

Abstract
Pathogenic mutations in cyclin-dependent kinase-like 5 (CDKL5) result in CDKL5 deficiency disorder (CDD), a rare disease marked by early-life seizures, autistic behaviors, and intellectual disability. Although mouse models of CDD exhibit dendritic instability and alterations in synaptic scaffolding proteins, studies of glutamate receptor levels and function are limited. Here we used a novel mouse model of CDD, the Cdkl5R59X knock-in mouse (R59X), to investigate changes in synaptic glutamate receptor subunits and functional consequences. Male mice were used for all experiments to avoid the confounding effects of X-inactivation that would be present in female heterozygous mice. We showed that adult male R59X mice recapitulated the behavioral outcomes observed in other mouse models of CDD, including social deficits and memory and learning impairments, and exhibited decreased latency to seizure upon pentylenetetrazol administration. Furthermore, we observed a specific increase in GluA2-lacking α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid)-type glutamate receptors (AMPARs) in the adult R59X hippocampus, which is accompanied electrophysiologically by increased rectification ratio of AMPAR EPSCs and elevated early-phase long term potentiation (LTP). Finally, we showed that acute treatment with the GluA2-lacking AMPAR blocker IEM-1460 decreased AMPAR currents, and rescued social deficits, working memory impairments, and seizure behavior latency in R59X mice.
SIGNIFICANCE STATEMENT CDKL5 deficiency disorder (CDD) is a rare disease marked by autistic-like behaviors, intellectual disability, and seizures. While synaptic dysfunction has been observed in mouse models of CDD, there is limited information on how synaptic alterations contribute to behavioral and functional changes in CDD. Here we reveal elevated hippocampal GluA2-lacking AMPAR expression in a novel mouse model of CDD that is accompanied by changes in synaptic AMPAR function and plasticity. We also show, for the first time, that acutely targeting GluA2-lacking AMPAR dysregulation rescues core synaptic and neurobehavioral deficits in CDD.
Keywords: autism, behavior, CDKL5, excitability, learning and memory
Introduction
CDKL5 deficiency disorder (CDD) is a severe neurological disease caused by pathogenic mutations in the X-linked gene for cyclin-dependent kinase-like 5 (CDKL5), a serine-threonine kinase that is highly expressed in the brain (Montini et al., 1998; Kilstrup-Nielsen et al., 2012). Patients with CDD exhibit intellectual disability (ID), autistic-like behaviors, and early-life epilepsy, neurological deficits that often co-occur and share underlying mechanisms of synaptic dysfunction. Indeed, mouse models of CDD exhibit structural and functional changes at excitatory dendritic spines, which are accompanied by behavioral alterations, such as social interaction deficits, and impaired learning and memory (Wang et al., 2012; Amendola et al., 2014; Tang et al., 2017). Furthermore, CDKL5 itself has been found to localize to synapses and interact with postsynaptic density protein 95 (PSD-95), netrin-G1 ligand (NGL-1), and Rac1 (Chen et al., 2010; Ricciardi et al., 2012; Zhu et al., 2013), proteins that are involved with clustering and transport of excitatory glutamate receptors at the synapse. However, there is limited information regarding the link between alterations in synaptic glutamate receptors and neurobehavioral deficits in CDD mouse models.
Glutamate receptors, which include AMPA, NMDA, and kainate receptors, are heteromeric structures that mediate excitatory neurotransmission in the brain. Receptor subunits are dynamically expressed, altering receptor composition and biophysical properties to regulate the excitatory-inhibitory balance throughout the lifetime (Luján et al., 2005; Rakhade and Jensen, 2009). Dysregulated glutamate receptor subunit expression is often associated with epilepsy, autism, and ID (Loddenkemper et al., 2014; Mignogna et al., 2015; Lippman-Bell et al., 2016). In particular, even modest changes in the tightly regulated GluA2 AMPAR subunit have a profound effect on excitability and signal transduction due to the distinct properties of GluA2 (Lippman-Bell et al., 2013; Uzunova et al., 2014; Stephenson et al., 2017). Most notably, GluA2-lacking AMPARs are Ca2+-permeable (CP-AMPARs) and play a significant role in regulating dendritic spine enlargement as well as downstream signaling and gene transcription (Plant et al., 2006; Fortin et al., 2010; Henley and Wilkinson, 2016). Indeed, we have previously observed decreased GluA2 expression following neonatal hypoxic seizures in wild type (WT) rats, which contributes to social interaction deficits, elevated activity-dependent Ca2+ influx, altered downstream signaling, and increased epileptogenesis (Sanchez et al., 2001; Rakhade et al., 2008; Lippman-Bell et al., 2013, 2016; Rosenberg et al., 2018). Finally, altered GluA2 subunit expression has been observed in several neurodevelopmental disorders, such as Rett syndrome, Fragile X syndrome, tuberous sclerosis, and RAB39B-associated mental retardation (Talos et al., 2008; Mignogna et al., 2015; Li et al., 2016; Achuta et al., 2018), pointing to a critical role of GluA2 in disease pathophysiology.
Given that elevated GluA2-lacking AMPARs have been previously linked to neurobehavioral deficits and Cdkl5 knockdown causes dysregulated GluA2 in neurons in vitro (Tramarin et al., 2018), we investigated levels of AMPAR subunits GluA1 and GluA2 in a novel mouse model of CDD, the Cdkl5R59X knock-in mouse (R59X). The R59X mutation is based on the human mutation that has been observed to cause severe seizures and ID (Castrén et al., 2011) and occurs in the catalytic domain of the CDKL5 gene, resulting in premature truncation of the mRNA transcript and absence of functional CDKL5 protein. Our comprehensive behavioral assessment showed that R59X mice replicated behavioral alterations observed in prior Cdkl5 KO mouse models (Wang et al., 2012; Fuchs et al., 2014), and exhibited decreased latency to seizure. At the cellular level, we found increased hippocampal GluA2-lacking AMPARs accompanied by functional changes at CA1 pyramidal neurons. Furthermore, several behavioral alterations and latency to seizure behavior were rescued in R59X mice by acute administration of a GluA2-lacking AMPAR blocker, IEM-1460. Finally, as proof of principle, we showed increased GluA2-lacking AMPARs in postmortem hippocampal tissue from 2 human CDD patients. Together, these data reveal a GluA2-lacking AMPAR-associated mechanism of excitatory-inhibitory imbalance that may underlie functional alterations in CDD, and that targeting GluA2-lacking AMPAR dysregulation in CDD is potentially a viable therapeutic strategy for patients.
Materials and Methods
Animals
All procedures were performed in accordance with the guidelines of the National Institutes of Health Guide for the care and use of laboratory animals and approval of Institution of Animal Care and Use Committee at the University of Pennsylvania (Philadelphia). All studies were performed on Cdkl5R59X mice and age-matched WT littermates on a congenic sv129:C57BL/6 background, available at The Jackson Laboratory (IMSR, catalog #JAX:028856, RRID:IMSR_JAX:028856). Mice were originally generated and obtained from the laboratory of Dr. Zhaolan Zhou at the University of Pennsylvania. To produce R59X mice, a targeting vector was designed to insert a frt-flanked neomycin resistance (neo) cassette downstream of exon 5, and a single nucleotide change of C to T, leading to a nonsense mutation at arginine 59 (R59X) of the cyclin-dependent kinase-like 5 (Cdkl5) gene. The construct was electroporated into C57BL/6N embryonic stem cells. Correctly targeted embryonic stem cells were injected into BALB/c blastocysts, and resulting chimeric mice were bred with B6.Cg-Tg(ACTFLPe)9205Dym/J to remove the neo cassette. Resulting offspring were bred to C57BL/6 mice for at least 10 generations to establish a colony of R59X mice.
Male mice were used for all experiments to avoid confounding effects of X-inactivation that would be present in heterozygous female mice. All efforts were made to minimize animal suffering and numbers. Mice were genotyped using a PCR protocol designed to detect the R59X mutation. The genotyping primers (5′-GCTGCTTACATTAGGAGAGACTGC-3′ and 5′-GTCACATGACCAGCCAGCGT-3′) give rise to a 204 bp product from WT mice and a 347 bp product from the knock-in mutation.
Human postmortem tissue
Human postmortem tissue samples were obtained from the National Institutes of Health NeuroBioBank. The CDKL5 specimens were from the Harvard Brain Tissue Resource Center, Boston. Case AN06796 was from a 5.7 year old female and Case AN11848 was from a 30 year old female. Both specimens had confirmed CDKL5 gene mutations. Control samples were obtained from the Brain and Tissue Repository at the University of Maryland, Baltimore. The study was approved by the Institutional Review Board. Tissue processing for both cases was performed as described for the animal tissue, and the same antibodies were used to probe for GluA1 and GluA2 (for full tissue preparation procedure and antibody information, see western blotting).
Animal behavior
All behavioral studies were performed blinded to genotype. Mice were allowed habituate in the testing room at least 30 min before each test. All tests were performed at the same time of day. Behavioral tests were performed on male R59X mice and WT littermates at 12–16 weeks of age.
Open-field assay.
Home cage-like activity was monitored by beam breaks in a photobeam frame. Mice were placed into a clean environment in a photobeam frame. The number of beam breaks over 10 min was quantified as a measure of locomotor activity, with the first 3 min denoted as the habituation period.
Y maze.
Mice were placed at the end of one arm of a Y-shaped apparatus where each arm was labeled. Arm entries were recorded as they occurred. The mouse was considered to enter an arm when all four paws were in the arm. The mouse was allowed to explore the maze for 8 min. Percentage spontaneous alternation behavior (% SAB) was calculated using the following formula: % SAB = [(number of alternations/(total arm entries − 2))] × 100.
Three-chambered social approach test.
The social approach assay was performed in a three-chambered arena consisting of a center chamber and two end chambers, as previously described (Brodkin, 2007). One end chamber was designated the “social chamber,” whereas the other was designated the “nonsocial” chamber. Two Plexiglas cylinders with air holes were placed in each end chamber. The test mouse was placed in the arena for a 10 min habituation phase, during which the amount time spent in each chamber was measured (baseline). For the social phase, a stimulus mouse (adult gonadectomized A/J mouse; The Jackson Laboratory) was placed in the cylinder in the social chamber, and a novel object was placed in the cylinder in the nonsocial chamber. The test mouse was again allowed to explore the arena for 10 min. Time spent in each chamber over the 10 min test phase was measured.
Context-dependent fear conditioning.
In the training phase, mice were placed in conditioning chambers for 3 min terminated with a 1.5 mA foot shock. The uniqueness of the chamber was considered as the conditioned stimulus. Mice were left in the chamber for 1 min after the foot shock. Mice were returned to the conditioning chamber 24 h later for a 5 min test phase. Time spent freezing (no motion except for respiratory movements) during the 5 min test phase was measured with Freezescan software by Clever Systems.
Accelerating rotarod assay.
