

Summary
Following fertilization, totipotent cells undergo asymmetric cell divisions, resulting in three distinct cell types in the late pre-implantation blastocyst: epiblast (Epi), primitive endoderm (PrE), and trophectoderm (TE). Here, we aim to understand whether these three cell types can be induced from fibroblasts by one combination of transcription factors. By utilizing a sophisticated fluorescent knockin reporter system, we identified a combination of five transcription factors, Gata3, Eomes, Tfap2c, Myc, and Esrrb, that can reprogram fibroblasts into induced pluripotent stem cells (iPSCs), induced trophoblast stem cells (iTSCs), and induced extraembryonic endoderm stem cells (iXENs), concomitantly. In-depth transcriptomic, chromatin, and epigenetic analyses provide insights into the molecular mechanisms that underlie the reprogramming process toward the three cell types. Mechanistically, we show that the interplay between Esrrb and Eomes during the reprogramming process determines cell fate, where high levels of Esrrb induce a XEN-like state that drives pluripotency and high levels of Eomes drive trophectodermal fate.
Keywords: reprogramming, induced pluripotent stem cells, induced trophoblast stem cells, induced extraembryonic endoderm stem cells, early embryonic development, trophectoderm, inner cell mass, primitive endoderm, Esrrb, Eomes
Graphical Abstract
Highlights
-
•
Gata3, Eomes, Tfap2c, Myc, and Esrrb convert fibroblasts into iPSCs, iTSCs, and iXENs
-
•
Esrrb, but not other pluripotency genes, can shift the TSC fate into pluripotency
-
•
Esrrb induces pluripotency by the activation of a unique XEN-like state
-
•
The interplay between Eomes and Esrrb determines cell fate decision
Benchetrit and colleagues identified a combination of factors that can produce the three in vitro equivalent cell types of the blastocyst, iPSCs, iTSCs, and iXENs, from fibroblasts. They found that Esrrb was most potent in inducing pluripotency by the activation of a XEN-like state and Eomes most potent in promoting trophectoderm fate.
Introduction
After fertilization, a rapid reprogramming of the chromosomal content of the newly formed cell occurs, resulting in a totipotent zygote. Upon division, totipotency is gradually lost and an early blastocyst is formed, containing two compartments that are more committed: the inner cell mass (ICM), which will generate the embryo proper (epiblast-Epi) and primitive endoderm (PrE), and an outer layer of trophectoderm (TE), which will give rise to extraembryonic tissues, such as the placenta (Chen et al., 2010). Several models explain the first cell fate decision in the embryo (Jaber et al., 2017, Wu and Schöler, 2016), but it remains to be elucidated which are the key factors that derive each of the two lineages (i.e., ICM and TE). In embryonic stem cells (ESCs), reciprocal repression between the pluripotency master regulator, Oct4, and the TE key gene, Cdx2, has been suggested to determine cell lineage specification (Niwa et al., 2005). Accordingly, knockout of Oct4 or overexpression of Cdx2 leads to transdifferentiation of ESCs into trophoblast stem-like cells (Niwa et al., 2000, Niwa et al., 2005). Single-cell RNA sequencing throughout mouse pre-implantation development identified targets of the master pluripotency regulators Oct4 and Sox2 as being highly heterogeneously expressed within 4-cell stage embryos, with Sox21 showing one of the most heterogeneous expression profiles that drives cell fate commitment (Goolam et al., 2016).
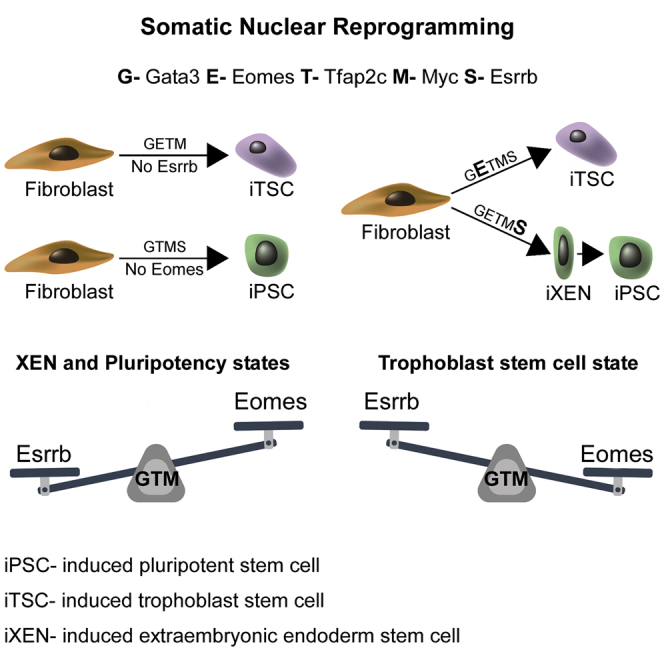
The induction of pluripotency from somatic cells by a small number of defined factors (Takahashi and Yamanaka, 2006) opened a new avenue in basic research (Buganim and Jaenisch, 2012), in which cell-type-specific combinations of key master regulators are identified by demonstrating their capability to impose a stable alternative cell fate (Xu et al., 2015). Recently, we and others have shown that the introduction of Gata3, Eomes, Tfap2c, and Myc (GETM) (Benchetrit et al., 2015) or Ets2 (Kubaczka et al., 2015) in fibroblasts can initiate a reprogramming process that leads to the formation of stable and fully functional induced trophoblast stem cells (iTSCs). The success in inducing pluripotent stem cell (PSC) and TSC states by ectopic expression of transcription factors led us to search for a combination of factors that would hold the capacity to convert fibroblasts into both iPSCs and iTSCs. We hypothesized that identifying such a combination would help to elucidate the counteracting forces that drive each lineage.
Results
Ectopic Expression of Esrrb Drives the TSC Reprogramming Combination toward Pluripotency
To distinguish between PSC and TSC fates, we established a fluorescent knockin reporter system harboring 4 unique reporters: (1) Nanog-2A-EGFP, a cytoplasmic reporter that specifically marks PSCs; (2) Elf5-2A-EYFP-NLS, a nuclear reporter that is specific for TSCs; and (3) Utf1-2A-tdTomato and (4) Esrrb-2A-TagBFP, cytoplasmic reporters that mark both cell types. Although Utf1 levels were reduced and a small fraction of the Nanog-2A-EGFP protein was not cleaved (Figure S1A), the targeted engineered KH2-ESC line (termed BYKE hereinafter) gave rise to chimeras with germline transmission following blastocyst injection (Figure S1B), indicating full developmental potential.
To test whether the BYKE system is suitable for reprogramming studies, we injected BYKE ESCs into host blastocysts and selected for BYKE mouse embryonic fibroblasts (MEFs) at 13.5 days post-coitum (DPC).
BYKE-MEFs were reprogrammed either into iPSCs by Oct4, Sox2, Klf4, and Myc (OSKM) or into iTSCs by GETM (Figure 1A). Reprogramming efficiency ranged between 2% and 20% in OSKM-iPSC reprogramming, and between 0.3% and 3% in GETM-iTSC reprogramming, depending on infection efficiency and duration of reprogramming. BYKE-iPSC clones were triple positive for the 3 PSC reporters, and BYKE-iTSC clones were triple positive for the 3 TSC reporters (Figure S1C). Taken together, these data demonstrate that the BYKE system is adequate for reprogramming and all 4 reporters are active.
Figure 1.

Esrrb Drives TSC Reprogramming with GETM to Pluripotency
(A) Schematic representation, bright field and fluorescence images of the 3 PSC reporters (left), 3 TSC reporters (right), and FACS analysis of the 4 reporters in BYKE-MEFs undergoing reprogramming for 20 days with OSKM (left) or GETM (right) followed by 10 days of dox removal.
(B) BYKE-MEFs were infected with GETM plus individual factors as indicated and tested for iPSC formation. The graph summarizes FACS analysis of the 3 PSC reporters in BYKE-MEFs undergoing reprogramming for 20 days with GETM plus the indicated single factor, followed by 10 days of dox removal. A typical experiment out of 3 independent experiments is shown.
(C) BYKE-MEFs were reprogrammed by GETM + Esrrb (GETMS) for 20 days with dox followed by 2 days of dox removal. Subsequently, different populations of cells (Utf1-2A-tdTomato+/Esrrb-2A-TagBFP-/Nanog-2A-EGFP- and Utf1-2A-tdTomato+/Esrrb-2A-TagBFP+/Nanog-2A-EGFP+) were sorted and tested for expression of ESC-specific (Oct4 and Nanog), TSC-specific (Elf5 and Cdx2), and trophoblast differentiation-specific (Gcm1, Ctsq, and Tpbpa) genes. mRNA levels were normalized to Gapdh. Error bars presented as a mean ± SD of 2 duplicate runs from a typical experiment out of 3 independent experiments.
(D and E) FACS analyses for the 4 reporters on BYKE-MEFs undergoing reprogramming with GETMS for 20 days followed by 10 days of dox removal under (D) ESC (GETMS-ESM) or (E) TSC (GETMS-TSM) culture conditions.
(F) (Left) Reprogramming strategy for producing both iPSCs and iTSCs with GETMS factors. (Right) FACS analysis of the 4 reporters in BYKE-MEFs undergoing reprogramming with GETMS following the aforementioned reprogramming strategy.
(G) qPCR of the GETMS transgenes normalized to Gapdh in BYKE-GETMS#10 2°MEFs after 72 h of dox induction. Error bars presented as a mean ± SD of 2 duplicate runs from a typical experiment out of 3 independent experiments.
(H and I) Graphs summarizing FACS analyses for all 4 reporters (H) and representative bright field and fluorescence images (I) after 20 days of dox followed by 10 days of dox removal of BYKE-GETMS#10 2°MEFs infected with additional single factor as depicted in ESC or TSC medium. Error bars presented as a mean ± SD of 2 duplicate runs from a typical experiment out of 3 independent experiments.
Related to Figure S1.
We assumed that GETM would be a good initial core reprogramming combination to produce both iPSCs and iTSCs, as these factors are expressed at early embryonic developmental stages (Auman et al., 2002, Home et al., 2009, McConnell et al., 2005).
We screened for factors that would shift the TSC fate, dictated by GETM, to pluripotency and generate iPSCs. We infected BYKE-MEFs with doxycycline (dox)-inducible lentiviruses encoding for the GETM proteins together with individual factors that are expressed at early developmental stages, such as Klf5, Tead4, and Sox15; factors that drive pluripotency, like Klf4, Oct4, and Nanog; or factors that are expressed in both compartments or shared between iPSCs and iTSCs, such as Sall4, Lin28, Utf1, Sox2, and Esrrb (Figure 1B).
iPSC formation was scored by fluorescence-activated cell sorting (FACS) after 20 days of dox exposure followed by 10 days of dox removal. Intriguingly, the only factor that could shift the GETM fate and form iPSCs was Esrrb (S). The resulting iPSCs were positive for the 3 PSC reporters (Figure 1B) and showed tight dome morphology similar to ESCs. Interestingly, all factor combinations resulted in a small fraction of Utf1-2A-tdTomato-only-positive cells (Figure 1B). qPCR analysis on sorted cells revealed that these cells are trophoblast differentiated cells (Figure 1C). These results suggest that Esrrb harbors a unique property that allows it to shift TSC fate, driven by GETM, into PSC fate.
