

A novel immunotherapy targeting the Mucorales unique CoH3 invasin is highly protective against murine mucormycosis.
Abstract
Mucorales are fungal pathogens that cause mucormycosis, a lethal angioinvasive disease. Previously, we demonstrated that Rhizopus, the most common cause of mucormycosis, invades endothelial cells by binding of its CotH proteins to the host receptor GRP78. Loss of CotH3 renders the fungus noninvasive and attenuates Rhizopus virulence in mice. Here, we demonstrate that polyclonal antibodies raised against peptides of CotH3 protected diabetic ketoacidotic (DKA) and neutropenic mice from mucormycosis compared to mice treated with control preimmune serum. Passive immunization with anti-CotH3 antibodies enhanced neutrophil inlfux and triggered Fc receptor-mediated enhanced opsonophagocytosis killing of Rhizopus delemar. Monoclonal antibodies raised against the CotH3 peptide also protected immunosuppressed mice from mucormycosis caused by R. delemar and other Mucorales and acted synergistically with antifungal drugs in protecting DKA mice from R. delemar infection. These data identify anti-CotH3 antibodies as a promising adjunctive immunotherapeutic option against a deadly disease that often poses a therapeutic challenge.
INTRODUCTION
Mucormycosis is a fungal infection with an often fatal prognosis (1, 2). Treatment options for the disease are limited, frequently involving extensive disfiguring surgical debridement and toxic antifungal therapy that, even if effective, frequently lead to a dismal quality of life (3). Even with aggressive and optimal current treatment, mortality rates for mucormycosis range from 50 to 100%. Higher mortalities are seen in patients with prolonged neutropenia, disseminated disease, or brain infection (1, 2).
Mucormycosis is caused by several fungi that belong to the order Mucorales. These organisms typically cause acute, aggressive, and predominantly angioinvasive infections in immunocompromised hosts with hematologic malignancy and/or stem cell transplantation (1). Other high-risk patients include those with poorly controlled hyperglycemia, diabetic ketoacidosis (DKA), and other forms of acidosis (1, 2). Rhizopus, Mucor, and Lichtheimia are the leading genera of fungi that cause mucormycosis, accounting for up to 90% of all cases of this disease (1, 2), whereas Apophysomyces, Cunninghamella, and Rhizomucor, besides others, cause between 1 and 5% of all reported cases (4). These Mucorales can also cause life-threatening infections in immunocompetent individuals suffering from severe trauma due to natural disasters (5), blast injuries during combat operation (6), or motor vehicle accidents (7).
There has been an alarming rise in the incidence of mucormycosis at major U.S. transplant centers (8). Mucormycosis is the third most common invasive fungal infection in hematopoietic stem cell transplant patients in the United States (9), with prevalence rates of 8% in autopsied leukemia patients (10). Similarly, a 70% increase in mucormycosis cases has been reported in France over a 9-year period (between 1997 and 2006) (11). Notably, a study reviewing cases in India for the past five decades predicted a prevalence of 0.14 cases per 1000 population, which equates to ~200,000 cases per year (12). Most recently, the number of mucormycosis patients per 10,000 hospital admissions increased substantially between 2008 and 2017 from 0.47 to 1.18 in a hospital in Lebanon (13).
The treatment of mucormycosis is compromised by the limited arsenal of approved effective antifungal drugs, coupled with an inherent tendency for these fungi to be drug resistant (14). Only three antifungal drugs are currently approved for the treatment of mucormycosis; they are lipid formulations of amphotericin B, posaconazole, and isavuconazole (15). In the absence of surgical debridement of necrotic tissues at infected foci, antifungal therapy alone is rarely curative (16). Thus, alternative strategies to prevent and treat mucormycosis are needed.
Previously, we determined that Rhizopus strains adhere to and invade human umbilical vein endothelial cells (HUVECs) in vitro by induced endocytosis (17). This fungal-host cell interaction is mediated by the fungal cell surface protein CotH (specifically CotH3), which binds to the glucose-regulated protein 78 (GRP78) on the endothelial cell surface (17, 18). Elevated concentrations of glucose, iron, and ketone bodies, which occur in patients with hyperglycemia and DKA, enhance GRP78 and CotH3 expression, leading to augmented fungal invasion and damage of HUVECs (18, 19). Correspondingly, repression of CotH3 expression by RNA interference abrogates HUVEC invasion and Rhizopus-mediated host cell injury and results in significantly reduced virulence in mice (18). CotH proteins are highly conserved within the order Mucorales, making it a potential therapeutic target (18, 20).
To address the unmet need for novel treatment alternatives against mucormycosis, we developed antibodies against peptide regions of CotH3, which are conserved among Mucorales and in the binding site to GRP78 (18). We sought to evaluate the potential use of passive immunotherapy against Mucorales in mice, using polyclonal and monoclonal antibodies directed at this 16-mer peptide. Last, we examined the mechanism of action of the antibodies in mediating protection and evaluated their potential utility as adjunctive therapy with clinically relevant antifungal drugs. Overall, this work illustrates the potential of a new immunotherapeutic modality to combat mucormycosis based on a unique protein universally present in Mucorales fungi.
RESULTS
Human sera have low natural antibody titers against CotH3 protein
We compared the presence of natural anti-CotH3 antibodies in sera collected from healthy volunteers who are routinely exposed to Rhizopus (member from our laboratory) to sera from patients with mucormycosis using enzyme-linked immunosorbent assay (ELISA) plates coated with either recombinant CotH3 protein (rCotH3p) or the antigenic, surface-exposed,16-mer peptide (MGQTNDGAYRDPTDNN). In general, low and similar titers of anti-CotH3 antibodies were detected in healthy individuals and patients with mucormycosis ranging from 1:200 to 1:800 dilution. A single patient had a titer of 1:1600 (Fig. 1A). Similarly, there was no difference in the low anti–16-mer peptide antibody titers between the two subject groups. As expected, the titers against the rCotH3p were higher than those against the 16-mer peptide (Fig. 1A). Concordant with the low antibody titers in healthy volunteers, samples taken from the same patient over 2 to 8 weeks following diagnosis did not show a significant increase in antibody titers (Fig. 1A, colored symbols). The results of low titers of natural anti-CotH3 antibodies among healthy individuals and the lack of increase in anti-CotH3 titers following infection suggested that treatment with antibodies targeting an invasin required for mucormycosis pathogenesis is likely to be effective in attenuating infections by Mucorales organisms by blocking adherence and invasion of the organisms to host cells.
Fig. 1. CotH antibody titers in mucormycosis patients are not different from those in healthy subjects, while rabbit anti-CotH3 polyclonal antibodies specifically bind to Mucorales.
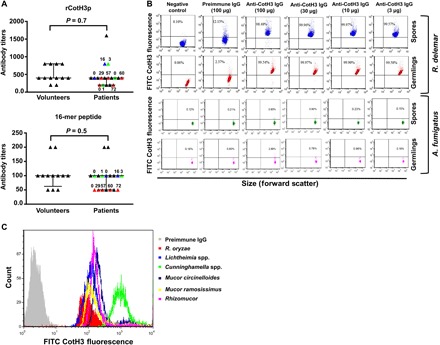
(A) ELISA plates coated with rCotH3 from Escherichia coli or the 16-mer peptide showing low and similar anti-CotH3 antibody titers in sera collected from mucormycosis patients (11 total) or healthy volunteers (12 total). Colored symbols indicate sera collected from the same patient at different days (numbers) following diagnosis (day 0). (B) Flow cytometry analysis revealed that different concentrations of anti-CotH3 polyclonal antibody (and not the preimmune IgG antibody) bound both R. delemar spores and germlings with high consistency but not with A. fumigatus. The different doses of antibodies ranged from the highest (100 μg/ml) to the lowest (3 μg/ml). (C) The polyclonal antibodies also bound to a number of other fungi belonging to the order Mucorales.
Polyclonal anti-CotH3 antibodies are promising novel therapy against Rhizopus delemar infections
We generated rabbit polyclonal antibodies against two peptides of R. delemar CotH3 that are predicted to be antigenic and reside in the binding site of the host GRP78 protein (18). In our previous study, the CotH3 antibodies prevented R. delemar–mediated invasion and subsequent damage of human endothelial cells in vitro and prolonged survival of DKA mice with pulmonary mucormycosis due to R. delemar. This attenuation of pathogenesis required a high dose of antibody, 1 mg per mouse (18). This dose translates to ~2.4 g of antibody per patient, an amount that is clinically unprecedented and impractical. Consequently, in the current study, we intended to further determine the efficacy of the polyclonal antibodies, define their mechanism of action in vitro and in vivo, and examine their potential transferability as novel therapeutics against mucormycosis.
We began by testing the binding of the polyclonal anti-CotH3 antibodies to R. delemar spores and germlings. This member of Mucorales expresses high levels of CotH mRNA (18, 20). Immunostaining of spores and germlings with anti-CotH3 polyclonal antibodies, followed by flow cytometry, showed consistent immunoglobulin G (IgG) binding to the fungus (Fig. 1B). The lowest (3 μg/ml) and the highest (100 μg/ml) concentrations of the antibodies had almost similar total binding to the organism. In contrast, negligible binding was observed in negative control conditions (secondary antibody only) or when fungi were treated with preimmune IgG (Fig. 1B). Furthermore, the anti-CotH3 antibodies did not bind to Aspergillus fumigatus spores or germlings, a mold that has no CotH orthologs. However, the anti-CotH3 antibodies had no effect on the growth or germination of R. delemar, as indicated by the lack of effect of anti-CotH3 antibodies on germ tube length or metabolic activity of the fungus (fig. S1).