Adapted from Rothwell et al. (2014) a five station rotarod treadmill (IITC) was modified with a pulley and belt purchased from the vendor to increase the standard of acceleration, allowing the rotarod to attain speeds of up to 80 rpm while maintaining a constant rate of acceleration over a 300 s trial. Mice were placed on the rotarod for 3 trials per day over 4 consecutive days with at least a 15 min intertrial interval. On day 1, mice were allowed to acclimate to the stationary rod for 1 min before beginning the first trial. Days 1 and 2 consisted of 4–40 rpm trials, and days 3 and 4 consisted of 8–80 rpm trials. Trials were terminated after a mouse fell off the rod, made one complete revolution while clinging to the rod, or after 300 s (maximum speed).
Hindlimb clasping test.
Mice were picked up by the base of the tail and lifted clear of all surrounding objects. Hindlimb position was observed for 10 s. If the hindlimbs were consistently splayed outward, away from the abdomen, the mouse was assigned a score of 0. If one hindlimb was retracted toward the abdomen for more the 50% of the time, the score was 1. If both hindlimbs were partially retracted for >50% of the time, the score was 2. Finally, if hindlimbs were entirely retracted and touching the abdomen for >50% of the time suspended, the mouse received a score of 3.
Olfactory test.
Mice were tested on whether or not they could detect and differentiate odors. Mice were exposed to cotton-tipped wooden applicators dipped in water, vanilla, or swiped across the bottom of an unfamiliar social cage. Each applicator was presented for 2 min with a 1 min interval between applicators. Time spent sniffing was defined as when the mouse had its nose within 2 cm of the cotton tip.
Pentylenetetrazol (PTZ) seizure threshold test.
Eighteen-week-old male R59X mice and WT littermates were allowed to habituate in the procedure room for 30 min. Mice were injected with a subthreshold dose of PTZ (40 mg/kg, i.p.) and placed in a Plexiglas grid of 12 individual chambers. Each chamber had fresh bedding. Mice were video-monitored for 2 h. Videos were scored by blinded observer using a modified Racine scale (Lüttjohann et al., 2009) as follows: 0, normal behavior; 1, freezing or immobility for at least 5 s; 2, myoclonic jerks; 3, forelimb clonus; 4, rearing, forelimb clonus; 5, tonic-clonic, loss of posture.
Western blotting
To compare developmental expression patterns of AMPA receptor subunits GluA1 and GluA2, R59X and WT littermates were killed at P7, P14, P21, P30, P50, and P125. Brains were rapidly dissected from the skull, and hippocampal and cortical regions were separated. Tissue was flash-frozen in chilled ethanol and stored at −80°C until homogenization. Membrane and whole-cell protein extraction from the anterior two-thirds of the cortex, and the entire hippocampal tissue was performed as described previously (Wenthold et al., 1992; Talos et al., 2006; Rakhade et al., 2012). Halt Protease and Phosphatase Inhibitor Cocktail EDTA-free (Thermo Fisher Scientific) and PMSF (Thermo Fisher Scientific) were added to inhibit proteases and phosphatases. Total protein concentrations were measured by the Bradford protein assay (Bio-Rad), and samples were diluted to obtain equal amounts of protein for each sample (1 μg/μl). Samples were separated by gel electrophoresis on Criterion TGX 4–20% precast gels and transferred to PVDF membranes (Immobilon-FL). Primary antibodies used in this study were GluA2 (1:1000; Millipore, catalog #MAB397, RRID:AB_2113875), GluA1 (1:1000; Abcam, catalog #ab31232, RRID:AB_2113447), GluN2A (1:1000, Sigma-Aldrich, catalog #M264, RRID:AB_260485), GluN2B (1:1000, Thermo Fisher Scientific, catalog #MA1-2014, RRID:AB_560347) CDKL5 (1:1000, Millipore, catalog #MABS1132), and β-actin (1:3000; Sigma-Aldrich, catalog #A5441, RRID:AB_476744). Secondary antibodies used were rabbit IRDye 800CW (LI-COR Biosciences, catalog #926-32213, RRID:AB_621848) and mouse IRDye 680LT (LI-COR Biosciences, catalog #926-68024, RRID:AB_10706168). The Odyssey Infrared Imaging System protocols were used for blots and quantification of protein expression. Protein levels were normalized to age-matched WT littermates and the P50 time point for developmental curves.
qRT-PCR.
P50 mice were killed, and whole hippocampus was collected. RNA was isolated using QIAGEN RNeasy kit and then converted to cDNA with Invitrogen SuperScript III First-Strand Synthesis System. qRT-PCR was run using TaqMan optimized primer probe sets (for GRIA1, GRIA2, ACTB, and HPRT) and TaqMan Master Mix.
Immunohistochemistry
P50 WT and R59X mice were perfused transcardially with 4% PFA. Brains were dissected from the skull and postfixed for 1 h at room temperature before being transferred to 30% sucrose. Once saturated with sucrose, the brains were embedded in OCT and frozen; 16 μm sections were collected using a cryostat and mounted on slides. Sections containing the middle one-third of the hippocampus were blocked with 10% normal goat serum in PBS and incubated with either anti-GluA2 (extracellular, 1:500; Millipore, catalog #MAB397, RRID:AB_2113875) or anti-GluA1 (extracellular, 1:500, NeuroMab, catalog #75-327, RRID:AB_2315840) overnight, then incubated with AlexaFluor secondary (1:1000, Thermo Fisher Scientific, catalog #A-11001, RRID:AB_2534069) for 1 h at room temperature. Sections were then permeabilized with 0.2% Triton X-100 in PBS for 5 min and then blocked with 10% NGS with 0.1% Triton in PBS for 1 h. Finally, the sections were incubated with anti-synapsin (1:1000, Millipore, catalog #574777, RRID:AB_2200124) and anti-MAP2 (1:1000, Millipore, catalog #AB5543, RRID:AB_571049) diluted in 0.1% NGS and 0.1% Triton in PBS overnight at 4°C, washed with PBS, and incubated with AlexaFluor secondary (1:1000, Thermo Fisher Scientific, catalog #R37119, RRID:AB_2556547) for 1 h at room temperature. Fluoromount with DAPI was used to mount the sections with a coverslip. Sections incubated without primary and only with secondary antibody were imaged to verify antibody specificity. We have previously confirmed the surface specificity of the GluA2 antibody used here in permeabilized and nonpermeabilized sections incubated with an antibody to MAP2 in addition to GluA2 (Lippman-Bell et al., 2016). Blinded sections were placed on an LSM710 confocal microscope (Carl Zeiss) with an oil-immersion 63 × 1.7 NA objective. z stacks for all images were 0.25 μm.
Analysis of z stacks was performed blindly using ImageJ software. First, a mask was created from the MAP2 staining to define an ROI so that only puncta on the surface of dendrites would be incorporated in our analysis. To avoid a thresholding artifact, colocalization was first measured at the maximum threshold incorporating all GluA2 puncta; then the GluA2 channel was thresholded at multiple intervals of the max threshold (95%, 90%, 85%, 80%, 75%, and 70%), and colocalization was determined again at each threshold (Nie et al., 2010; Zhou et al., 2011; Lippman-Bell et al., 2016). After thresholding each channel, the overlap of GluA2 with synapsin puncta was quantified using the plugin coloc2 to determine the Manders coefficient (Manders et al., 1993).
Electrophysiology
Hippocampal slice preparation.
P28-P32 R59X and WT animals were decapitated and brains rapidly dissected from the skull and placed for section in ice-cooled cutting solution containing ACSF as follows (in mm): 220 sucrose, 3 KCl, 1.25 NaH2PO4, 0.5 CaCl2, 1 MgCl2, 26.19 NaHCO3, and 10 d-glucose, pH 7.4, 295–305 mOsm bubbled with 95%O2/5%CO2 at 4°C. Coronal hippocampal slices (350 μm thickness) were sectioned from the middle one-third of hippocampus with a vibratome (VT1000S, Leica Microsystems) in cutting solution. Slices were incubated for 30 min at 32°C followed by at least 30 min at room temperature in oxygenated ACSF, containing the following (in mm): 130 NaCl, 3 KCl, 1.25 NaH2PO4, 1 MgCl2, 2 CaCl2, 26 NaHO2, 10 d-glucose, pH 7.4, 295–305 mOsm, bubbled with 95%O2/5%CO2.
Extracellular field recordings.
Slices were transferred to the recording chamber with ACSF heated to 32°C and Schaffer collaterals from CA3 to CA1 were stimulated at 30 s intervals to obtain input-output curves. Stimulus intensity (0.1 ms in duration) that evoked 60%–70% maximal slope response was used for paired-pulse ratio and LTD and LTP induction. Paired-pulse stimuli were delivered at interstimulus intervals ranging from 6 to 500 ms, and the degree of paired-pulse inhibition or facilitation was calculated for each interval. For paired-pulse analysis, a ratio of the second response amplitude to that of the first was used. At a given interstimulus interval, a ratio >1 indicated paired-pulse facilitation and a ratio <1 indicated paired-pulse inhibition. Finally, baseline responses were recorded every 30 s for 10–20 min. After a stable baseline of ≥10 min, either LTD or LTP was induced. LTP was induced by high-frequency stimulation (HFS; 4 stimuli at 100 Hz, 1 s duration, repeated 20 s apart) (Jensen et al., 1998). After tetanus, single half-maximal stimuli were applied every 30 s for 60 min in all slices. The percentage change in fEPSP 10%–90% slope was calculated after tetanus compared with pretetanus baseline. To induce LTD low-frequency stimulation (LFS), the protocol consisted of 1800 shocks at 2 Hz for 15 min (Lippman-Bell et al., 2016).
Whole-cell electrophysiology.
Whole-cell patch-clamp recordings from hippocampal CA1 pyramidal neurons in brain slices using infrared differential interference contrast microscope as described previously (Rakhade et al., 2008). The patch-pipette internal solution contained the following (in mm): 110 Cs-methanesulfonate, 10 TEA-Cl, 4 NaCl, 2 MgCl2, 10 EGTA, 10 HEPES, 4 ATP-Mg, and 0.3 GTP, pH 7.25, 5 QX-314, 7 phosphocreatine, creatine phosphokinase (17 unit/ml), and 0.1 spermine, 280–290 mOsm. Filled electrodes had resistances of 2–5 mΩ. eEPSCs were elicited at 10 s intervals. Picrotoxin (100 μm) or AP5 (50 μm) was added to block GABA receptors or NMDA receptors, respectively, for the I–V recordings. To collect evoked AMPA receptor currents for I–V curves, cells were held from −80 mV to 40 mV and evoked by stimulating the Schaffer collaterals. The data were obtained from the average peak amplitude from 3 or 4 recordings per holding voltage (Vh). For GluA2-lacking AMPAR-selective antagonist slice application of 100 μm 1-naphthyl acetyl spermine trihydrochloride (NASPM, Abcam #146810) or 100 μm IEM-1460 (Abcam #141507) were added to the ACSF after control eEPSCs were evoked by stimulating the Schaffer collaterals. The data were obtained from the average peak amplitude from 10 consecutive recordings with Vh = −60 mV (Magazanik et al., 1997; Noh et al., 2005). Data were collected using an Axopatch200B amplifier (Molecular Devices) and Clampex 9.2 software (Molecular Devices) with compensation for series resistance (70%) and cell capacitance, filtered at 2 kHz, and digitized at 20 kHz using a Digidata 1320A analog to digital converter (Molecular Devices).
