. 2016.
- 11.Waggoner JJ and Pinsky BA, Zika Virus: Diagnostics for an Emerging Pandemic Threat. J Clin Microbiol, 2016. 54(4): p. 860–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Balmaseda A, et al. , Antibody-based assay discriminates Zika virus infection from other flaviviruses. Proc Natl Acad Sci U S A, 2017. 114(31): p. 8384–8389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steinhagen K, et al. , Serodiagnosis of Zika virus (ZIKV) infections by a novel NS1-based ELISA devoid of cross-reactivity with dengue virus antibodies: a multicohort study of assay performance, 2015 to 2016. Euro Surveill, 2016. 21(50). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee AJ, et al. , Identification of diagnostic peptide regions that distinguish Zika virus from related mosquito-borne Flaviviruses. PLoS One, 2017. 12(5): p. e0178199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hayes EB, Zika virus outside Africa. Emerg Infect Dis, 2009. 15(9): p. 1347–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mackay IM, Arden KE, and Nitsche A, Real-time PCR in virology. Nucleic Acids Res, 2002. 30(6): p. 1292–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Watzinger F, et al. , Real-time quantitative PCR assays for detection and monitoring of pathogenic human viruses in immunosuppressed pediatric patients. J Clin Microbiol, 2004. 42(11): p. 5189–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Faye O, et al. , Quantitative real-time PCR detection of Zika virus and evaluation with field-caught mosquitoes. Virol J, 2013. 10: p. 311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tien WP, et al. , SYBR green-based one step quantitative real-time polymerase chain reaction assay for the detection of Zika virus in field-caught mosquitoes. Parasit Vectors, 2017. 10(1): p. 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schrader C, et al. , PCR inhibitors - occurrence, properties and removal. J Appl Microbiol, 2012. 113(5): p. 1014–26. [DOI] [PubMed] [Google Scholar]
- 21.Costafreda MI, Bosch A, and Pinto RM, Development, evaluation, and standardization of a real-time TaqMan reverse transcription-PCR assay for quantification of hepatitis A virus in clinical and shellfish samples. Appl Environ Microbiol, 2006. 72(6): p. 3846–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Konet DS, et al. , Inhibitors of RT-PCR in serum. J Virol Methods, 2000. 84(1): p. 95–8. [DOI] [PubMed] [Google Scholar]
- 23.Tonry JH, et al. , West Nile virus detection in urine. Emerg Infect Dis, 2005. 11(8): p. 1294–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barzon L, et al. , Excretion of West Nile virus in urine during acute infection. J Infect Dis, 2013. 208(7): p. 1086–92. [DOI] [PubMed] [Google Scholar]
- 25.Barzon L, et al. , Isolation of West Nile virus from urine samples of patients with acute infection. J Clin Microbiol, 2014. 52(9): p. 3411–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gourinat AC, et al. , Detection of Zika virus in urine. Emerg Infect Dis, 2015. 21(1): p. 84–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Agbulos DS, et al. , Zika Virus: Quantification, Propagation, Detection, and Storage. Curr Protoc Microbiol, 2016. 43: p. 15d.4.1–15d.4.16. [DOI] [PubMed] [Google Scholar]
- 28.Pickett BE, et al. , ViPR: an open bioinformatics database and analysis resource for virology research. Nucleic Acids Res, 2012. 40(Database issue): p. D593–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakamura T, et al. , Parallelization of MAFFTfor large-scale multiple sequence alignments. Bioinformatics, 2018. 34(14): p. 2490–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pickett BE, et al. , Metadata-driven comparative analysis tool for sequences (meta-CATS): an automated process for identifying significant sequence variations that correlate with virus attributes. Virology, 2013. 447(1–2): p. 45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Altschul SF, et al. , Basic local alignment search tool. J Mol Biol, 1990. 215(3): p. 403–10. [DOI] [PubMed] [Google Scholar]