Next, we determined whether polyclonal antibodies were protective in neutropenic mice at lower and clinically translational doses. Neutropenic mice were intratracheally infected with R. delemar and then, 24 hours later, treated intraperitoneally with a single dose of 30, 100, or 300 μg of purified rabbit anti-CotH3 IgG or isotype-matched IgG purified from preimmune serum (control). All mice treated with preimmune purified IgG expired by day 8 after infection, resulting in a median survival time of 5 days. In contrast, mice treated with 30, 100, or 300 μg of anti-CotH3 IgG had prolonged survival of 70, 30, and 40%, respectively (P < 0.05 for anti-CotH3 IgG versus preimmune IgG at all doses; Fig. 2A). The differences in survival among the different doses of anti-CotH3 IgG-treated mice were not significant (P = 0.07 for 30 μg versus 100 μg and P = 0.17 for 30 μg versus 300 μg). Surviving mice were healthy by day 21 when the experiment was terminated.
Fig. 2. Anti-CotH3 polyclonal antibodies protect mice from R. delemar intratracheal infection.

Intraperitoneal treatment with a single dose of the polyclonal antibody (Ab), 24 hours after intratracheal infection of neutropenic mice (A to C) or DKA mice (D and E), protected them from R. delemar infection. Survival of neutropenic (A) or DKA mice (D) was conducted using 10 mice per group with confirmed inhaled inoculum of 1.2 × 104 for (A) and 6.3 × 103 for (D). *P < 0.05 versus mice treated with preimmune IgG. A single dose of 30 μg of anti-CotH3 IgG resulted in a significant reduction of lung and brain fungal burden in neutropenic (B) (inhaled inoculum of 3.2 × 103) and DKA mice (E) (inhaled inoculum of 3.6 × 103) when compared to isotype-matched control IgG at the same dose. Mice were euthanized 72 hours after infection. Hematoxylin and eosin staining of lung and brain sections harvested from neutropenic mice (C) revealed extensive tissue edema and hemorrhage in the lungs of mice treated with isotype-matched IgG, whereas mice treated with anti-CotH3 IgG had normal architecture (lower magnification). Abundant fungal hyphae invading lungs of mice treated with isotype-matched IgG, but not anti-CotH3 IgG, could be detected (higher magnification). Brains from isotype-matched IgG-treated, but not anti-CotH3 IgG-treated, mice showed mucormycosis angioinvasion. Scale bars, 300 μm (C).
In a separate study, neutropenic mice were infected and treated as above and then euthanized 48 hours after treatment, and their target organs (lungs and brain) were collected for tissue fungal burden analysis. Anti-CotH3 antibodies reduced the lung fungal burden by >1 log and almost completely abrogated the ability of R. delemar to hematogenously disseminate to the brain [a secondary target organ in this model (21)] (Fig. 2B). Consistent with these results, histopathological examination of the lungs revealed extensive hemorrhage and tissue edema in mice infected with R. delemar and treated with control IgG, while the lungs harvested from infected mice treated with the anti-CotH3 IgG had a more normal organ architecture (Fig. 2C, top). On a higher magnification, numerous broad, ribbon-like, and aseptate hyphae with 90° angle bifurcation were present in mice treated with control preimmune IgG, whereas very few fungal elements were present in the lungs of mice treated with anti-CotH3 IgG (Fig. 2C, middle). Furthermore, in mice treated with control IgG, fungal cells were present in thrombosed cerebral blood vessels with hyphae egressing to seed the tissue parenchyma (Fig. 2C, bottom). In contrast, the brains harvested from mice treated with anti-CotH3 IgG had little evidence of infection and no angioinvasion.
To further confirm the therapeutic potential of anti-CotH3 antibodies, we tested the lower and most protective dose of the antibodies in the DKA model of pulmonary mucormycosis. Infection with R. delemar, followed by treatment with anti-CotH3 IgG (30 μg), was conducted as above. While DKA mice treated with preimmune IgG had 100% mortality by day 13 after infection, 60% of mice treated with anti-CotH3 IgG survived the infection. The surviving mice appeared healthy by day 21 (Fig. 2D). Last, anti-CotH3 IgG treatment caused reduction of at least 1.5 log when compared to mice treated with isotype-matched control IgG (Fig. 2E). Overall, the protective effect of low doses of these antibodies in animal models confirms their translational potential as a novel treatment for R. delemar–associated pneumonia in different hosts (neutropenic and diabetic). Further, anti-CotH3 antibodies prevented host cell invasion and hematogenous dissemination to secondary target organs.
Anti-CotH3 antibodies protect against mucormycosis caused by other Mucorales
Querying peptide MGQTNDGAYRDPTDNN against 38 Mucorales genomes [sequenced by us and others (all raw sequencing data are from the National Center for Biotechnology Information Sequence Read Archive database)] (20) revealed the universal presence of CotH among these fungi with a high degree of predicted amino acid identity (73 to 100% with one to seven copy numbers) (20). This peptide was also found to be highly conserved among Mucorales with 71 to 100% amino acid identity (table S1). Consequently, we hypothesized that the polyclonal antibodies should recognize CotH proteins on Mucorales fungi other than R. delemar. Immunostaining of Rhizopus oryzae, Lichtheimia corymbifera, Cunninghamella bertholletiae, Mucor circinelloides, Mucor ramosissimus, and Rhizomucor spp. showed robust binding of the anti-CotH3 antibodies to the germlings of these highly pathogenic Mucorales, while the preimmune IgG did not (Fig. 1C).
Given that the polyclonal anti-CotH3 antibodies bound several members of the Mucorales order, we posited that these antibodies would also protect mice from Mucorales other than R. delemar. Thus, we infected neutropenic mice intratracheally with five different Mucorales and, 24 hours after infection, treated them with a single intraperitoneal dose of 30 μg of purified anti-CotH3 IgG or the same amount of preimmune IgG. The antibodies significantly reduced mortality in mice infected with fungi tested, with the highest level of protection observed in mice infected with Rhizomucor and Apophysomyces (40 to 60% survival) (Fig. 3). Therefore, the anti-CotH3 antibodies protect against >95% of organisms causing mucormycosis.
Fig. 3. Anti-CotH3 antibodies protect neutropenic mice from infection by five other Mucorales.
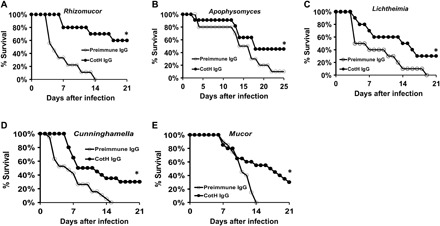
Neutropenic mice were infected intratracheally with Rhizomucor (A), Apophysomyces (B), Lichtheimia (C), Cunninghamella (D), or M. circinelloides (E) and then, 24 hours later, treated with 30 μg of anti-CotH3 IgG or isotype-matched control IgG. Survival of mice served as an end point. Inhaled inocula were 1.6 × 103, 3.7 × 103, 4.6 × 103, 2.2 × 103 (average inoculum of two experiments), and 2.2 × 103 (average inoculum of two experiments) for Rhizomucor (A), Apophysomyces (B), Lichtheimia (C), Cunninghamella (D), or M. circinelloides (E), respectively. Ten mice per group were used, except for (D) and (E), in which 20 mice per group were used. *P < 0.05.
Anti-CotH3 antibodies augment polymorphonuclear leukocyte–mediated damage of R. delemar via opsonophagocytosis
We next performed a series of experiments to investigate whether anti-CotH3 antibodies enhance polymorphonuclear leukocyte (PMN)–mediated killing of R. delemar in vivo. We first examined the role of PMNs in the antibody-mediated clearance of the fungus. Recruitment and activation of PMNs reflects a primary immunological response to invading pathogens and is a hallmark of vascular inflammation. One of the key enzymes released upon PMN activation is myeloperoxidase (MPO), a heme protein that neutralizes nitric oxide–dependent signaling cascades within the vasculature (22). Thus, we measured the effects of treatment with control or anti-CotH3 antibodies on the levels of MPO in organs of DKA mice (because these mice are not neutropenic) infected with R. delemar. The anti-CotH3 IgG significantly increased the MPO content of the lungs and spleen relative to the control IgG. As expected, the greatest increase in MPO occurred in the lungs, which are the primary target organ and spleen, which contains larger numbers of PMNs (23), with a 10-fold increase in both organs as compared to mice treated with control (Fig. 4A). Treatment with the anti-CotH3 antibodies did not significantly increase the MPO levels in the brain, likely because the antibodies prevented hematogenous dissemination, leading to a very low brain fungal burden (Fig. 2).
Fig. 4. Anti-CotH3 antibodies stimulate immune recognition of R. delemar in vivo and in vitro through opsonophagocytosis.
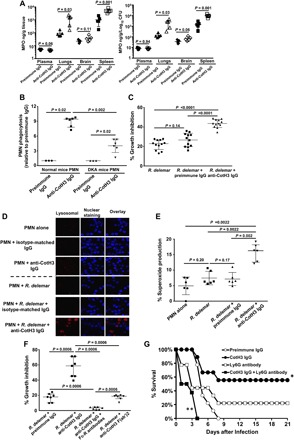
(A) Higher MPO levels detected in lungs and spleens harvested from DKA mice (n = 5 per group) infected with R. delemar (inhaled inoculum of 1.6 × 103 spores) and treated with 30 μg of anti-CotH3 IgG versus preimmune IgG. Treatment with the antibodies started 24 hours after infection, and the mice were euthanized 72 hours after infection. Data are presented as raw MPO values in nanograms per gram of tissue or normalized to burden of infection in each individually marked organ in nanograms per gram per log10 colony-forming unit (CFU). (B) Anti-CotH3 IgG enhanced the ability of PMNs harvested from normal or DKA mice in phagocytizing R. delemar ex vivo. (C) Incubation of PMNs harvested from DKA with anti-CotH3 IgG, but not preimmune IgG, enhanced their ability to damage R. delemar ex vivo as determined using the XTT assay. (D) Confocal imaging using LysoTracker Red staining confirms the activation of PMN by anti-CotH3 IgG showing acidification and maturation of the phagolysosome after phagocytosis of the fungal spores. (E) Anti-CotH3 IgG-activated PMNs harvested from DKA mice generated significantly higher amounts of ROS compared to control conditions including PMNs alone, PMNs + R. delemar, or PMNs + R. delemar + preimmune IgG. Blocking of Fc receptors (Fc-R) of PMNs harvested from DKA mice with Fc receptor antibodies abrogates the ability of PMNs to damage R. delemar even in the presence of anti-CotH3 IgG. (F) Conversely, F(ab)′2 anti-CotH3 IgG fragments fail to enhance the DKA mouse PMN–mediated damage activity of R. delemar. (G) Neutrophil depletion by Ly6G antibody completely reverses the enhanced survival of DKA mice (n = 9 per group except for mice treated with anti-CotH3 IgG + Ly6G antibody, which had 8) infected with R. delemar and treated with anti-CotH3 IgG (given at 30 μg 24 hours after infection). *P < 0.05 versus all groups, **P < 0.001 versus preimmune IgG, and P = 0.1 versus Ly6G antibody–treated mice. Data in (A) to (C), (E), and (F) are expressed as median ± interquartile changes.