Drug treatment
IEM-1460 effect on working memory, long-term memory, and social interaction.
Twelve- to 16-week-old mice were administered 10 mg/kg IEM-1460 (Tocris Bioscience, catalog #1636, i.p.) or 0.9% saline vehicle (i.p.). Mice were tested 1 h later for each behavioral test. For fear conditioning experiments, mice were administered IEM-1460 1 h before training and again 24 h later, 1 h before testing.
IEM-1460 effect on seizure threshold.
Eighteen-week-old mice were administered 10 mg/kg IEM-1460 or 0.9% saline vehicle (i.p.) followed 1 h later by 40 mg/kg PTZ (i.p.). Mice were placed in a Plexiglas grid for video-monitoring. Videos were scored by blinded observer using a modified Racine scale as before (Lüttjohann et al., 2009).
Experimental design and statistical analysis
For behavioral tests, we chose sample sizes based on previously published results in the CDD literature (Wang et al., 2012). For western blots, immunohistochemistry, and electrophysiology, we determined sample size and power based on previously published work in our laboratory and using the StatMate software. The number of mice used in each experiment was predetermined before the start of the experiment, and mice were randomly assigned to receive either vehicle or drug for treatment experiments. Mice were excluded if their weight was >2 SDs from the mean weight of age-matched mice.
Statistical analysis was completed using Prism (GraphPad, RRID:SCR_002798). p values, n values, and mean ± SEM are reported in Results. All datasets were analyzed using the D'Agostino-Pearson test for normality. Datasets with normal distributions were analyzed for significance using unpaired Student's two-tailed t test. Two-way repeated-measures ANOVA was conducted for the appropriate datasets with Holm–Sidak's multiple-comparison test.
Results
Absence of full-length CDKL5 protein in R59X whole-brain lysates
To ensure that no functional CDKL5 protein was present in mutant mice, we performed western blots with whole-cell lysates of R59X and WT whole brains. No CDKL5 protein was observed in lysates from R59X mice (Fig. 1), and no smaller peptide fragments were observed (data not shown), suggesting that the R59X mutation results in complete knockdown of CDKL5 protein.
Figure 1.
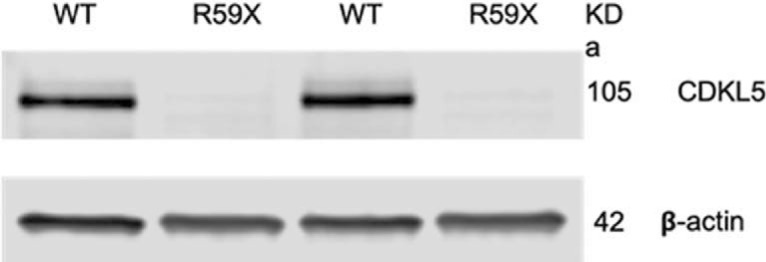
Western blot showing absence of CDKL5 protein in R59X whole-brain lysates compared with WT tissue with CDKL5 expression. β-actin is the loading control.
R59X mice demonstrate hyperactivity and deficits in motor coordination and control
We aimed to determine whether R59X mice exhibited alterations in activity and motor coordination because hyperactivity and motor deficits are frequently observed in patients with ASD and in ASD murine models (Crawley, 2004, 2007; Brodkin, 2007; Moy et al., 2008; Wang et al., 2012). In the open field assay, R59X mice exhibited greater motor activity by increased number of beam breaks over time compared with WT controls (Fig. 2a; two-way ANOVA, F(9,280) = 15.86, interaction, p < 0.0001). Additionally, mutant mice spent more time in the periphery of the arena (Fig. 2b; unpaired t test, R59X: 69.04 ± 2.307 vs WT: 58.72 ± 2.900, p = 0.0095), suggesting an elevated anxiety-like phenotype. In the rotarod assay, R59X mice displayed decreased latency to fall, demonstrating impaired motor coordination (Fig. 2c; two-way ANOVA, F(1,336) = 30.13, main effect of genotype, p < 0.0001). Despite this impairment, latency to fall in mutant mice increased similarly across trials to WT, suggesting that the rate of motor learning was not altered. Finally, R59X mice had a significantly greater hindlimb clasping score than littermate controls (Fig. 2d; unpaired t test, R59X: 2.200 ± 0.1447 vs WT: 0.6667 ± 0.1869, p < 0.0001), a phenotype observed in other mouse models of autism and CDD that is interpreted as impaired motor neuron function (Guy et al., 2001; Wang et al., 2012).
Figure 2.

R59X mice display behavioral deficits consistent with mouse models of autism and intellectual disability, and lowered seizure threshold. a, Ten minute open field locomotion test measuring number of infrared beam breaks over time. R59X mice exhibit increased activity compared with WT littermates, after a 3 min habituation period. p < 0.0001 (two-way ANOVA; interaction). b, R59X spend significantly more time in periphery of the open field arena, indicating elevated anxiety compared with WT. p = 0.0095 (unpaired t test). c, Accelerating rotarod assay measuring latency to fall. R59X mice show comparable motor learning but decreased motor coordination compared with WT mice. p < 0.0001 (two-way ANOVA; main effect of genotype). d, Five second hindlimb clasping test assessing motor neuron function: with a score of 0 representing no hindlimb clasping and a score of 4 representing both hindlimbs clasped in the middle during the entire trial period. R59X mice display increased clasping compared with WT littermates. p < 0.0001 (unpaired t test). e, Three-chambered social choice assay measuring social interaction. WT and R59X mice spent comparable amounts of time sniffing the nonsocial object (NS), but R59X mice spent significantly less time sniffing the stimulus mouse (S). p < 0.0001 (two-way ANOVA with Sidak correction for multiple comparisons; interaction), p < 0.0001. f, Five minute direct interaction period with freely moving stimulus mouse. R59X mice spent less time interacting with stimulus mouse than WT littermates. p < 0.0001 (unpaired t test). g, Olfactory test measuring time spent sniffing neutral, vanilla, and social scents. R59X mice and WT controls spend comparable amounts of time sniffing each scent. p = 0.9499 (two-way ANOVA). h, Y maze assay testing working memory by measuring percentage of SAB. R59X mice alternate arms of the Y maze significantly less than WT controls, suggesting impaired working memory. p < 0.0001 (unpaired t test). i, Context-dependent fear conditioning assay measuring freezing behavior. R59X mice freeze similar amounts after a mild foot shock compared with WT mice (after shock) but show decreased freezing upon returning to the testing chamber 24 h later (context). p = 0.0309 (two-way ANOVA with Sidak correction for multiple comparisons; interaction). a–i, N = 15 for all groups. j, Seizure threshold test measuring latency to first behavioral seizure after PTZ injection (40 mg/kg, i.p.). R59X (N = 10) reach the first behavioral seizure significantly faster than WT littermates (N = 10). p = 0.0117 (unpaired t test). *p < 0.05, **p < 0.01, ****p < 0.0001.
R59X mice exhibit social interaction deficits and learning and memory impairments
We next investigated sociability in R59X mice in the three-chambered social approach test, given the hallmark social interaction deficits in CDD (Bahi-Buisson et al., 2008; Fehr et al., 2015). As expected, neither WT nor R59X mice showed a chamber preference during the initial habituation phase. However, upon introduction of a novel mouse, R59X mice spent significantly less time interacting with the novel mouse compared with WT littermates (Fig. 2e; two-way ANOVA, F(1,47) = 115.6, interaction, p < 0.0001, Sidak's multiple comparisons test R59X: 16.653 ± 2.536 vs WT: 61.903 ± 3.3737, p < 0.0001). Furthermore, during the 5 min direct interaction period, R59X mice spent significantly less time sniffing and interacting with the novel mouse than WT littermates (Fig. 2f; unpaired t test, R59X: 26.43 ± 5.493 vs WT: 64.85 ± 5.002, p < 0.0001). The deficit in social interaction in R59X mice was not due to impaired olfactory function, given there were no differences in time spent sniffing neutral, vanilla, and social scents between mutant mice and controls in the olfactory test (Fig. 2g).
We also tested whether R59X mice also have deficits in working and long-term memory, given that ID is another core feature of CDD (Fehr et al., 2016). The Y maze assay was used to test hippocampal-dependent working memory (Murray and Ridley, 1999), where mice were allowed to freely explore a maze with three arms. The number of alternation events was measured, where one alternation event was when the mouse entered all three arms without repeating one. We found that R59X mice had significantly decreased percentage of SAB compared with WT littermates, indicating a deficit in working memory (Fig. 2h; unpaired t test, R59X: 44.03 ± 2.061 vs WT: 57.49 ± 1.329, p < 0.0001). We then performed context-dependent fear conditioning to examine potential changes in hippocampal-dependent long-term memory (Phillips and LeDoux, 1992). Fear conditioning was performed over 2 d: a training day (day 1) and testing day (day 2). On day 1, both R59X mice and WT littermates showed similar levels of exploratory behavior during the habituation phase as well as comparable levels of freezing after the foot shock in the acquisition phase, indicating similar sensitivity to the foot shock. However, when placed back inside the shock-box 24 h later (day 2) to test for long-term fear memory, the R59X mice froze significantly less than WT littermates (Fig. 2i; two-way ANOVA, F(2,84) = 3.624, interaction, p < 0.0309, Sidak's multiple comparisons test, R59X: 40.05 ± 4.865 vs WT 61.936 ± 4.114, p < 0.001). Together, these data demonstrate impaired hippocampal-dependent working and contextual memory in the R59X mice compared with WT littermates.
R59X mice show decreased latency to seizure behavior
Epilepsy has high prevalence in CDD patients, and many patients are refractory to conventional antiepileptic drugs (Bahi-Buisson et al., 2008). There were no observed spontaneous seizures during both behavioral testing and routine animal husbandry interactions with R59X mice, consistent with other CDD mouse models to date (Wang et al., 2012; Okuda et al., 2017; Tang et al., 2017). Given the lack of evidence of a severe spontaneous seizure phenotype, we next aimed to determine whether R59X mice had elevated network hyperexcitability by evaluating whether mutant mice had increased seizure susceptibility. We therefore tested sensitivity to a single dose of PTZ (40 mg/kg), a GABAA antagonist (Squires et al., 1984), which is well tolerated and causes mild seizure phenotypes (Racine Stages 1–3) in WT mice. Mice were videotaped for 2 h following a single dose of PTZ, and videos were scored for latency to onset of seizure activity. R59X mice exhibited significantly decreased latency to Racine Stage 1 compared with WT mice (Fig. 2j; unpaired t test, R59X: 79.00 ± 6.869 vs WT: 123.9 ± 14.46, p = 0.0117). Seizure activity lasted on average 30 min after injection for both R59X and WT mice, and no significant changes were found in total duration or overall severity of seizures (data not shown). Increased sensitivity to a subthreshold dose of PTZ suggests that, although R59X mice do not exhibit spontaneous seizures, they do possess underlying hyperexcitability that may be contributing to their behavioral alterations.
