

Abstract
Bacterial toxin-antitoxin systems are important factors implicated in growth inhibition and plasmid maintenance. Type II toxin-antitoxin pairs are regulated at the transcriptional level by the antitoxin itself. Here, we examined how the HigA antitoxin regulates the expression of the Proteus vulgaris higBA toxin-antitoxin operon from the Rts1 plasmid. The HigBA complex adopts a unique architecture suggesting differences in its regulation as compared to classical type II toxin-antitoxin systems. We find that the C-terminus of the HigA antitoxin is required for dimerization and transcriptional repression. Further, the HigA structure reveals that the C terminus is ordered and does not transition between disorder-order states upon toxin binding. HigA residue Arg40 recognizes a TpG dinucleotide in higO2, an evolutionary conserved mode of recognition among prokaryotic and eukaryotic transcriptional factors. Comparison of the HigBA and HigA-higO2 structures reveals the distance between helix-turn-helix motifs of each HigA monomer increases by ~4 Å in order to bind to higO2. Consistent with these data, HigBA binding to each operator is two-fold less tight than HigA alone. Together, these data show the HigB toxin does not act as a co-repressor suggesting potential novel regulation in this toxin-antitoxin system.
Keywords: toxin-antitoxin complex, transcriptional repressor, DNA-binding protein, plasmid maintenance, X-ray crystallography
Graphical Abstract
INTRODUCTION
Nearly all bacteria encode a diverse set of toxin-antitoxin gene pairs with the capacity to limit cell growth in response to environmental stress (Moyed and Bertrand 1983; Gerdes et al. 2005; Gonzalez Barrios et al. 2006; Harrison et al. 2009; Wang et al. 2011). These systems consist of a toxin that inhibits cell growth by blocking essential cellular processes during stress and an antitoxin that neutralizes the toxin during normal growth. Under conditions of stress, the labile antitoxins are proteolytically degraded and the more stable toxins are liberated to inhibit growth (Van Melderen et al. 1994; Christensen et al. 2004). In order for toxin-antitoxins to function as effective governors of cell growth in response to stress, several system parameters must be regulated. Prior to the appearance of stress, a sufficient amount of antitoxin needs to be expressed to maintain toxin quiescence, allowing for normal growth. When stress is encountered, toxins need to be freed from their cognate antitoxins to enable cells to halt their growth (through toxin activity). Since toxins are activated by antitoxin proteolysis, too much antitoxin may dampen the responsiveness of the system and delay growth inhibition. Moreover, too much toxin (an amount that exceeds the capacity of antitoxin neutralization) also needs to be avoided, as this would slow or halt growth in the absence of stress. Therefore, the expression of toxin and antitoxins must be finely regulated for a response to stress.
To establish and maintain the optimal levels of their two system components, bacterial antitoxins typically serve as transcriptional autorepressors that constitute a negative feedback loop (Page and Peti 2016). When the appropriate amount of antitoxin is reached, the antitoxin binds upstream of the toxin-antitoxin operon and represses new transcription. Antitoxins can weakly repress transcription in the absence of their toxin partners ensuring continued transcription until the appropriate amount of toxin is reached (Afif et al. 2001; Overgaard et al. 2008; Garcia-Pino et al. 2010). Often the toxin can serve as a transcriptional co- and anti-repressor depending on its abundance relative to the antitoxin (Afif et al. 2001; Overgaard et al. 2008; Garcia-Pino et al. 2010). Once a particular ratio of the toxin and antitoxin is exceeded, the toxin functions as an anti-repressor that severely decreases the antitoxin affinity for DNA allowing for transcription. For each toxin-antitoxin transcript, a disproportionate amount of antitoxin is translated because of the presence of a stronger ribosome binding site than the toxin’s (Li et al. 2014). In some well-studied toxin-antitoxin systems, toxins exert their co-repressor functions by influencing the structure of their antitoxins. Toxin binding can order intrinsically disordered regions of their antitoxins (Kamada et al. 2003; Garcia-Pino et al. 2010) and can promote and/or disrupt the formation of antitoxin complexes that are proficient for DNA binding (Afif et al. 2001; Kedzierska et al. 2007; Garcia-Pino et al. 2008; De Jonge et al. 2009; Garcia-Pino et al. 2010). Whether these regulatory mechanisms can be generally applied to understand other toxins-antitoxin systems is unknown.