Next, we assessed whether the anti-CotH3 antibodies enhanced the phagocytosis of R. delemar ex vivo. PMNs from normal or DKA mice were isolated and tested for their capacity to phagocytose R. delemar in the presence of control or the anti-CotH3 IgG. We found that the anti-CotH3 antibodies significantly enhanced the phagocytosis of R. delemar spores in both normal and DKA mice (Fig. 4B). The anti-CotH3 IgG also augmented the capacity of PMN of DKA mice to inhibit the metabolic activity of R. delemar, in contrast to the control IgG (Fig. 4C). Furthermore, the anti-CotH3 IgG enhanced the ability of murine peritoneal macrophages to kill R. delemar spores (5:1 multiplicity of infection, macrophages/R. delemar) ex vivo relative to the control IgG (P < 0.0002; fig. S2A).
We also investigated the mechanism by which the anti-CotH3 IgG enhanced R. delemar phagocytosis and growth inhibition. We stained murine PMNs with LysoTracker Red before incubating them with R. delemar in the presence or absence of the control or anti-CotH3 IgG. LysoTracker Red is a fluorescent dye that stains the acidic compartments in live cells, enabling tracking of phagolysosome maturation (24). Acidic organelles could only be detected when PMNs were incubated with R. delemar in the presence of anti-CotH3 IgG, indicating that enhanced phagocytosis of fungal spores in the presence of anti-CotH3 antibodies leads to maturation of the phagolysosome (Fig. 4D). Similar results occurred for peritoneal murine macrophage incubated with anti-CotH3 IgG (fig. S2B).
To determine whether the enhanced metabolic inhibition of R. delemar by murine PMN in the presence of the anti-CotH3 IgG was due to killing by reactive oxygen species (ROS), we measured ROS release using dihydrorhodamine (DHR) 123. This dye is oxidized by hydrogen peroxide formed from superoxide, resulting in the formation of a fluorescent product. A significant increase in ROS production by PMN incubated with R. delemar occurred in the presence of anti-CotH3 IgG when compared to PMN incubated with the fungus alone or with the fungus and preimmune IgG (Fig. 4E).
A pathogen is recognized by the F(ab′)2 fragment of an antibody, while the Fc portion of the antibody is, in turn, identified and bound by immune cells such as neutrophils via the Fc receptors, which triggers activation of phagocytosis. To confirm the opsonophagocytic potential of anti-CotH3 IgG, we investigated the role of the antibody F(ab′)2 fragment and the Fc receptor in the ability of murine PMN to kill the fungus. The impact of Fc receptor blocking or the F(ab′)2 fragment on R. delemar killing by PMNs was measured using the XTT [2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide] assay. While intact anti-CotH3 IgG enhanced murine PMN killing of R. delemar, blocking the PMN Fc receptor by antibodies completely abrogated fungal killing. Similarly, the use of F(ab′)2 fragments from CotH3 IgG did not enhance PMN-mediated killing of R. delemar over PMN incubated with the fungus in the presence of preimmune IgG (Fig. 4F). This finding shows that phagocytosis and subsequent killing of R. delemar spores by murine PMNs are triggered by anti-CotH3 IgG Fc fragment binding to the Fc receptor. These results also show that neutralization of CotH3 antigen by the F(ab′)2 fragments is not sufficient to trigger PMN-mediated killing.
Collectively, these data show that anti-CotH3 antibodies enhance phagocytosis of R. delemar, which leads to enhanced growth inhibition via maturation of the phagolysosome and ROS production. Therefore, the generated CotH3 IgGs are opsonophagocytic.
Because our in vitro and ex vivo data suggested an opsonophagocytic role for anti-CotH3 IgG, we investigated whether these antibodies protect by a similar mechanism in vivo. DKA mice were infected with R. delemar intratracheally. However, they were devoid of neutrophils by treatment with anti-Ly6G antibody (fig. S3). The lack of neutrophils in the DKA mouse model completely reversed the protective activity of anti-CotH3 antibodies (Fig. 4G), thus confirming the importance of their opsonophagocytic activity in vivo.
Monoclonal antibodies raised against CotH3 protect mice from pulmonary mucormycosis
Our encouraging results with polyclonal antibodies prompted us to generate mouse monoclonal antibodies against a 16-mer peptide of the CotH3 protein. Eight hybridoma cells were generated commercially, and their antibodies were screened for their capacity to block R. delemar–mediated damage to HUVEC using 51Cr-release assay. Previously, we have found that polyclonal anti-CotH3 IgG blocks fungal-induced damage to these cells (18). Of the eight hybridomas, the antibodies from four clones (C1 to C4) were found to block R. delemar–induced endothelial cell damage (fig. S4) and were thus selected for expansion. One of the four clones (C4) died after several passages, whereas clones C1 to C3 grew robustly. The purified monoclonal antibody from these three clones (all IgG1) was tested for their capacity to prevent pulmonary mucormycosis in the DKA and neutropenic mice infected with R. delemar. In the DKA mice, all monoclonal IgG1s tested protected mice similarly to the polyclonal anti-CotH3 IgG (Fig. 5A). Monoclonal antibodies C1 and C2 were tested in the neutropenic mice and significantly improved survival. Antibody C2 almost completely protected the neutropenic mice, significantly higher than the protection conferred by the polyclonal anti-CotH3 antibodies (Fig. 5B). Antibody C2 also recognized native CotH3 of R. delemar and the rCotH3p by Western blotting (fig. S5). It also bound to different Mucorales and the polyclonal antibody (fig. S6).
Fig. 5. Monoclonal antibodies against CotH3 protect DKA and neutropenic mice from R. delemar and other Mucorales.
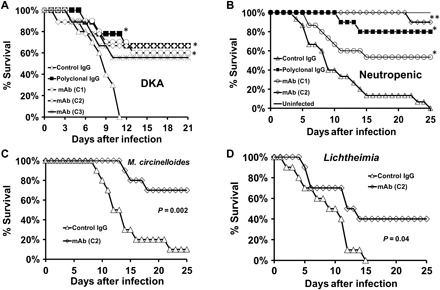
Treatment with 30 μg of three different clones of anti-CotH3 monoclonal antibodies (mAb) (C1 to C3), 24 hours after intratracheal R. delemar infection, protected DKA mice (n = 9 to 10 mice per group) (A) and neutropenic mice (n = 10 to 15 per group) (B) from lethal mucormycosis. *P < 0.05 versus isotype-matched control IgG; **P < 0.05 versus isotype-matched control IgG, C1 clone, and polyclonal IgG. C2 clone also protected neutropenic mice from mucormycosis due to M. circinelloides (C) and L. corymbifera (D). n = 10 per group for mice in (C) and (D). Confirmed inhaled inocula were 5.6 × 103, 2.2 × 103, 1.6 × 103, and 2.1 × 103 for (A) to (D), respectively.
Because of its superior activity and recognition of CotH on other Mucorales, the C2 antibody was chosen for further characterization of its protective activity against murine mucormycosis due to Mucorales other than Rhizopus. A single 30-μg dose of this antibody significantly reduced mortality in neutropenic mice infected with M. circinelloides and Lichtheimia (Fig. 5, C and D). These results support the development of the C2 antibody as a novel immunotherapeutic approach to treat mucormycosis.
C2 IgG1 is synergistic with antifungal drugs in vivo
Any immunotherapeutic treatment is likely to be applied as an adjunctive therapy to antifungal drugs. Thus, we investigated the protective activity of combining the C2 antibody with two antifungal drugs that are commonly used to treat mucormycosis. We infected DKA mice intratracheally and treated them 48 hours later (to replicate clinically advanced disease) with a single 30-μg dose of C2 with or without posaconazole (30 mg/kg) twice daily for 7 days. Posaconazole is often used for mucormycosis salvage therapy (25). As expected, the C2 antibody alone resulted in a reduced but significant protection when compared to mice given the monoclonal antibody 24 hours after infection (compare 50% survival in Fig. 6A to 70% survival in Fig. 5B). Posaconazole trended to enhance survival of DKA mice (33% survival) versus mice treated with isotype-matched control IgG1 (14% survival; P = 0.07). Impressively, all mice treated with the C2 antibody combined with posaconazole survived the infection (Fig. 6A) and appeared healthy at the termination of the study, and the lungs and brains harvested from surviving mice had no residual fungi as demonstrated by organ culturing. Concordant with these results, combination therapy significantly reduced the lung and brain fungal burden by 1 to 1.5 logs when harvested 72 hours after infection (Fig. 6, B and C).