Ectopic Expression of GETMS Produces Both iPSCs and iTSCs
We examined whether the 5 factor combination (5F or GETMS) is capable of also producing iTSCs or whether the addition of Esrrb to GETM abrogates this capability. MEFs were infected with GETMS (Figure S1D) and then reprogrammed in ESC or in TSC medium. Strikingly, infected BYKE-MEFs that were cultured in ESC medium produced iPSCs and trophoblast differentiated cells, and infected BYKE-MEFs grown in TSC medium formed mostly iTSCs, with few iPSC colonies (Figures 1D and 1E). Cultivating the infected cells for 2 weeks in 10% fetal bovine serum (FBS) in DMEM with dox, followed by 1 week in TSC reprogramming medium, and subsequently removing dox for 14 days in TSC culture medium produced both stable iPSC and iTSC colonies in the same dish (Figures 1F and S1E).
6/7 examined iPSC clones and 4/7 iTSC clones were positive for all 5 transgenes in their genome (Figure S1F), indicating that the GETMS combination holds the potential to produce both iPSCs and iTSCs.
The Interplay between Esrrb and Eomes Determines Cell Identity during Reprogramming with GETMS
We asked whether a specific stoichiometry of reprogramming factors within the GETMS combination is favorable for the production of iPSCs versus iTSCs or whether GETMS produce transient bi-potential cells. Thus, we examined the expression levels of GETMS in various secondary (2°) MEF systems. 2°MEF systems were obtained by infecting MEFs with dox-inducible lentiviruses encoding for GETMS, and then “primary” iPSC colonies were isolated and injected into host blastocysts to derive “secondary” MEFs. This allowed us to generate multiple 2°MEF systems in which each system captures only one stoichiometry of factors and can be immediately activated by the addition of dox. We then examined the correlation between stoichiometry and reprogramming outcome.
In a 2°MEF system (2° BYKE-GETMS#10), which expressed low levels of all 5 factors (Figure 1G) and which following dox addition resulted in proliferation and differentiated trophoblasts, additional infection of Esrrb, but not any of the other 4 factors, produced iPSCs under ESC culture conditions (Figures 1H, upper, and 1I, left). In contrast, under TSC culture conditions, the addition of Eomes, but not any of the other factors, produced iTSCs (Figures 1H, lower, and 1I, right).
When a 2°MEF system (2° KH2-NgUrEb-GETMS#1) with very high levels of Eomes was examined (Figure S1G), no iPSC formation was noted, even when ESC conditions were used and Esrrb was overexpressed in the cells (Figure S1H). Overexpression of Eomes in this 2° system under TSC culture conditions facilitated further formation of iTSCs, but excessive levels of Esrrb or Gata3 hindered iTSC production (Figure S1I). According to its main role in inducing a TSC fate, removing Eomes from the 5F combination (Gata3, Tfap2c, Myc, and Esrrb [GTMS]) still allowed the production of iPSCs under ESC culture conditions (Figure S1J).
Lastly, to determine whether the ratio between Esrrb and Eomes can predict the outcome of resulting colonies early in reprogramming, we infected Oct4-GFP MEFs with GTM together with Eomes-2A-tdTomato and Esrrb-2A-EGFP. At day 6 of reprogramming, an early time point before Oct4-GFP signal can be detected, two cell populations were sorted, Esrrb-2A-EGFP positive with high Eomes-2A-tdTomato expression (Esrrb+/EomesHigh) and Esrrb-2A-EGFP positive with low Eomes-2A-tdTomato expression (Esrrb+/EomesLow). Reprogramming toward iPSCs was continued for each population separately. Notably, Esrrb+/EomesLow cells gave rise to 4-fold more Oct4-GFP iPSC colonies compared to Esrrb+/EomesHigh cells (Figure 2A).
Figure 2.

GETMS-iPSCs and GETMS-iTSCs Are Fully Functional
(A) (Left) Oct4-GFP MEFs were infected with GETMS factors, with Esrrb marked by EGFP and Eomes marked by tdTomato. 6 days post-dox induction, 16,000 of Esrrb-EGFP-positive and Eomes-tdTomato-high cells or 16,000 of Esrrb-EGFP-positive and Eomes-tdTomato-low cells were sorted and plated on feeder cells. Reprogramming continued for 20 more days in ESC medium followed by 10 days of dox removal. Left: number of Oct4-GFP-positive iPSC colonies is shown. Right: FACS analysis of Oct4-GFP-positive cells is shown. (Right) Oct4-GFP MEFs were infected with GETMS factors, with Eomes marked by EGFP and Esrrb marked by tdTomato. 6 days post-dox induction, 80,000 of Eomes-EGFP-positive and Esrrb-tdTomato-high cells or 80,000 of Eomes-EGFP-positive and Esrrb-tdTomato-low cells were sorted and plated on feeder cells. Reprogramming continued for 20 more days in TSC medium followed by 10 days of dox removal and one split for colony stabilization. Left: number of iTSC colonies is shown. Right: FACS analysis for the trophoblast-specific cell surface marker, Cd40, is shown. Asterisk indicates p ≤ 0.05 using Student’s t test.
(B) tdTomato-marked ESC, OSKM-iPSC, GETMS-iPSC, and EGFP-marked bdTSC and GETMS-iTSC clones were injected into blastocysts. The resulting embryos and placentas were analyzed at 13.5 DPC. Bright field, red and green channel images of the chimeric embryos and placentas are depicted.
(C) Immunofluorescent and immunohistochemical staining images of 13.5-DPC chimeric placenta showing a clear EGFP signal in the central and marginal areas (marked by blue and orange rectangles, respectively) following GETMSEGFP-iTSCs injection.
(D) (Left) Hemorrhagic lesions 6 to 7 days following subcutaneous injection of the indicated GETMS-iTSC and bdTSC lines. (Right) H&E staining of paraffin sections of hemorrhagic lesions shows necrotic tissue with blood and scattered trophoblastic cells.
(E) PCA of global gene expression profiles from RNA-seq data of two biological replicates of 2 MEF, 2 OSKM-iPSC, 2 GETMS-iPSC, 2 ESC, 2 GETM-iTSC, 2 GETMS-iTSC, and 2 bdTSC lines.
Related to Figure S2.
A reciprocal experiment resulted in enrichment for iTSC colonies in cells with Eomes-2A-EGFP-positive and low Esrrb-2A-tdTomato levels compared to high Esrrb-2A-tdTomato expression, as assessed by the TSC-specific cell surface marker Cd40 (Figure 2A). Taken together, these results indicate that a bi-potential state is not achieved during reprogramming with GETMS but rather that the interplay between Eomes and Esrrb, along with culture conditions, are the factors that determine cellular identity.
GETMS-iPSCs and GETMS-iTSCs Are Fully Functional
To examine whether GETMS-iPSCs and GETMS-iTSCs are fully functional and that their developmental potential resembles ESCs and blastocyst-derived TSCs (bdTSCs), respectively, we injected GETMS-tdTomato-marked iPSCs and GETMS-EGFP-marked iTSCs into blastocysts and monitored their contribution to 13.5-DPC embryos and placentas. All injected GETMS-iPSC clones produced high-grade chimeras with a contribution that was comparable to their ESC parental clone (Figures 2B and S2A). Of note, contribution to the extraembryonic mesoderm or endoderm, which originate from the ICM, was noted in the placenta as well (shown as tdTomato signal in the blood vessels of the placenta; Figure S2B).
In contrast, GETMS-iTSC clones contributed solely to the trophoblast compartment of the placenta, with no signal in the embryo (Figures 2B, 2C, and S2A). Moreover, GETMS-iTSCs formed transient hemorrhagic lesions with large lacunas when injected subcutaneously into nude mice and produced all trophoblastic cell types (i.e., giant multinucleated trophoblast cells, spongiotrophoblast cells, and syncytiotrophoblast cells) within the lesions (Figures 2D, S2C, and S2D). GETMS-iTSCs showed the same developmental potential in vitro, following removal of Fgf4 and heparin from culture medium, as depicted by qPCR for the relevant markers and cell cycle FACS analysis (Figures S2E and S2F). Lastly, whole-transcriptome analysis of 2 clones from each GETMS-iPSC, OSKM-iPSC, ESC, GETMS-iTSC, GETM-iTSC, bdTSC, and MEF clustered all PSCs together and all TSCs together and far from MEFs (Figure 2E). These data indicate that GETMS can produce high-quality iPSCs and iTSCs, which resemble ESCs and bdTSCs, respectively, in their function and transcription.
In-Depth Transcriptomic, Chromatin, and Epigenetic Analyses of GETMS Reprogramming
Our next goal was uncovering the molecular mechanisms underlying GETMS reprogramming. We performed RNA sequencing (RNA-seq) and the assay for transposase-accessible chromatin (ATAC-seq) on cells in reprogramming with various combinations of factors: OSKM and GTMS for iPSC-only reprogramming, GETM for iTSC-only reprogramming, and GETMS in ESC medium (GETMS-ESM) or in TSC medium (GETMS-TSM). We compared the reprogramming cells to MEF, ESC, iPSC, and bdTSC controls following 3 days of transgene expression, an early time point where changes are relatively homogeneous among all reprogramming cells (Buganim et al., 2013). Principal-component analysis (PCA) of the transcriptional profiles of the various conditions clustered ESCs and bdTSCs far from all reprogramming combinations and MEF controls, as expected (Figure 3A). Plotting only MEFs and MEFs undergoing reprogramming showed a clear difference between the transcriptome of OSKM-expressing MEFs, control MEFs, and all GTM-containing combinations (Figure 3B).
Figure 3.

Unbiased Comparative Transcriptome and Chromatin Analysis of the Various Reprogramming Combinations
(A) PCA of global gene expression profiles from RNA-seq data of two biological replicates of MEFs, bdTSCs, ESCs, and the indicated reprogramming combinations after 72 h of dox induction.
(B) PCA of the same samples as in (A), excluding bdTSCs and ESCs.
(C) Clustering and heatmap of transcriptional differences detected by RNA-seq for the indicated groups, showing 13,309 expressed genes (FPKM > 2 in at least one condition) grouped into 19 clusters. Blue (0) denotes no expression, and yellow (1) denotes maximal expression.
(D) Clustering and heatmap of all (n = 188,432) intronic and distal ATAC-seq peaks of the indicated samples in 18 clusters exhibiting variation across all samples. Heatmap colors correspond to maximal ATAC-seq signal for every peak across 8 conditions, ranging from 0 (purple) to 20 (green) reads (normalized).
