

Unprecedented reorganization in a butterfly genome sheds light on the enigma of holocentric chromosome evolution.
Abstract
Chromosome evolution presents an enigma in the mega-diverse Lepidoptera. Most species exhibit constrained chromosome evolution with nearly identical haploid chromosome counts and chromosome-level gene collinearity among species more than 140 million years divergent. However, a few species possess radically inflated chromosomal counts due to extensive fission and fusion events. To address this enigma of constraint in the face of an exceptional ability to change, we investigated an unprecedented reorganization of the standard lepidopteran chromosome structure in the green-veined white butterfly (Pieris napi). We find that gene content in P. napi has been extensively rearranged in large collinear blocks, which until now have been masked by a haploid chromosome number close to the lepidopteran average. We observe that ancient chromosome ends have been maintained and collinear blocks are enriched for functionally related genes suggesting both a mechanism and a possible role for selection in determining the boundaries of these genome-wide rearrangements.
INTRODUCTION
While the role of genomic evolution in the micro- and macroevolutionary dynamics of metazoans has received renewed attention, via the growing number of studies revealing large-scale chromosomal inversions associated with adaptive phenotypes (1, 2), our understanding of the selection dynamics acting upon chromosome number and gene order remains limited. Butterflies and moths comprise nearly 10% of all described species (3) and inhabit diverse niches with varied life histories, yet they exhibit notable conservation in their genome architecture despite 140 million generations (4) of divergence. The vast majority have a haploid chromosome number between 28 and 32 (4–6) with a mode of 31 in Ditrysia (4, 7, 8), suggesting that this is the ancestral count of the clade. Within chromosomes, the gene content and order are remarkably similar among divergent species as adduced by three previous chromosome level assemblies (4, 7), bacterial artificial chromosome sequencing, and chromosomal structure analyses (9, 10). Within this highly conserved gene order, a small number of chromosomal fusion events have been identified, while at most 6% of orthologs have been identified as translocated between synteny blocks (4, 7). Complicating this picture of conservation, haploid chromosome counts in species of Lepidoptera, as compared to all nonpolyploid animals, exhibit the highest variance in number between species within a genus [n = 5 to 226 (11–13)], the highest single count (n = 226) (14), and polymorphism in counts that do not affect fertility in crosses (15–17). Lepidoptera tolerate such chromosomal variation owing to their holokinetic chromosomes, which facilitate the successful inheritance of novel fission or fusion fragments (17, 18). Thus, while Lepidoptera can avoid the deleterious consequences of large-scale bouts of chromosomal fission and fusion, in the vast majority of cases, these events are only found in young clades, suggesting an evolutionary cost of chromosomal rearrangement.
RESULTS
Genome assembly and rearrangement identification
We generated our Pieris napi (Linnaeus, 1758) (Pieridae) genome using DNA from inbred siblings from Sweden, variable fragment size libraries [180 base pairs (bp) to 100 kb, N50 length of 4.2 Mb, and a total length of 350 Mb; notes S1 and S2], and a high-density linkage map (https://github.com/JasonAnthonyHill/Piers_napi) computed using 275 full-sib larvae, which placed 122 scaffolds into 25 linkage groups, consistent with previous karyotyping of P. napi (19, 20) The total chromosome level assembly was 299 Mb, comprising 85% of the total assembly size and 114% of the k-mer estimated haploid genome size, with 2943 scaffolds left unplaced (note S1). Unplaced scaffolds either were too small to have informative markers or were alternatively assembled haplotypes whose sequence was contained within a larger scaffold (note S1). Subsequent annotation predicted 13,622 gene models with 9346 functional predictions (note S4), and 94% of expected single-copy genes [Benchmarking Universal Single-Copy Orthologs (BUSCOs)] were found complete (note S1).
The content and structure of the P. napi genome were compared with the available chromosome level assemblies of the Lepidoptera: the silk moth Bombyx mori (Bombycidae), the postman butterfly Heliconius melpomene (Nymphalidae), and the Glanville fritillary Melitaea cinxia (Nymphalidae). These latter three species exhibit extensive gene collinearity along their chromosomes that is maintained even after chromosomal fusion and fission events, readily reflecting the history of these few events (7) (Fig. 1). After identifying the shared single-copy orthologs (SCOs) among these four species (note S5), we placed our results into this comparative chromosomal context. Unexpectedly, nearly every P. napi chromosome was uniquely reorganized on the scale of megabase in what appeared to be the result of a massive bout of translocation events (Fig. 1A). A detailed comparison of the size and number of these rearrangements was then made between P. napi and B. mori, as the latter has a high-quality genome and a haploid chromosome count (n = 28) close to the lepidopteran mode of n = 31 (8). Using shared SCOs between these two species, 99 well-defined blocks of collinear gene order (hereafter referred to as “collinear blocks”) were identified, with each collinear block having an average of 69 SCOs. Each P. napi chromosome contained an average of 3.96 (SD = 1.67) collinear blocks, which were derived from an average of 3.5 different B. mori chromosomes. In P. napi, the average collinear block length was 2.82 Mb (SD = 1.97 Mb) and contained an average of 118 genes in our annotation (SD = 87). Some regions of the genome contained no identifiable syntenic relationship to B. mori, and 2037 of the 13,622 genes in our annotation lie in regions not assigned to a collinear block. Even the Z chromosome of P. napi (chromosome 1), which contains the complete B. mori Z chromosome, has undergone rearrangement, resulting in a fusion between the ancestral Z chromosome and a fragment of the autosome corresponding to chromosome 2 in B. mori. The Z chromosome is especially conserved within Lepidoptera, but sex chromosome autosome fusions have been observed before (21, 22).
Fig. 1. Chromosomal mapping of the moth B. mori (Bombycoidea) to the butterflies P. napi (Pieridae), H. melpomene, and M. cinxia (Nymphalidae).

(A) SCOs connecting B. mori to P. napi (n = 2354), M. cinxia (n = 2086), and H. melpomene (n = 2771). The Z chromosome is chromosome 1 in B. mori and P. napi and chromosome 21 in H. melpomene. Links between orthologs originate from B. mori and are colored by their chromosome of origin, and then extend to P. napi (colored green), M. cinxia (colored orange), and H. melpomene chromosomes (colored blue). Links are clustered into blocks of synteny, forming colored ribbons that represent a contiguous block of genes spanning a region in both species. Chromosomes 2 to 25 in P. napi are ordered in size from largest to smallest. (B) SCOs between the two largest autosomes of P. napi and the other Lepidoptera highlight the former’s derived chromosomal translocation events. Here, band width for each species is proportional to the length of the inferred chromosomal region of orthology with white space indicating regions with homology to other chromosomes not shown. Individual chromosomes are not to scale.
This unanticipated genomic reorganization was validated using four complementary but independent approaches. First, since recombination is suppressed in females and thus all maternally derived markers within a chromosome will show complete linkage with each other in her offspring, we generated a maternal marker map for P. napi, using a separate family from that used in the linkage map (note S6). This maternal marker map revealed extensive linkage among all the scaffolds assigned to a given chromosome, but not with any of the other scaffolds, thereby validating the placement of scaffolds within linkage groups (Fig. 2, B and C, and fig. S1). Second, the relative order of scaffolds within chromosomes can be assessed by quantifying support for the regions where scaffolds are joined together, end to end along chromosomes in our assembly (these are hereafter referred to as scaffold joins). Since our mate pair (MP) reads with 7- and 40-kb insert sizes should cross many of these scaffolds joins, we quantified the number of uniquely mapped and properly oriented MP reads spanning each base pair position along chromosomes. Of the 97 scaffold join regions within chromosomes, 74 were crossed by at least 50 MP reads each (Fig. 2A, fig. S1, and note S7) and thereby validating their ordering and orientation along chromosomes. Third, these scaffold joins were further assessed by aligning the scaffolds of a recently published, high-quality genome of the closely related species Pieris rapae (23) to our P. napi genome. By identifying P. rapae scaffolds that crossed the aforementioned scaffold joins within the chromosomes of P. napi, we found support for 71 of the 97 scaffold joins (fig. S2 and note S8). Fourth, we investigated the identified collinear blocks of SCO genes between P. napi and B. mori in relation to the scaffold join regions along chromosomes. The P. napi scaffold joins are not associated with the boundaries of the reorganized B. mori collinear blocks. After identifying the B. mori collinear blocks that spanned scaffold joins within P. napi chromosomes, we found support for 62 of the 97 scaffold joins in P. napi (Fig. 2D, fig. S1, and note S9). In sum, our linkage map assignment of scaffolds to chromosomes was confirmed by maternal markers, and the assignment of relative ordering and orientation of scaffolds along chromosomes was confirmed by investigating the 97 regions where these scaffolds were joined together. Of these 97 scaffold joins, 96 were supported by at least two of the following: MP spanning of scaffold joins, aligned P. rapae scaffolds spanning scaffold joins of P. napi, and collinear blocks of SCO genes between B. mori and P. napi extending across scaffold joins. The one exception was the join between scaffold 2 and the small scaffold 84 at the end of chromosome 20, which only had linkage map and maternal marker support but contained no identified SCOs, MP spanning support, or genome alignment results.