Fig. 6. C2 monoclonal IgG1 synergistically protects DKA mice from R. delemar when combined with antifungal drugs.
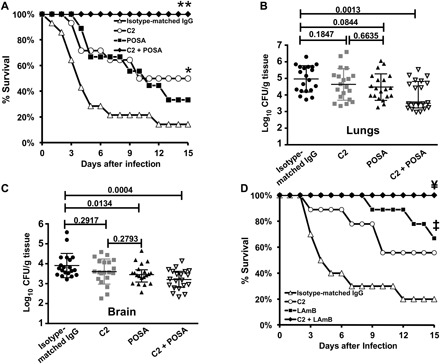
DKA mice were infected with R. delemar (confirmed inhaled inoculum of 3.9 × 103) and, 48 hours later, treated with either a single dose of 30 μg of C2 monoclonal IgG1, posaconazole (POSA) (30 mg/kg, twice daily) for 7 days, or a combination of both. End points were survival (A) (n = 9 to 14 per group) or tissue fungal burden in lungs (B) or brain (C) as determined by quantitative polymerase chain reaction (PCR). (D) The same model was used with similar treatment except that posaconazole treatment was substituted with LAmB (10 mg/kg per day for 4 days) (n = 9 to 10 per group). *P < 0.05 versus isotype-matched IgG, **P < 0.008 versus all other arms, ‡P < 0.006 versus isotype-matched IgG, ¥P < 0.03 versus isotype-matched IgG or C2, and P = 0.05 versus LAmB.
We also evaluated the efficacy of combination therapy of the C2 antibody with liposomal amphotericin B (LAmB), a drug that is the first-line therapy for mucormycosis (15). When therapy was initiated 48 hours after infection, the mice receiving either the C2 antibody or LAmB as monotherapy had improved survival, which was statistically significant for the mice treated with LAmB (56%; P = 0.005) but not the C2 antibody (P = 0.07). Mice treated with both the C2 antibody and LAmB had 100% long-term survival, which was significantly superior to that of mice receiving the control IgG (P = 0.0002) and the C2 antibody (P = 0.02), and trended to be better than the mice treated with LAmB (P = 0.05; Fig. 6D). Collectively, these results demonstrated that the C2 antibody significantly enhances the efficacy of currently used antifungals for treating mucormycosis.
DISCUSSION
We previously determined that Mucorales, including R. delemar, bind mammalian GRP78 via its cell surface fungal ligand CotH3, leading to HUVEC invasion and damage (18, 26). Repression of expression of either protein abrogates fungal-mammalian cell interaction and blocks fungal invasion and endothelial cell damage in vitro (18, 26). Inhibition of Rhizopus CotH3 expression prevents invasion even in mammalian cells overexpressing GRP78, highlighting the crucial role of this fungal protein in pathogenicity (18). In silico docking studies demonstrate that CotH3 conforms to a bimodal secondary structure that affords a complimentary and energetically favorable facet for binding to GRP78 (18). An important domain within this GRP78-binding region of CotH3 is predicted to be highly immunogenic and conserved among all medically important Mucorales, giving it the potential to be used for immunotherapeutic target. However, natural antibodies against CotH3 in healthy volunteers and in patients with mucormycosis are low. Thus, we reasoned that antibodies targeting CotH3 would be beneficial in treating murine mucormycosis, given the high importance of CotH proteins in the pathogenesis of mucormycosis (18, 19). Polyclonal antibodies raised against two peptides residing in the GRP78-binding region of CotH3 (18) not only prevented fungal adhesion/invasion to endothelial cells in vitro but also protected DKA mice from R. oryzae–associated pulmonary mucormycosis.
In this study, we confirmed that the anti-CotH3 polyclonal antibodies protected by prevention of invasion and subsequent hematogenous dissemination, as indicated by the almost negligible spread of infection from the initial site of infection (lungs) to the secondary target organ (brain) in anti-CotH3 antibody-treated mice. This lack of dissemination was also apparent by lack of angioinvasion in organs collected from anti-CotH3 IgG-treated mice as early as 2 days after infection. The fact that polyclonal antibodies alone can interrupt hematogenous spread to the brain supported its further development as a potential immunotherapeutic drug. Among immunocompromised hosts, DKA and neutropenic patients represent the patients who are the most susceptible to mucormycosis (1, 3). Therefore, any novel therapy must demonstrate efficacy in both patient populations to be clinically and commercially viable. Here, we show that the protective effect of the anti-CotH3 antibodies extends to neutropenic mice, suggesting that antibody-based therapy is likely to be efficacious in both DKA and neutropenic patients. We found that the anti-CotH3 IgG was highly protective when administered as a single dose of 30 μg per mouse. This low dose of anti-CotH3 IgG is about 1.2 mg/kg, which is within the dosage range of 1 to 15 mg/kg of most monoclonal antibodies that are approved for human use (27, 28).
Despite concordant studies showing an increase in mucormycosis around the world (8–13), it is still a relatively rare disease. Therefore, any commercially viable therapy must target all Mucorales especially in the absence of a reliable rapid assay to identify the species of the infecting fungus in patients with mucormycosis. CotH proteins are universally present in Mucorales but absent in other medically important organisms. This property makes them a desirable target for the development of specific therapy for mucormycosis. In addition, a 16-mer peptide sequence of CotH3 that was used for developing monoclonal antibody–based therapy is highly conserved among Mucorales with >70% amino acid identity, is surface-exposed, and, importantly, resides in the binding functional facet of the protein. Because of these desirable attributes, antibodies raised against this region recognized all Mucorales strains tested in vitro and demonstrated protective efficacy in mice. Although treatment with anti-CotH3 antibodies alone prolonged survival in mice infected with all strains of Mucorales tested, the extent of protection varied with the strain of Mucorales and was never 100%. However, when the lead C2 monoclonal antibody was combined with conventional antifungal agents, 100% of the mice survived with no evidence of residual infection, possibly leading to sterilizing immunity. These promising results suggest that combination therapy with the C2 antibody and conventional antifungal agents will likely be effective against multiple members of the Mucorales family.
The anti-CotH3 antiserum protects mice against mucormycosis by multiple mechanisms. Previously, we found that it inhibited the capacity of Mucorales to invade and damage host cells in vitro. Antibodies protect against infection by neutralizing virulence factors (28), directly inhibiting and/or killing the targeted organism (29), and/or enhancing clearance of the infecting organism by phagocytes (30). Here, we found that treatment of DKA mice with the anti-CotH3 antibodies resulted in an 8- to 20-fold increase in MPO content in the spleen and lungs, indicating that the antibody also enhances phagocyte recruitment. In addition, the polyclonal anti-CotH3 antibodies augmented neutrophil and peritoneal macrophage killing of R. delemar ex vivo in an Fc-dependent manner. As expected, the enhanced phagocytosis of R. delemar in the presence of anti-CotH3 antibodies was accompanied by increased acidification of phagolysosome, enhanced ROS production, and augmented killing of the fungus. These properties may be especially beneficial in patients who are in DKA, whose phagocytes have reduced capacity to recognize and clear Mucorales (31). Reducing the number of circulating neutrophil by anti-Ly6G antibody in the DKA mice reversed the protective effect seen with the anti-CotH3 antibodies to a level that is even worse than mice treated with preimmune IgG. These data emphasize the role of anti-CotH3 in enhancing neutrophil function in the DKA host.
A notable finding was that the anti-CotH3 antibodies also protected neutropenic mice from mucormycosis. One possible explanation for this result is that the antiserum enhanced the capacity of fungicidal activity of macrophages in vivo. Although administration of cyclophosphamide to mice results in complete pancytopenia, it has a lesser effect on tissue macrophages, which are usually derived from an embryonic, and not from hematopoietic, origin (32). These results suggest that the anti-CotH3 antibodies would likely be effective in neutropenic patients. Future studies to decipher the role of phagocytes in protection afforded by anti-CotH3 antibodies using transgenic mice (e.g., p47phox−/− CD11c-depleted mice) are underway.
The use of monoclonal antibodies in clinical application is superior to polyclonal antibodies owing to providing unlimited supply of antibodies with consistency in efficacy, production, as well as pharmacokinetic-pharmacodynamic and toxicity profiles (33). We were able to isolate several monoclonal antibodies that prevented invasion and damage of HUVECs in vitro and showed similar or superior activity to polyclonal anti-CotH3 antibodies in protecting DKA and neutropenic mice. Our lead monoclonal IgG1 (C2) performed better against other Mucorales such as M. circinelloides and Lichtheimia, with higher survival rates of mice compared to survival rates of infected mice treated with polyclonal antibodies or control IgG. Of critical clinical importance is the remarkable effectiveness of treatment of the C2 antibody in combination with either posaconazole or LAmB, even when therapy was initiated after 48 hours of infection. The capacity of the C2 antibody to enhance the efficacy of posaconazole was especially notable as this drug has only modest activity (10 to 30% survival) in the DKA mouse model when used as a single agent (21).
In summary, we show that antibodies targeting CotH proteins represent an effective and reliable novel therapy, especially when combined with currently approved drugs for the treatment of mucormycosis. The mechanism of action of these antibodies includes prevention of invasion, enhanced phagocyte recruitment, and increased fungal clearance via enhanced opsonophagocytosis. Our future studies are focused on producing an equally efficacious humanized version of the C2 monoclonal antibodies and developing a viable cell line that will produce the humanized version of the antibody in commercial quantities. Achieving this goal would enable manufacturing, toxicity studies, and clinical trials to determine the efficacy of the humanized anti-CotH3 antibody as adjunctive therapy.