(E and F) UCSC genome browser visualization of normalized ATAC-seq profiles (top) and RNA-seq levels in FPKM (bottom) at the Gdf3 (E) or Elf5 (F) locus in the indicated samples. Data from two merged biological replicates are shown. Colored rectangles indicate condition(s) (cell type[s] or reprogramming combination[s])-specific regions.
(G) Transcription factor binding motifs that were identified by comparing the top distal and intronic ATAC-seq peaks in GETMS and GTMS to those in GETM.
(H) Venn diagram showing the overlaps between differentially expressed genes (upregulated) in the present study (GETMS versus MEFs), 8 lines of chemically induced XENs versus MEFs (Li et al., 2017), 4 lines of OSKM-induced XENs grown in ESC medium versus MEFs (Parenti et al., 2016), and intermediate cells on days 26 and 28 during chemical reprogramming versus MEFs (Zhao et al., 2015). The significance of the overlap between the present study and the respective study is indicated.
(I) Heatmap visualization of 96 differentially expressed genes shared between the present study and at least one of the 3 studies mentioned above.
Related to Figure S3.
Clustering of 13,309 expressed genes revealed 19 clusters with unique signatures (Figure 3C): MEF-specific genes (clusters nos. 1, 2, and 6; e.g., Thy1 and Col5a1), OSKM-specific genes (cluster no. 3; e.g., Cd34 and Aldh3a1), GETM, GETMS, and GTMS-specific genes (cluster no. 9; e.g., Pcsa and Prl3d2), GTMS, GETM, GETMS, and bdTSC-specific genes (cluster no. 11; e.g., Gatsl13 and Mgat4b), and ESC and bdTSC or ESC- or TSC-specific genes (clusters nos. 10, 12, 15, 16, 17, 18, and 19; e.g., Gdf3 and Elf5).
Similar cluster analysis was performed for 188,432 intronic and distal ATAC-seq peaks in all 8 conditions, yielding 18 distinct clusters with a chromatin-reshaping pattern that closely resembles that of the transcriptome (Figure 3D).
Using GREAT, we allocated distal and intronic peaks to their adjacent genes and ran gene ontology (GO) annotation for the ATAC-seq and RNA-seq clusters (Tables S1 and S2). Although TSC- and iPSC-specific clusters were enriched for blastocyst formation (false discovery rate [FDR] = 2.7E−16), TSC-specific clusters for placenta development (FDR = 5.9E−23), GETM-, GETMS-, and bdTSC-specific cluster for focal cell adhesion (FDR = 7.8E−14), and labyrinthine layer development (FDR = 9.7E−14) as expected, OSKM-specific cluster was enriched also for genes that cause placental defects (Table S1; Figure S3A), suggesting a role of OSKM in repressing placental genes early on. The other reprogramming clusters were enriched for processes involved in MET, proliferation and metabolic shift, all known to take place at early time points of reprogramming with OSKM (Tables S1 and S2).
The strongest 1,000 ATAC-seq peaks were located at promoters (40%–50%); however, although ATAC-seq peaks at the TSS region (±2.5 Kb) of the 19 RNA clusters could not identify unique signatures (Figure S3B), distal and intronic peaks showed a clear overlap with the transcriptome (Figure 3D). These data suggest that intronic and distal regions are more specifically arranged in each cell type and are reshaped early in reprogramming. Indeed, unique intronic and distal peaks were observed in OSKM-specific markers, such as Gdf3 (Buganim et al., 2012), and for iTSC-specific markers, such as Elf5 (Benchetrit et al., 2015, Kubaczka et al., 2015), in the various combination of factors (Figures 3E and 3F).
We compared global ATAC-seq peaks located near active genes (FPKM > 1) to those located near inactive genes (FPKM < 1) in OSKM and GETM reprogramming. Interestingly, although peaks near active genes showed a significant enrichment for Theiler stage 1, 2, and 4 (FDR = 6.8E−10) for OSKM and TE (FDR = 2.3E−22) for GETM, no specific enrichment was found for ATAC-seq peaks located near inactive genes (Table S3).
In contrast, GETMS-specific peaks near active genes were enriched for genes regulating PrE (p ≤ 0.00003) and TE (p ≤ 0.0001), and peaks located near inactive genes were enriched for genes regulating gonad primordium (p ≤ 0.00003; Table S3). These results suggest that OSKM, GETM, and GETMS bind and activate genomic loci involved in early embryogenesis already at day 3 of reprogramming and imply that OSKM and GETMS combinations produce iPSCs by reshaping distinct genomic loci.
Esrrb Drives GETM Reprogramming toward Pluripotency by Inducing a XEN-like State
To understand how Esrrb in conjunction with GETM drives iPSC formation, we initially searched for DNA binding motifs that are enriched within Esrrb-specific ATAC-seq peaks (GETMS and GTMS) compared to controls (GETM, MEFs, bdTSCs). The most significant binding motifs within Esrrb-specific peaks were shown to be enriched in regulatory elements of germ cells (Elk1, Elk4, Nrf1, YY1, Sp1, and Zfx; Hammoud et al., 2014, Luoh et al., 1997, Thomas et al., 2007, Wu et al., 2009), XENs or PrE (Elf1, Nr5a2, and Klf5; Cho et al., 2012, Lin et al., 2010, McDonald et al., 2014), and PSCs (Nr5a2, Klf4, and Zfx; Table S4; Figures 3G and S3C).
Given that a XEN-like state is achieved during chemical reprogramming just before pluripotency (Li et al., 2017, Zhao et al., 2015) and the above binding motifs analysis, we sought to determine whether such a state is induced in reprogramming with GETMS. We performed independent per-study differential gene expression analyses of RNA-seq data from the present study and from 3 different studies describing a XEN-like signature (Li et al., 2017, Parenti et al., 2016, Zhao et al., 2015). We revealed a statistically significant XEN-like signature at day 3 of factor induction in combinations that include Esrrb with an overall 225 genes that were upregulated by GETMS and GTMS combinations, but not in controls. 96 of them were shared with one or more of the studies describing a XEN-like signature (Figures 3H and 3I).
Chromatin immunoprecipitation (ChIP)-seq for H3K4me2 and H3K27ac was conducted on MEFs overexpressing GETMS, GTMS, or Esrrb alone (S) as control, following 3 days of dox. Differential binding analysis of H3K4me2 ChIP-seq data, contrasting GETMS and GTMS against the control samples, MEFs and MEFs +S, found 116 genes that were intersected with the 225 XEN-signature genes (p ≤ 2.7E−08; Figure S3D). Indeed, activation signals were accumulated in XEN-specific loci, such as Myh6, as well as in Oct4 in GETMS and GTMS compared to MEFs + S control (Figure S3E).
Accordingly, we were able to isolate stable XEN-like stem cells (GETMS-iXENs) after 20 days of GETMS expression by applying XEN culture conditions. GETMS-iXENs exhibited a similar morphology to that of blastocyst-derived XENs (bdXENs) and expressed XEN-specific markers (Figures 4A and S4A). Importantly, higher levels of Oct4 and Sall4 were observed in stable GETMS-iXENs compared to bdXENs, explaining how a XEN-like state might lead to pluripotency (Figure S4A).
Figure 4.

GETMS Combination or Esrrb Overexpression during ESC-TSC Transdifferentiation Produces Stable iXENs
(A) Bright field and immunostaining images for Gata6 (magenta), Sox17 (green), and Dapi (blue) in GETMS-derived induced XEN stem cells (GETMS-iXENs) grown on feeder cells.
(B) FACS analysis showing the percentage of GFP-positive cells for Oct4-GFP MEFs that were reprogrammed for 20 days, followed by 6 days of dox removal, with GETMS together with either empty vector (EV), Gata6, Sox17, or both.
(C and D) Oct4-GFP MEFs were reprogrammed for 20 days, followed by 10 days of dox removal, with GETMS together with either an empty vector (EV), Sox17, Gata6, or Sox17 and Gata6. Subsequently, 100,000 GFP-positive cells were sorted from each group and analyzed. qPCR analysis for PSC-specific genes, Nanog and Fgf4 (C), and XEN-specific genes, Pdgfr and Sra6 (D), normalized to Gapdh in the indicated samples is shown. Error bars presented as a mean ± SD of 2 duplicate runs from a typical experiment out of 3 independent experiments.
(E) qPCR of the indicated genes normalized to Gapdh during transdifferentiation of ZHBTc4 ESCs into TS-like cells at the indicated time points after infection with either constitutively active Eomes or Esrrb. Day 0 refers to pre-dox induction. Error bars presented as a mean ± SD of 2 duplicate runs from a typical experiment out of 3 independent experiments.
(F) Western blot analysis detecting Esrrb and actin in ZHBTc4 cell lines harboring Esrrb-KD (EsrrbHypomorph), Esrrb overexpression (EsrrbOE), or control (EsrrbWT).
(G) ChIP-qPCR for Esrrb in ZHBTc4 cells harboring WT (EsrrbWT), knockdown (EsrrbHypomorph), or overexpression (EsrrbOE) of Esrrb. A graph depicting the binding intensity of Esrrb to the indicated loci in all conditions as measured by the fold change of DNA enrichment between control antibody (Igg) and Esrrb antibody is shown. All samples were normalized to their corresponding input. Error bars presented as a mean ± SD of 2 duplicate runs from a typical experiment out of 3 independent experiments.
Related to Figure S4.
To examine whether XEN-specific genes are enriched in GETMS-reprogrammed cells just before the acquisition of pluripotency, we reprogrammed Oct4-GFP cells with either GETMS or with OSKM, and sorted Oct4-GFP-positive cells at day 12 of dox induction from both groups. qPCR analysis showed a clear induction of the XEN-specific gene, Myh6, in GETMS reprogrammable Oct4-GFP-positive cells compared to Oct4-GFP-negative or OSKM reprogrammable cells (Figure S4B). Myh6 was also higher in cells of GETMS reprogramming in a combination with high Esrrb expression sorted at day 6 (Figure S4C).
To understand whether a XEN-like state with high Oct4 expression can be boosted at the expense of the PSC state during GETMS reprogramming, we infected Oct4-GFP MEFs with GETMS together with either an empty vector or with key XEN genes, Sox17 and Gata6. A dramatic increase in Oct4-GFP-positive cells was noted in GETMS combinations harboring XEN-specific transgenes (Figure 4B). qPCR analysis for PSC and XEN-specific markers on sorted Oct4-GFP cells following 20 days of dox induction and 10 days of dox removal revealed increased percentage of Oct4-GFP cells in GETMS plus Sox17 and Gata6 due to increased number of iXENs at the expense of iPSCs (Figures 4C and 4D). The elevation in Oct4-GFP cells was mostly attributed to Esrrb, as GETM combination together with Gata6 and Sox17 gave rise to very few Oct4-GFP cells (Figure S4D). These results suggest that Esrrb, in conjunction with GETM, is a potent inducer of a unique XEN-like state that leads to pluripotency.