Decreased membrane-bound GluA2:GluA1 protein in R59X hippocampus
Given that previous studies support a role for CDKL5 in stabilizing excitatory synapses (Ricciardi et al., 2012; Della Sala et al., 2016) and that dysregulated GluA2 has been observed in an in vitro model of CDD (Tramarin et al., 2018), we hypothesized that R59X mice may have molecular and functional alterations in AMPAR subunits associated with the aforementioned behavioral deficits and seizure susceptibility. To investigate whether R59X mice show AMPAR subunit dysregulation, we performed western blot analysis in cortical and hippocampal tissue of membrane-bound AMPAR subunits GluA1 and GluA2, the predominant AMPAR subunits in the brain (Lu et al., 2009; Reimers et al., 2011). Although levels of GluA1 in R59X mice were unaltered compared with WT littermates at all time points (Fig. 3a), R59X mice showed significant decreases in hippocampal membrane-bound GluA2 beginning at P30 and persisting to P125 (Fig. 3b; two-way ANOVA, F(5,118) = 2.365, interaction, p = 0.0438, Sidak's multiple comparisons test: P30, R59X: 109.10 ± 9.31 vs WT: 145.02 ± 15.66, p < 0.01; P50, R59X: 73.52 ± 4.76 vs WT: 100 ± 5.32, p < 0.01; P125, R59X: 69.60 ± 5.16 vs WT: 94.32 ± 4.96, p < 0.05). Importantly, when considering the ratio of subunits GluA2:GluA1, which is a measure of GluA2-lacking AMPARs, R59X mice had significantly decreased GluA2:GluA1 in the hippocampus at P30, P50, and P125 (Fig. 3c; two-way ANOVA, F(5,127) = 2.337, interaction, p = 0.0456, Sidak's multiple comparisons test: P30, R59X: 90.430 ± 3.090 vs WT: 109.019 ± 5.030, p < 0.05; P50, R59X: 74.54 ± 2.892 vs WT: 100 ± 4.368, p < 0.01; P125, R59X: 83.050 ± 2.181 vs WT: 99.75 ± 4.805, p < 0.05), suggesting reduced GluA2-containing AMPARs or, in other words, increased GluA2-lacking AMPARs. GluA2-lacking AMPARs are Ca2+-permeable and are key regulators of synaptic function and plasticity that are particularly implicated in neurodevelopmental disease pathophysiology (Isaac et al., 2007; Uzunova et al., 2014; Lippman-Bell et al., 2016). Consistent with the hippocampal behavioral deficits, this finding was specific to the hippocampus; no significant changes in GluA1 and GluA2 expression were observed across the lifespan in the cortex of R59X mice (Fig. 3d–f). Therefore, decreased GluA2:GluA1 in R59X hippocampus may contribute to hyperexcitability and hippocampal-dependent learning and memory observed in the whole animal.
Figure 3.

AMPAR subunit ratio GluA2:GluA1 is significantly decreased from P30 onwards in R59X mice. a, Developmental expression of GluA1 is not altered in R59X hippocampus compared with WT controls (two-way ANOVA, p = 0.99). b, GluA2 expression is significantly decreased in R59X hippocampus at P30, P50, and P125 (two-way ANOVA with Sidak's multiple comparisons, interaction, p = 0.0438). c, GluA2:GluA1 is significantly decreased at P30, P50, and P125 in R59X hippocampus (two-way ANOVA with Sidak's multiple comparisons, interaction, p = 0.0456). d–f, Expression of GluA1, GluA2, and GluA2:GluA1 is not significantly altered at any time point in R59X cortex. g, Representative western blots of GluA1 and GluA2 in hippocampus compared with actin loading control. h, Representative western blots of GluA1 and GluA2 in cortex compared with actin loading control. All time points normalized to WT P50. N = 10–12 for all time points. *p < 0.05, **p < 0.01.
Prior reports suggest that CDKL5 shuttles from the cytoplasm to the nucleus where it may affect gene regulation (Rusconi et al., 2008), and the observed changes in GluA2 protein expression may be secondary to transcriptional changes. Therefore, we next used qRT-PCR to measure the relative mRNA transcript levels of Gria1 or Gria2 (for GluA1 and GluA2, respectively) in hippocampal tissue from P50 R59X mice and WT littermates. We found that R59X mice do not have significantly altered mRNA transcript levels of Gria1 or Gria2 (Table 1). Furthermore, in whole-cell hippocampal homogenates, there were no changes in total protein levels of GluA1 or GluA2 in R59X mice compared with controls (Table 2), suggesting that decreased membrane-bound GluA2 in the R59X hippocampus is more likely due to alterations in membrane trafficking or localization.
Table 1.
Transcript levels of GluA1 and GluA2 (Gria1 and Gria2, respectively) are not significantly altered in P50 R59X mice compared with WT littermatesa
| mRNA | WT | R59X | n | p (unpaired t test) |
|---|---|---|---|---|
| Gria1 | 1.00 ± 0.046 | 1.03 ± 0.069 | 8 | 0.788 |
| Gria2 | 1.00 ± 0.054 | 1.07 ± 0.087 | 8 | 0.587 |
aValues expressed as fold change normalized to WT controls.
Table 2.
Protein levels of total GluA1, GluA2, and GluA2:GluA1 in whole-cell prepped hippocampal tissue and GluA2S880/total in membrane prepped hippocampal tissuea
| Protein | Tissue preparation | WT | R59X | n | p |
|---|---|---|---|---|---|
| GluA1 (total) | Whole-cell | 100.000 ± 8.46 | 98.74 ± 8.48 | 8 | 0.918 |
| GluA2 (total) | Whole-cell | 100.000 ± 9.22 | 97.27 ± 8.37 | 8 | 0.830 |
| GluA2:GluA1 | Whole-cell | 100.000 ± 2.13 | 99.13 ± 2.66 | 8 | 0.803 |
| GluA2S880/total | Membrane | 100.000 ± 4.21 | 113.920 ± 6.76 | 10 | 0.215 |
aAll tissue obtained from P50 R59X mice and WT littermates.
Given the decrease in membrane-associated GluA2 without a concomitant decrease in transcription or total protein levels, we next determined whether altered post-translational modification of GluA2, which regulates protein trafficking and membrane insertion, could be responsible for the observed differences. We thus examined phosphorylation levels of GluA2 at Ser880 (GluA2S880), which normally causes internalization of GluA2. However, we observed no significant change in GluA2S880 in the P50 R59X hippocampus (Table 2).
Finally, due to a recent study showing that NMDAR subunit NR2B is elevated in the hippocampus of a Cdkl5 KO mouse model of CDD (Okuda et al., 2017), we checked for alterations in NMDAR subunits in R59X mice. Interestingly, no significant alterations were observed in membrane-bound GluN2A or GluN2B, or the ratio of GluN2B:2A, at any developmental time point in the hippocampus or cortex of R59X mice compared with controls (Fig. 4), suggesting a specific decrease of hippocampal membrane-bound GluA2 in adult R59X mice.
Figure 4.

NMDAR subunit expression is not altered in the hippocampus or cortex in R59X mice. a–c, Developmental expression of GluN2A, GluN2B, and GluN2B:GluN2A is not altered in R59X hippocampus compared with WT controls. d–f, Developmental expression of GluN2A, GluN2B, and GluN2B:GluN2A is not altered in R59X cortex compared with WT controls. g, Representative western blots of GluN2A and GluN2B in hippocampus compared with actin loading control. h, Representative western blots of GluN2A and GluN2B in cortex compared with actin loading control. All time points normalized to WT P50.
Decreased synaptic GluA2 and AMPAR-mediated inward rectification in CA1
To determine whether the decrease in membrane-associated GluA2 was occurring at intact synapses, we performed immunohistochemistry of surface GluA2 or GluA1 staining colocalized with presynaptic marker synapsin as performed previously (Lippman-Bell et al., 2016). The fraction of synapsin puncta that colocalized with surface GluA2 was calculated using the Manders coefficient (Manders et al., 1993). The Manders coefficient has previously been used to quantify association of GABAA subunits with inhibitory synaptic anchoring protein gephyrin (Gao and Heldt, 2016). We found that the fraction of synapses containing GluA2 was significantly decreased in R59X CA1 stratum radiatum (Fig. 5a,b; multiple t tests, p < 0.05), whereas the fraction of CA1 synapses containing GluA1 was unaltered compared with WT littermates (Fig. 5c,d). Together, our results show that R59X mice have an increased population of synaptic GluA2-lacking AMPARs in hippocampal CA1.
Figure 5.

Evidence of decreased synaptic GluA2 in immunohistochemistry. a, Immunohistochemistry of GluA2 (green) and presynaptic marker synapsin (red) in CA1. b, Quantification shows a significant decrease in the fraction of synapsin puncta that colocalize with GluA2, representative of decreased synaptic GluA2 in CA1. p = 0.003 (unpaired t test). c, d, Synaptic GluA1 is not significantly altered in R59X CA1 stratum radiatum. p = 0.24 (unpaired t test). Circles represent colocalization. Arrowheads indicate synapsin without GluA2. **p < 0.01.
To more directly examine the functional consequence of GluA2 deficiency in CA1, we performed whole-cell patch-clamp recordings in CA1 pyramidal neurons to observe whether the responses in the AMPAR I–V relationship in R59X mice showed inward rectification, characteristic of an increased population of GluA2-lacking AMPARs (Kamboj et al., 1995; Washburn et al., 1997; Lippman-Bell et al., 2016; Ha et al., 2018). Cells were held at potentials from −80 to 40 mV and the observed current at each holding potential were recorded. R59X mice displayed significantly greater inward rectification in the I–V plot at 20 and 40 mV holding potentials (Fig. 6c,d; two-way ANOVA, F(6,105) = 7.074, interaction, p < 0.000, Sidak's multiple-comparison test: 20 mV, R59X: 0.176 ± 0.03 vs WT: 0.367 ± 0.029; 40 mV, R59X: 0.299 ± 0.025 vs WT: 0.614 ± 0.058) and a significantly greater rectification index (40/−60 mV) of AMPAR-mediated EPSCs in slices from mutant mice compared with controls (Fig. 6e; unpaired t test, R59X: 2.55 ± 0.194 vs WT: 1.46 ± 0.136).
Figure 6.

Whole-cell recordings from CA1 primary cells confirm an increase in GluA2 lacking AMPARs. Representative AMPA evoked EPSC traces in both WT (a) and R59X (b) acute hippocampal slices; 100 μm spermine was added to the internal solution to maintain rectification. c, d, I–V plot relationship of peak responses from eEPSCs from holding potentials of −80 mV to 40 mV normalized to −80 mV shows a linear relationship of AMPA currents in WT animals and an inwardly rectifying relationship in R59X mice. e, The ratio of current evoked current at −60 mV to 40 mV is significantly higher in R59X mice showing a rectification indicative of GluA2 lacking AMPARs (n = 8 or 9 cells per group, two-way ANOVA with Sidak's multiple comparisons, interaction, p < 0.0001). f, Representative traces of eEPSCs at Vh = −60 mV after 100 μm IEM-1460 (top) and 100 μm NASPM (bottom), showing a partial inhibition of the AMPA current after both applications. g, Percentage decrease of the eEPSC at −60 mV by IEM-1460 (n = 5, p < 0.01, paired t test) and NASPM (n = 5, p < 0.05, paired t test), which is significantly decreased from control conditions. Calibration: y axis, 100 pA; x axis, 25 ms. *p < 0.05, **p < 0.01, ***p < 0.001.