MATERIALS AND METHODS
Organisms and culture conditions
R. delemar 99-880 (formerly classified as R. oryzae), R. oryzae 99-892, M. circinelloides f. jenssenii UTHSCSA DI15-131, and M. ramosissimus 97-1192 are clinical isolates obtained from the Fungus Testing Laboratory at the University of Texas Health Sciences Center at San Antonio (UTHSCSA). C. bertholletiae 182, also a clinical isolate, was a gift from T. Walsh (Weill Cornell Medical College of Cornell University, New York, NY, USA). L. corymbifera is another clinical isolate obtained from the DEFEAT Mucor clinical study (34). Apophysomyces ATCC 90757 was obtained from the American Type Culture Collection (ATCC; Manassas, VA), while Rhizomucor was obtained from a patient seen at Harbor-UCLA (University of California at Los Angeles) Medical Center. A. fumigatus AF293 was used for CotH3 antibody binding studies. All organisms were grown on potato dextrose agar (PDA; BD Diagnostic, NJ) plates by sprinkling silica beads that have been absorbed with spore suspension of each organism. The plates were incubated at 37°C for 4 to 7 days for Mucorales and 14 days for A. fumigatus until sporulation is apparent on confluent growth plates.
Spores were collected in endotoxin-free Dulbecco’s phosphate-buffered saline (PBS) containing 0.01 or 0.2% Tween 80 for Mucorales or Aspergillus, respectively. Collected spores were washed with PBS and counted with a hemocytometer to prepare the final inocula. To form germlings, spores were incubated in liquid yeast extract–peptone–dextrose [YPD; 1% yeast extract (Difco Laboratories, Detroit), 2% Bacto Peptone (Difco Laboratories), and 2% glucose (Sigma-Aldrich, St. Louis)] medium at 37°C with shaking for 3 hours.
Saccharomyces cerevisiae BJ5464 (ATCC) was used for rCotH3p production and was routinely cultured on YPD medium. When transformed with CotH3, it was grown on yeast nitrogen base (YNB) medium supplemented with ammonium sulfate without uracil.
Antibody binding assay
Rabbit polyclonal antibodies targeting two peptides (MGQTNDGAYRDPTDNN and GAGKKHNNAKQSWNW) or mouse monoclonal antibodies against the CotH3 peptide MGQTNDGAYRDPTDNN were commercially obtained from ProMab Biotechnologies (Richmond, CA). R. delemar spores or germlings (100 μl of 5 × 105 cells/ml) were incubated for an hour on ice with CotH3 purified IgG (3 to 100 μg/ml), monoclonal antibodies (100 μg/ml), or preimmune IgG (100 μg/ml). For the polyclonal antibodies, the cells were counterstained with anti-rabbit IgG–fluorescein isothiocyanate (FITC) conjugated or control IgG–FITC (eBioscience) for another hour on ice. For the monoclonal antibody binding, the cells were counterstained similar to polyclonal antibodies as previously, with the exception of using anti-mouse IgG–FITC or its respective control IgG–FITC (BD Biosciences). The cells were rinsed twice with staining buffer [2% fetal bovine serum (FBS) (Gibco, Thermo Fisher Scientific, Waltham, MA) in PBS] before determining the binding capacity of the antibodies using a FACSCalibur (Becton Dickinson) instrument equipped with an argon laser emitting at 488 nm. Fluorescence emission was read with a 515/40 band-pass filter. Fluorescence data were collected with logarithmic amplifiers. The population percent fluorescence of 104 events was calculated using the CellQuest software.
Polyclonal and monoclonal antibody preparation
Rabbit IgG antibodies were purified from rabbits before (preimmune IgG) or after immunization with the CotH3 peptides (anti-CotH3 IgG) by protein A/G spin column (Thermo Fisher Scientific) according to the manufacturer’s instructions. Monoclonal CotH3 hybridoma cells were propagated in WHEATON CELLine bioreactor 350 using protein-free hybridoma medium 1× (Gibco) for 5 to 7 days at 37°C in 5% CO2. The supernatant containing monoclonal antibody was collected and purified using protein G spin column (Thermo Fisher Scientific). The antibodies were dialyzed in PBS using a dialysis cassette (Thermo Fisher Scientific), and the purity of the antibody was confirmed by SDS–polyacrylamide gel electrophoresis (PAGE) before determining the concentration using the Bradford protein assay (Bio-Rad, Hercules, CA). Endotoxin level measured by a Limulus Amebocyte Lysate (LAL) kit (Charles River Laboratories) was determined to be <0.8 endotoxin units (EU)/ml for polyclonal antibodies and <0.4 EU/ml for monoclonal antibodies, which are below the body weight (5 EU/kg) set for intraperitoneal injection (35).
rCotH3p production and Western blot
The Flag tag–CotH3 construct was created using the yeast plasmid pXW55-URA3. To generate the pXW55-FLAG-TEV-CotH3-URA3 [8063–base pair (bp)] construct, 1705-bp CotH3 complementary DNA (cDNA) was synthesized from R. delemar 99-880 using an RNA extraction kit (Qiagen, Hilden, Germany) and a cDNA synthesis kit (Promega, Madison, WI) according to the manufacturers’ instructions. The synthesized CotH3 cDNA was flanked by Flag-TEV sequences at the 5′ end using polymerase chain reaction (PCR) and then subcloned into the Spe I and Pml I cloning site. The constructed vector was transformed into S. cerevisiae BJ5464, and the positive colonies were selected on uracil-free YNB medium. The positive yeast colonies containing the right constructs appeared after incubation for 3 days at 30°C, which were then confirmed by PCR and sequencing.
To produce the rCotH3p, a clone was grown in YPD medium at 30°C for 3 days with shaking at 200 rpm. The cells were centrifuged and the pellet was washed with tris-buffered saline (TBS) before suspending in a small volume of cold TBS. Suspended yeast cells were then disrupted by sonication for 30 min with 1-min intervals interrupted with 1-min storing on ice. The lysate was then centrifuged at 10,000g for 30 min. The collected supernatant was filtered through a 0.20-μm filter, followed by purification using anti-Flag magnetic beads (Sigma-Aldrich) according to the manufacturer’s instructions. The purity of CotH3 protein was measured by SDS-PAGE gel, followed by Western blotting using a mouse CotH3 monoclonal antibody (1:1000) and an anti-mouse IgG–horseradish peroxidase (HRP) antibody (1:3000) as a secondary antibody. Endotoxin level was measured by LAL as above. The use of yeast as an expression system provided endotoxin-free protein.
To detect the expression of CotH3 on R. delemar, spores (5 × 107) were grown in YPD medium overnight at 37°C. Mycelia were collected by filtration, washed briefly with PBS, and then ground thoroughly in liquid nitrogen using pestle and mortar for 3 min. The ground powder was immediately transferred to a microfuge tube containing 500 μl of extraction buffer that consisted of 50 mM tris-HCl (pH 7.5), 150 mM NaCl, and 10 mM MgCl2. The extraction buffer was supplemented with 1× Halt Protease Inhibitor Cocktails (Thermo Fisher Scientific). The sample was vortex-mixed vigorously for 1 min and then centrifuged for 5 min at 21,000g at 4°C. The supernatant was transferred to a new tube, and the protein concentration was determined using the Bradford method.
Human anti-CotH3 antibodies
To determine the level of natural anti-CotH3 antibodies, we collected sera from 12 healthy volunteers who routinely work with Mucorales and 11 patients diagnosed with definitive mucormycosis by either histology or cultures. The volunteer and patient samples were obtained at the Los Angeles Biomedical Research Institute at Harbor-UCLA Medical Center or Stanford University under an institutional review board (IRB)–approved protocol. Some of the patient samples were sequentially taken from the same patient following initial diagnosis (1 to 72 days after diagnosis). Anti-CotH3 antibody titers were measured using ELISA in 96-well plates. Each well was coated with either rCotH3p (100 μl of 5 μg/ml) in PBS or the 16-mer antigenic peptide dissolved in carbonate-bicarbonate buffer. Human sera were incubated for 1 hour at room temperature following a blocking step with PBS containing 3% bovine serum albumin. The wells were washed three times with PBS containing 0.05% Tween 20, followed by another three washes with PBS. Goat anti-human secondary antibody conjugated with HRP (Sigma-Aldrich) was added at a final dilution of 1:5000, and the plate was further incubated for 1 hour at room temperature. Wells were washed with PBS and incubated with the substrate containing 0.1 M citrate buffer (pH 5.0), 50 mg of o-phenylenediamine (Sigma-Aldrich), and 10 μl of 30% H2O2. The color was allowed to develop for 30 min, after which the reaction was terminated by addition of 3 N HCl and the optical density (OD) at 490 nm was determined in a microtiter plate reader. Negative control wells received only diluent, and background absorbance was subtracted from the test wells to obtain final OD readings. The ELISA titer was taken as the reciprocal of the last serum dilution that gave a positive OD reading (i.e., more than the mean OD of negative control samples plus two SDs).
Endothelial cell propagation and damage assay
Human umbilical vein endothelial cells (HUVEC) were collected by the method of Jaffe et al. (36). Cells were harvested using collagenase and grown in M-199 (Gibco) enriched with 10% FBS, 10% defined bovine calf serum, l-glutamine, penicillin, and streptomycin (all from Gemini Bio-Products, Sacramento, CA). Second-passage cells were grown to confluence in 96-well tissue culture plates (Corning Costar, NY) on fibronectin (BD Biosciences). All incubations were in 5% CO2 at 37°C. All propagations were conducted under endotoxin-free conditions [concentration of <0.01 IU/ml as determined by a chromogenic LAL assay (BioWhittaker Inc., Walkersville, MD)].