Esrrb Regulates XEN Genes in Other Systems
To examine whether the activation of XEN-specific genes by Esrrb is a general phenomenon, we utilized ZHBTc4 ESCs, which are capable of transdifferentiating into TS-like cells by elimination of Oct4 expression using a Tet-OFF system (Niwa et al., 2000). We infected ZHBTc4 ESCs with Esrrb-2A-EGFP or Eomes-2A-EGFP and isolated EGFP-positive ESC clones that overexpressed either Esrrb or Eomes. As a control, we used the parental ZHBTc4 ESC line. All clones were grown under ESC culture conditions, and dox was added for 4 days to allow transdifferentiation. Each day, cells were collected and tested for expression of TSC-specific genes (Cdx2, Gata3, Tfap2c, Elf5, and Tead4), XEN-specific genes (Sox17, Gata6, Gata4 Sox7, and Stra6), and PSC-specific genes (Zfp42 and Nanog). As expected, Eomes-expressing cells showed a stronger induction of Elf5 and Tead4 compared to control and Esrrb-expressing cells (Figure S4E). On the other hand, a delay in the activation of TSC-specific genes was noted in Esrrb-expressing cells, which was accompanied by a strong upregulation of XEN-specific genes (Figures 4E and S4E). Accordingly, stable XEN-like colonies and a smaller number of stable TS-like colonies were observed in Esrrb-expressing cells compared to control cells that exhibited only stable TS-like colonies (Figures S4F–S4I).
In agreement with that, ChIP-qPCR for Esrrb in ZHBTc4 cells overexpressing Esrrb revealed enhanced binding of Esrrb to XEN-specific and TSC-specific genes compared to wild-type (WT) and CRISPR-Cas9-mediated knockdown (KD) control cells (Figures 4F and 4G). Taken together, these data imply that Esrrb drives pluripotency in conjunction with GETM via activation of a XEN-like state and by controlled repression of TSC-specific genes.
Discussion
In this study, we asked whether one combination of transcription factors is capable of producing cells that are the in vitro equivalent of the ICM-Epi (i.e., iPSCs) and TE (i.e., iTSCs).
We identified a combination of 5 factors, GETMS, that can reprogram MEFs into both iPSCs and iTSCs. Although 3 or 4 factors out of these 5F combinations were sufficient to produce either iPSCs (GTMS or ETMS) or iTSCs (GET or GETM), the only combination that could produce both cell types was GETMS. We show that, out of the 3 TSC key master regulators, Gata3, Tfap2c, and Eomes, the expression of Eomes is the most influential on the outcome of the induced cells, where high levels of Eomes drive cells toward the TSC state. In contrast, Esrrb was the only factor that could shift the TSC reprogramming process exerted by GETM toward pluripotency. This is in accordance with the observation that Esrrb was shown to act downstream to Sox21, a protein that was shown to initiate the pluripotency program at the 4-cell stage (Goolam et al., 2016). These results are extremely surprising, given that Esrrb is highly expressed in the ICM and TE compartments of the embryo (Deng et al., 2014) and is essential for TSC self-renewal (Latos et al., 2015).
Our molecular analyses show that GETMS triggered pluripotency via a distinct pathway and chromatin dynamics different than OSKM. Pluripotency markers and genomic loci, such as Gdf3, that were activated by OSKM in the reprogramming process were not expressed or marked by GETMS reprogramming and vice versa.
In contrast, Esrrb in conjunction with GETM, or when overexpressed in ESC that lost Oct4 expression, activated XEN cell-specific genes that could produce stable iXENs. However, in contrast to bdXENs, GETMS-iXENs expressed very high levels of Oct4. Although the activation of a XEN-like state with high levels of Oct4 by Esrrb can explain how pluripotency is initiated, we believe that the role of Esrrb in repressing the TSC state when cells start to acquire pluripotency is at least equally important for the stabilization process. These two parallel functions of Esrrb distinguish it from other pluripotency genes and might explain why Esrrb was the only factor that could shift the TSC fate into PSC fate.
Because all five factors have been shown to be expressed to some degree in the embryo before specification (Auman et al., 2002, Home et al., 2009, McConnell et al., 2005), it is tempting to speculate that the interplay between Esrrb and Eomes is also relevant for the first cell fate decision in the embryo.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Esrrb | Perseus Proteomics | PP-H6705-00; RRID: AB_1964232 |
| H3K4me2 | Millipore | 07-030; RRID: AB_310342 |
| H3K27ac | Abcam | ab4729; RRID: AB_2118291 |
| CD-40 | Abcam | ab22469; RRID: AB_447074 |
| Nanog | Bethyl | A300-379A; RRID: AB_2632098 |
| Elf5 | Santa cruz biotechnology | sc-9645; RRID: AB_640106 |
| Gata6 | Abcam | ab22600; RRID: AB_732529 |
| Sox17 | Santa cruz biotechnology | sc-130295; RRID: AB_2286667 |
| Utf1 | Abcam | ab24273; RRID: AB_778767 |
| Cdx2 | Biogenex | CDX2-88; RRID: AB_2650531 |
| Eomes | Abcam | ab23345; RRID: AB_778267 |
| Tfap2c | Abcam | ab110635; RRID: AB_10858471 |
| Gata3 | Abcam | ab106625; RRID: AB_10887935 |
| Igg | Santa cruz biotechnology | sc-2025; RRID: AB_737182 |
| EGFP | Abcam | Ab6556; RRID: AB_305564 |
| Myc | Abcam | ab32072; RRID: AB_731658 |
| Actin | Santa cruz biotechnology | sc-1616; RRID: AB_630836 |
| Bacterial and Virus Strains | ||
| Biological Samples | N/A | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| mFgf4 | In-house production | N/A |
| mLIF | In-house production | N/A |
| Heparin | Sigma-Aldrich | H3149 |
| Recombinant human-TGFβ | PeproTech | 100-21c-10UG |
| PD0325901 | Biogems | 3911091-25MG |
| CHIR99021 | Biogems | 2520691-10MG |
| Protease inhibitor cocktail | Sigma-Aldrich | P8340 |
| SYBR green Fast qPCR Mix | Applied Biosystems | A25742 |
| Transfection Reagent | Mirus Bio | TransIT-LT1 |
| Propidium Iodide | Sigma-Aldrich | P4864 |
| β-mercaptoethanol | GIBCO | 31350-010 |
| RNaseA | Sigma-Aldrich | R6513-10MG |
| PMS hormone | AIP veterinary | 59450-02 |
| hCG hormone | CalbioChem | mbs230734 1mg |
| FHM medium | Zenith Biotech | ZEHP-050 |
| Puromycin | TOKU-E | P001 |
| Zeocin | invivoGen | ant-zn-1 |
| Neomycin | Mercury | 345812-20ml |
| Hygromycin | Invitrogen | 10687-010 |
| NP-40 | Mercury | 492018 |
| Critical Commercial Assays | ||
| adaptor-loaded Nextera Tn5 | Illumina, Fc-121-1030 | 15028212 |
| SENSE mRNA-Seq library prep kit V2 | Lexogen | k00124 |
| Deposited Data | ||
| ATAC-Seq | This study | GEO: GSE98124 |
| RNA-Seq | This study | GEO: GSE98124 |
| ChIP-Seq | This study | GEO: GSE98124 |
| Experimental Models: Cell Lines | ||
| KH2 ESCs | (Beard et al., 2006) | N/A |
| V6.5 ESCs | (Beard et al., 2006) | N/A |
| R26-tdTomato ESCs | (Cohen et al., 2018) | N/A |
| Oct4-GFP MEFs | (Lengner et al., 2007) | N/A |
| BYKE ESCs | This study | N/A |
| ZHBTc4 ESCs | (Niwa et al., 2005) | N/A |
| Recombinant DNA | ||
| FUW-TetO-Sox15 | This study | N/A |
| FUW-TetO-Klf5 | This study | N/A |
| FUW-TetO-Sox17 | This study | N/A |
| FUW-TetO-Gata6 | This study | N/A |
| FUW-TetO-Esrrb-2A-EGFP | This study | N/A |
| FUW-TetO-Eomes-2A-EGFP | This study | N/A |
| FUW-Esrrb-2A-EGFP | This study | N/A |
| FUW-Eomes-2A-EGFP | This study | N/A |
| FUW-TetO-Esrrb-2A-tdTomato | This study | N/A |
| FUW-TetO-Eomes-2A-tdTomato | This study | N/A |
| FUW-TetO-mOKSM STEMCCA | (Sommer et al., 2009) | N/A |
| FUW-TetO-Gata3 | (Benchetrit et al., 2015) | N/A |
| FUW-TetO-Tfap2c | (Benchetrit et al., 2015) | N/A |
| FUW-TetO-Eomes | (Benchetrit et al., 2015) | N/A |
| FUW-TetO-Tead4 | (Benchetrit et al., 2015) | N/A |
| FUW-TetO-Nanog | (Buganim et al., 2012) | Addgene #40800 |
| FUW-TetO-Sall4 | (Buganim et al., 2012) | N/A |
| FUW-TetO-Utf1 | (Buganim et al., 2012) | N/A |
| FUW-TetO-Lin28 | (Buganim et al., 2012) | Addgene #60345 |
| FUW-TetO-Esrrb | (Buganim et al., 2012) | Addgene #40798 |
| FUW-TetO-Oct4 | (Brambrink et al., 2008) | Addgene #20323 |
| FUW-TetO-Sox2 | (Brambrink et al., 2008) | Addgene #20326 |
| FUW-TetO-Klf4 | (Brambrink et al., 2008) | Addgene #20322 |
| FUW-TetO-Myc | (Brambrink et al., 2008) | Addgene #20324 |
| FUW-2A-EGFP | This study | N/A |
| Software and Algorithms | ||
| MATLAB | MathWorks | R2015b |
| Cufflinks (v2.0.2) | (Trapnell et al., 2010) | https://cole-trapnell-lab.github.io/cufflinks/releases/v2.0.2/ |
| Cutadapt | https://doi.org/10.14806/ej.17.1.200 | https://cutadapt.readthedocs.io/ |
| TopHat (v2.0.14) | (Trapnell et al., 2009) | https://ccb.jhu.edu/software/tophat/ |
| HtSeq-count (v0.6.0) | (Anders et al., 2015) | https://htseq.readthedocs.io/en/release_0.11.1/ |
| DESeq2 (v1.16.1) | (Love et al., 2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Bowtie | (Langmead, 2010) | http://bowtie-bio.sourceforge.net/index.shtml |
| HOMER’s annotatePeaks.pl program | (Heinz et al., 2010) | http://homer.ucsd.edu/homer/ngs/annotation.html |
| deepTools | (Ramírez et al., 2016) | https://github.com/deeptools/deepTools |
| GREAT | (McLean et al., 2010) | http://great.stanford.edupublic/html |
| MACS | (Zhang et al., 2008) | http://liulab.dfci.harvard.edu/MACS/ |
| DiffBind | (Stark and Brown, 2011) | https://bioconductor.org/packages/release/bioc/html/DiffBind.html |
| ChIPseeker | (Yu et al., 2015) | http://bioconductor.org/packages/release/bioc/html/ChIPseeker.html |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Yosef Buganim (yossibu@ekmd.huji.ac.il)
Experimental Model and Subject Details
Mice and Cell Culture
MEFs were grown in DMEM supplemented with 10%FBS, 2mM L-glutamine, and antibiotics. ESCs and iPSCs (male sex) were grown in DMEM supplemented with 10%FBS, 1%non-essential amino acids, 2mM L-glutamine, in-house mouse Leukemia inhibitory factor (mLif), 0.1mM β-mercaptoethanol (Sigma), and antibiotics with or without 2i- PD0325901 (1 μM) and CHIR99021 (3 μM) (PeproTech). BYKE ESCs or 5F-iPSCs were injected into blastocysts and at 13.5dpc embryos were isolated and MEFs were extracted and used for primary infection. All infections were performed on MEFs (passage 0 or 1) that were seeded at 60%–80% confluency two days prior to the first infection. Blastocyst-derived TSC (bdTSC) lines were isolated previously (Benchetrit et al., 2015). bdTSCs and stable iTSCs were grown in TSC culturing medium, combined of 30%TSC medium containing RPMI supplemented with 20%FBS, 0.1mM β-mercaptoethanol (Sigma), 2mML-glutamine, in-house mouse FGF4 (equivalent to 25ng/ml) and 1 μg/ml heparin (Sigma-Aldrich), and 70%MEF conditioned TSC media (MEF-CM) with the same supplements. For culturing bdTSCs and stable iTSCs in defined medium, cells were grown on GFR-Matrigel-coated dishes in TX medium as described previously (Kubaczka et al., 2014). Extraembryonic endoderm (XEN) cells and induced XENs were grown in RPMI supplemented with 15%FBS, 2mM L-glutamine, 1mM b-mercaptoethanol (sigma), in-house mouse FGF4 (equivalent to 25ng/ml) and 1 μg/ml heparin (Sigma-Aldrich). For differentiation experiment DMEM, without Fgf4 and heparin, supplemented with 10%FBS, 2mML-glutamine and antibiotics was used. The joint ethics committee (IACUC) of the Hebrew University and Hadassah Medical Center approved the study protocol for animal welfare. The Hebrew University is an AAALAC international accredited institute.