Elevated early-phase LTP
Given the increased functional GluA2-lacking AMPARs observed at CA1 synapses, we next aimed to determine whether R59X mice exhibit alterations in network function and synaptic plasticity by performing extracellular field recordings in ex vivo hippocampal slices. First, we observed no change in input-output function (Fig. 7a) or paired-pulse ratio (Fig. 7b), suggesting that both basal synaptic function and presynaptic function were unaltered at Schaffer collateral-CA1 synapses. Next, we tested synaptic plasticity by evaluating LTD and LTP. LTD was induced using LFS and found to be unaltered in R59X mice (Fig. 7c). However, in the LTP paradigm, R59X mice responded significantly differently to HFS compared with controls (Fig. 7d; two-way ANOVA, main effect of genotype, p < 0.0001); mutant mice displayed significantly elevated early-phase potentiation 15 min after HFS (unpaired t test, R59X: 188.7 ± 14.88, vs WT: 136.8 ± 9.911, p = 0.0125) followed by a steeper decay slope (linear regression, R59X: −1.192 ± 0.1071 vs WT: −0.1192 ± 0.08017 p < 0.0001), resulting in R59X activity approximately reaching WT levels of potentiation by 60 min after HFS.
Figure 7.

Extracellular recordings in R59X mice show elevated early-phase LTP. a, R59X mice have normal input-output function (N = 16). b, Paired-pulse ratio, a conventional measure of presynaptic function, is not altered in R59X mice (N = 22–27). c, LTD is normal in R59X mice (N = 10). d, R59X display abnormal LTP. Two-way ANOVA, main effect of genotype, p < 0.0001, with significantly elevated potentiation at 15 min after HFS and steeper decay slope. *p < 0.05 (unpaired t test). p < 0.0001 (linear regression). N = 6 or 7.
The specific GluA-2 lacking AMPAR antagonists IEM-1460 and NASP selectively block neuronal AMPAR currents in hippocampal CA1in CDKL5 mouse but not WT
To confirm that a portion of the AMPA current at −60 mV was contributed by GluA2-lacking AMPAR, we used two selective blockers of GluA2-lacking AMPARs, the adamantane derivative IEM-1460 and NASPM (Magazanik et al., 1997; Noh et al., 2005). Both IEM-1460 and NASPM at 100 μm decreased the current at −60 mV by ∼20% (Fig. 6g; IEM-1460: 23 ± 2%, p < 0.01; and NASPM: 24.1 ± 5%, p < 0.05, paired t test; Fig. 6f,g). These results suggest that there is a significant contribution of GluA2-lacking AMPARs to the AMPA current in R59X mice, and that the specific AMPAR antagonist used in the in vivo experiments also blocked these currents. Notably, there was no effect of either blocker on WT currents, and CNQX abolished all evoked currents (data not shown). Input resistance was not altered between R59X mice and WT littermates (data not shown), suggesting that cell size does not vary significantly between genotypes. Together, these electrophysiological data demonstrate a physiologically relevant increase in synaptic GluA2-lacking AMPARs in R59X mice, consistent with the immunohistochemical results and results from published cell culture studies (Tramarin et al., 2018).
IEM-1460 rescues core features of CDD in R59X mice
Because GluA2-lacking lacking AMPARs are involved in plasticity and implicated in autism, ID, and hyperexcitability (Isaac et al., 2007; Mignogna et al., 2015; Lippman-Bell et al., 2016), we sought to determine whether increased hippocampal synaptic GluA2-lacking AMPARs contribute to hippocampal-associated behavioral alterations and hyperexcitability in R59X mice. Mutant mice and WT littermates were treated with IEM-1460 (10 mg/kg, i.p.) 1 h before behavioral tests (Magazanik et al., 1997; Szczurowska and Mares, 2015). In the social choice assay, drug treatment rescued R59X duration of time sniffing to saline-treated WT levels (Fig. 8a; two-way ANOVA F(3,76) = 8.947, interaction p < 0.0001), while also increasing the time spent directly interacting with the novel mouse (Fig. 8b; one-way ANOVA, F(3,38) = 10.71, p < 0.0001). In the Y maze, IEM-1460 rescued working memory in R59X mice to WT levels (Fig. 8d; one-way ANOVA, F(3,33) = 14.06, interaction p < 0.0001), suggesting that blocking GluA2-lacking AMPARs acutely has therapeutic effects on short-term working memory deficits. While IEM-1460 improved context-dependent fear memory in mutant mice, the effect was not significant enough to rescue it to WT-saline levels (Fig. 8e; two-way ANOVA, F(6,129) = 2.819, interaction p = 0.013). Although we did not observe a significant improvement in long-term fear memory in this study, it is possible that the effects of IEM-1460 on long-term memory will be more pronounced with a chronic treatment paradigm. Finally, IEM-1460 rescued latency to seizure in mutant mice to WT saline levels, indicating that targeting GluA2-lacking AMPARs in CDD may attenuate hyperexcitability in the brain (Fig. 8f; one-way ANOVA, F(3,36) = 4.621, p = 0.0078). These data provide evidence to support the role of elevated GluA2-lacking AMPARs underlying deficits in memory, social behavior, and hyperexcitability in R59X mice.
Figure 8.

Effect of GluA2-lacking AMPAR-specific blocker IEM-1460 on hippocampal-associated behaviors in R59X mice. a, b, IEM-1460 rescues social behavior deficits in R59X mice and increases direct interaction time. p < 0.0001 (two-way ANOVA with Sidak's multiple comparisons test, interaction). c, Olfactory function is not altered in R59X mice treated with IEM-1460. p = 0.806 (two-way ANOVA). d, IEM-1460 rescues percentage of SAB in R59X mice to WT levels. p < 0.0001 (ordinary one-way ANOVA with Sidak's multiple comparisons test). e, Drug treatment partially attenuates deficits in long-term fear memory. p = 0.013 (two-way ANOVA with Sidak's multiple comparisons test, interaction). f, Latency to first behavioral seizure after PTZ injection is rescued with pretreatment of IEM-1460. p = 0.0078 (ordinary one-way ANOVA, with Sidak's multiple-comparison's test). N = 8–14 for all groups. *p < 0.05. **p < 0.01. ***p < 0.001. ****p < 0.0001.
Postmortem hippocampal tissue from 2 CDD patients shows decreased GluA2:GluA1
We obtained postmortem hippocampal tissue samples from the only 2 postmortem CDD patient samples available worldwide and sought to determine whether a similar trend of decreased GluA2:GluA1 could be observed in the human hippocampus, adding some clinical relevance to the results we obtained in the R59X model. Indeed, although statistical analysis could not be performed with just 2 cases, we see a trend toward decreased GluA2:GluA1 in CDD hippocampus (Fig. 9). This is an intriguing finding given the varied ages of the cases (5 and 30 years old) and the likelihood that they were on different antiepileptic drug regimens that could differentially affect levels of neurotransmitter receptors.
Figure 9.

Membrane preparation of postmortem hippocampal tissue from 2 CDD patients shows a trend toward decreased GluA2 and GluA2:GluA1. a, Case 1 was a female, 5.7 years of age at death, and was compared with male and female neurotypical controls (NTC) aged 3–7 years. b, Case 2 was a female, 30 years of age at death, and was compared with male and female NTCs 20–33 years old. c, Representative blots of CDD cases and NTCs.
Discussion
Autism, ID, and seizures are key features of CDD and frequently co-occur in neurodevelopmental disorders, suggesting common disease mechanisms of dysregulated synaptic structure and function (Eichler and Meier, 2008; Zoghbi and Bear, 2012). There is currently no therapy that targets the core symptoms of CDD, which drove us to ask whether there are targetable synaptic alterations in the novel R59X mouse model of CDD that may contribute to disease pathogenesis.
Our western blot analyses show a significant decrease in AMPAR subunit GluA2 in the R59X hippocampus beginning at P30. Regulation of AMPAR subunit ratio GluA2:GluA1 determines levels of GluA2-lacking, CP-AMPARs at the synapse and resulting Ca2+ influx (Jonas and Burnashev, 1995), which has long-lasting effects on synaptic plasticity, neuronal growth, and gene transcription (Anggono and Huganir, 2012; Chater and Goda, 2014). GluA2-lacking AMPARs are normally present postnatally during the critical period but decrease by adulthood. In contrast, the R59X mice exhibited unexpected persistence of GluA2-lacking AMPARs from P30 onwards, suggesting that the normal developmental switch from GluA2-lacking to GluA2-containing AMPARs does not occur. GluA2-lacking AMPAR dysregulation has been implicated in numerous disorders of excitatory-inhibitory imbalance and autism (Lippman-Bell et al., 2013; Mignogna et al., 2015), and indeed the GluA2 deficits in R59X mice were accompanied by neurobehavioral alterations and seizure susceptibility.
Although membrane-associated GluA2 is decreased in R59X mice, we did not observe a similar trend in relative mRNA expression of Gria2, suggesting that loss of CDKL5 results in disrupted trafficking or localization of GluA2 to the membrane (Geiger et al., 1995). These findings are similar to those reported in an in vitro cell culture model of CDD, where silencing Cdkl5 by shRNA decreased surface levels of GluA2:GluA1 (Tramarin et al., 2018). However, contrary to our results showing no change in total GluA2 or GluA2S880, the cell culture model exhibited significantly decreased total GluA2, which was accompanied by increased GluA2S880, suggesting increased internalization of the subunit (Tramarin et al., 2018). The differences in these results may be due to the heterogenous cell population of tissue samples used for our western blotting experiments versus the pure neuronal population studied in the cell culture model. However, another mechanism for impaired synaptic localization of GluA2 may be through dysregulation of PSD95, a molecule involved in clustering of AMPARs through interaction with scaffolding protein stargazin/TARP (Coley and Gao, 2018). A reduction of PSD95 puncta has been observed in neurons after Cdkl5 knockdown (Ricciardi et al., 2012; Tramarin et al., 2018), suggesting that anchoring and stability of glutamate receptors at the synapse may be compromised in this disorder. Interestingly, palmitoylation of PSD95 recruits CDKL5 to synapses (Zhu et al., 2013) and has also been found to increase surface GluA2 levels (Jeyifous et al., 2016), suggesting a possible link between CDKL5 function and GluA2 localization at the synapse. Indeed, hippocampal GluA2 dysregulation in R59X mice begins at P30, a time at which Cdkl5 expression normally peaks in WT hippocampus (Schroeder et al., 2018). However, a deeper understanding of CDKL5-interacting partners and phosphorylation substrates is necessary to identify the primary cause of dysregulated synaptic GluA2 and its functional consequences.