To screen for protective monoclonal antibodies, the HUVEC damage assay using radiolabeled chromium (51Cr) was used (37). Briefly, HUVECs grown in 96-well tissue culture plates containing detachable wells were incubated with Na251CrO4 (1 μCi per well) in M-199 medium for 16 hours. Excess 51Cr was aspirated, and wells were washed twice with prewarmed Hanks’ balanced salt solution (HBSS; ScienCell Research Laboratories, Carlsbad, CA). Cells were infected with fungal germlings (1.0 × 106 ml germinated for 1 hour at 37°C with 50 to 100 μg of the tested antibody) suspended in 200 μl of dextrose minimal essential medium (DMEM; Thermo Fisher Scientific) supplemented with 50 or 100 μg of the tested monoclonal antibody, purified CotH3 polyclonal IgG [as a positive control because we reported that these polyclonal antibodies block HUVEC damage (18)], or isotype-matched control IgG. Spontaneous 51Cr release was determined by incubating HUVECs in DMEM without R. delemar but with each of the tested antibody (to determine toxicity). After 4 hours of incubation at 37°C in a 5% CO2 incubator, 50% of the medium was aspirated from each well and transferred to glass tubes, and cells were manually detached and placed into another set of tubes. The amount of 51Cr in the aspirate and the detached well was determined by gamma counting. The total amount of 51Cr incorporated by endothelial cells in each well was calculated as the sum of radioactive counts per minute of the aspirated medium and radioactive counts of the corresponding detached wells. After data were corrected for variations in the amount of tracer incorporated in each well, the percentage of specific endothelial cell release of 51Cr was calculated as follows: [(experimental release × 2) − (spontaneous release × 2)]/[total incorporation − (spontaneous release × 2)]. Each experimental condition was tested at least in triplicate, and the experiment was repeated at least once.
Mouse models
Two mouse models were used in these studies: (i) the DKA and (ii) the neutropenic mice. Both models used male ICR mice (20 to 23 g) obtained from Envigo (NJ). For the DKA model, mice were injected with a single intraperitoneal dose of streptozotocin (210 mg/kg; Sigma-Aldrich) in 0.2 ml of Na citrate buffer 10 days before infection, as described previously (21). Glycosuria and ketonuria were confirmed in all mice 7 days after streptozotocin treatment. In addition, on days −2 and +3 relative to infection, DKA mice were given a dose of cortisone acetate (250 mg/kg) by subcutaneous injection. For the neutropenic model, mice were injected with cyclophosphamide (200 mg/kg, given intraperitoneally) and cortisone acetate (500 mg/kg, given subcutaneously) on days −2 and +3 relative to infection (21).
Mouse PMNs were isolated from peripheral blood cells of DKA or normal mice after heparinization using dextran sedimentation followed by Ficoll-Paque (GE Healthcare, Chicago, IL), as described (38). After removing contaminating erythrocytes by hypotonic lysis, the neutrophils were washed, counted with a hemocytometer, and resuspended at the appropriate concentration with downstream assay solution or medium. Neutrophils were kept on ice until use, within an hour of collection.
For the DKA mice depleted of neutrophils, mice were injected intraperitoneally with anti-mouse Ly6G (1A8 clone) antibody (4 mg/kg) (BioLegend) on days −2, 0, +2, and +4 relative to intratracheal challenge with R. delemar as above. Neutrophil depletion was confirmed by microscopically counting circulating neutrophils in ~50 μl of samples collected from the mice tail veins using Wright-Giemsa staining. The Ly6G antibody treatment resulted in neutropenia for at least days +4 to +6 (fig. S3).
Peritoneal macrophage collection
ICR mice (6 weeks old) were euthanized by cervical dislocation. Each mouse was cleaned with 70% alcohol, and the abdominal skin was removed with sterile scissors to expose the intact peritoneal wall. The peritoneum was then injected with 10 ml of cold PBS without calcium and magnesium with 5 mM EDTA using a 20-gauge needle syringe. The mouse was slightly shaken for 1 min, followed by harvesting the peritoneal fluid slowly with the same syringe. The peritoneal fluid was then transferred into a 50-ml conical tube, which was kept on ice while washing three times with cold PBS buffer. The cells were resuspended in RPMI 1640 with 10% FBS at a concentration of 2 × 105 cells/ml before transferring them to a six-well plate (5 ml per well). The plate was incubated for 6 hours at 37°C with 5% CO2 to allow macrophages to adhere to the surface of the plate. Nonadherent cells were removed, and the remaining cells represented >90% macrophages. The cell concentration was adjusted in HBSS before performing the ex vivo assay.
Infection and treatment
Immunosuppressed mice were infected intratracheally with 25 μl of PBS containing 2.5 × 105 spores of each Mucorales (inoculum for M. circinelloides was 2.5 × 106 spores). Immediately after challenge, two mice from each group infected with different Mucorales were euthanized and their lungs were homogenized in PBS and quantitatively cultured on PDA plates containing 0.1% Triton X-100. Colonies were counted after 24-hour incubation period at 37°C. After infection, mice were treated daily with 5 mg in 0.2 ml of irrigation water of ceftazidime subcutaneously to prevent bacterial superinfection (21). Antibody treatment with a single dose of 30, 100, or 300 μg of polyclonal CotH3 purified IgG, preimmune polyclonal purified IgG, CotH3 monoclonal antibody, or isotype-matched control mouse IgG (Bio X Cell) started 24 or 48 hours after infection and was administered by intraperitoneal injection. Time to moribundity (equated with survival) served as the primary end point.
As a secondary end point and in some experiments, fungal burden in the lungs and brains (primary and secondary target organs) (21) was determined 72 hours after infection by quantitative PCR assay, as we previously described (39). Values were expressed as log10 spore equivalent per gram of tissue. Histopathological examination was carried out on sections of the harvested organs after fixing in 10% zinc formalin. The fixed organs were embedded in paraffin, and 5-mm sections were stained with hematoxylin and eosin (40).
For determining the protective activity of the monoclonal antibodies with antifungal drugs, DKA mice were infected as described and treated with the monoclonal antibody posaconazole (Merck and Co., Kenilworth, NJ), LAmB (Gilead Sciences, Foster City, CA), or a combination of the monoclonal antibody and either antifungal drug started 48 hours after infection. Monoclonal antibody was given once by intraperitoneal injection at 30 μg, posaconazole was administered by oral gavage at 30 mg/kg twice daily for 7 days, and LAmB was given intravenously at 10 mg/kg per day for 4 days. Time to moribund state and tissue fungal burden served as end points as above.
Mouse MPO assay
To determine the level of PMN recruitment to site of infection and the effect of antibody treatment on this recruitment, we used the MPO ELISA Kit (Abcam, Cambridge, UK). Tissue homogenate collected from organs processed for tissue fungal burden as described (to correlate the data of PMN recruitment to tissue fungal burden and reduce the number of mice used) was processed according to the manufacturer’s instructions. Results were expressed as the amount of MPO per gram of tissue or MPO per gram per colony-forming unit (CFU) (41).
PMN phagocytosis assay
To investigate the phagocytic activity of mouse PMNs collected from normal or DKA mice and the effect of anti-CotH3 polyclonal IgG on this process, fungal spores were stained with Alexa Fluor 488 carboxylic acid (Thermo Fisher Scientific) for 60 min on ice and then incubated with PMNs (1:20, PMNs/R. delemar) at 37°C for 30 min before the cells were quantified by flow cytometry (42). PMNs containing phagocytized organisms were identified by size and fluorescence as described.
Growth inhibition assay
Inhibition of growth of R. delemar by PMN from DKA mice was conducted using the XTT assay (38). R. delemar spores were collected and washed twice with HBSS. Fifty-eight microliters of PMNs (3.4 × 105 cells/ml) and 100 μl of R. delemar (1 × 105 cells/ml) were cocultured in 96-well flat-bottom plates at 37°C for 2 hours with 10% heat-inactivated mouse serum (2:1, PMNs/R. delemar) with or without preimmune IgG or anti-CotH3 IgG. Next, the plate was stored overnight at 4°C to lyse all the PMNs. Forty microliters of XTT (1 mg/ml; Sigma-Aldrich) and 2 μl of 0.625 mM menadione solution (Sigma-Aldrich) were added to the 96-well plates to allow the conversion of the XTT to its formazan derivative (43). The plate was shaken for 1 to 2 min at 100 rpm until complete dissolution of the formazan derivatives. The plate was incubated at 37°C for additional 6 hours before the XTT conversion was measured using a Dynex MRX Revelation microtiter plate reader (BioSurplus, San Diego, CA) at OD at 450 nm. XTT conversion by metabolically active cells was calculated after subtracting the background OD of simultaneously incubated wells containing media and reagents without spores.
Fc receptor and F(ab′)2 blocking assay
To determine the role of the Fc receptor in promoting anti-CotH3 antibody-mediated PMN phagocytosis and growth inhibition of R. delemar spores, we conducted the growth inhibition assay as described above but with inclusion of antibodies (3 μg/ml; BD Biosciences) targeting the Fc receptor. We also evaluated the role of F(ab′)2 fragments purified from anti-CotH3 IgG in inducing the inhibition of R. delemar spores by PMNs collected from DKA mice (44). F(ab′)2 fragments from anti-CotH3 IgG were purified with the Pierce F(ab′)2 Preparation Kit (Thermo Fisher Scientific) according to the manufacturer’s instruction. The inhibition assay was conducted as above using the same assay parameters.
Lysosome staining
A LYSO-ID Red lysosomal detection kit (Enzo Life Sciences, Farmingdale, NY) was used for the lysosome detection in PMNs or macrophages. Lysosomal staining was performed according to the manufacturer’s recommendations. Briefly, PMNs or macrophages collected from DKA mice were suspended in 250 μl of HBSS with 10% mouse heat-inactivated serum at a concentration of 2 × 106 cells/ml. The phagocytes were incubated with or without 250 μl of R. delemar spores and R. delemar spores + either preimmune IgG or anti-CotH3 IgG (3 μg/ml) for 30 min at 37°C with gentle shaking (1 rpm). A LYSO-ID Red detection reagent (0.5 μl) (Enzo Life Sciences) and Hoechst 33342 nuclear stain (0.5 μl) were added to 500 μl of cells. The samples were then incubated for an additional 30 min at 37°C and washed with 100 μl of 1× assay buffer. The stained cells were imaged with a Leica confocal microscope using a Texas Red filter set for lysosomal imaging and a 4′,6-diamidino-2-phenylindole (DAPI) filter set for Hoechst 33342 staining nuclear imaging.