Primary MEFs and secondary MEF system production
Mouse embryonic fibroblasts (MEFs) were isolated as previously described (Wernig et al., 2008). Briefly, BYKE-ESCs, Oct4-GFP or OSKM/GETMS-iPSCs were injected into blastocysts and chimeric embryos were isolated at E13.5 (see Chimeric embryo and placenta contribution section for more details). For MEF production, embryos were dissected under the binocular removing internal organs and heads. The remaining body was chopped thoroughly by scalpels and exposed to 1ml Tripsin-EDTA (0.25%, GIBCO) for 30 minutes at 37°C. Following that, 10 mL of DMEM medium containing 10%FBS was added to the plate and the chopped tissue was subjected to thorough and intensive pipetting resulting in a relatively homogeneous mix of cells. Each chopped embryo was seeded in 15cm plate and cells were cultured in DMEM supplemented with 10%FBS, 2mM L-glutamine, and antibiotics until the plate was full. Puromycin (2 μg/ml) was added to each 15cm plate for positive selection for BYKE or Oct4-GFP MEFs (the M2rtTA cassette that resides inside the Rosa26 locus of the injected cells contains a resistance gene for puromycin), killing only the host cells.
Method Details
BYKE Quadruple Reporter System
To establish dox-inducible quadruple fluorescent knock-in reporter system, the mouse ESC line KH2 containing the dox-inducible activator M2rtTA in the Rosa26 locus and a flip-in system in the collagen (Col1a1) locus (Beard et al., 2006) was Sequentially targeted with targeting constructs either containing RBGpA or Neomycin selection cassette. RBGpA was introduced into pNTKV-frt-loxP (Dawlaty and van Deursen, 2006) vector using HpaI and HindII restriction sites using specific primers (For Sequences see Methods S1). Four different fluorescent reporters (Nanog-2A-EGFP, Utf1-2A-tdTomato, Esrrb-2A-TagBFP and Elf5-2A-EYFP-NLS) were introduced into the 3′UTR of the Nanog, Utf1, Esrrb or Elf5 genes and targeted clones were isolated either by FACS or Neomycin selection. The targeting vectors were designed by cloning the 5′arms and 3′arms using HpaI and SacII or SmaI respectively. 2A-tdTomato for Utf1 and 2A-TagBFP for Esrrb were introduced into pNTKV-RBGpA using HpaI and HindII restriction sites. gRNA for all loci was synthesized and cloned into pX330 (addgene #42230) using BbsI sites. The targeting constructs together with corresponding gRNA were transfected into the targeting cells using TransIT-LT1 Transfection Reagent (Mirus Bio) according to manufacturer’s instructions. Correctly targeted clones for Utf1 and Esrrb were selected by FACS and verified by Southern blot and PCR, respectively. For Southern blot (Utf1 locus), DNA was digested with NheI and probed for the 3′ region and PCR for Esrrb was done using primer pair A (Figures S1F and S1G). LacIq gene together with constitutively active Ubiquitin promoter (Ubb) were amplified by PCR (for primer Sequence see Methods S1) and cloned into pBS31-RBGpA (Beard et al., 2006) plasmid, using EcoRI site. The resulting construct was co-transfected together with flippase containing plasmid into KH2-NgUrEb ESCs and LacIq expressing clones were isolated following Hygromycin selection as previously described (Beard et al., 2006). The targeting construct for Elf5 locus was designed by cloning the 5′arm and 3′arm with HpaI and SacII/SmaI respectively and 2A-EYFP-NLS with HpaI and BglII into pNTKV-frt-loxP (Dawlaty and van Deursen, 2006). The positive colonies for Elf5 correctly targeted locus were picked after 10-12 day of G418 selection at 500μg/ml (Millipore) and verified by primer pair B (Figures S1J–S1L). The correctly targeted clone was transfected with Cre-expressing vector (FUW-zeo-Cre) for 10-12 days of selection of Zeocin (Invivogen) at 500μg/ml and correctly targeted clones were verified by primer pair C (Figures S1J–S1L) and Sequenced.
Molecular Cloning, Lentiviral Infection, and iTSC and iPSC reprogramming
All dox-inducible factors were generated by cloning the open reading frame of each factor, obtained by reverse transcription with specific primers (for primer Sequences see Methods S1), into the pMINI vector (NEB) and then restricted with EcoRI or MfeI and inserted into the FUW-TetO expression vector. The mouse Eomes and Esrrb-2A-EGFP or 2A-tdTomato constructs were generated by cloning the ORF of the genes (without stop codon) into FUW-TetO-2A-EGFP or FUW-TetO-2A-tdTomato expression vector with EcoRI site (for primer Sequences see Methods S1). For infection, replication-incompetent lentiviruses containing the various reprogramming factors and rations (GETM 3:3:3:1, GTMS: 3:3:1:3, GETMS 2:2.5:2:1:2.5, OSKM 3:3:3:1 or STEMCCA cassette) were packaged with a lentiviral packaging mix (7.5 μg psPAX2 and 2.5 μg pGDM.2) in 10cm plate of 293T cells and collected 48, 60, and 72 hr after transfection. The supernatants were filtered through a 0.45μm filter, supplemented with 8 μg/ml of polybrene (Sigma), and then used to infect MEFs. Six hours following the third infection, medium was replaced with fresh DMEM containing 10%FBS. Eighteen hours later, medium was replaced to either TSC reprogramming medium (RPMI supplemented with 20%FBS, 0.1mM β-mercaptoethanol, 2mM L-glutamine, in house mouse recombinant FGF4 (equivalent to 25ng/ml), 1 μg/ml heparin (Sigma-Aldrich), and 2 μg/ml doxycycline) or ESC reprogramming medium (DMEM supplemented with 10%FBS, 0.1mM β-mercaptoethanol, 2mM L-glutamine, 1%non-essential amino acids, in-house mouse Leukemia inhibitory factor (mLif), and 2 μg/ml doxycycline). TSC or ESC reprogramming medium was replaced every other day for 20 days, followed by 10 days in TSC culturing medium or 2i/L culturing medium-respectively. Ten days (or as indicated in the figure) after dox removal, plates were screened for primary iTSC or iPSC colonies. For OSKM reprogramming, excluding Figure 1 that shows reprogramming to iPSCs with separate OSKM plasmids, all OSKM reprogramming experiments were performed using STEMCCA cassette. Reprogramming efficiency was measured by FACS and colony number. For iPSC or iTSC clone isolation, a single iPSC/iTSC colony was trypsinized (0.25%), and plated in a separate well in a 6-well plate on feeder cells. Wells were followed and medium was replaced every other day for five to ten passages, until stable colonies developed.
FACS analysis
Cells were washed twice with PBS and trypsinized (0.25%) and filtered through mesh paper. Flow cytometry analysis was performed on a Beckman Coulter and analyzed using Kaluza Software. All FACS experiments were repeated at least three times, and the bar graph results are presented as a mean ± standard deviation of two biological duplicate from a typical experiment. For cell cycle analysis, GETMS-iTSCs grown in differentiation media for the indicated time points were trypsinized and fixed with 95% ice-cold ethanol. Cells were stained with propidium iodide (PI) staining solution (50 μg/ml PI [BD]; 0.1% [v/v] Triton X-100 [Sigma-Aldrich]; 0.2mg/ml RNaseA [Sigma-Aldrich] in PBS) for 30 min at room temperature. Flow cytometry analysis was performed on a Beckman Coulter and analyzed using Kaluza Software.
Chimeric embryo and placenta contribution
Blastocyst injections were performed using CB6F1 host embryos. After priming with PMSG (M.I.P. Veterinary) and hCG (Merck) hormones and mating with CB6F1 males, embryos were obtained at 3.5dpc (blastocyst stage), and then injected with 10–20 ES/iPS/iTS cells with a flat tip microinjection pipette with an internal diameter of 16 mm (Origio) in a drop of FHM medium (Zenith Biotech, ZEHP-050) covered by mineral oil. Shortly after injection, blastocysts were transferred to 2.5dpc pseudopregnant CD1/ICR females (10-15 blastocysts per female). Chimeric embryos and placentas were isolated at E13.5 and observed by fluorescent microscope (Nikon Eclipse T!).
ZHBTc4 ESCs transdifferentiation into TS-like cells
ZHBTc4 ESCs were kindly provided by Professor Austin Smith. Cells were infected with either Esrrb-2A-EGFP or Eomes-2A-EGFP and bright EGFP-positive clones were isolated and treated according to Niwa et al., (2000) to induce transdifferentiation. Briefly, cells were cultured on feeder cells in ESC medium until formation of colonies. Doxycycline (2 μg/ml) was added, and medium was changed to 70:30% MEF conditioned TSC media (MEF-CM) on day 4 after dox induction. Pellets were collected every 24 hours for 5 days, and mRNA was purified for qPCR analysis. To validate formation of stable TS-like or XEN-like colonies, the cells were passaged five times after the transdifferentiation experiment.