Fig. 2. Validation of the largest four P. napi chromosomes.
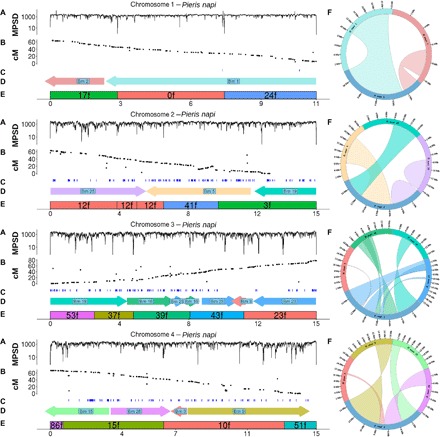
Within each, the x axis indicates physical distance in megabase. (A) Mate pair spanning depth (MPSD) is shown across each chromosome, summed from the 3-, 7-, and 40-kb libraries (genome average = 1356). Of the scaffold join positions, 74 of 97 were each spanned by >50 properly paired reads (mean = 117.8; SD = 298.7), while the remaining 23 scaffold joins had 0 MP spans. (B) Black dots represent the linkage map markers [restriction site-associated DNA sequencing (RAD-seq) data] and their recombination distance in centimorgans (cM) along chromosomes (i.e., this is the linkage map). (C) Results from the female marker map of maternally inherited markers (RNA sequencing and whole-genome data), where all markers within a chromosome are completely linked owing to suppressed recombination in females (thus, recombination distance is not shown in the y axis). (D) B. mori collinear blocks, colored and labeled by their chromosomal origin, as in Fig. 1A, along with orientation in B. mori indicated by an arrow with its head toward the nearest telomere. (E) P. napi scaffolds comprising each chromosome, labeled to indicate scaffold number and orientation. (F) To the right of each P. napi chromosome is a Circos plot showing the location and orientation of the collinear blocks from each B. mori donor chromosome that comprise a given P. napi chromosome, colored as in Fig. 1A. A twist in the ribbon indicates a reversal of the 5′ to 3′ orientation of the B. mori relative to the P. napi chromosomes. Ribbon width on the P. napi chromosome is relative to the size of the collinear block. The remaining chromosomes are shown in fig. S1.
Comparisons of genome organization within Lepidoptera
We next compared the P. napi chromosomal structure to the additional genomes available in the Lepidoptera (n = 20) to assess when the genome reorganization of P. napi might have taken place. Like most eukaryotic genomes, most lepidopteran genome assemblies are not at the chromosomal level, complicating comparative assessments of chromosomal structure (Fig. 3A). To overcome this limitation, we queried each scaffold of each of these genomes for SCOs that were shared with B. mori and P. napi and then quantified whether the scaffold supported the chromosomal structure of B. mori or P. napi, or both (which is expected when scaffolds are short and within the identified collinear blocks; fig. S3). While all non-Pieris species have many scaffolds supporting B. mori–like chromosomes and rarely supporting P. napi–like chromosomes, two recently published, independent assemblies of the congener P. rapae (23, 24) exhibited many P. napi–like scaffolds (Fig. 3B), suggesting that this rearrangement may be shared across Pieris. We further examined how different stages of our P. napi assembly captured these rearrangements and found an increase in number of rearrangements observed with increasing assembly contiguity (N50; Fig. 3B). We note that our analysis here only interrogated genomes for their support for either B. mori– or P. napi–like chromosomal structure, as future work will investigate to what extent similar approaches can be used upon these datasets to detect other types and rates of genome reorganization.
Fig. 3. Comparative assessment of genome assemblies and chromosomal evolution across the Lepidoptera.
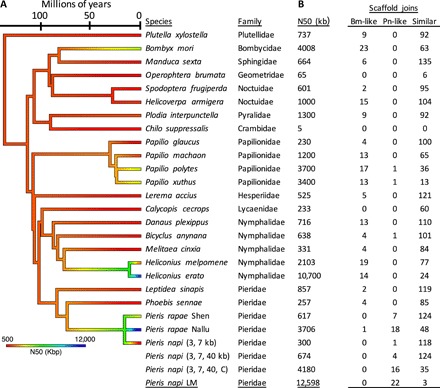
(A) A time-calibrated phylogeny of the currently available lepidopteran genomes (n = 24), with branches colored by the N50 of their assemblies and time in million years ago. (B) Table of the genome assembly N50 for each species, followed by estimates of their chromosomal similarity relative to B. mori versus P. napi. In each scaffold of each genome, SCOs were identified that were shared with B. mori and P. napi. Then, we quantified the number of times a scaffold contained SCOs derived from >1 chromosome of B. mori, but from a single P. napi chromosome (Pnapi-like scaffold), or vice versa (Bmori-like scaffold), or both from one chromosome (similar) (fig. S3 and note S9). Three additional P. napi genomes are included, representing the original Allpaths assembly that used only 3- and 7-kb MP libraries [Pieris napi (3, 7 kb)], the Allpaths assembly after an additional round of scaffolding with 40-kb MP libraries [Pieris napi (3, 7, and 40 kb)], and then the 40-kb scaffolded assembly scaffolded a third time and error-corrected with HiC libraries and the HiRise pipeline [Pieris napi (3, 7, and 40 kb)]. Higher Pn-like values indicate support for a P. napi–like chromosomal structure (see note S11 for more details). For a complementary BLAST-like approach, see fig. S9.
Last, we expected the extensive genomic reorganization observed within Pieris to be associated with taxa recently experiencing chromosomal instability, such as fission and fusion events. However, the evolutionary history of haploid chromosome count in the family Pieridae is unknown. Therefore, we integrated for the first time nearly 200 species-level observations of haploid chromosomal count (6, 25) with a temporally calibrated evolutionary phylogeny (25). The haploid chromosomal count of P. napi and P. rapae, which ranges from 25 to 28, is essentially identical to all but one species in their clade going back to a common ancestor ~30 million years divergent; all species have counts between 24 and 29, with the vast majority at 26 with only one species markedly different, Pieris brassicae, at n = 14 (26) (Fig. 4). Thus, chromosome number alone suggests a similar chromosome structure of P. napi to other known lepidopteran genomes, but this similarity masks the underlying extensive rearrangement that we observe. In addition, within collinear blocks, the synteny of genes is conserved, also masking the rearrangement that occurs at the megabase scale. Only by looking for gene collinearity in a chromosomal context is this previously unobserved pattern of genomic rearrangement revealed.
Fig. 4. Haploid chromosomal evolution across Pieridae.
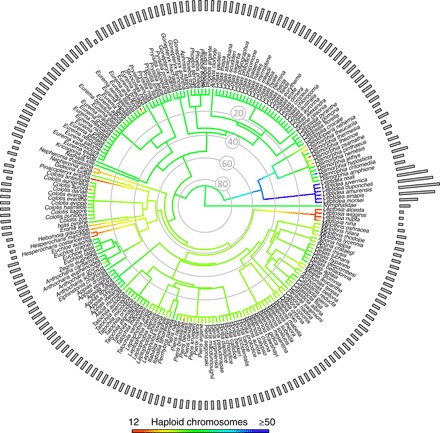
Shown in the center is a genus-level phylogeny for Pieridae, colored by the ancestral state reconstruction of the chromosomal fusion and fission events giving rise to the haploid chromosome counts of the taxa, whose values are represented as a bar plot (n = 201 species). As only a time-calibrated genus-level phylogeny exists for Pieridae, all genera with >1 species were set to an arbitrary polytomy at 5 million years ago, while deeper branches reflect calibrated nodes. The outgroup is set to n = 31, reflecting the butterfly chromosomal mode. Note that while the color scale stops at 50, members of the Leptidea clade exceed this value.
Functional enrichment within collinear blocks
To further assess this unexpected chromosomal reorganization, we inspected the ordering and content of the collinear blocks that constitute the P. napi chromosomes. To investigate the most likely mechanism of genomic reorganization, we tested whether the extensive chromosomal reshuffling in P. napi maintained the traditional chromosomal ends of Lepidoptera as would be expected if the underlying mechanism was reciprocal translocations, or if the chromosomal ends of P. napi were novel, which would be the result of a process of random fission and fusion (Fig. 5A). We found that 38 of the 56 chromosomal ends in B. mori are located within the interior of P. napi chromosomes, which would remarkably require the generation of novel telomeres in the reorganized chromosomes for all but 18 chromosomal ends. We compared this observation of 18 ancestral ends to an expectation distribution generated by randomly combining the observed collinear blocks into simulated chromosomes of approximate size to the observed genome and counting the conserved chromosomal ends relative to B. mori. This revealed evidence of significantly more chromosomal ends in common between B. mori and P. napi in the observed genome than random chance (P < 0.01, two tailed t test; Fig. 5C). We also identified a significant enrichment for SCOs in B. mori and P. napi located at a roughly similar distance from the end of their respective chromosomes (fig. S4). We also reconstructed optimal scenarios of rearrangement events among the 99 collinear blocks identified in B. mori and P. napi, with estimated optimal scenarios between them finding between 47 and 57 translocations, three to six inversions, and always four fission events, depending on scenario settings (note S12). These three findings are consistent with the ongoing use of ancestral chromosome ends, suggesting that the chromosomal reorganization proceeded via multiple rounds of reciprocal translocations rather than extensive fission and fusion events.