Our whole-cell patch-clamp recordings levels expand upon our western blot data and indicate that there is a physiologically relevant, increased population of GluA2-lacking AMPARs at hippocampal synapses. We observed inward rectification in the AMPAR I–V plot in R59X mice, a hallmark characteristic of increased GluA2-lacking AMPARs (Jonas et al., 1994), due to their susceptibility to intracellular spermine block at high voltages (Rozov et al., 2012). These inwardly rectifying currents can be reversed by the addition of NASPM (Koike et al., 1997, Noh et al., 2005) and also IEM-1460 (Chen et al., 2013; Twomey et al., 2018), which was used in the in vivo model in this study. Elevated GluA2-lacking, CP-AMPARs may cause hyperexcitability through elevated Ca2+ influx as well as increased NMDAR activation. Additionally, increased Ca2+ influx through dysregulated GluA2-lacking AMPARs may underlie enhanced early-phase LTP in R59X mice observed in our extracellular electrophysiology recordings. Normally, GluA2-lacking AMPARs are crucial for LTP induction and are inserted into the synaptic membrane immediately following HFS (Park et al., 2016). Upon Ca2+ influx through GluA2-lacking AMPARs, GluA2 translocates to the synaptic membrane via a PICK1-dependent mechanism (Jaafari et al., 2012) to form GluA2-containing AMPARs, which gradually replace GluA2-lacking AMPARs at the synapse during the consolidation phase of LTP (Plant et al., 2006; Hanley, 2014). The elevated potentiated response in R59X mice 15 min after HFS is likely a result of increased Ca2+ influx due to (1) elevated basal levels of GluA2-lacking AMPARs at the synapse and (2) additional insertion of GluA2-lacking AMPARs that normally occurs during the induction phase of LTP. Furthermore, given that our earlier data suggest impaired transport of GluA2 to the membrane in R59X mice, it is possible that GluA2-containing AMPARs are hindered from being inserted into the membrane during the late-phase of LTP, causing the observed decay in the response. Our data from extracellular field recordings align with studies performed in hippocampal slices from GluA2 KO mice, showing that mice lacking the GluA2 subunit exhibited normal input-output function, paired-pulse ratio, and LTD along with elevated LTP (Jia et al., 1996). However, our data differ significantly from a study performed in cortex layer II-III in Cdkl5 KO mice where LTP was observed to be significantly decreased (Della Sala et al., 2016). We did not investigate LTP in this region as our molecular analysis focused on hippocampal alterations. However, it is possible that loss of CDKL5 affects LTP differentially in the cortex and hippocampus due to variations in cell populations and circuit dynamics. Indeed, more recently in a different Cdkl5 KO mouse, LTP induced by tetanic stimulation was observed to be significantly elevated in the hippocampus (Okuda et al., 2017).
The behavioral effects observed in the R59X mice are similar to a Cdkl5 KO mouse model that also exhibited hyperexcitability, deficits in motor coordination and social interaction, and impaired working and long-term memory (Wang et al., 2012). Furthermore, hippocampal-dependent learning and memory impairments were observed in conditional KO mice where CDKL5 was selectively depleted from forebrain excitatory neurons, emphasizing the importance of normal function of CDKL5 in excitatory hippocampal neurons (Tang et al., 2017). Our observed increase in GluA2-lacking AMPARs at hippocampal synapses combined with the functional and plasticity alterations at hippocampal synapses in R59X mice suggest an underlying mechanism for altered behavior at the whole animal level. Indeed, changes in GluA2 subunit levels and signaling have previously been linked to social interaction deficits (Lippman-Bell et al., 2013), learning and memory impairments (Wang et al., 2018; Zhou et al., 2018), and early-life seizures (Lippman-Bell et al., 2016). Additionally, our laboratory has observed increased phosphorylation of calcium calmodulin-dependent protein kinase II (CaMKII) at Thr286 and downstream genome-wide transcription factor methyl CPG binding protein 2 (MeCP2) at Ser421 as a result of Ca2+ influx through GluA2-lacking AMPARs (Rosenberg et al., 2018). Although there is no change in total MeCP2 protein levels in mouse models of CDD (Amendola et al., 2014), elevated phosphorylation of CaMKII and MeCP2 may be a downstream signaling mechanism by which increased GluA2-lacking AMPARs result in behavioral deficits and hyperexcitability observed in R59X mice. Indeed, CaMKII has been implicated in fear conditioning and spatial memory (Wang et al., 2017), and increased CaMKII Thr286 phosphorylation has been observed to cause seizures and ID in humans (Akita et al., 2018). Furthermore, mutations in MeCP2 are known to cause autism and Rett syndrome (Amir et al., 1999) and MeCP2 Ser421 phosphorylation controls ability of MeCP2 to regulate dendritic growth and spine maturation (Zhou et al., 2006). In R59X mice, these effector molecules may be causing chronic alterations in plasticity, excitability, and gene transcription due to increased Ca2+ influx through GluA2-lacking AMPARs, which we aim to investigate in future experiments.
The GluA2-lacking AMPAR blocker IEM-1460 is not currently available for clinical use in humans, but there are a number of pharmacological agents that broadly inhibit AMPARs in humans (Russo et al., 2012). The demonstration that IEM-1460 is effective in vivo but also attenuated AMPAR currents in vitro strongly supports the clinical significance of the blockade of GluA2-lacking receptors in this CDD model. While our human tissue data are limited, they are supportive of the clinical potential of targeting GluA2-lacking AMPARs in CDD. Our findings showing therapeutic potential of IEM-1460 on seizure threshold, social interaction, and working memory deficits align with our previous work showing that the general AMPAR antagonist NBQX attenuates deficits in social preference following seizure-induced elevation of GluA2-lacking AMPARs in rats (Lippman-Bell et al., 2013). Additionally, prior reports show that IEM-1460 has age-specific therapeutic effects against seizures coinciding with peak levels of GluA2-lacking AMPARs during development (Szczurowska and Mares, 2015), and prevents seizure-induced phosphorylation of CaMKII and MeCP2 in immature rodents (Rosenberg et al., 2018). Finally, we previously showed a significant increase in synaptic GluA2-lacking AMPARs 48 h posthypoxic seizures in rats that resulted in enhanced epileptogenesis later in life (Sanchez et al., 2001; Rakhade et al., 2008; Lippman-Bell et al., 2013), suggesting that increased hippocampal GluA2-lacking AMPARs in R59X mice may be a targetable contributor to hyperexcitable circuit formation and behavioral alterations, even in the absence of visible seizures, and that blocking GluA2-lacking AMPARs with IEM-1460 may mitigate this process.
Footnotes
This work was supported by National Institutes of Health Grants NS 031718, NS 080565, and DP1 OD003347 to F.E.J., Hope4Harper Foundation, and the Lou Lou Foundation to M.Y. and F.E.J. We thank the Neurobehavior Testing Core at University of Pennsylvania and Intellectual and Developmental Disabilities Research Center at Children's Hospital of Philadelphia/University of Pennsylvania U54 HD086984 for assistance with behavior procedures; Dr. Zhaolan Zhou (Professor of Genetics, University of Pennsylvania) for guidance and providing CDKL5R59X knock-in mice prior to publication; Dr. Delia Talos (Assistant Professor of Neurology, University of Pennsylvania) for advice with human tissue analysis; Dr. Chengwen Zhou (Assistant Professor of Neurology, Vanderbilt University) for help with electrophysiology analysis; and Sydney Zebrowitz for work assessing animal behavior.
The authors declare no competing financial interests.
References
- Achuta VS, Möykkynen T, Peteri UK, Turconi G, Rivera C, Keinänen K, Castrén ML (2018) Functional changes of AMPA responses in human induced pluripotent stem cell-derived neural progenitors in fragile X syndrome. Sci Signal 11:eaan8784. 10.1126/scisignal.aan8784 [DOI] [PubMed] [Google Scholar]
- Akita T, Aoto K, Kato M, Shiina M, Mutoh H, Nakashima M, Kuki I, Okazaki S, Magara S, Shiihara T, Yokochi K, Aiba K, Tohyama J, Ohba C, Miyatake S, Miyake N, Ogata K, Fukuda A, Matsumoto N, Saitsu H (2018) De novo variants in CAMK2A and CAMK2B cause neurodevelopmental disorders. Ann Clin Transl Neurol 5:280–296. 10.1002/acn3.528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amendola E, Zhan Y, Mattucci C, Castroflorio E, Calcagno E, Fuchs C, Lonetti G, Silingardi D, Vyssotski AL, Farley D, Ciani E, Pizzorusso T, Giustetto M, Gross CT (2014) Mapping pathological phenotypes in a mouse model of CDKL5 disorder. PLoS One 9:e91613. 10.1371/journal.pone.0091613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY (1999) Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 23:185–188. 10.1038/13810 [DOI] [PubMed] [Google Scholar]
- Anggono V, Huganir RL (2012) Regulation of AMPA receptor trafficking and synaptic plasticity. Curr Opin Neurobiol 22:461–469. 10.1016/j.conb.2011.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahi-Buisson N, Nectoux J, Rosas-Vargas H, Milh M, Boddaert N, Girard B, Cances C, Ville D, Afenjar A, Rio M, Héron D, N′guyen Morel MA, Arzimanoglou A, Philippe C, Jonveaux P, Chelly J, Bienvenu T (2008) Key clinical features to identify girls with CDKL5 mutations. Brain 131:2647–2661. 10.1093/brain/awn197 [DOI] [PubMed] [Google Scholar]
- Brodkin ES. (2007) BALB/c mice: low sociability and other phenotypes that may be relevant to autism. Behav Brain Res 176:53–65. 10.1016/j.bbr.2006.06.025 [DOI] [PubMed] [Google Scholar]
- Castrén M, Gaily E, Tengström C, Lähdetie J, Archer H, Ala-Mello S (2011) Epilepsy caused by CDKL5 mutations. Eur J Paediatr Neurol 15:65–69. 10.1016/j.ejpn.2010.04.005 [DOI] [PubMed] [Google Scholar]