Hemorrhagic lesion formation
A total of 5x106 GETMS-iTSCs were resuspended in 100 μL CM containing in house mouse recombinant Fgf4 (equivalent to 25ng/ml) and 1 μg/ml heparin (Sigma-Aldrich) and injected subcutaneously into male athymic nude mice. 6-7 days later, lesions were dissected, fixed in 4% paraformaldehyde overnight, embedded in paraffin, and sectioned (4mm). Sections were stained with H&E and analyzed by a certified pathologist.
Generation of ZHBTc4 ESCs Esrrb hypomorph using CRISPR/cas9
ZHBTc4 ESCs were plated in ESC medium (without CHIR & PD). The next day, cells were transfected with 2.5μg PX330 vector containing Esrrb gRNA (for gRNA Sequences see Methods S1), and 0.5μg Puromycin resistance plasmid, using TransIT-LT1 transfection reagent (Mirus). 48 hours later, medium was changed to selective ESC medium with CHIR, PD and Puromycin (2μg/μl). Resistant colonies were picked, and Esrrb knockdown was validated by Sequencing, immunostaining, sm-FISH and Western Blot.
Quantitative PCR
Total RNA was isolated using the Macherey-Nagel kit (Ornat). 500–2000 ng of total RNA was reverse transcribed using iScript cDNA Synthesis kit (Bio-Rad). Quantitative PCR analysis was performed in duplicates using 1/100 of the reverse transcription reaction in a StepOnePlus (Applied Biosystems) with SYBR green Fast qPCR Mix (Applied Biosystems). Specific primers flanking an intron were designed for the different genes (for primer Sequences see Methods S1). All quantitative real-time PCR experiments were repeated at least three times, and the results were normalized to the expression of GAPDH and presented as a mean ± standard deviation of two duplicate runs from a typical experiment.
Immunostaining and western blot
Cells were fixed in 4% paraformaldehyde (in PBS) for 20 minutes. The cells were rinsed 3 times with PBS and blocked for 1hr with PBS containing 0.1% Triton X-100 and 5% FBS. The cells were incubated overnight with primary antibodies (1:200) in 4°C. The antibodies are: anti-Gata6 (Abcam, ab22600), anti-sox17 (Santa Cruz, sc-130295), anti-Esrrb (Perseus Proteomics, PP-H6705-00), anti-Eomes (Abcam, ab23345), anti-Cdx2 (Biogenex, CDX2-88), anti-Nanog (Bethyl, A300-379A) and anti-Elf5 (Santa Cruz, SC-9645) in PBS containing 0.1% Triton X-100 and 1%FBS (1:200 dilution). The next day, the cells were washed 3 times and incubated for 1hr with relevant (Alexa) secondary antibody in PBS containing 0.1% Triton X-100 and 1% FBS (1:500 dilution). DAPI (1:1000 dilution) was added 10 minutes before end of incubation. For western blot, cell pellets were lysed on ice in lysis buffer (20 mM Tris-HCl, pH 8, 1mM EDTA pH 8, 0.5% Nonidet P-40, 150mM NaCl, 10% glycerol, 1mM, protease inhibitors (Roche Diagnostics)) for 10 min, supernatant were collected and 40μg protein were suspended with sample buffer and boiled for or 5 min at 100Co, and subjected to western blot analysis. Primary antibodies: anti-Gata3 (Abcam, ab106625), anti-Tfap2c (Abcam, ab110635,), anti-Esrrb (Perseus Proteomics, PP-H6705-00), anti-Myc (Abcam, ab32072), anti-Eomes (Abcam, ab3345), and anti-actin (Santa cruz, Sc-1616). Blots were probed with anti-mouse, anti-goat, or anti-rabbit IgG-HRP secondary antibody (1:10,000) and visualized using ECL detection kit.
Chromatin immunoprecipitation (ChIP)
Chromatin immunoprecipitation (ChIP) assay was performed as previously described (Mendenhall et al., 2013). Briefly, cells were fixed for 10min at RT with a final formaldehyde concentration of 0.8%. Formaldehyde was quenched with glycine at a final concentration of 125mM. The cells were then lysed with lysis buffer (100mM Tris-HCl, 300mM NaCl, 2% Triton® X-100, 0.2%v sodium deoxycholate and 10mM Cacl2) supplemented with EDTA-free protease inhibitor (Roche, 11873580001) for 20min in Ice. The chromatin was digested by adding MNase (Thermo Scientific, 88216) for 20min at 37°C and MNase was inactivated by adding 20mM EGTA. The fragmented chromatin was added to pre-bounded Dynabeads (A and G mix, Invitrogen, 10004D/ 10002D) using H3K27ac antibody (Abcam, ab4729), H3K4me2 antibody (Millipore, 07-030), Esrrb antibody (Perseus Proteomics, PP-H6705-00) or Igg (santa cruz biotechnology, sc-2025). Samples were then washed twice with RIPA buffer, twice with RIPA high salt buffer (NaCl 360mM), twice with LiCl wash buffer (10mM Tris-Hcl, 250mM LiCl, 0.5% DOC, 1mM EDTA, 0.5% IGEPAL) and twice with 10mM Tris-HCl pH = 8. DNA was purified by adding RNase A (Thermo Scientific, EN0531) and incubated for 30 min at 37°C and then with Proteinase K (Invitrogen, 25530049) for 2h. The DNA was eluted by adding 2X concentrated elution buffer (10mM Tris-HCl, 300mM NaCl, 1% SDS, 2mM EDTA) and reverse crosslinked overnight at 65°C. The DNA was then extracted using AMPure XP beads (Beckman Coulter Genomics, A63881). Chip sample libraries were prepared according to Illumina Genomic DNA protocol.
RNA libraries and Sequencing
Total RNA was isolated using the QIAGEN RNeasy kit. mRNA libraries were prepared using the SENSE mRNA-Seq library prep kit V2 (Lexogen), and pooled libraries were Sequenced on an Illumina NextSeq 500 platform to generate 75-bp single-end reads.
ATAC libraries and Sequencing
ATAC-Seq library preparation was performed as previously described (Buenrostro et al., 2013). Briefly, 50,000 cells per replicate (two biological replicates per line) were incubated with 0.1% NP-40 to isolate nuclei. Nuclei were then transposed for 30 min at 37°C with adaptor-loaded Nextera Tn5 (Illumina, Fc-121-1030). Transposed fragments were directly PCR amplified and Sequenced on an Illumina NextSeq 500 platform to generate 2 × 36-bp paired-end reads.
Mapping and analysis of RNA-Seq data
Reads were mapped to the mouse genome (mm9) using TopHat (Trapnell et al., 2009) with default parameters. Expression levels were estimated in six conditions (MEFs, OSKM, GTMS, GETM, GETMS_ESM, and GETMS_TSM, in biological duplicates) using cufflinks (Trapnell et al., 2010) with default parameters. ESC and bdTSC RNA-Seq data were similarly reanalyzed from (Benchetrit et al., 2015) (GEO accession, database: GSE64684).
Principal component analysis
Gene expression levels (FPKM) for 23,309 genes were analyzed to remove genes with very low (< 10) or very high (> 1000) variance, retaining 7,446 genes. These were then standard normalized and PCA was applied using MATLAB (R2015b, “pca” function). For a closer examination of the three-day reprogramming dynamics, PCA was rerun on the same genes, using six libraries (MEFs, OSKM, GTMS, GETM, and GETMS in ESC and TSC media, in duplicates).
For transcriptional analysis of the various iPSC/ESC clones, raw reads (fastq files) were quality-trimmed and adapters removed with cutadapt (version 1.12). The processed fastq files were mapped to the mouse transcriptome and genome using TopHat (v2.0.14). The genome version was GRCm38, with annotations from Ensembl release 84. Quantification was done using htSeq-count (version 0.6.0). Genes with a sum of counts less than 10 over all samples were filtered out, retaining 21568 genes. Normalization was done with the DESeq2 package (version 1.16.1). PCA was applied and visualized in R (version 3.4.1).
Cluster analysis
Cluster analysis. Gene expression levels (FPKM) for 23,309 genes in eight RNA-Seq libraries were analyzed. Genes with FPKM lower than 2 in all eight conditions were removed, remaining with 13,309 genes. These were log2 transformed, and then multiplicatively normalized to maximal expression of 1 per gene. Genes were then clustered using spectral clustering algorithm with K = 19 (Ng et al., 2001). Briefly, this clustering algorithm computes the distance between each pair of genes, and embeds the data in a graph, whose nodes corresponds to genes and edges correspond to the similarity (or adjacency) of their expression patterns. Then, K densely connected components ( = K clusters of similarly expressed genes) are identified (using K-means, with 100 random starting points). K was selected as an optimal tradeoff between generalization (fewer clusters) and specificity – as shown in Figure 4C each cluster offers a unique expression profile.
Mapping and analysis of ATAC-Seq data
Paired-end reads were mapped using Bowtie (Langmead, 2010) to the mouse genome (mm9), using max insert size of 2000. Only unique hits with up to 3 mismatches were retained. Genome-wide paired-end read coverage was then calculated and normalized to a total primary data size of 150M bases (using UCSC’s bigWigInfo program). ATAC-Seq peaks were called using the MACS2 function bdgpeakcall (Zhang et al., 2008), with min-length and max-gap parameters set to 500 bp. The top 50K peaks were then selected for each experiment, and annotated using HOMER’s annotatePeaks.pl program (Heinz et al., 2010), using default parameters. Metagene plot was created from the ChIP-Seq bigwig files using deepTools plotProfile.
Cluster analysis of ATAC-Seq peaks
For each peak, we used UCSC’s bigWigAverageOverBed program with -minMax option to compute the maximal ATAC-Seq signal for each of the eight conditions. These peaks were then united and cluster analysis was performed as described above, with K = 18. Metagene and heatmap plots were created from the ATAC-Seq bigwig files using deepTools (Ramírez et al., 2016).
Motif analyses and functional annotations
The clustered ATAC-Seq peaks (Intronic+distal peaks among top 50K ATAC-Seq peaks, Figure 4D) were systematically analyzed for enriched transcription factor binding sites using HOMER (Heinz et al., 2010). Specifically, we compared the genomics Sequences at GETMS accessible regions against accessible Sequences from the GETM ATAC-Seq peaks as background. This was done using HOMER’s findMotifsGenome.pl program. In addition, we compared the motif enrichment of each cluster versus the overall enrichment in all other classes of intronic and distal ATAC-Seq peaks. This analysis highlighted several motifs, associated with various transcription factors including Esrrb (1e-128) and others (Table S4). We then identified the ATAC-Seq peaks that contain the Esrrb motif (HOMER’s “findMotifsGenome.pl –find esrrb.motif” option), and used GREAT to analyze the specific annotations (GO and others) enriched with those genes.