Fig. 5. Comparison of gene content and chromosomal location of collinear blocks between P. napi and B. mori in observed and randomly permuted genomes.
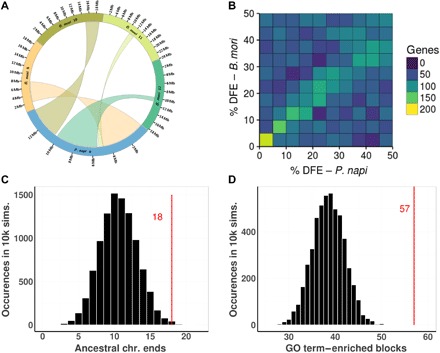
(A) Observed pattern of conserved collinear block location within P. napi chromosome 9, wherein telomere-facing and interior origins of the syntenic blocks are conserved between species despite their reshuffling. (B) Percentage distance from the end (DFE) of SCOs in P. napi versus B. mori chromosomes. Counts binned on the color axis. (C) Comparisons of the number of collinear blocks that are terminal in both P. napi and B. mori chromosomes (n = 18, red line), compared to 10,000 simulated genomes composed of randomly joined collinear blocks that would be expected in a fission/fusion scenario (histogram: average = 10.7; SD = 6.8). (D) Distribution of number of syntenic blocks with GO term enrichment in 10,000 simulated genomes. Simulated syntenic blocks were constructed by breaking the P. napi genome into blocks of the same size as observed but in a random order. The mean number of GO-enriched fragments in each of the simulated 10,000 genomes was 38.8 with a variance of 46.6 and maximum of 52. This is significantly lower than the observed 57 enriched regions in P. napi (P < 0.0001).
Second, we tested the hypothesis that the boundaries of the rearranged collinear blocks were determined randomly relative to the function of the genes in a given region. While there are few examples of polycistronic operons in eukaryotes (27), gene order is nonrandom across diverse eukaryotes, though the effect appears weak and diffuse (28) and thus some degree of functional enrichment is expected within any arbitrary collinear block. We generated an expectation distribution by randomly partitioning the genome into collinear blocks of the same size as in the observed genome but at random places, followed by testing of gene set functional enrichment within these collinear blocks. We used the full set of annotated P. napi genes and found, on average, 38.8 (variance of 46.6 and maximum of 52) collinear blocks in each of the 10,000 simulated genomes that contained at least three genes with a shared gene ontology (GO) term that was significantly less frequent in the rest of the genome (P < 0.01, Fisher’s exact test). The observed number of these GO-enriched collinear blocks (57 of the 99) was significantly higher than in the simulated genomes (P < 0.0001) (Fig. 5D). Since the actual P. napi genome and each of the 10,000 simulated genomes underwent the same number of tests resulting in the same false discovery rate of GO enrichment, false discovery rate correction was not used, nor was it necessary.
Lack of repetitive element enrichment
Transposable elements such as retrotransposons are known to affect chromosomal reorganization (29, 30). To assess their possible role in the Pieris genome reorganization, we surveyed the distribution of different repeat element classes across the genome, looking for enrichment of specific categories near the borders of collinear blocks (i.e., shuffled chromosomal fragments). However, no repeat elements were enriched at the boundary of B. mori collinear regions (fig. S5). We therefore investigated whether any repeat element classes had expanded within Pieris compared to other sequenced genomes by assessing the distribution of repeat element classes and genome size among sequenced Lepidoptera genomes. Consistent with previous findings (31), we observe a strong relationship between genome size and repetitive element content in Pieris species, though no element classes appear enriched within this lineage. Thus, the lack of evidence for any enrichment of repeats at the boundaries between the collinear blocks suggests that the translocation events may be old and their signal decayed, though detecting these signals is also notoriously difficult.
DISCUSSION
The chromosomal organization of P. napi and P. rapae reported here implies that their ancestral lineage underwent a rampant series of chromosomal rearrangements, likely dominated by reciprocal translocation events. Although changes in haploid chromosomal count via fission and fusion events are well documented in Lepidoptera (5, 6, 10, 14, 32), and the Pieridae in particular (10, 15), translocation events appear to be rare and dominated by observations of autosomal fusions to sex chromosomes (10, 21, 33). However, this likely reflects a bias in karyotype detection, as reciprocal translocations can remain cryptic in cytological spreads while fissions and fusions can be readily detected when they result in a change in chromosome size and count. To what extent chromosomal rearrangements via reciprocal translocations exist among additional taxa and lineages will have to be addressed by future studies, as the vast majority of lepidopteran genomes lack sufficient resolution to address these issues. Of perhaps greater interest, the rearranged chromosomal fragments do not appear to have been entirely random, as we detect an excess of functional gene clusters as compared to random fragments. This suggests that these clusters might have a potential fitness advantage, perhaps arising because of the benefits of shared chromatin dynamics for similar gene expression patterns (34). While it is tempting to envision nearly neutral selection pressures acting upon these aspects of chromosomal structure, varying over time because of changes in effective population size, such speculation awaits future study as understanding these patterns and selection dynamics is in its infancy. It must also be emphasized that the models of randomness that were used to detect the significant deviations of GO enrichment and chromosome end conservation may not account for biological processes that constrain the unknown mechanism of genomic restructuring observed here in P. napi. Nevertheless, our results further document the enigma of lepidopteran chromosome evolution, highlighting the need to further investigate the factors shaping and maintaining the genomic organization in this ecologically important and diverse clade of holocentic species.
MATERIALS AND METHODS
DNA sequencing and genome scaffold assembly
Pupal DNA was isolated from a fourth-generation inbred cohort that originated from a wild-caught female collected in Skåne, Sweden, using standard salt extraction (35). Illumina sequencing was used for all data generation used in genome construction. One paired end (PE) and the two MP libraries were constructed at the Science for Life Laboratory (SciLifeLab), the National Genomics Infrastructure, Sweden, using one polymerase chain reaction–free PE DNA library (180 bp) and two Nextera MP libraries (3 and 7 kb) all from a single individual. All sequencing was done on Illumina HiSeq 2500 High Output mode, PE 2 × 100-bp by SciLifeLab. Two additional 40-kb MP fosmid jumping libraries were constructed from a sibling used in the previous library construction. Genomic DNA, isolated as above, was shipped to Lucigen Co. (Middleton, WI, USA) for the fosmid jumping library construction, and sequencing was performed on an Illumina MiSeq using 2 × 250-bp reads (36). Last, a variable insert size library of 100 to 100,000 bp in length was generated using the Chicago and HiRise methods (37). Genomic DNA was again isolated from a sibling of those used in previous library construction. The genomic DNA was isolated as above and shipped to Dovetail Co. (Santa Cruz, CA, USA) for library construction, sequencing, and scaffolding. These library fragments were sequenced by Centrillion Biosciences Inc. (Palo Alto, CA, USA) using the Illumina HiSeq 2500 High Output mode, PE 2 × 100 bp. Nearly 500 million read pairs of data were generated, providing ~285× genomic coverage (table S1). The 3- and 7-kb MP pair libraries were filtered for high-confidence true MPs using NextClip v0.8 (38). All read sets were then quality-filtered, and the ends were trimmed of adapters and low-quality bases and screened of common contaminants using BBDuk v37.51 (39). Insert size distributions were plotted to assess library quality, which was high (fig. S6). The 180-bp, 3-kb, and 7-kb read datasets were used as input for AllpathsLG r50960 (40) for initial contig generation and scaffolding (note S1). AllpathsLG was run with haploidify = true to compensate for the high degree of heterozygosity. A further round of superscaffolding using the 40-kb library alongside the 3- and 7-kb libraries was done using SSPACE v2 (41). Last, the assembly was scaffolded a third time and error-corrected using the Chicago read libraries and the HiRise software pipeline. These steps produced a final assembly of 3005 scaffolds with an N50 length of 4.2 Mb and a total length of 350 Mb (note S1), and a 14,945-bp mitochondrial DNA (mtDNA) genome (note S1 and fig. S7). The final assembly’s complete and SCO content was 94% for P. napi as assessed by BUSCO v3.0.2 (42) (for more details, see note S1). Haploid status of the chromosome level assembly was further explored using self-alignments and HaploMerger2 (43). See note S1 for more details.