- Chater TE, Goda Y (2014) The role of AMPA receptors in postsynaptic mechanisms of synaptic plasticity. Front Cell Neurosci 8:401. 10.3389/fncel.2014.00401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Zhu YC, Yu J, Miao S, Zheng J, Xu L, Zhou Y, Li D, Zhang C, Tao J, Xiong ZQ (2010) CDKL5, a protein associated with Rett syndrome, regulates neuronal morphogenesis via Rac1 signaling. J Neurosci 30:12777–12786. 10.1523/JNEUROSCI.1102-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SR, Zhou HY, Byun HS, Pan HL (2013) Nerve injury increases GluA2-lacking AMPA receptor prevalence in spinal cords: functional significance and signaling mechanisms. J Pharmacol Exp Ther 347:765–772. 10.1124/jpet.113.208363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coley AA, Gao WJ (2018) PSD95: A synaptic protein implicated in schizophrenia or autism? Prog Neuro-psychopharmacology Biological Psychiatry 82:187–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawley JN. (2004) Designing mouse behavioral tasks relevant to autistic-like behaviors. Ment Retard Dev Disabil Res Rev 10:248–258. 10.1002/mrdd.20039 [DOI] [PubMed] [Google Scholar]
- Crawley JN. (2007) Mouse behavioral assays relevant to the symptoms of autism. Brain Pathol 17:448–459. 10.1111/j.1750-3639.2007.00096.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Della Sala G, Putignano E, Chelini G, Melani R, Calcagno E, Michele Ratto G, Amendola E, Gross CT, Giustetto M, Pizzorusso T (2016) Dendritic spine instability in a mouse model of CDKL5 disorder is rescued by insulin-like growth factor 1. Biol Psychiatry 80:302–311. 10.1016/j.biopsych.2015.08.028 [DOI] [PubMed] [Google Scholar]
- Eichler SA, Meier JC (2008) E-I balance and human diseases: from molecules to networking. Front Mol Neurosci 1:2. 10.3389/neuro.02.002.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehr S, Leonard H, Ho G, Williams S, de Klerk N, Forbes D, Christodoulou J, Downs J (2015) There is variability in the attainment of developmental milestones in the CDKL5 disorder. J Neurodev Disord 7:2. 10.1186/1866-1955-7-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehr S, Downs J, Ho G, de Klerk N, Forbes D, Christodoulou J, Williams S, Leonard H (2016) Functional abilities in children and adults with the CDKL5 disorder. Am J Med Genet A 170:2860–2869. 10.1002/ajmg.a.37851 [DOI] [PubMed] [Google Scholar]
- Fortin DA, Davare MA, Srivastava T, Brady JD, Nygaard S, Derkach VA, Soderling TR (2010) Long-term potentiation-dependent spine enlargement requires synaptic Ca2+-permeable AMPA receptors recruited by CaM-kinase I. J Neurosci 30:11565–11575. 10.1523/JNEUROSCI.1746-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs C, Trazzi S, Torricella R, Viggiano R, De Franceschi M, Amendola E, Gross C, Calzà L, Bartesaghi R, Ciani E (2014) Loss of CDKL5 impairs survival and dendritic growth of newborn neurons by altering AKT/GSK-3β signaling. Neurobiol Dis 70:53–68. 10.1016/j.nbd.2014.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Heldt SA (2016) Enrichment of GABAA Receptor α-Subunits on the Axonal Initial Segment Shows Regional Differences. Front Cell Neurosci 10:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger JR, Melcher T, Koh DS, Sakmann B, Seeburg PH, Jonas P, Monyer H (1995) Relative abundance of subunit mRNAs determines gating and Ca2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron 15:193–204. 10.1016/0896-6273(95)90076-4 [DOI] [PubMed] [Google Scholar]
- Guy J, Hendrich B, Holmes M, Martin JE, Bird A (2001) A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet 27:322–326. 10.1038/85899 [DOI] [PubMed] [Google Scholar]
- Ha HT, Leal-Ortiz S, Lalwani K, Kiyonaka S, Hamachi I, Mysore SP, Montgomery JM, Garner CC, Huguenard JR, Kim SA (2018) Shank and zinc mediate an AMPA receptor subunit switch in developing neurons. Front Mol Neurosci 11:405. 10.3389/fnmol.2018.00405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanley JG. (2014) Subunit-specific trafficking mechanisms regulating the synaptic expression of Ca(2+)-permeable AMPA receptors. Semin Cell Dev Biol 27:14–22. 10.1016/j.semcdb.2013.12.002 [DOI] [PubMed] [Google Scholar]
- Henley JM, Wilkinson KA (2016) Synaptic AMPA receptor composition in development, plasticity and disease. Nat Rev Neurosci 17:337–350. 10.1038/nrn.2016.37 [DOI] [PubMed] [Google Scholar]
- Isaac JT, Ashby MC, McBain CJ (2007) The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron 54:859–871. 10.1016/j.neuron.2007.06.001 [DOI] [PubMed] [Google Scholar]
- Jaafari N, Henley JM, Hanley JG (2012) PICK1 mediates transient synaptic expression of GluA2-lacking AMPA receptors during glycine-induced AMPA receptor trafficking. J Neurosci 32:11618–11630. 10.1523/JNEUROSCI.5068-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen FE, Wang C, Stafstrom CE, Liu Z, Geary C, Stevens MC (1998) Acute and chronic increases in excitability in rat hippocampal slices after perinatal hypoxia in vivo. J Neurophysiol 79:73–81. 10.1152/jn.1998.79.1.73 [DOI] [PubMed] [Google Scholar]
- Jeyifous O, Lin EI, Chen X, Antinone SE, Mastro R, Drisdel R, Reese TS, Green WN (2016) Palmitoylation regulates glutamate receptor distributions in postsynaptic densities through control of PSD95 conformation and orientation. Proc Natl Acad Sci U S A 113:E8482–E8491. 10.1073/pnas.1612963113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Z, Agopyan N, Miu P, Xiong Z, Henderson J, Gerlai R, Taverna FA, Velumian A, MacDonald J, Carlen P, Abramow-Newerly W, Roder J (1996) Enhanced LTP in mice deficient in the AMPA receptor GluR2. Neuron 17:945–956. 10.1016/S0896-6273(00)80225-1 [DOI] [PubMed] [Google Scholar]
- Jonas P, Burnashev N (1995) Molecular mechanisms controlling calcium entry through AMPA-type glutamate receptor channels. Neuron 15:987–990. 10.1016/0896-6273(95)90087-X [DOI] [PubMed] [Google Scholar]
- Jonas P, Racca C, Sakmann B, Seeburg PH, Monyer H (1994) Differences in Ca2+ permeability of AMPA-type glutamate receptor channels in neocortical neurons caused by differential GluR-B subunit expression. Neuron 12:1281–1289. 10.1016/0896-6273(94)90444-8 [DOI] [PubMed] [Google Scholar]
- Kamboj SK, Swanson GT, Cull-Candy SG (1995) Intracellular spermine confers rectification on rat calcium-permeable AMPA and kainate receptors. J Physiol 486:297–303. 10.1113/jphysiol.1995.sp020812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilstrup-Nielsen C, Rusconi L, La Montanara P, Ciceri D, Bergo A, Bedogni F, Landsberger N (2012) What we know and would like to know about CDKL5 and its involvement in epileptic encephalopathy. Neural Plast 2012:728267. 10.1155/2012/728267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike M, Iino M, Ozawa S (1997) Blocking effect of 1-naphthyl acetyl spermine on Ca2+-permeable AMPA receptors in cultured rat hippocampal neurons. Neurosci Res 29:27–36. 10.1016/S0168-0102(97)00067-9 [DOI] [PubMed] [Google Scholar]
- Li W, Xu X, Pozzo-Miller L (2016) Excitatory synapses are stronger in the hippocampus of Rett syndrome mice due to altered synaptic trafficking of AMPA-type glutamate receptors. Proc Natl Acad Sci U S A 113:E1575–E1584. 10.1073/pnas.1517244113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippman-Bell JJ, Rakhade SN, Klein PM, Obeid M, Jackson MC, Joseph A, Jensen FE (2013) AMPA receptor antagonist NBQX attenuates later-life epileptic seizures and autistic-like social deficits following neonatal seizures. Epilepsia 54:1922–1932. 10.1111/epi.12378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippman-Bell JJ, Zhou C, Sun H, Feske JS, Jensen FE (2016) Early-life seizures alter synaptic calcium-permeable AMPA receptor function and plasticity. Mol Cell Neurosci 76:11–20. 10.1016/j.mcn.2016.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loddenkemper T, Talos DM, Cleary RT, Joseph A, Sánchez Fernández I, Alexopoulos A, Kotagal P, Najm I, Jensen FE (2014) Subunit composition of glutamate and gamma-aminobutyric acid receptors in status epilepticus. Epilepsy Res 108:605–615. 10.1016/j.eplepsyres.2014.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Shi Y, Jackson AC, Bjorgan K, During MJ, Sprengel R, Seeburg PH, Nicoll RA (2009) Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron 62:254–268. 10.1016/j.neuron.2009.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luján R, Shigemoto R, López-Bendito G (2005) Glutamate and GABA receptor signalling in the developing brain. Neuroscience 130:567–580. 10.1016/j.neuroscience.2004.09.042 [DOI] [PubMed] [Google Scholar]
- Lüttjohann A, Fabene PF, van Luijtelaar G (2009) A revised Racine's scale for PTZ-induced seizures in rats. Physiol Behav 98:579–586. 10.1016/j.physbeh.2009.09.005 [DOI] [PubMed] [Google Scholar]
- Magazanik LG, Buldakova SL, Samoilova MV, Gmiro VE, Mellor IR, Usherwood PN (1997) Block of open channels of recombinant AMPA receptors and native AMPA/kainate receptors by adamantane derivatives. J Physiol 505:655–663. 10.1111/j.1469-7793.1997.655ba.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manders EMM, Verbeek FJ, Aten JA (1993) Measurement of co-localization of objects in dual-colour confocal images. Journal of Microscopy 169:375–382. [DOI] [PubMed] [Google Scholar]
- Mignogna ML, Giannandrea M, Gurgone A, Fanelli F, Raimondi F, Mapelli L, Bassani S, Fang H, Van Anken E, Alessio M, Passafaro M, Gatti S, Esteban JA, Huganir R, D'Adamo P (2015) The intellectual disability protein RAB39B selectively regulates GluA2 trafficking to determine synaptic AMPAR composition. Nat Commun 6:6504. 10.1038/ncomms7504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montini E, Andolfi G, Caruso A, Buchner G, Walpole SM, Mariani M, Consalez G, Trump D, Ballabio A, Franco B (1998) Identification and characterization of a novel serine-threonine kinase gene from the Xp22 region. Genomics 51:427–433. 10.1006/geno.1998.5391 [DOI] [PubMed] [Google Scholar]
- Moy SS, Nadler JJ, Poe MD, Nonneman RJ, Young NB, Koller BH, Crawley JN, Duncan GE, Bodfish JW (2008) Development of a mouse test for repetitive, restricted behaviors: relevance to autism. Behav Brain Res 188:178–194. 10.1016/j.bbr.2007.10.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray TK, Ridley RM (1999) The effect of excitotoxic hippocampal lesions on simple and conditional discrimination learning in the rat. Behav Brain Res 99:103–113. 10.1016/S0166-4328(98)00077-1 [DOI] [PubMed] [Google Scholar]