Antitoxin proteins have two major functions during normal growth. They inhibit toxin function by direct binding and repress transcription to limit expression (Loris and Garcia-Pino 2014; Page and Peti 2016). Antitoxins are structurally diverse and contain either a ribbon-helix-helix (RHH) (De Jonge et al. 2009; Boggild et al. 2012), Phd/YefM (Garcia-Pino et al. 2010), SpoVT/AbrB (Dienemann et al. 2011), or a helix-turn-helix (HTH) DNA-binding motifs (Brown et al. 2009; Schumacher et al. 2009; Schureck et al. 2014). These distinct antitoxin architectures raise the possibility that the regulatory mechanisms that establish and maintain the appropriate amounts of antitoxin proteins may be distinct from those that have been established for more traditional systems including CcdAB, RelBE, and bacteriophage P1 Phd-Doc (Afif et al. 2001; Overgaard et al. 2008; Garcia-Pino et al. 2010).
The host inhibition of growth BA (higBA) operon was identified on the Rts1 plasmid associated with Proteus vulgaris in a post-operative urinary tract infection that displayed resistance to antibiotics (Tian et al. 1996). higBA is classified as a bacterial toxin-antitoxin pair whereby the HigB toxin protein inhibits protein synthesis by cleaving adenosine-rich mRNA transcripts on actively translating ribosomes (Hurley and Woychik 2009; Schureck et al. 2015; Schureck et al. 2016a; Schureck et al. 2016b). Homologues of HigBA are also chromosomally distributed although their structure and regulation suggest their mechanism may be distinct from P. vulgaris HigBA (Christensen-Dalsgaard et al. 2010; Kirkpatrick et al. 2016; Hadzi et al. 2017). Structures of the heterotetrameric P. vulgaris HigBA complex (two HigAs and two HigBs) reveal that the HigB toxin is a member of the RelE family of bacterial toxins while the HigA antitoxin contains a HTH DNA-binding motif that recognizes its operator for transcriptional autorepression (Schureck et al. 2014). The structure also indicated a number of differences between the HigBA complex and other RelE toxin-antitoxin family members. For example, other antitoxins that recognize RelE toxin family members typically contain ribbon-helix-helix (RHH) DNA-binding motifs that require dimerization to form a single DNA-binding motif (Kamada and Hanaoka 2005; Boggild et al. 2012; Ruangprasert et al. 2014). HigA antitoxin contains a HTH motif that is a complete DNA-binding motif; therefore, the two HigA antitoxins in the HigBA complex contain two DNA-binding motifs in contrast to other RelE family members that contain a single DNA-binding motif. Another distinction of the P. vulgaris HigBA complex is that the HigA antitoxin does not wrap around the HigB toxin for inactivation but, instead, has a C-terminal extension that engages the adjacent HigA. We previously determined that this C-terminus is critical for HigA dimerization and the HigA dimer is required for binding to its operator (Schureck et al. 2014). In this study, we sought to understand the molecular basis of HigA interaction with its DNA operator.
RESULTS
Dimerization is critical for HigA to repress transcription of hig.