Linkage maps
RAD-seq data of 5463 single-nucleotide polymorphism (SNP) markers from 275 full-sib individuals, without parents, were used as input into Lep-MAP2 (44). The RAD-seq data were generated from nextRAD technology by SNPsaurus (Oregon, USA) (note S10). To obtain genotype data, the RAD-seq data were mapped to the reference genome using BWA mem (45), and SAMtools (46) was used to produce sorted bam files of the read mappings. On the basis of read coverage (SAMtools depth), Z chromosomal regions were identified from the genome, and the sex of offspring was determined. Custom scripts (47) were used to produce genotype likelihoods (called posteriors in Lep-MAP) from the output of SAMtools mpileup.
The parental genotypes were inferred with the Lep-MAP2 ParentCall module using parameters “ZLimit=2 and ignoreParentOrder=1,” first calling Z markers and second calling the parental genotypes by ignoring which way the parents are informative (the parents were not genotyped, so we could not separate maternal and paternal markers at this stage). Scripts provided with Lep-MAP2 were used to produce a linkage file from the output of ParentCall, and all single-parent informative markers were converted to paternally informative markers by swapping parents, when necessary. Filtering by segregation distortion was performed using the Filtering module.
Following this, the SeparateChromosomes module was run on the linkage file, and 25 chromosomes were identified using a logarithm of the odds (LOD) score limit of 39. Then, the JoinSingles module was run twice to add more markers on the chromosomes with an LOD score limit of 20. Then, SepareteChromosomes was run again but only on markers informative on single parents with an LOD limit of 10 to separate paternally and maternally informative markers. Fifty-one linkage groups were found, and all were ordered using the OrderMarkers module. On the basis of likelihood improvement of marker ordering, paternal and maternal linkage groups were determined. This was possible as there is no recombination in females (achiasmatic meiosis), and thus the order of the markers does not improve likelihood on the female map. The markers on the corresponding maternal linkage groups were converted to maternally informative, and OrderMarkers was run on the resulting data twice for each of the 25 chromosomes (without allowing recombination in female). The final marker order was obtained as the order with the higher likelihood from the two runs.
A maternal marker map was constructed from a different family of P. napi in which a female from Abisko, Sweden, was crossed with a male from Catalonia, Spain. Genomic DNA libraries were constructed for the mother, father, and four offspring (two males and two females). RNA libraries were constructed for an additional six female and six male offspring. All sequencing was performed on an Illumina HiSeq 2500 platform using High Output mode, with PE 2 × 100-bp reads at SciLifeLab (Stockholm, Sweden). Both DNA and RNA reads were mapped to the genome assembly with bbmap using default settings. SAMtools was used to sort read mappings and merge them into an mpileup file (note S6). Variants were called with BCFtools (48) and filtered with VCFtools (49). Linkage between SNPs was assessed with PLINK (50). A custom script was used to assess marker density and determine sex-specific heterozygosity.
Chromosomal assembly
The 5463 markers that composed the linkage map were mapped to the P. napi HiRise assembly using bbmap (39) with sensitivity = slow. Reads that mapped uniquely were used to identify misassemblies in the HiRise scaffolds and subsequently rearrange those fragments into the correct chromosomal order. Fifty-four misassemblies were identified, and a total of 115 fragments were joined together into 25 chromosomes using a series of custom R scripts and the R package Biostrings (51). Scaffold joins and misassembly corrections were validated by comparing the number of correctly mapped MPs spanning a join between two scaffolds. MP reads from the 3-, 7-, and 40-kb libraries were mapped to their respective assemblies with bbmap (po = t, ambig = toss, kbp = t). SAM output was filtered for quality (mapq > 20 and properly paired), and a custom R script was used to tabulate read spanning counts for each base pair in the assembly.
Synteny comparisons within Lepidoptera
A list of 3100 SCOs occurring in the Lepidoptera lineage curated by OrthoDB v9.1 (52) was used to extract gene names and protein sequences of SCOs in B. mori from KAIKObase (53) (note S5) using a custom script. Reciprocal best hits (RBHs) between gene sets of P. napi, B. mori, M. cinxia, and H. melpomene SCOs were identified using BLASTP (54) and custom scripts. Gene sets of H. melpomene v2.5 and M. cinxia v1 were downloaded from Lepbase v4 (55). Coordinates from BLAST tables were converted to chromosomal locations and visualized using Circos (56) and custom R scripts.
To investigate the extent of chromosomal reorganization among the existing lepidopteran genomes, with reference to having either a B. mori– or a P. napi–like organization, two separate bioinformatic analyses were conducted. Data used were genome assemblies and annotated protein sets downloaded for 24 species of Lepidoptera from Lepbase v4 (57) and other sources (table S2). For the first analysis, we used a BLAST-like approach, with each target species protein set aligned to its own genome as well as to the P. napi protein set using Diamond v0.9.10 (58) with default options. The protein-genome comparison was used to assign each target species gene to one of its assembled scaffolds, while the protein-protein comparison was used to identify RBHs between the protein of each species and its ortholog in P. napi and B. mori. Using this information, we used a custom R script to examine each assembly scaffold for evidence of synteny to either P. napi or B. mori. First, each scaffold of the target species genome was assigned genes based on the protein-genome BLAST results, using its own protein set and genome. A gene was assigned to a scaffold if at least three HSPs of less than 200 bp from a gene aligned with ≥95% identity. Second, if any of these scaffolds then contained five genes whose orthologs resided on a single B. mori chromosome but two P. napi chromosomes, and those same two P. napi chromosome segments were also joined in the B. mori assembly, that was counted as a “mori-like scaffold” (see note S13 for cutoff selection criteria). Conversely, if a target species scaffold contained five genes whose orthologs resided on a single P. napi chromosome but two B. mori chromosomes, and those same two B. mori chromosome segments were also joined in the P. napi assembly, that was counted as a “napi-like scaffold” (see fig. S9 for results using this approach).
The second analysis differed from the first by using an intron-aware aligner of proteins to genomes and used B. mori SCOs rather than the P. napi protein set for alignment. In addition, this analysis used a much more restricted set of proteins that had been filtered extensively for high-quality orthology assignment, which reduced detection of SCOs while increasing accuracy. First, P. napi proteins were extracted from the genome using the GFF annotation file, via the program gffread from the cufflinks software package v. 2.2.1 (59). The resulting proteins were then filtered for those having a start, stop, and no internal stop codons. Then, a reciprocal best BLAST (RBB) was conducted upon these P. napi proteins against the B. mori SCOs, with BLASTP via Diamond v.0.9.10, with results filtered (eval <0.00001, bitscore >100) before RBB comparison using custom scripts. This resulted in n = 1835 SCO proteins. The chromosomal locations and start positions of each protein were then added to the resulting RBB table for the two species. Then, for each species genome, these B. mori SCOs were aligned to the genome using SPALN, a high-performing protein to genome alignment software (60). These results were then filtered to only consider scaffolds where >3 SCOs per B. mori chromosome were identified (e.g., scaffolds having five B. mori SCOs from five different chromosomes were ignored). Then, scaffolds were queried for instances where they had SCOs from only 1 B. mori chromosome, with >1 P. napi chromosome, which were then considered B. mori–like scaffolds. Those with one P. napi chromosome and >1 B. mori chromosome were considered P. napi like. If scaffolds only contained SCOs from one chromosome of B. mori and P. napi, they were considered to be supporting the same chromosomal region (see note S11 for more information).
Pieridae chromosomal evolution
Chromosomal fusions and fissions were reconstructed across the family Pieridae by placing previously published karyotype studies of haploid chromosomal counts into their evolutionary context. There are approximately 1000 species in the 85 recognized genera of Pieridae, and we recently reconstructed a robust fossil-calibrated chronogram for this family at the genus level (25, 26). We placed the published chromosomal counts for 201 species (6, 61) on this time-calibrated phylogeny, using the maximal reported haploid chromosomal count per species when more than one was recorded, with ancestral chromosomal reconstructions for chromosome count, treated as a continuous character, using the contMap function of the phytools R package (62). For instances where there was more than one species per genus, all species per genus were treated as a polytomy 5 million years old. This approach allowed us to incorporate the extensive within-genus data that exist in the literature, along with the genus-level and high time-calibrated phylogenetic information.
Lepidopteran chronogram showing relationships among species with genomes
The topology and node ages of the phylogenetic hypothesis were based on the consensus of relationships within Lepidoptera summarized by Mitter et al. (63), with the relationships between families being largely derived from Kawahara and Breinholt (64), and within butterflies from Heikkilä et al. (65). These external estimates of evolutionary relationships (fig. S8) and divergence times were used (table S3), as they were far more robust given their density of taxonomic sampling and used fossil calibrations than any analysis of our limited taxon set.