Assessment of ROS
The ability of anti-CotH3 purified polyclonal IgG to stimulate superoxide production by PMNs collected from DKA mice was assessed ex vivo using DHR123 dye. This technique relies on the induction of fluorescence of DHR in the presence of ROS. Briefly, PMNs incubated with R. delemar (1:2, PMN/R. delemar spores) with anti-CotH3 IgG or isotype-matched IgG (3 μg/ml) were incubated in the dark at 37°C for 25 min in the presence and absence of 100 μM DHR123 (Molecular Probes, Eugene, OR) in prewarmed HBSS with Ca/Mg (ScienCell Research Laboratories, Carlsbad, CA). The cells were then analyzed for size and fluorescence with a FACSCalibur instrument (Becton Dickinson) within 30 min after DHR staining, as described above.
Statistical analysis
Statistical analysis was carried out by the nonparametric log-rank test for survival studies or Wilcoxon rank sum test for the rest of the comparisons. For all comparisons, a P value of <0.05 was considered significant.
Study approval
Animal studies were approved by the Institutional Animal Care and Use Committee of the Los Angeles Biomedical Research Institute at Harbor-UCLA Medical Center, according to the NIH guidelines for animal housing and care. Human endothelial cell collection was approved by the IRB at Los Angeles Biomedical Research Institute at Harbor-UCLA Medical Center. Sera from healthy volunteers or mucormycosis patients were collected using an IRB-approved protocol and tested for CotH3 antibody titers at LA BioMed (protocol #11671).
Supplementary Material
Acknowledgments
We acknowledge the technical assistance of the perinatal nurses of the Harbor-UCLA Medical Center for collection of umbilical cords. Research described in this manuscript was conducted at the research facilities of the Los Angeles Biomedical Research Institute at Harbor-UCLA Medical Center. Funding: This work was supported by Public Health Service grants R01 AI063503 and R43 AI138904 to A.S.I. Author contributions: T.G. helped in the evaluation of the efficacy of the antibodies, conducted screening for the protective monoclonal antibodies in vitro and in vivo, and edited the paper. S.A. purified polyclonal antibodies, propagated and maintained the monoclonal antibodies, and helped in the in vivo testing of the monoclonal antibodies. S.S.M.S. designed and produced the rCotH3p. Y.G. conducted protein extractions and Western blot analysis. H.H.J. produced the rCotH3p. L.Z. determined the antibody titers using ELISA. S.W.F. conducted the histopathological studies. D.A.S. provided sera from patients with mucormycosis. J.E.E., S.G.F., and P.U. advised on experimental design and helped with manuscript preparation. A.S.I. conducted the overall design and implementation of the study, helped with the murine models, analyzed the data, and wrote the manuscript. All authors reviewed the manuscript. Competing interests: A.S.I. owns shares in Vitalex Biosciences, a start-up company that is developing immunotherapies and diagnostics for mucormycosis. A.S.I., T.G., S.G.F., and J.E.E. are inventors on a patent related to this work (USA: no. US 2013/0108642, filed on 2 May 2013; Europe: no. 12 831 998.5-1401, filed on 13 December 2017). The authors declare no other competing interests. Data and materials availability: The anti-CotH3 antibodies (polyclonal or monoclonal) can be provided by Los Angeles Biomedical Research Institute pending scientific review and a completed material transfer agreement. Requests for the antibodies should be submitted to A.S.I. at Ibrahim@labiomed.org. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/6/eaaw1327/DC1
Table S1. The R. delemar MGQTNDGAYRDPTDNN peptide is highly conserved among CotH proteins from other Mucorales.
Fig. S1. Effect of anti-CotH3 antibodies on the metabolic activity and germination of R. delemar.
Fig. S2. Anti-CotH3 antibodies enhance murine macrophage killing of R. delemar ex vivo through maturation of the phagolysosome.
Fig. S3. Induction of neutropenia in DKA mice by treatment with anti-Ly6G antibody.
Fig. S4. Screening for protective monoclonal anti-CotH3 antibodies using 51Cr-release assay.
Fig. S5. Recognition of native and recombinant R. delemar CotH3 protein by monoclonal anti-CotH3 antibody.
Fig. S6. Percent binding (relative to negative control without antibodies) of monoclonal C2 and polyclonal anti-CotH3 antibodies to different Mucorales.
REFERENCES AND NOTES
- 1.Spellberg B., Edwards J. Jr., Ibrahim A., Novel perspectives on mucormycosis: Pathophysiology, presentation, and management. Clin. Microbiol. Rev. 18, 556–569 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ribes J. A., Vanover-Sams C. L., Baker D. J., Zygomycetes in human disease. Clin. Microbiol. Rev. 13, 236–301 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.A. M. Sugar, Agents of mucormycosis and related species, in Principles and Practice of Infectious Diseases, G. L. Mandell, J. E. Bennett, R. Dolin, Eds. (Elsevier Churchill Livingstone, 2005), vol. 2, pp. 2973–2984. [Google Scholar]
- 4.Gomes M. Z. R., Lewis R. E., Kontoyiannis D. P., Mucormycosis caused by unusual mucormycetes, non-Rhizopus, -Mucor, and -Lichtheimia species. Clin. Microbiol. Rev. 24, 411–445 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neblett Fanfair R., Benedict K., Bos J., Bennett S. D., Lo Y.-C., Adebanjo T., Etienne K., Deak E., Derado G., Shieh W.-J., Drew C., Zaki S., Sugerman D., Gade L., Thompson E. H., Sutton D. A., Engelthaler D. M., Schupp J. M., Brandt M. E., Harris J. R., Lockhart S. R., Turabelidze G., Park B. J., Necrotizing cutaneous mucormycosis after a tornado in Joplin, Missouri, in 2011. N. Engl. J. Med. 367, 2214–2225 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Tribble D. R., Rodriguez C. J., Combat-related invasive fungal wound infections. Curr. Fungal Infect. Rep. 8, 277–286 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ingram P. R., Suthananthan A. E., Rajan R., Pryce T. M., Sieunarine K., Gardam D. J., Heath C. H., Cutaneous mucormycosis and motor vehicle accidents: Findings from an Australian case series. Med. Mycol. 52, 819–825 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Kontoyiannis D. P., Wessel V. C., Bodey G. P., Rolston K. V. I., Zygomycosis in the 1990s in a tertiary-care cancer center. Clin. Infect. Dis. 30, 851–856 (2000). [DOI] [PubMed] [Google Scholar]
- 9.Kontoyiannis D. P., Marr K. A., Park B. J., Alexander B. D., Anaissie E. J., Walsh T. J., Ito J., Andes D. R., Baddley J. W., Brown J. M., Brumble L. M., Freifeld A. G., Hadley S., Herwaldt L. A., Kauffman C. A., Knapp K., Lyon G. M., Morrison V. A., Papanicolaou G., Patterson T. F., Perl T. M., Schuster M. G., Walker R., Wannemuehler K. A., Wingard J. R., Chiller T. M., Pappas P. G., Prospective surveillance for invasive fungal infections in hematopoietic stem cell transplant recipients, 2001–2006: Overview of the Transplant-Associated Infection Surveillance Network (TRANSNET) Database. Clin. Infect. Dis. 50, 1091–1100 (2010). [DOI] [PubMed] [Google Scholar]
- 10.Greenberg R. N., Scott L. J., Vaughn H. H., Ribes J. A., Zygomycosis (mucormycosis): Emerging clinical importance and new treatments. Curr. Opin. Infect. Dis. 17, 517–525 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Bitar D., Van Cauteren D., Lanternier F., Dannaoui E., Che D., Dromer F., Desenclos J.-C., Lortholary O., Increasing incidence of zygomycosis (mucormycosis), France, 1997–2006. Emerg. Infect. Dis. 15, 1395–1401 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chakrabarti A., Singh R., Mucormycosis in India: Unique features. Mycoses 57 (suppl. 3), 85–90 (2014). [DOI] [PubMed] [Google Scholar]
- 13.El Zein S., El-Sheikh J., Zakhem A., Ibrahim D., Bazarbachi A., Kanj S. S., Mucormycosis in hospitalized patients at a tertiary care center in Lebanon: A case series. Infection 46, 811–821 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Alastruey-Izquierdo A., Hoffmann K., de Hoog G. S., Rodriguez-Tudela J. L., Voigt K., Bibashi E., Walther G., Species recognition and clinical relevance of the zygomycetous genus Lichtheimia (syn. Absidia Pro Parte, Mycocladus). J. Clin. Microbiol. 48, 2154–2170 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Skiada A., Lanternier F., Groll A. H., Pagano L., Zimmerli S., Herbrecht R., Lortholary O., Petrikkos G. L., Diagnosis and treatment of mucormycosis in patients with hematological malignancies: Guidelines from the 3rd European Conference on Infections in Leukemia (ECIL 3). Haematologica 98, 492–504 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rapidis A. D., Orbitomaxillary mucormycosis (zygomycosis) and the surgical approach to treatment: Perspectives from a maxillofacial surgeon. Clin. Microbiol. Infect. 15, 98–102 (2009). [DOI] [PubMed] [Google Scholar]