The P. vulgaris Rts1 plasmid encodes the toxin higB gene followed by the antitoxin higA antitoxin gene, an inverted gene organization compared as to other RelE family members (Fig. 1A) (Gerdes et al. 2005). The hig promoter (Phig) controls the transcription of both genes and is regulated by two operator sites (higO1 and higO2) that overlap with portions of the Phig −35 and −10 promoter sites. The HigA protein binds both operators and represses transcription from Phig. We previously purified a HigA lacking its C-terminal 21 amino acids (Δ84–104) that disrupts HigA dimerization in the context of the HigBA complex (Schureck et al. 2014). Although each monomeric HigA variant contained a complete HTH DNA-binding motif, the HigA monomer was unable to bind to its DNA operator as assessed by EMSA. Additionally, disruption of the HigA dimer had no impact on the ability of a monomer of HigB to bind in vitro. We next wanted to test that the HigA C-terminal truncation specifically disrupted the transcriptional repression function of HigA in vivo while preserving its toxin neutralization function. The toxin higB gene, without or with either the full-length or truncated antitoxin higA gene, was placed under the control of the arabinose-inducible expression pBAD promoter. The expression plasmids were introduced into E. coli BW25113 and grown to mid-exponential growth phase in rich medium, serially diluted, and spotted onto agar plates without or with 0.2% arabinose (to induce gene expression). Cell growth was inhibited when expression of the higB toxin gene was induced in the absence of higA and normal cell growth was restored when HigB and full-length HigA were co-expressed, as previously observed (Tian et al. 1996) (Fig. 1B). Importantly, normal cell growth was also observed when HigB and the truncated HigA were co-expressed, indicating that the C-terminal portion of HigA is dispensable for its toxin neutralization function in vivo (Fig. 1B). Thus, dimerization appears to be feature of HigA that is dedicated to its transcriptional repression function rather than toxin neutralization.
Figure 1. HigA dimerization is dispensable for toxin inhibition but necessary for transcriptional repression.
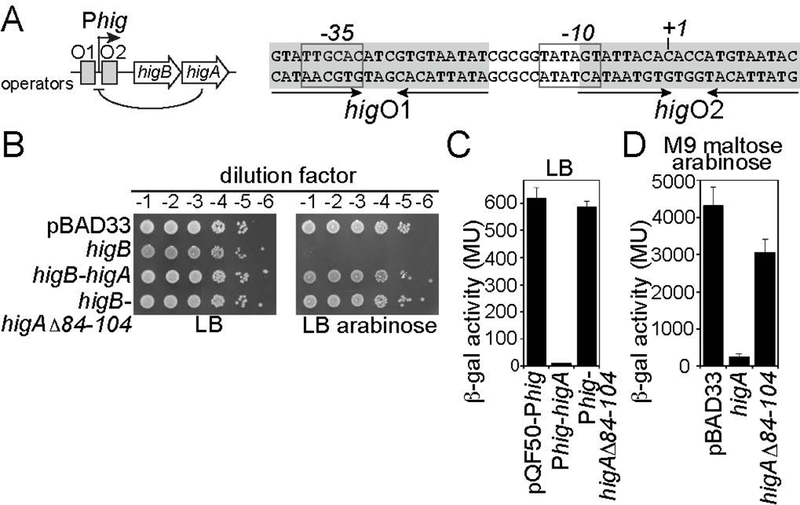
A. Organization of the hig operon with the regions HigA recognizes shown in grey shading and the −35 and −10 promoter regions boxed. B. Spot dilution assay of E. coli BW25113 transformed with indicated plasmids, overexpressed and the indicated amounts were plated on LB and LB in the presence of 0.2% arabinose. C. β-gal assays of E. coli BW25113 transformed with indicated plasmids in LB medium. D. β-gal assays of E. coli BW25113 transformed with indicated plasmids in M9 maltose media and 0.2% arabinose.