Mapping and analysis of ChIP-Seq
Paired-end reads were mapped to the genome (mm9) using Bowtie (Langmead, 2010) using max insert size of 1000, mapping unique hits with up to 3 mismatches. Genome-wide paired-end read coverage was calculated and normalized to 20M reads. ChIP-Seq peaks were called using MACS (Zhang et al., 2008), using a p value threshold of 1e-3, shift size of 2Kb, short local window of 10Kb and long local window of 50Kb. Differential peaks were identified using DiffBind version 2.4.8 (Stark and Brown, 2011). Annotation was performed using ChIPseeker version 1.12.1 (Yu et al., 2015). Data for MEF control were taken from database: GSE36292 (Chang et al., 2014).
Venn diagram and heatmap of XEN signature
Reads from our study and three studies (Li et al., 2017, Parenti et al., 2016, Zhao et al., 2015) describing a XEN-like signature (database: GSE73631, GSE77550, and GSE97721) were aligned to the Ensembl mouse genome version mm9 (NCBI37) using STAR 2.5.2b (default parameters). Uniquely aligned reads in BAM format were annotated against the protein-coding mRNA regions using SeqMonk v 1.38.2 platform (Babraham Bioinformatics, Cambridge UK). Raw counts per mRNA, strand-specific, merging isoforms were quantified using SeqMonk. Differential expression analysis was using R (version 3.4.1) and packages DESeq2 and EdgeR. The Venn diagram and heatmap were generated by comparing day 3 RNA-Seq data of MEFs, GETMS, GETM and GTMS induced cells and aligning them with genes related to OSKM-induced XEN cells (Parenti et al., 2016) or chemically-induced XEN cells. Each study was processed independently to obtain upregulated differentially expressed genes of MEF and chemically-induced XEN cells or MEF and OSKM-induced XEN cells. In our study we tried to exclude all genes that were upregulated in GETM-induced cells. For each study, upregulated differentially expressed genes identified using DESeq2 and EdgeR packages were merged as one list. A Venn diagram was constructed from the four lists to explore the overlap between the four studies. We identified 96 genes that were upregulated in our study and shared at least once with another study. The significance of the overlaps between our study and the other three studies were explored by the hypergeometric test and showed p value < 2.834E-16, 2.874E-07, and 7.835E-11.
Quantification and Statistical Analysis
For experiments comparing differences between two groups, we used unpaired Student’s t test. Differences were considered significant when p value < 0.05. All experiments were repeated at least 3 times. For quantitative PCR experiments the results were normalized to the expression of the housekeeping control gene, Gapdh and presented as a mean ± standard deviation of two duplicate runs from a typical experiment. Unless indicated otherwise a representative experiment is shown for each Figure. The statistical analyses for the high throughput analyses are depicted in the Method Details section. All
Identification of a XEN-like signature
The significance of the overlaps between our study and the other three studies were explored by the hypergeometric test and showed p value < 2.834E-16, 2.874E-07, and 7.835E-11.
P values for overlap between any two studies across all four studies.
| Our Study | Zhao et al. (2015) | Parenti et al. (2016) | Li et al. (2017) | |
|---|---|---|---|---|
| The present Study | ||||
| Zhao et al. (2015) | 2.83E-16 | |||
| Parenti et al. (2016) | 2.87E-07 | 0 | ||
| Li et al. (2017) | 7.84E-11 | 0 | 0 |
Data and Software Availability
All ATAC-Seq, ChIP-Seq and RNA-Seq were deposited in the Gene Expression Omnibus database (GEO) under accession number database: GSE98124. The authors declare no competing financial interests. Correspondence and requests for materials should be addressed to Y.B. (yossibug@ekmd.huji.ac.il).
Acknowledgments
Y.B. is supported by the European Research Council (ERC) (no. 676843), the Israeli Center of Research Excellence (I-CORE) program (center no. 41/11), the Israel Science Foundation (ISF) (no. 823/14), EMBO Young Investigator Programme (YIP), DKFZ-MOST (CA 177), and Howard Hughes Medical Institute (HHMI) (no. 55008727). A.R. is supported by BIRAX (030-5187). T.K. is supported by I-CORE program (centers no. 41/11 and no. 1796/12) and ISF, no. 913/15.
Author Contributions
Y.B. conceived the study, wrote the manuscript, and prepared the figures. H.B., M.J., and Y.B. designed the experiments. H.B. and M.J. performed cloning, reprogramming, infection, iTSC and iPSC isolation, FACS, ChIP-qPCR, RNA preparation, and immunostaining. S.S. generated RNA and ATAC libraries. T.K. analyzed all RNA-seq, ATAC-seq, and ChIP-seq data. M.J. and O.R. performed ChIP-seq. Y.B. and V.Z. built the BYKE system. K.M. performed iTSC and iPSC injections. N.M. validated Esrrb KD. N.R. cloned factors. Z.Z. characterized GETMS-iXENs. A.R. analyzed the XEN-like signature. A.P. and H.B. performed the experiments with ZHBTc4. M.I. analyzed some of the ChIP-seq and RNA-seq data.
Declaration of Interests
The authors declare no competing interests.
Published: April 25, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.stem.2019.03.018.
Supporting Citations
The following reference appears in the Supplemental Information: Faddah et al. (2013).
Supplemental Information
A table summarizing the eighteen ATAC-Seq clusters (Figure 3D) that were grouped into 9 blocks, based on their similarity, and tested for functional annotations using GREAT. The 9 groups include: (1) MEF-specific clusters (clusters #1 and #5); (2) OSKM-specific cluster (cluster #4); (3) Reprogramming and MEF-specific (cluster #2); (4) GETM/GETMS/GTMS-specific cluster (clusters #6, # 8 and #13); (5) GTMS/GETM/GETMS/bdTSC-specific cluster (cluster #10); (6) GETM/GETMS/bdTSC-specific cluster (cluster #15); (7) iPSC-specific cluster (clusters #7 and #18); (8) TSC-specific cluster (cluster #17); and (9) iPSC and TSC-specific (clusters #14 and #16).
A table summarizing the nineteen RNA-Seq clusters (Figure 3C) that were grouped into 8 blocks, based on their similarity, and tested for functional enrichments using GOrilla. The 8 blocks include: (1) MEF-specific clusters (clusters #1, #2 and #6); (2) OSKM-specific cluster (cluster #3); (3) Reprogramming and MEF-specific (cluster #4); (4) GTMS/GETM/GETMS-specific clusters (cluster #9); (5) GTMS/GETM/GETMS/bdTSC-specific cluster (cluster #11); (6) ESC-specific clusters (clusters #12 and #19); (7) TSC-specific cluster (cluster #10 and 17); and (8) ESC and TSC-specific (clusters #15, #16 and #18).
A table summarizing the functional annotation and enrichment of peaks that are located near active or inactive genes in OSKM, GETM and GETMS reprogramming combinations. Active genes are defined by FPKM value > 1 and inactive genes are defined by FPKM value < 1.
A table depicting the enriched binding motifs within GETMS-specific accessible regions against accessible Sequences from the GETM ATAC-Seq peaks as background. Enriched annotations for GETMS ATAC-Seq peaks containing the Esrrb motif (HOMER and GREAT) are shown as well.
References
- Anders S., Pyl P.T., Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auman H.J., Nottoli T., Lakiza O., Winger Q., Donaldson S., Williams T. Transcription factor AP-2gamma is essential in the extra-embryonic lineages for early postimplantation development. Development. 2002;129:2733–2747. doi: 10.1242/dev.129.11.2733. [DOI] [PubMed] [Google Scholar]
- Beard C., Hochedlinger K., Plath K., Wutz A., Jaenisch R. Efficient method to generate single-copy transgenic mice by site-specific integration in embryonic stem cells. Genesis. 2006;44:23–28. doi: 10.1002/gene.20180. [DOI] [PubMed] [Google Scholar]
- Benchetrit H., Herman S., van Wietmarschen N., Wu T., Makedonski K., Maoz N., Yom Tov N., Stave D., Lasry R., Zayat V. Extensive nuclear reprogramming underlies lineage conversion into functional trophoblast stem-like cells. Cell Stem Cell. 2015;17:543–556. doi: 10.1016/j.stem.2015.08.006. [DOI] [PubMed] [Google Scholar]
- Brambrink T., Foreman R., Welstead G.G., Lengner C.J., Wernig M., Suh H., Jaenisch R. Sequential expression of pluripotency markers during direct reprogramming of mouse somatic cells. Cell Stem Cell. 2008;2:151–159. doi: 10.1016/j.stem.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro J.D., Giresi P.G., Zaba L.C., Chang H.Y., Greenleaf W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buganim Y., Jaenisch R. Transdifferentiation by defined factors as a powerful research tool to address basic biological questions. Cell Cycle. 2012;11:4485–4486. doi: 10.4161/cc.22665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buganim Y., Faddah D.A., Cheng A.W., Itskovich E., Markoulaki S., Ganz K., Klemm S.L., van Oudenaarden A., Jaenisch R. Single-cell expression analyses during cellular reprogramming reveal an early stochastic and a late hierarchic phase. Cell. 2012;150:1209–1222. doi: 10.1016/j.cell.2012.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buganim Y., Faddah D.A., Jaenisch R. Mechanisms and models of somatic cell reprogramming. Nat. Rev. Genet. 2013;14:427–439. doi: 10.1038/nrg3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang G., Gao S., Hou X., Xu Z., Liu Y., Kang L., Tao Y., Liu W., Huang B., Kou X. High-throughput sequencing reveals the disruption of methylation of imprinted gene in induced pluripotent stem cells. Cell Res. 2014;24:293–306. doi: 10.1038/cr.2013.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Wang D., Wu Z., Ma L., Daley G.Q. Molecular basis of the first cell fate determination in mouse embryogenesis. Cell Res. 2010;20:982–993. doi: 10.1038/cr.2010.106. [DOI] [PubMed] [Google Scholar]
- Cho L.T., Wamaitha S.E., Tsai I.J., Artus J., Sherwood R.I., Pedersen R.A., Hadjantonakis A.K., Niakan K.K. Conversion from mouse embryonic to extra-embryonic endoderm stem cells reveals distinct differentiation capacities of pluripotent stem cell states. Development. 2012;139:2866–2877. doi: 10.1242/dev.078519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen M.A., Markoulaki S., Jaenisch R. Matched developmental timing of donor cells with the host is crucial for chimera formation. Stem Cell Reports. 2018;10:1445–1452. doi: 10.1016/j.stemcr.2018.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawlaty M.M., van Deursen J.M. Gene targeting methods for studying nuclear transport factors in mice. Methods. 2006;39:370–378. doi: 10.1016/j.ymeth.2006.06.009. [DOI] [PubMed] [Google Scholar]