- 17.Ibrahim A. S., Spellberg B., Avanessian V., Fu Y., Edwards J. E. Jr., Rhizopus oryzae adheres to, is phagocytosed by, and damages endothelial cells in vitro. Infect. Immun. 73, 778–783 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gebremariam T., Liu M., Luo G., Bruno V., Phan Q. T., Waring A. J., Edwards J. E. Jr., Filler S. G., Yeaman M. R., Ibrahim A. S., CotH3 mediates fungal invasion of host cells during mucormycosis. J. Clin. Invest. 124, 237–250 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gebremariam T., Lin L., Liu M., Kontoyiannis D. P., French S., Edwards J. E. Jr., Filler S. G., Ibrahim A. S., Bicarbonate correction of ketoacidosis alters host-pathogen interactions and alleviates mucormycosis. J. Clin. Invest. 126, 2280–2294 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chibucos M. C., Soliman S., Gebremariam T., Lee H., Daugherty S., Orvis J., Shetty A. C., Crabtree J., Hazen T. H., Etienne K. A., Kumari P., O’Connor T. D., Rasko D. A., Filler S. G., Fraser C. M., Lockhart S. R., Skory C. D., Ibrahim A. S., Bruno V. M., An integrated genomic and transcriptomic survey of mucormycosis-causing fungi. Nat. Commun. 7, 12218 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luo G., Gebremariam T., Lee H., French S. W., Wiederhold N. P., Patterson T. F., Filler S. G., Ibrahim A. S., Efficacy of liposomal amphotericin B and posaconazole in intratracheal models of murine mucormycosis. Antimicrob. Agents Chemother. 57, 3340–3347 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van der Veen B. S., de Winther M. P. J., Heeringa P., Myeloperoxidase: Molecular mechanisms of action and their relevance to human health and disease. Antioxid. Redox Signal. 11, 2899–2937 (2009). [DOI] [PubMed] [Google Scholar]
- 23.Puga I., Cols M., Barra C. M., He B., Cassis L., Gentile M., Comerma L., Chorny A., Shan M., Xu W., Magri G., Knowles D. M., Tam W., Chiu A., Bussel J. B., Serrano S., Lorente J. A., Bellosillo B., Lloreta J., Juanpere N., Alameda F., Baró T., de Heredia C. D., Torán N., Català A., Torrebadell M., Fortuny C., Cusí V., Carreras C., Diaz G. A., Blander J. M., Farber C.-M., Silvestri G., Cunningham-Rundles C., Calvillo M., Dufour C., Notarangelo L. D., Lougaris V., Plebani A., Casanova J.-L., Ganal S. C., Diefenbach A., Aróstegui J. I., Juan M., Yagüe J., Mahlaoui N., Donadieu J., Chen K., Cerutti A., B cell–helper neutrophils stimulate the diversification and production of immunoglobulin in the marginal zone of the spleen. Nat. Immunol. 13, 170–180 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Viegas M. S., Estronca L. M. B. B., Vieira O. V., Comparison of the kinetics of maturation of phagosomes containing apoptotic cells and IgG-opsonized particles. PLOS ONE 7, e48391 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Greenberg R. N., Mullane K., van Burik J.-A.-H., Raad I., Abzug M. J., Anstead G., Herbrecht R., Langston A., Marr K. A., Schiller G., Schuster M., Wingard J. R., Gonzalez C. E., Revankar S. G., Corcoran G., Kryscio R. J., Hare R., Posaconazole as salvage therapy for zygomycosis. Antimicrob. Agents Chemother. 50, 126–133 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu M., Spellberg B., Phan Q. T., Fu Y., Lee A. S., Edwards J. E. Jr., Filler S. G., Ibrahim A. S., The endothelial cell receptor GRP78 is required for mucormycosis pathogenesis in diabetic mice. J. Clin. Invest. 120, 1914–1924 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hendrikx J. J. M. A., Haanen J. B. A. G., Voest E. E., Schellens J. H. M., Huitema A. D. R., Beijnen J. H., Fixed dosing of monoclonal antibodies in oncology. Oncologist 22, 1212–1221 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilcox M. H., Gerding D. N., Poxton I. R., Kelly C., Nathan R., Birch T., Cornely O. A., Rahav G., Bouza E., Lee C., Jenkin G., Jensen W., Kim Y.-S., Yoshida J., Gabryelski L., Pedley A., Eves K., Tipping R., Guris D., Kartsonis N., Dorr M.-B.; MODIFY I and MODIFY II Investigators , Bezlotoxumab for prevention of recurrent Clostridium difficile Infection. N. Engl. J. Med. 376, 305–317 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Torosantucci A., Chiani P., Bromuro C., De Bernardis F., Palma A. S., Liu Y., Mignogna G., Maras B., Colone M., Stringaro A., Zamboni S., Feizi T., Cassone A., Protection by Anti-β-glucan antibodies is associated with restricted β-1,3 glucan binding specificity and inhibition of fungal growth and adherence. PLOS ONE 4, e5392 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Monari C., Casadevall A., Retini C., Baldelli F., Bistoni F., Vecchiarelli A., Antibody to capsular polysaccharide enhances the function of neutrophils from patients with AIDS against Cryptococcus neoformans. AIDS 13, 653–660 (1999). [DOI] [PubMed] [Google Scholar]
- 31.Chinn R. Y., Diamond R. D., Generation of chemotactic factors by Rhizopus oryzae in the presence and absence of serum: Relationship to hyphal damage mediated by human neutrophils and effects of hyperglycemia and ketoacidosis. Infect. Immun. 38, 1123–1129 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yona S., Kim K.-W., Wolf Y., Mildner A., Varol D., Breker M., Strauss-Ayali D., Viukov S., Guilliams M., Misharin A., Hume D. A., Perlman H., Malissen B., Zelzer E., Jung S., Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38, 79–91 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dimitrov D. S., Marks J. D., Therapeutic Antibodies: Current state and future trends – Is a paradigm change coming soon? Methods Mol. Biol. 525, 1–27 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spellberg B., Ibrahim A. S., Chin-Hong P. V., Kontoyiannis D. P., Morris M. I., Perfect J. R., Fredricks D., Brass E. P., The Deferasirox-AmBisome Therapy for Mucormycosis (DEFEAT Mucor) study: A randomized, double-blinded, placebo-controlled trial. J. Antimicrob. Chemother. 67, 715–722 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Malyala P., Singh M., Endotoxin limits in formulations for preclinical research. J. Pharm. Sci. 97, 2041–2044 (2008). [DOI] [PubMed] [Google Scholar]
- 36.Jaffe E. A., Nachman R. L., Becker C. G., Minick C. R., Culture of human endothelial cells derived from umbilical veins. Identification by morphologic and immunologic criteria. J. Clin. Invest. 52, 2745–2756 (1973). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ibrahim A. S., Gebremariam T., Liu M., Chamilos G., Kontoyiannis D., Mink R., Kwon-Chung K. J., Fu Y., Skory C. D., Edwards J. E. Jr., Spellberg B., Bacterial endosymbiosis is widely present among zygomycetes but does not contribute to the pathogenesis of mucormycosis. J. Infect. Dis. 198, 1083–1090 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsuchimori N., Sharkey L. L., Fonzi W. A., French S. W., Edwards J. E. Jr., Filler S. G., Reduced virulence of HWP1-deficient mutants of Candida albicans and their interactions with host cells. Infect. Immun. 68, 1997–2002 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ibrahim A. S., Bowman J. C., Avanessian V., Brown K., Spellberg B., Edwards J. E. Jr., Douglas C. M., Caspofungin inhibits Rhizopus oryzae 1,3-β-d-glucan synthase, lowers burden in brain measured by quantitative PCR, and improves survival at a low but not a high dose during murine disseminated zygomycosis. Antimicrob. Agents Chemother. 49, 721–727 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ibrahim A. S., Gebermariam T., Fu Y., Lin L., Husseiny M. I., French S. W., Schwartz J., Skory C. D., Edwards J. E. Jr., Spellberg B. J., The iron chelator deferasirox protects mice from mucormycosis through iron starvation. J. Clin. Invest. 117, 2649–2657 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin L., Ibrahim A. S., Xu X., Farber J. M., Avanesian V., Baquir B., Fu Y., French S. W., Edwards J. E. Jr., Spellberg B., Th1-Th17 cells mediate protective adaptive immunity against Staphylococcus aureus and Candida albicans infection in mice. PLOS Pathog. 5, e1000703 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tóth E. J., Boros É., Hoffmann A., Szebenyi C., Homa M., Nagy G., Vágvölgyi C., Nagy I., Papp T., Interaction of THP-1 monocytes with conidia and hyphae of different Curvularia strains. Front. Immunol. 8, 1369 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meshulam T., Levitz S. M., Christin L., Diamond R. D., A simplified new assay for assessment of fungal cell damage with the tetrazolium dye, (2,3)-bis-(2-methoxy-4-nitro-5-sulphenyl)-(2H)-tetrazolium-5-carboxanilide (XTT). J. Infect. Dis. 172, 1153–1156 (1995). [DOI] [PubMed] [Google Scholar]
- 44.Luo G., Ibrahim A. S., French S. W., Edwards J. E. Jr., Fu Y., Active and passive immunization with rHyr1p-N protects mice against hematogenously disseminated candidiasis. PLOS ONE 6, e25909 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/6/eaaw1327/DC1
Table S1. The R. delemar MGQTNDGAYRDPTDNN peptide is highly conserved among CotH proteins from other Mucorales.
Fig. S1. Effect of anti-CotH3 antibodies on the metabolic activity and germination of R. delemar.
Fig. S2. Anti-CotH3 antibodies enhance murine macrophage killing of R. delemar ex vivo through maturation of the phagolysosome.
Fig. S3. Induction of neutropenia in DKA mice by treatment with anti-Ly6G antibody.
Fig. S4. Screening for protective monoclonal anti-CotH3 antibodies using 51Cr-release assay.
Fig. S5. Recognition of native and recombinant R. delemar CotH3 protein by monoclonal anti-CotH3 antibody.
Fig. S6. Percent binding (relative to negative control without antibodies) of monoclonal C2 and polyclonal anti-CotH3 antibodies to different Mucorales.