Estimation of chromosomal rearrangements
A table of the identified SCOs between B. mori and P. napi, including their start locations and chromosomal location, was filtered to remove potential false positives (i.e., any translocated SCO that appeared once within a given collinear block set of four genes) and then collapsed to remove any local chromosome inversions, so that we would detect only the evolutionary dynamics among the collinear blocks. We then further simplified the dataset to have only one gene represent each collinear block (again, to ensure that we were only detecting the large-scale events). The resulting table was then used as input for the software GRIMM v.2.01 (66) to detect an optimal scenario of rearrangements between the two genomes with genes unsigned (options “-s -u”). We also generated predictions of gene signs using an approximation algorithm with a range of iterations from 1000 to 200,000 (option “-U”) and then ran analyses on the resulting datasets.
Annotation of P. napi genome
Genome annotation was carried out by the Bioinformatics Short-term Support and Infrastructure (BILS, Sweden). BILS was provided with the chromosomal assembly of P. napi, and 45 RNA sequencing read sets representing three different tissues (head, fat body, and gut) of seven male and eight female larvae from laboratory lines that were separate from the ones used for the initial sequencing. Sequence evidence for the annotation was collected in two complementary ways. First, we queried the UniProt database (67) for protein sequences belonging to the taxonomic group of Papilionoidea (2516 proteins). To be included, proteins gathered in this way had to be supported on the level of either proteomics or transcriptomics and could not be fragments. In addition, we downloaded the UniProt/Swiss-Prot reference dataset (downloaded on 15 May 2014) (545,388 proteins) for a wider taxonomic coverage with high-confidence proteins. In addition, 493 proteins were used that were derived from a P. rapae expressed sequence tag library that was Sanger-sequenced.
Permutation test of collinear block position within chromosomes
Collinear blocks were identified as interior versus terminal within a chromosome, and the ends of terminal blocks were marked as inward or outward facing (i.e., telomere facing). In total, of the 198 ends of the 99 collinear blocks, 50 of those ends are required to be outward facing since each of the 25 chromosomes has two telomeres. Collinear blocks were reshuffled into 25 random chromosomes of one to four collinear blocks with each collinear block in a random orientation to generate a simulated genome of approximate chromosome size distribution to the actual P. napi genome, and the number of times that 1 of the 50 original outward-facing ends of a terminal block appeared again as an outward-facing terminal block in the simulated genome was counted. This was repeated 10,000 times to generate a random distribution expectation of how often an ancestral telomere would remain a telomere if a genome was randomly rearranged in collinear blocks of the observed size, in a random way, that preserved chromosome size. The number of terminal outward-facing collinear blocks in B. mori that were also terminal and outward facing in P. napi was compared to this random distribution to derive the significance of our observation. To test the randomness of gene location within chromosomes, the previously identified SCOs were numbered by their position along each chromosome in both B. mori and P. napi. We then generated 10,000 random genomes as above. Distance from the end of the new chromosome and distance from the end of B. mori chromosome were calculated for each ortholog, and the results were binned. P values were determined by comparing the number of orthologs in a bin to the expected distribution of genes in a bin from the random genomes. All tests were done using a custom R script.
Gene set enrichment analysis of collinear blocks
GO set enrichment was initially tested within collinear blocks of the P. napi genome using topGO (68) with 11,585 of the 13,622 gene models generated from the annotation that occur within the collinear blocks. For each collinear block within the genome, each GO term of any level within the hierarchy that had at least three genes belonging to it was analyzed for enrichment. If a GO term was overrepresented in a collinear block compared to the rest of the genome at a P value of <0.01 by Fisher’s exact test, then that block was counted as enriched. Fifty-seven of the 99 collinear blocks in the P. napi genome were enriched in this way (table S4). The number of enriched blocks increased or decreased on the basis of the P value cutoff used, or if false discovery rate correction was applied, but our interest was in evaluating relatively enriched blocks compared to an expected distribution computed from a permutation test. Given that we expect gene duplication to have generated some clusters of functionally related genes, we wished to test if the observed level of gene enrichment within collinear blocks was to be expected for blocks of the size observed in any random configuration. The permutation test used to address this fragmented the P. napi assembly into 99 collinear blocks of the same size as those observed in our synteny analysis but from random chromosomes, thereby capturing collinear blocks of genes at random. We used the same size blocks as observed in our genome, because breaking up a genome and testing for GO enrichment can yield results that are dependent on the distribution of the sizes used. This resulted in a random genome of 99 fragments that in total contained the entire genome, but the content of a given fragment was of a random genomic region. This random genome was tested for GO enrichment of the fragments in the same way as the collinear blocks in the original genome, and the number of enriched blocks was counted. This was then repeated 10,000 times to generate a distribution of expected enrichment in genome fragments of the same size as the observed P. napi collinear blocks. The P value cutoff for determining GO term enrichment influenced the number of enriched blocks found in the P. napi genome and each simulated genome, but for all tested P values at the GO term enrichment step (P < 0.001 up to P < 0.5), the P. napi genome contained significantly more GO term–enriched blocks than expected from the permutation test (P < 0.001).
Supplementary Material
Acknowledgments
We acknowledge support from SciLifeLab, the National Genomics Infrastructure, NGI, and Uppmax for providing assistance in massive parallel sequencing and computational infrastructure. We also thank SNPsaurus for support in nextRAD sequencing for linkage map construction. Funding: Funding was provided by the Swedish Research Council [VR grant numbers 2012-3715 (to S.N.), 2010-5341 (to S.N.), and 621-2012-4001 (to C.W.W.)], the Academy of Finland [grant number 131155 (to C.W.W.)], the Knut and Alice Wallenberg Foundation (grant number 2012.0058), and MINECO-FEDER [project number CGL2016-78093-R (to J.C.C.)]. Author contributions: C.W.W., S.N., and J.H. designed the project. C.W. collected and inbred the material. J.H. led the genome assembly. J.H. and C.W.W. were responsible for the bioinformatic analysis of the genome. P.R. led the linkage mapping, with help from J.H. Manual annotations were performed by R.N., N.C., N.M., M.d.l.P.C.-M., J.C.C., H.D., C.M., N.K., P.P., K.S., M.V., H.V., A.W., C.L.B., and S.N. J.H. and C.W.W. wrote the manuscript with input from the other authors. All authors approved the manuscript before submission. Competing interests: The authors declare that they have no competing interests. Data and materials availability: Illumina reads were submitted to the European Nucleotide Archive (ENA) under accession no. PRJEB24862. The assembly and annotation are available at Lepbase http://ensembl.lepbase.org/Pieris_napi_pnv1x1/. Pierid phylogenetic tree, linkage map, and analysis scripts are available at https://github.com/JasonAnthonyHill/Piers_napi. All other data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/6/eaau3648/DC1
Note S1. Genome assembly, mtDNA assembly, HaploMerger results, and BUSCO analysis.
Note S2. HiRise scaffolding and misassembly correction.
Note S3. Linkage group correction of misassemblies.
Note S4. Functional annotation summary.
Note S5. RBH of orthologs.
Note S6. Whole-genome sequencing and RNA-based linkage map.
Note S7. MP spanning.
Note S8. Alignment between P. napi and P. rapae.
Note S9. Syntenic block support of scaffold joins.
Note S10. SNP generation from nextRAD library sequence.
Note S11. Comparison of P. napi versus B. mori collinear blocks within Lepidoptera genomes.
Note S12. Estimation of genomic rearrangements.
Note S13. Lepidopteran synteny cutoff selection.
Table S1. Read data used in assembly and analysis.
Table S2. Genomic data from Lepbase used in the comparative assessment of genome assemblies.
Table S3. Lepidopteran phylogeny calibrations.
Table S4. GO term enrichment in syntenic blocks.
Fig. S1. Chromosomal assembly validation figures.
Fig. S2. Alignment of P. rapae scaffolds to P. napi chromosomes.
Fig. S3. Example of criteria used to determine napi-like and mori-like joins.
Fig. S4. Permutation analysis of chromosomal terminal ends.
Fig. S5. Repetitive element distribution across chromosomes.
Fig. S6. Insert size distribution.
Fig. S7. The mtDNA of P. napi.
Fig. S8. Chronogram of lepidopteran genomes with node labels.
Fig. S9. Comparative assessment of genome assemblies and chromosomal evolution across, using a BLAST-like approach.