- Nie D, Nardo A, Han JM, Baharanyi H, Kramvis I, Huynh T, Dabora S, Codeluppi S, Pandolfi P, Pasquale EB, Sahin M (2010) Tsc2-Rheb signaling regulates EphA-mediated axon guidance. Nat Neurosci 13:163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noh KM, Yokota H, Mashiko T, Castillo PE, Zukin RS, Bennett MV (2005) Blockade of calcium-permeable AMPA receptors protects hippocampal neurons against global ischemia-induced death. Proc Natl Acad Sci U S A 102:12230–12235. 10.1073/pnas.0505408102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda K, Kobayashi S, Fukaya M, Watanabe A, Murakami T, Hagiwara M, Sato T, Ueno H, Ogonuki N, Komano-Inoue S, Manabe H, Yamaguchi M, Ogura A, Asahara H, Sakagami H, Mizuguchi M, Manabe T, Tanaka T (2017) CDKL5 controls postsynaptic localization of GluN2B-containing NMDA receptors in the hippocampus and regulates seizure susceptibility. Neurobiol Dis 106:158–170. 10.1016/j.nbd.2017.07.002 [DOI] [PubMed] [Google Scholar]
- Park P, Sanderson TM, Amici M, Choi SL, Bortolotto ZA, Zhuo M, Kaang BK, Collingridge GL (2016) Calcium-permeable AMPA receptors mediate the induction of the protein kinase A-dependent component of long-term potentiation in the hippocampus. J Neurosci 36:622–631. 10.1523/JNEUROSCI.3625-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips RG, LeDoux JE (1992) Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci 106:274–285. 10.1037/0735-7044.106.2.274 [DOI] [PubMed] [Google Scholar]
- Plant K, Pelkey KA, Bortolotto ZA, Morita D, Terashima A, McBain CJ, Collingridge GL, Isaac JT (2006) Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat Neurosci 9:602–604. 10.1038/nn1678 [DOI] [PubMed] [Google Scholar]
- Rakhade SN, Jensen FE (2009) Epileptogenesis in the immature brain: emerging mechanisms. Nat Rev Neurol 5:380–391. 10.1038/nrneurol.2009.80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakhade SN, Zhou C, Aujla PK, Fishman R, Sucher NJ, Jensen FE (2008) Early alterations of AMPA receptors mediate synaptic potentiation induced by neonatal seizures. J Neurosci 28:7979–7990. 10.1523/JNEUROSCI.1734-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakhade SN, Fitzgerald EF, Klein PM, Zhou C, Sun H, Huganir RL, Jensen FE (2012) Glutamate receptor 1 phosphorylation at serine 831 and 845 modulates seizure susceptibility and hippocampal hyperexcitability after early life seizures. J Neurosci 32:17800–17812. 10.1523/JNEUROSCI.6121-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimers JM, Milovanovic M, Wolf ME (2011) Quantitative analysis of AMPA receptor subunit composition in addiction-related brain regions. Brain Res 1367:223–233. 10.1016/j.brainres.2010.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricciardi S, Ungaro F, Hambrock M, Rademacher N, Stefanelli G, Brambilla D, Sessa A, Magagnotti C, Bachi A, Giarda E, Verpelli C, Kilstrup-Nielsen C, Sala C, Kalscheuer VM, Broccoli V (2012) CDKL5 ensures excitatory synapse stability by reinforcing NGL-1-PSD95 interaction in the postsynaptic compartment and is impaired in patient iPSC-derived neurons. Nat Cell Biol 14:911–923. 10.1038/ncb2566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg EC, Lippman-Bell JJ, Handy M, Soldan SS, Rakhade S, Hilario-Gomez C, Folweiler K, Jacobs L, Jensen FE (2018) Regulation of seizure-induced MeCP2 Ser421 phosphorylation in the developing brain. Neurobiol Dis 116:120–130. 10.1016/j.nbd.2018.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell P, Fuccillo M, Maxeiner S, Hayton S, Gokce O, Lim B, Fowler S, Malenka R, Südhof T (2014) Autism-Associated Neuroligin-3 Mutations Commonly Impair Striatal Circuits to Boost Repetitive Behaviors. Cell 158:198–212. 10.1016/j.cell.2014.04.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozov A, Sprengel R, Seeburg PH (2012) GluA2-lacking AMPA receptors in hippocampal CA1 cell synapses: evidence from gene-targeted mice. Front Mol Neurosci 5:22. 10.3389/fnmol.2012.00022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusconi L, Salvatoni L, Giudici L, Bertani I, Kilstrup-Nielsen C, Broccoli V, Landsberger N (2008) CDKL5 expression is modulated during neuronal development and its subcellular distribution is tightly regulated by the C-terminal tail. J Biol Chem 283:30101–30111. 10.1074/jbc.M804613200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo E, Gitto R, Citraro R, Chimirri A, Sarro G (2012) New AMPA antagonists in epilepsy. Expert Opin Investig Drug 21:1371–1389. 10.1517/13543784.2012.705277 [DOI] [PubMed] [Google Scholar]
- Sanchez RM, Koh S, Rio C, Wang C, Lamperti ED, Sharma D, Corfas G, Jensen FE (2001) Decreased glutamate receptor 2 expression and enhanced epileptogenesis in immature rat hippocampus after perinatal hypoxia-induced seizures. J Neurosci 21:8154–8163. 10.1523/JNEUROSCI.21-20-08154.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder E, Yuan L, Seong E, Ligon C, DeKorver N, Gurumurthy CB, Arikkath J (2018) Neuron-Type Specific Loss of CDKL5 Leads to Alterations in mTOR Signaling and Synaptic Markers. Mol Neurobiol. Advance online publication. Retrieved October 4, 2018. doi: 10.1007/s12035-018-1346-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squires RF, Saederup E, Crawley JN, Skolnick P, Paul SM (1984) Convulsant potencies of tetrazoles are highly correlated with actions on GABA/benzodiazepine/picrotoxin receptor complexes in brain. Life Sci 35:1439–1444. 10.1016/0024-3205(84)90159-0 [DOI] [PubMed] [Google Scholar]
- Stephenson JR, Wang X, Perfitt TL, Parrish WP, Shonesy BC, Marks CR, Mortlock DP, Nakagawa T, Sutcliffe JS, Colbran RJ (2017) A novel human CAMK2A mutation disrupts dendritic morphology and synaptic transmission, and causes ASD-related behaviors. J Neurosci 37:2216–2233. 10.1523/JNEUROSCI.2068-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczurowska E, Mares P (2015) An antagonist of calcium permeable AMPA receptors, IEM-1460: anticonvulsant action in immature rats? Epilepsy Res 109:106–113. 10.1016/j.eplepsyres.2014.10.020 [DOI] [PubMed] [Google Scholar]
- Talos DM, Fishman RE, Park H, Folkerth RD, Follett PL, Volpe JJ, Jensen FE (2006) Developmental regulation of alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor subunit expression in forebrain and relationship to regional susceptibility to hypoxic/ischemic injury: I. Rodent cerebral white matter and cortex. J Comp Neurol 497:42–60. 10.1002/cne.20972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talos DM, Kwiatkowski DJ, Cordero K, Black PM, Jensen FE (2008) Cell-specific alterations of glutamate receptor expression in tuberous sclerosis complex cortical tubers. Ann Neurol 63:454–465. 10.1002/ana.21342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S, Wang IJ, Yue C, Takano H, Terzic B, Pance K, Lee JY, Cui Y, Coulter DA, Zhou Z (2017) Loss of CDKL5 in glutamatergic neurons disrupts hippocampal microcircuitry and leads to memory impairment in mice. J Neurosci 37:7420–7437. 10.1523/JNEUROSCI.0539-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tramarin M, Rusconi L, Pizzamiglio L, Barbiero I, Peroni D, Scaramuzza L, Guilliams T, Cavalla D, Antonucci F, Kilstrup-Nielsen C (2018) The antidepressant tianeptine reverts synaptic AMPA receptor defects caused by deficiency of CDKL5. Hum Mol Genet 27:2052–2063. 10.1093/hmg/ddy108 [DOI] [PubMed] [Google Scholar]
- Twomey EC, Yelshanskaya MV, Vassilevski AA, Sobolevsky AI (2018) Mechanisms of channel block in calcium-permeable AMPA receptors. Neuron 99:956–968.e4. 10.1016/j.neuron.2018.07.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uzunova G, Hollander E, Shepherd J (2014) The role of ionotropic glutamate receptors in childhood neurodevelopmental disorders: autism spectrum disorders and fragile x syndrome. Curr Neuropharmacol 12:71–98. 10.2174/1570159X113116660046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IT, Allen M, Goffin D, Zhu X, Fairless AH, Brodkin ES, Siegel SJ, Marsh ED, Blendy JA, Zhou Z (2012) Loss of CDKL5 disrupts kinome profile and event-related potentials leading to autistic-like phenotypes in mice. Proc Natl Acad Sci U S A 109:21516–21521. 10.1073/pnas.1216988110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Mei F, Hu J, Zhu M, Qi H, Chen X, Li R, McNutt MA, Yin Y (2017) PTENα modulates CaMKII signaling and controls contextual fear memory and spatial learning. Cell Rep 19:2627–2641. 10.1016/j.celrep.2017.05.088 [DOI] [PubMed] [Google Scholar]
- Wang W, Tan T, Yu Y, Huang Z, Du Y, Han H, Dong Z (2018) Inhibition of AMPAR endocytosis alleviates pentobarbital-induced spatial memory deficits and synaptic depression. Behav Brain Res 339:66–72. 10.1016/j.bbr.2017.11.020 [DOI] [PubMed] [Google Scholar]
- Washburn MS, Numberger M, Zhang S, Dingledine R (1997) Differential dependence on GluR2 expression of three characteristic features of AMPA receptors. J Neurosci 17:9393–9406. 10.1523/JNEUROSCI.17-24-09393.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenthold RJ, Yokotani N, Doi K, Wada K (1992) Immunochemical characterization of the non-NMDA glutamate receptor using subunit-specific antibodies: evidence for a hetero-oligomeric structure in rat brain. J Biol Chem 267:501–507. [PubMed] [Google Scholar]
- Zhou C, Lippman-Bell JJ, Sun H, Jensen F (2011) Hypoxia-Induced Neonatal Seizures Diminish Silent Synapses and Long-Term Potentiation in Hippocampal CA1 Neurons. The Journal of Neuroscience 31:18211–18222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Hong EJ, Cohen S, Zhao WN, Ho HY, Schmidt L, Chen WG, Lin Y, Savner E, Griffith EC, Hu L, Steen JA, Weitz CJ, Greenberg ME (2006) Brain-specific phosphorylation of MeCP2 regulates activity-dependent BDNF transcription, dendritic growth, and spine maturation. Neuron 52:255–269. 10.1016/j.neuron.2006.09.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Liu A, Xia S, Leung C, Qi J, Meng Y, Xie W, Park P, Collingridge GL, Jia Z (2018) The C-terminal tails of endogenous GluA1 and GluA2 differentially contribute to hippocampal synaptic plasticity and learning. Nat Neurosci 21:50–62. 10.1038/s41593-017-0030-z [DOI] [PubMed] [Google Scholar]
- Zhu YC, Li D, Wang L, Lu B, Zheng J, Zhao SL, Zeng R, Xiong ZQ (2013) Palmitoylation-dependent CDKL5-PSD-95 interaction regulates synaptic targeting of CDKL5 and dendritic spine development. Proc Natl Acad Sci U S A 110:9118–9123. 10.1073/pnas.1300003110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY, Bear MF (2012) Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb Perspect Biol 4:a009886. [DOI] [PMC free article] [PubMed] [Google Scholar]