- Deng Q., Ramsköld D., Reinius B., Sandberg R. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science. 2014;343:193–196. doi: 10.1126/science.1245316. [DOI] [PubMed] [Google Scholar]
- Faddah D.A., Wang H., Cheng A.W., Katz Y., Buganim Y., Jaenisch R. Single-cell analysis reveals that expression of nanog is biallelic and equally variable as that of other pluripotency factors in mouse ESCs. Cell Stem Cell. 2013;13:23–29. doi: 10.1016/j.stem.2013.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goolam M., Scialdone A., Graham S.J.L., Macaulay I.C., Jedrusik A., Hupalowska A., Voet T., Marioni J.C., Zernicka-Goetz M. Heterogeneity in Oct4 and Sox2 targets biases cell fate in 4-cell mouse embryos. Cell. 2016;165:61–74. doi: 10.1016/j.cell.2016.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammoud S.S., Low D.H., Yi C., Carrell D.T., Guccione E., Cairns B.R. Chromatin and transcription transitions of mammalian adult germline stem cells and spermatogenesis. Cell Stem Cell. 2014;15:239–253. doi: 10.1016/j.stem.2014.04.006. [DOI] [PubMed] [Google Scholar]
- Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Home P., Ray S., Dutta D., Bronshteyn I., Larson M., Paul S. GATA3 is selectively expressed in the trophectoderm of peri-implantation embryo and directly regulates Cdx2 gene expression. J. Biol. Chem. 2009;284:28729–28737. doi: 10.1074/jbc.M109.016840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaber M., Sebban S., Buganim Y. Acquisition of the pluripotent and trophectoderm states in the embryo and during somatic nuclear reprogramming. Curr. Opin. Genet. Dev. 2017;46:37–43. doi: 10.1016/j.gde.2017.06.012. [DOI] [PubMed] [Google Scholar]
- Kubaczka C., Senner C., Araúzo-Bravo M.J., Sharma N., Kuckenberg P., Becker A., Zimmer A., Brüstle O., Peitz M., Hemberger M., Schorle H. Derivation and maintenance of murine trophoblast stem cells under defined conditions. Stem Cell Reports. 2014;2:232–242. doi: 10.1016/j.stemcr.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubaczka C., Senner C.E., Cierlitza M., Araúzo-Bravo M.J., Kuckenberg P., Peitz M., Hemberger M., Schorle H. Direct induction of trophoblast stem cells from murine fibroblasts. Cell Stem Cell. 2015;17:557–568. doi: 10.1016/j.stem.2015.08.005. [DOI] [PubMed] [Google Scholar]
- Langmead B. Aligning short sequencing reads with Bowtie. Curr. Protoc. Bioinformatics. 2010;Chapter 11 doi: 10.1002/0471250953.bi1107s32. Unit 11.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latos P.A., Goncalves A., Oxley D., Mohammed H., Turro E., Hemberger M. Fgf and Esrrb integrate epigenetic and transcriptional networks that regulate self-renewal of trophoblast stem cells. Nat. Commun. 2015;6:7776. doi: 10.1038/ncomms8776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengner C.J., Camargo F.D., Hochedlinger K., Welstead G.G., Zaidi S., Gokhale S., Scholer H.R., Tomilin A., Jaenisch R. Oct4 expression is not required for mouse somatic stem cell self-renewal. Cell Stem Cell. 2007;1:403–415. doi: 10.1016/j.stem.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Liu D., Ma Y., Du X., Jing J., Wang L., Xie B., Sun D., Sun S., Jin X. Direct reprogramming of fibroblasts via a chemically induced XEN-like state. Cell Stem Cell. 2017;21:264–273.e7. doi: 10.1016/j.stem.2017.05.019. [DOI] [PubMed] [Google Scholar]
- Lin S.C., Wani M.A., Whitsett J.A., Wells J.M. Klf5 regulates lineage formation in the pre-implantation mouse embryo. Development. 2010;137:3953–3963. doi: 10.1242/dev.054775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luoh S.W., Bain P.A., Polakiewicz R.D., Goodheart M.L., Gardner H., Jaenisch R., Page D.C. Zfx mutation results in small animal size and reduced germ cell number in male and female mice. Development. 1997;124:2275–2284. doi: 10.1242/dev.124.11.2275. [DOI] [PubMed] [Google Scholar]
- McConnell J., Petrie L., Stennard F., Ryan K., Nichols J. Eomesodermin is expressed in mouse oocytes and pre-implantation embryos. Mol. Reprod. Dev. 2005;71:399–404. doi: 10.1002/mrd.20318. [DOI] [PubMed] [Google Scholar]
- McDonald A.C., Biechele S., Rossant J., Stanford W.L. Sox17-mediated XEN cell conversion identifies dynamic networks controlling cell-fate decisions in embryo-derived stem cells. Cell Rep. 2014;9:780–793. doi: 10.1016/j.celrep.2014.09.026. [DOI] [PubMed] [Google Scholar]
- McLean C.Y., Bristor D., Hiller M., Clarke S.L., Schaar B.T., Lowe C.B., Wenger A.M., Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendenhall E.M., Williamson K.E., Reyon D., Zou J.Y., Ram O., Joung J.K., Bernstein B.E. Locus-specific editing of histone modifications at endogenous enhancers. Nat. Biotechnol. 2013;31:1133–1136. doi: 10.1038/nbt.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng A.Y., Jordan M.I., Weiss Y. Proceedings of the 14th International Conference on Neural Information Processing Systems: Natural and Synthetic. MIT; 2001. On spectral clustering: analysis and an algorithm; pp. 849–856. [Google Scholar]
- Niwa H., Miyazaki J., Smith A.G. Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat. Genet. 2000;24:372–376. doi: 10.1038/74199. [DOI] [PubMed] [Google Scholar]
- Niwa H., Toyooka Y., Shimosato D., Strumpf D., Takahashi K., Yagi R., Rossant J. Interaction between Oct3/4 and Cdx2 determines trophectoderm differentiation. Cell. 2005;123:917–929. doi: 10.1016/j.cell.2005.08.040. [DOI] [PubMed] [Google Scholar]
- Parenti A., Halbisen M.A., Wang K., Latham K., Ralston A. OSKM induce extraembryonic endoderm stem cells in parallel to induced pluripotent stem cells. Stem Cell Reports. 2016;6:447–455. doi: 10.1016/j.stemcr.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F., Ryan D.P., Grüning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dündar F., Manke T. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44(W1):W160–W165. doi: 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer C.A., Stadtfeld M., Murphy G.J., Hochedlinger K., Kotton D.N., Mostoslavsky G. Induced pluripotent stem cell generation using a single lentiviral stem cell cassette. Stem Cells. 2009;27:543–549. doi: 10.1634/stemcells.2008-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark R., Brown G. 2011. DiffBind: differential binding analysis of ChIP-Seq peak data.http://bioconductor.org/packages/release/bioc/vignettes/DiffBind/inst/doc/DiffBind.pdf.other [Google Scholar]
- Takahashi K., Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Thomas K., Wu J., Sung D.Y., Thompson W., Powell M., McCarrey J., Gibbs R., Walker W. SP1 transcription factors in male germ cell development and differentiation. Mol. Cell. Endocrinol. 2007;270:1–7. doi: 10.1016/j.mce.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Trapnell C., Pachter L., Salzberg S.L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J., Salzberg S.L., Wold B.J., Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernig M., Lengner C.J., Hanna J., Lodato M.A., Steine E., Foreman R., Staerk J., Markoulaki S., Jaenisch R. A drug-inducible transgenic system for direct reprogramming of multiple somatic cell types. Nat. Biotechnol. 2008;26:916–924. doi: 10.1038/nbt1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G., Schöler H.R. Lineage segregation in the totipotent embryo. Curr. Top. Dev. Biol. 2016;117:301–317. doi: 10.1016/bs.ctdb.2015.10.014. [DOI] [PubMed] [Google Scholar]
- Wu S., Hu Y.C., Liu H., Shi Y. Loss of YY1 impacts the heterochromatic state and meiotic double-strand breaks during mouse spermatogenesis. Mol. Cell. Biol. 2009;29:6245–6256. doi: 10.1128/MCB.00679-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Du Y., Deng H. Direct lineage reprogramming: strategies, mechanisms, and applications. Cell Stem Cell. 2015;16:119–134. doi: 10.1016/j.stem.2015.01.013. [DOI] [PubMed] [Google Scholar]
- Yu G., Wang L.G., He Q.Y. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics. 2015;31:2382–2383. doi: 10.1093/bioinformatics/btv145. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y., Zhao T., Guan J., Zhang X., Fu Y., Ye J., Zhu J., Meng G., Ge J., Yang S. A XEN-like state bridges somatic cells to pluripotency during chemical reprogramming. Cell. 2015;163:1678–1691. doi: 10.1016/j.cell.2015.11.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A table summarizing the eighteen ATAC-Seq clusters (Figure 3D) that were grouped into 9 blocks, based on their similarity, and tested for functional annotations using GREAT. The 9 groups include: (1) MEF-specific clusters (clusters #1 and #5); (2) OSKM-specific cluster (cluster #4); (3) Reprogramming and MEF-specific (cluster #2); (4) GETM/GETMS/GTMS-specific cluster (clusters #6, # 8 and #13); (5) GTMS/GETM/GETMS/bdTSC-specific cluster (cluster #10); (6) GETM/GETMS/bdTSC-specific cluster (cluster #15); (7) iPSC-specific cluster (clusters #7 and #18); (8) TSC-specific cluster (cluster #17); and (9) iPSC and TSC-specific (clusters #14 and #16).
A table summarizing the nineteen RNA-Seq clusters (Figure 3C) that were grouped into 8 blocks, based on their similarity, and tested for functional enrichments using GOrilla. The 8 blocks include: (1) MEF-specific clusters (clusters #1, #2 and #6); (2) OSKM-specific cluster (cluster #3); (3) Reprogramming and MEF-specific (cluster #4); (4) GTMS/GETM/GETMS-specific clusters (cluster #9); (5) GTMS/GETM/GETMS/bdTSC-specific cluster (cluster #11); (6) ESC-specific clusters (clusters #12 and #19); (7) TSC-specific cluster (cluster #10 and 17); and (8) ESC and TSC-specific (clusters #15, #16 and #18).
A table summarizing the functional annotation and enrichment of peaks that are located near active or inactive genes in OSKM, GETM and GETMS reprogramming combinations. Active genes are defined by FPKM value > 1 and inactive genes are defined by FPKM value < 1.
A table depicting the enriched binding motifs within GETMS-specific accessible regions against accessible Sequences from the GETM ATAC-Seq peaks as background. Enriched annotations for GETMS ATAC-Seq peaks containing the Esrrb motif (HOMER and GREAT) are shown as well.
Data Availability Statement
All ATAC-Seq, ChIP-Seq and RNA-Seq were deposited in the Gene Expression Omnibus database (GEO) under accession number database: GSE98124. The authors declare no competing financial interests. Correspondence and requests for materials should be addressed to Y.B. (yossibug@ekmd.huji.ac.il).