REFERENCES AND NOTES
- 1.Lamichhaney S., Fan G., Widemo F., Gunnarsson U., Thalmann D. S., Hoeppner M. P., Kerje S., Gustafson U., Shi C., Zhang H., Chen W., Liang X., Huang L., Wang J., Liang E., Wu Q., Lee S. M.-Y., Xu X., Höglund J., Liu X., Andersson L., Structural genomic changes underlie alternative reproductive strategies in the ruff (Philomachus pugnax). Nat. Genet. 48, 84–88 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Joron M., Frezal L., Jones R. T., Chamberlain N. L., Lee S. F., Haag C. R., Whibley A., Becuwe M., Baxter S. W., Ferguson L., Wilkinson P. A., Salazar C., Davidson C., Clark R., Quail M. A., Beasley H., Glithero R., Lloyd C., Sims S., Jones M. C., Rogers J., Jiggins C. D., ffrench-Constant R. H., Chromosomal rearrangements maintain a polymorphic supergene controlling butterfly mimicry. Nature 477, 203–206 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mora C., Tittensor D. P., Adl S., Simpson A. G. B., Worm B., How many species are there on earth and in the ocean? PLOS Biol. 9, e1001127 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahola V., Lehtonen R., Somervuo P., Salmela L., Koskinen P., Rastas P., Välimäki N., Paulin L., Kvist J., Wahlberg N., Tanskanen J., Hornett E. A., Ferguson L. C., Luo S., Cao Z., de Jong M. A., Duplouy A., Smolander O.-P., Vogel H., McCoy R. C., Qian K., Chong W. S., Zhang Q., Ahmad F., Haukka J. K., Joshi A., Salojärvi J., Wheat C. W., Grosse-Wilde E., Hughes D., Katainen R., Pitkänen E., Ylinen J., Waterhouse R. M., Turunen M., Vähärautio A., Ojanen S. P., Schulman A. H., Taipale M., Lawson D., Ukkonen E., Mäkinen V., Goldsmith M. R., Holm L., Auvinen P., Frilander M. J., Hanski I., The Glanville fritillary genome retains an ancient karyotype and reveals selective chromosomal fusions in Lepidoptera. Nat. Commun. 5, 4737 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lukhtanov V. A., Sex chromatin and sex chromosome systems in nonditrysian Lepidoptera (Insecta). J. Zool. Syst. Evol. Res. 38, 73–79 (2000). [Google Scholar]
- 6.R. Robinson, Lepidoptera Genetics (Pergamon Press, 1971). [Google Scholar]
- 7.Davey J. W., Chouteau M., Barker S. L., Maroja L., Baxter S. W., Simpson F., Merrill R. M., Joron M., Mallet J., Dasmahapatra K. K., Jiggins C. D., Major improvements to the Heliconius melpomene genome assembly used to confirm 10 chromosome fusion events in 6 million years of butterfly evolution. G3 6, 695–708 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dasmahapatra K. K., Walters J. R., Briscoe A. D., Davey J. W., Whibley A., Nadeau N. J., Zimin A. V., Hughes D. S., Ferguson L. C., Martin S. H., Salazar C., Lewis J. J., Adler S., Ahn S. J., Baker D. A., Baxter S. W., Chamberlain N. L., Chauhan R., Counterman B. A., Dalmay T., Gilbert L. E., Gordon K., Heckel D. G., Hines H. M., Hoff K. J., Holland P. W., Jacquin-Joly E., Jiggins F. M., Jones R. T., Kapan D. D., Kersey P., Lamas G., Lawson D., Mapleson D., Maroja L. S., Martin A., Moxon S., Palmer W. J., Papa R., Papanicolaou A., Pauchet Y., Ray D. A., Rosser N., Salzberg S. L., Supple M. A., Surridge A., Tenger-Trolander A., Vogel H., Wilkinson P. A., Wilson D., Yorke J. A., Yuan F., Balmuth A. L., Eland C., Gharbi K., Thomson M., Gibbs R. A., Han Y., Jayaseelan J. C., Kovar C., Mathew T., Muzny D. M., Ongeri F., Pu L. L., Qu J., Thornton R. L., Worley K. C., Wu Y. Q., Linares M., Blaxter M. L., ffrench-Constant R. H., Joron M., Kronforst M. R., Mullen S. P., Reed R. D., Scherer S. E., Richards S., Mallet J., McMillan W., Jiggins C. D., Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature 487, 94–98 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yasukochi Y., Ohno M., Shibata F., Jouraku A., Nakano R., Ishikawa Y., Sahara K., A FISH-based chromosome map for the European corn borer yields insights into ancient chromosomal fusions in the silkworm. Heredity 116, 75–83 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Šíchová J., Ohno M., Dincă V., Watanabe M., Sahara K., Marec F., Fissions, fusions, and translocations shaped the karyotype and multiple sex chromosome constitution of the northeast-Asian wood white butterfly, Leptidea amurensis. Biol. J. Linn. Soc. 118, 457–471 (2016). [Google Scholar]
- 11.Brown K. S. Jr., Von Schoultz B., Suomalainen E., Chromosome evolution in Neotropical Danainae and Ithomiinae (Lepidoptera). Hereditas 141, 216–236 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Kandul N. P., Lukhtanov V. A., Pierce N. E., Karyotypic diversity and speciation in Agrodiaetus butterflies. Evolution 61, 546–559 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Saura A., Von Schoultz B., Saura A. O., Brown K. S. Jr., Chromosome evolution in Neotropical butterflies. Hereditas 150, 26–37 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Lukhtanov V. A., The blue butterfly Polyommatus (Plebicula) atlanticus (Lepidoptera, Lycaenidae) holds the record of the highest number of chromosomes in the non-polyploid eukaryotic organisms. Comp. Cytogenet. 9, 683–690 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Šíchová J., Voleníková A., Dincă V., Nguyen P., Vila R., Sahara K., Marec F., Dynamic karyotype evolution and unique sex determination systems in Leptidea wood white butterflies. BMC Evol. Biol. 15, 89 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagaraju J., Jolly M. S., Interspecific hybrids of Antheraea roylei and A. pernyi—a cytogenetic reassessment. Theor. Appl. Genet. 72, 269–273 (1986). [DOI] [PubMed] [Google Scholar]
- 17.Lukhtanov V. A., Dincă V., Friberg M., Šíchová J., Olofsson M., Vila R., Marec F., Wiklund C., Versatility of multivalent orientation, inverted meiosis, and rescued fitness in holocentric chromosomal hybrids. Proc. Natl. Acad. Sci. U.S.A. 115, E9610–E9619 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marec F., Tothova A., Sahara K., Traut W., Meiotic pairing of sex chromosome fragments and its relation to atypical transmission of a sex-linked marker in Ephestia kuehniella (Insecta: Lepidoptera). Heredity 87, 659–671 (2001). [DOI] [PubMed] [Google Scholar]
- 19.Lorkovic Z., The genetics and reproductive isolating mechanisms of the Pieris napi–bryoniae group. J. Lepid. Soc. 16, 5–19 (1955). [Google Scholar]
- 20.Maeki K., Kawazoé A., On the hybridization between two karyotype Lineages(n. 25*n. 26) of Pieris napi Linnaeus from Japan. Cytologia 59, 279–284 (1994). [Google Scholar]
- 21.Nguyen P., Sýkorová M., Šichová J., Kůta V., Daliková M., Čapková Frydrychová R., Neven L. G., Sahara K., Marec F., Neo-sex chromosomes and adaptive potential in tortricid pests. Proc. Natl. Acad. Sci. U.S.A. 110, 6931–6936 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mongue A. J., Nguyen P., Voleníková A., Walters J. R., Neo-sex chromosomes in the monarch butterfly, Danaus plexippus. G3 (Bethesda) 7, 3281–3294 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nallu S., Hill J. A., Don K., Sahagun C., Zhang W., Meslin C., Snell-Rood E., Clark N. L., Morehouse N. I., Bergelson J., Wheat C. W., Kronforst M. R., The molecular genetic basis of herbivory between butterflies and their host plants. Nat. Ecol. Evol. 2, 1418–1427 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen J., Cong Q., Kinch L. N., Borek D., Otwinowski Z., Grishin N. V., Complete genome of Pieris rapae, a resilient alien, a cabbage pest, and a source of anti-cancer proteins. F1000Res. 5, 2631 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wahlberg N., Rota J., Braby M. F., Pierce N. E., Wheat C. W., Revised systematics and higher classification of pierid butterflies (Lepidoptera: Pieridae) based on molecular data. Zool. Scr. 43, 641–650 (2014). [Google Scholar]
- 26.Edger P. P., Heidel-Fischer H. M., Bekaert M., Rota J., Glöckner G., Platts A. E., Heckel D. G., der J. P., Wafula E. K., Tang M., Hofberger J. A., Smithson A., Hall J. C., Blanchette M., Bureau T. E., Wright S. I., dePamphilis C. W., Eric Schranz M., Barker M. S., Conant G. C., Wahlberg N., Vogel H., Pires J. C., Wheat C. W., The butterfly plant arms-race escalated by gene and genome duplications. Proc. Natl. Acad. Sci. U.S.A. 112, 8362–8366 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andrews J., Smith M., Merakovsky J., Coulson M., Hannan F., Kelly L. E., The stoned locus of Drosophila melanogaster produces a dicistronic transcript and encodes two distinct polypeptides. Genetics 143, 1699–1711 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hurst L. D., Pál C., Lercher M. J., The evolutionary dynamics of eukaryotic gene order. Nat. Rev. Genet. 5, 299–310 (2004). [DOI] [PubMed] [Google Scholar]
- 29.González J., Petrov D. A., The adaptive role of transposable elements in the Drosophila genome. Gene 448, 124–133 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miller W. J., Capy P., Mobile genetic elements as natural tools for genome evolution. Methods Mol. Biol. 260, 1–20 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Drosophila 12 Genomes Consortium , Evolution of genes and genomes on the Drosophila phylogeny. Nature 450, 203–218 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Kandul N. P., Lukhtanov V. A., Dantchenko A. V., Coleman J. W. S., Sekercioglu C. H., Haig D., Pierce N. E., Phylogeny of Agrodiaetus Hübner 1822 (Lepidoptera: Lycaenidae) inferred from mtDNA sequences of COI and COII and nuclear sequences of EF1-α: Karyotype diversification and species radiation. Syst. Biol. 53, 278–298 (2004). [DOI] [PubMed] [Google Scholar]
- 33.J. R. Walters, A. J. Mongue, A neo-sex chromosome in the Monarch butterfly, Danaus plexippus bioRxiv, 036483 [Preprint] 12 January 2016. 10.1101/036483. [DOI] [PMC free article] [PubMed]
- 34.Eichler E. E., Sankoff D., Structural dynamics of eukaryotic chromosome evolution. Science 301, 793–797 (2003). [DOI] [PubMed] [Google Scholar]
- 35.Aljanabi S. M., Martinez I., Universal and rapid salt-extraction of high quality genomic DNA for PCR- based techniques. Nucleic Acids Res. 25, 4692–4693 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bentley D. R., Balasubramanian S., Swerdlow H. P., Smith G. P., Milton J., Brown C. G., Hall K. P., Evers D. J., Barnes C. L., Bignell H. R., Boutell J. M., Bryant J., Carter R. J., Keira Cheetham R., Cox A. J., Ellis D. J., Flatbush M. R., Gormley N. A., Humphray S. J., Irving L. J., Karbelashvili M. S., Kirk S. M., Li H., Liu X., Maisinger K. S., Murray L. J., Obradovic B., Ost T., Parkinson M. L., Pratt M. R., Rasolonjatovo I. M. J., Reed M. T., Rigatti R., Rodighiero C., Ross M. T., Sabot A., Sankar S. V., Scally A., Schroth G. P., Smith M. E., Smith V. P., Spiridou A., Torrance P. E., Tzonev S. S., Vermaas E. H., Walter K., Wu X., Zhang L., Alam M. D., Anastasi C., Aniebo I. C., Bailey D. M. D., Bancarz I. R., Banerjee S., Barbour S. G., Baybayan P. A., Benoit V. A., Benson K. F., Bevis C., Black P. J., Boodhun A., Brennan J. S., Bridgham J. A., Brown R. C., Brown A. A., Buermann D. H., Bundu A. A., Burrows J. C., Carter N. P., Castillo N., Chiara M., Catenazzi E., Chang S., Neil Cooley R., Crake N. R., Dada O. O., Diakoumakos K. D., Dominguez-Fernandez B., Earnshaw D. J., Egbujor U. C., Elmore D. W., Etchin S. S., Ewan M. R., Fedurco M., Fraser L. J., Fuentes Fajardo K. V., Scott Furey W., George D., Gietzen K. J., Goddard C. P., Golda G. S., Granieri P. A., Green D. E., Gustafson D. L., Hansen N. F., Harnish K., Haudenschild C. D., Heyer N. I., Hims M. M., Ho J. T., Horgan A. M., Hoschler K., Hurwitz S., Ivanov D. V., Johnson M. Q., James T., Huw Jones T. A., Kang G. D., Kerelska T. H., Kersey A. D., Khrebtukova I., Kindwall A. P., Kingsbury Z., Kokko-Gonzales P. I., Kumar A., Laurent M. A., Lawley C. T., Lee S. E., Lee X., Liao A. K., Loch J. A., Lok M., Luo S., Mammen R. M., Martin J. W., McCauley P. G., McNitt P., Mehta P., Moon K. W., Mullens J. W., Newington T., Ning Z., Ling Ng B., Novo S. M., O’Neill M. J., Osborne M. A., Osnowski A., Ostadan O., Paraschos L. L., Pickering L., Pike A. C., Pike A. C., Chris Pinkard D., Pliskin D. P., Podhasky J., Quijano V. J., Raczy C., Rae V. H., Rawlings S. R., Chiva Rodriguez A., Roe P. M., Rogers J., Rogert Bacigalupo M. C., Romanov N., Romieu A., Roth R. K., Rourke N. J., Ruediger S. T., Rusman E., Sanches-Kuiper R. M., Schenker M. R., Seoane J. M., Shaw R. J., Shiver M. K., Short S. W., Sizto N. L., Sluis J. P., Smith M. A., Ernest Sohna Sohna J., Spence E. J., Stevens K., Sutton N., Szajkowski L., Tregidgo C. L., Turcatti G., vandeVondele S., Verhovsky Y., Virk S. M., Wakelin S., Walcott G. C., Wang J., Worsley G. J., Yan J., Yau L., Zuerlein M., Rogers J., Mullikin J. C., Hurles M. E., McCooke N. J., West J. S., Oaks F. L., Lundberg P. L., Klenerman D., Durbin R., Smith A. J., Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456, 53–59 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Putnam N. H., O'Connell B. L., Stites J. C., Rice B. J., Blanchette M., Calef R., Troll C. J., Fields A., Hartley P. D., Sugnet C. W., Haussler D., Rokhsar D. S., Green R. E., Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 26, 342–350 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leggett R. M., Clavijo B. J., Clissold L., Clark M. D., Caccamo M., NextClip: An analysis and read preparation tool for Nextera Long Mate Pair libraries. Bioinformatics 30, 566–568 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.B. Bushnell, BBTools (2017); https://jgi.doe.gov/data-and-tools/bbtools/.
- 40.Gnerre S., MacCallum I., Przybylski D., Ribeiro F. J., Burton J. N., Walker B. J., Sharpe T., Hall G., Shea T. P., Sykes S., Berlin A. M., Aird D., Costello M., Daza R., Williams L., Nicol R., Gnirke A., Nusbaum C., Lander E. S., Jaffe D. B., High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl. Acad. Sci. U.S.A. 108, 1513–1518 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boetzer M., Henkel C. V., Jansen H. J., Butler D., Pirovano W., Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579 (2011). [DOI] [PubMed] [Google Scholar]
- 42.Simão F. A., Waterhouse R. M., Ioannidis P., Kriventseva E. V., Zdobnov E. M., BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Huang S., Kang M., Xu A., HaploMerger2: Rebuilding both haploid sub-assemblies from high-heterozygosity diploid genome assembly. Bioinformatics 33, 2577–2579 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rastas P., Calboli F. C. F., Guo B., Shikano T., Merilä J., Construction of ultradense linkage maps with Lep-MAP2: Stickleback F2 recombinant crosses as an example. Genome Biol. Evol. 8, 78–93 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.H. Li, Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997 [q-bio.GN] (16 March 2013).
- 46.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup , The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kvist J., Mattila A. L. K., Somervuo P., Ahola V., Koskinen P., Paulin L., Salmela L., Fountain T., Rastas P., Ruokolainen A., Taipale M., Holm L., Auvinen P., Lehtonen R., Frilander M. J., Hanski I., Flight-induced changes in gene expression in the Glanville fritillary butterfly. Mol. Ecol. 24, 4886–4900 (2015). [DOI] [PubMed] [Google Scholar]
- 48.Narasimhan V., Danecek P., Scally A., Xue Y., Tyler-Smith C., Durbin R., BCFtools/RoH: A hidden Markov model approach for detecting autozygosity from next-generation sequencing data. Bioinformatics 32, 1749–1751 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Danecek P., Auton A., Abecasis G., Albers C. A., Banks E., DePristo M. A., Handsaker R. E., Lunter G., Marth G. T., Sherry S. T., McVean G., Durbin R., 1000 Genomes Project Analysis Group , The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M. A. R., Bender D., Maller J., Sklar P., de Bakker P. I. W., Daly M. J., Sham P. C., PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.H. Pages, P. Aboyoun, R. Gentleman, S. DebRoy, Biostrings: String objects representing biological sequences, and matching algorithms. R Package version 2 (2008).
- 52.Zdobnov E. M., Tegenfeldt F., Kuznetsov D., Waterhouse R. M., Simão F. A., Ioannidis P., Seppey M., Loetscher A., Kriventseva E. V., OrthoDB v9.1: Cataloging evolutionary and functional annotations for animal, fungal, plant, archaeal, bacterial and viral orthologs. Nucleic Acids Res. 45, D744–D749 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shimomura M., Minami H., Suetsugu Y., Ohyanagi H., Satoh C., Antonio B., Nagamura Y., Kadono-Okuda K., Kajiwara H., Sezutsu H., Nagaraju J., Goldsmith M. R., Xia Q., Yamamoto K., Mita K., KAIKObase: An integrated silkworm genome database and data mining tool. BMC Genomics 10, 486 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J., Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kersey P. J., Allen J. E., Armean I., Boddu S., Bolt B. J., Carvalho-Silva D., Christensen M., Davis P., Falin L. J., Grabmueller C., Humphrey J., Kerhornou A., Khobova J., Aranganathan N. K., Langridge N., Lowy E., McDowall M. D., Maheswari U., Nuhn M., Ong C. K., Overduin B., Paulini M., Pedro H., Perry E., Spudich G., Tapanari E., Walts B., Williams G., Tello–Ruiz M., Stein J., Wei S., Ware D., Bolser D. M., Howe K. L., Kulesha E., Lawson D., Maslen G., Staines D. M., Ensembl Genomes 2016: More genomes, more complexity. Nucleic Acids Res. 44, D574–D580 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Krzywinski M., Schein J., Birol I., Connors J., Gascoyne R., Horsman D., Jones S. J., Marra M. A., Circos: An information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.R. J. Challi, S. Kumar, K. K. Dasmahapatra, C. D. Jiggins, M. Blaxter, Lepbase: The Lepidopteran genome database. bioRxiv, 056994 [Preprint] 6 June 2016. 10.1101/056994. [DOI]
- 58.Buchfink B., Xie C., Huson D. H., Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015). [DOI] [PubMed] [Google Scholar]
- 59.Trapnell C., Williams B. A., Pertea G., Mortazavi A., Kwan G., van Baren M. J., Salzberg S. L., Wold B. J., Pachter L., Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gotoh O., A space-efficient and accurate method for mapping and aligning cDNA sequences onto genomic sequence. Nucleic Acids Res. 36, 2630–2638 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lukhtanov V. A., Karyotype evolution and systematics of higher taxa of Pieridae (Lepidoptera) of the world. Ent. Obozr. 70, 619–636 (1991). [Google Scholar]
- 62.Revell L. J., phytools: An R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 3, 217–223 (2012). [Google Scholar]
- 63.Mitter C., Davis D. R., Cummings M. P., Phylogeny and evolution of Lepidoptera. Annu. Rev. Entomol. 62, 265–283 (2017). [DOI] [PubMed] [Google Scholar]
- 64.Kawahara A. Y., Breinholt J. W., Phylogenomics provides strong evidence for relationships of butterflies and moths. Proc. Biol. Sci. 281, 20140970 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heikkilä M., Kaila L., Mutanen M., Peña C., Wahlberg N., Cretaceous origin and repeated tertiary diversification of the redefined butterflies. Proc. Biol. Sci. 279, 1093–1099 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tesler G., GRIMM: Genome rearrangements web server. Bioinformatics 18, 492–493 (2002). [DOI] [PubMed] [Google Scholar]
- 67.The UniProt Consortium , UniProt: A hub for protein information. Nucleic Acids Res. 43, D204–D212 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.A. Alexa, J. Rahnenfuhrer, topGO: Enrichment Analysis for Gene Ontology. R Package version 2.26.0 (2016); http://bioconductor.org/packages/release/bioc/html/topGO.html.
- 69.Birney E., Clamp M., Durbin R., GeneWise and Genomewise. Genome Res. 14, 988–995 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lowe T. M., Eddy S. R., TRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vurture G. W., Sedlazeck F. J., Nattestad M., Underwood C. J., Fang H., Gurtowski J., Schatz M. C., GenomeScope: Fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ogata H., Goto S., Sato K., Fujibuchi W., Bono H., Kanehisa M., KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 27, 29–34 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Finn R. D., Bateman A., Clements J., Coggill P., Eberhardt R. Y., Eddy S. R., Heger A., Hetherington K., Holm L., Mistry J., Sonnhammer E. L. L., Tate J., Punta M., Pfam: The protein families database. Nucleic Acids Res. 42, D222–D230 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mitchell A., Chang H.-Y., Daugherty L., Fraser M., Hunter S., Lopez R., McAnulla C., McMenamin C., Nuka G., Pesseat S., Sangrador-Vegas A., Scheremetjew M., Rato C., Yong S.-Y., Bateman A., Punta M., Attwood T. K., Sigrist C. J. A., Redaschi N., Rivoire C., Xenarios I., Kahn D., Guyot D., Bork P., Letunic I., Gough J., Oates M., Haft D., Huang H., Natale D. A., Wu C. H., Orengo C., Sillitoe I., Mi H., Thomas P. D., Finn R. D., The InterPro protein families database: The classification resource after 15 years. Nucleic Acids Res. 43, D213–D221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gene Ontology Consortium , Gene Ontology Consortium: Going forward. Nucleic Acids Res. 43, D1049–D1056 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Caspi R., Billington R., Ferrer L., Foerster H., Fulcher C. A., Keseler I. M., Kothari A., Krummenacker M., Latendresse M., Mueller L. A., Ong Q., Paley S., Subhraveti P., Weaver D. S., Karp P. D., The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 44, D471–D480 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Morgat A., Coissac E., Coudert E., Axelsen K. B., Keller G., Bairoch A., Bridge A., Bougueleret L., Xenarios I., Viari A., UniPathway: A resource for the exploration and annotation of metabolic pathways. Nucleic Acids Res. 40, D761–D769 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fabregat A., Sidiropoulos K., Garapati P., Gillespie M., Hausmann K., Haw R., Jassal B., Jupe S., Korninger F., McKay S., Matthews L., May B., Milacic M., Rothfels K., Shamovsky V., Webber M., Weiser J., Williams M., Wu G., Stein L., Hermjakob H., D'Eustachio P., The reactome pathway knowledgebase. Nucleic Acids Res. 44, D481–D487 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jones P., Binns D., Chang H.-Y., Fraser M., Li W., McAnulla C., McWilliam H., Maslen J., Mitchell A., Nuka G., Pesseat S., Quinn A. F., Sangrador-Vegas A., Scheremetjew M., Yong S.-Y., Lopez R., Hunter S., InterProScan 5: Genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.R. Tate, B. Hall, T. DeRego, S. Geib, Annie: The ANNotation Information Extractor (Version 1.0) [Software] (2014); http://genomeannotation.github.io/annie.
- 81.Bolger A. M., Lohse M., Usadel B., Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Condamine F. L., Sperling F. A. H., Kergoat G. J., Global biogeographical pattern of swallowtail diversification demonstrates alternative colonization routes in the Northern and Southern hemispheres. J. Biogeogr. 40, 9–23 (2013). [Google Scholar]
- 83.Wahlberg N., Wheat C. W., Peña C., Timing and patterns in the taxonomic diversification of Lepidoptera (butterflies and moths). PLOS ONE 8, e80875 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kozak K. M., Wahlberg N., Neild A. F. E., Dasmahapatra K. K., Mallet J., Jiggins C. D., Multilocus species trees show the recent adaptive radiation of the mimetic heliconius butterflies. Syst. Biol. 64, 505–524 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mitchell A., Mitter C., Regier J. C., Systematics and evolution of the cutworm moths (Lepidoptera: Noctuidae): Evidence from two protein-coding nuclear genes. Syst. Entomol. 31, 21–46 (2006). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/6/eaau3648/DC1
Note S1. Genome assembly, mtDNA assembly, HaploMerger results, and BUSCO analysis.
Note S2. HiRise scaffolding and misassembly correction.
Note S3. Linkage group correction of misassemblies.
Note S4. Functional annotation summary.
Note S5. RBH of orthologs.
Note S6. Whole-genome sequencing and RNA-based linkage map.
Note S7. MP spanning.
Note S8. Alignment between P. napi and P. rapae.
Note S9. Syntenic block support of scaffold joins.
Note S10. SNP generation from nextRAD library sequence.
Note S11. Comparison of P. napi versus B. mori collinear blocks within Lepidoptera genomes.
Note S12. Estimation of genomic rearrangements.
Note S13. Lepidopteran synteny cutoff selection.
Table S1. Read data used in assembly and analysis.
Table S2. Genomic data from Lepbase used in the comparative assessment of genome assemblies.
Table S3. Lepidopteran phylogeny calibrations.
Table S4. GO term enrichment in syntenic blocks.
Fig. S1. Chromosomal assembly validation figures.
Fig. S2. Alignment of P. rapae scaffolds to P. napi chromosomes.
Fig. S3. Example of criteria used to determine napi-like and mori-like joins.
Fig. S4. Permutation analysis of chromosomal terminal ends.
Fig. S5. Repetitive element distribution across chromosomes.
Fig. S6. Insert size distribution.
Fig. S7. The mtDNA of P. napi.
Fig. S8. Chronogram of lepidopteran genomes with node labels.
Fig. S9. Comparative assessment of genome assemblies and chromosomal evolution across, using a BLAST-like approach.