To better understand how HigA controls higBA gene expression in vivo, we constructed a series of transcriptional reporters for the hig promoter. First, the hig promoter was cloned upstream of the lacZ gene (encoding the β-galactosidase enzyme) in the transcriptional reporter plasmid pQF50. Next, either the full-length or truncated higA gene (Δ84–104) was cloned between the hig promoter and the lacZ reporter gene, creating two different synthetic bicistronic operons where both higA and lacZ are expressed under the control of the hig promoter. The transcriptional reporter plasmids were introduced into E. coli BW25113 and grown to mid-exponential phase in rich medium and β-gal activity was measured (Fig. 1C). The strain containing the transcriptional reporter without higA (Phig-lacZ) produced ~600 Miller units (MU) of β-gal activity (Fig. 1C). In the strain containing the transcriptional reporter with the full-length higA (Phig-higA-lacZ), this activity was decreased nearly 60-fold (from ~630 to ~10 MU) through the transcriptional repression function of HigA. The strain bearing the transcriptional reporter with higA(Δ84–104) that disrupts HigA dimerization (Phig-higAΔ84–104-lacZ), however, had nearly the same amount of β-gal activity as the reporter without higA (~590 MU) (Fig. 1C). One possible interpretation could be that HigA(Δ84–104) expression is compromised. However, since coexpression of this variant with HigB leads to normal growth (Fig. 1B), this strongly suggests that HigA(Δ84–104) expression is sufficient to suppress HigB toxicity. Collectively, these data demonstrate that dimerization of HigA is necessary for transcriptional repression.
In the pQF50-Phig-higA-lacZ transcriptional reporter plasmid, the level of higA gene expression is a product of the strength of the hig promoter and HigA-mediated transcriptional repression. The configuration of this reporter system is simple and convenient but it lacks the ability to compare the transcriptional repression of HigA variants at defined expression levels. To perform this analysis, either the full-length or truncated higA gene (Δ84–104) was cloned into the arabinose-inducible expression plasmid pBAD33. The expression plasmids were then introduced into E. coli BW25113 bearing the Phig-lacZ transcriptional reporter (without higA). The strains were grown to mid-exponential phase in minimal medium (M9 maltose) supplemented with 0.2% arabinose and β-gal activity was measured. The strain with the Phig-lacZ transcriptional reporter and pBAD33 (without higA) produced ~4200 MU of β-gal activity (Fig. 1D). The β-gal activity in the strain containing pBAD33 with the full-length higA was decreased more than 20-fold (~150 MU), demonstrating that the amount of HigA produced at this expression level could effectively repress transcription from the hig promoter in trans (Fig. 1D). The strain containing pBAD33 with the truncated HigA(Δ84–104) was incapable of high levels of transcriptional repression as assessed by its high level of β-gal activity (~3000 MU). These results show that, at defined HigA expression levels, dimerization is necessary for HigA to repress transcription activity from the hig promoter. Additionally, the results show that HigA is capable of repressing transcription from the hig promoter in the absence of HigB, suggesting that HigB co-repressor function may not be necessary in the HigBA system. This seems to be in contrast to the prevailing models used to describe the transcriptional control of other toxin-antitoxin systems where the toxin is often necessary for effective transcriptional repression by the antitoxin (Afif et al. 2001; Overgaard et al. 2008; Garcia-Pino et al. 2010). This is not a trivial distinction between these systems, as conditional toxin co- and anti-repressor function is the central tenant used to understand how toxin-antitoxin systems control the expression of their own genes to be primed to respond to stress. These data strongly suggest that the molecular mechanisms that underpin the priming of HigBA are distinct from those that explain systems including RelBE, Phd-Doc, CcdAB, MqsRA, DinJ-YafQ and HicAB (Afif et al. 2001; Overgaard et al. 2008; Garcia-Pino et al. 2010; Brown et al. 2013; Ruangprasert et al. 2014; Turnbull and Gerdes 2017).
Distance between HigA HTH motifs increases in the absence of HigB.
The X-ray crystal structure of the heterotetrameric HigBA complex revealed how the antitoxin HigA inhibits HigB but whether the toxin HigB influences the overall conformation of HigA was unclear (Schureck et al. 2014). This is important because in other toxin-antitoxin systems, the toxin can profoundly influence the ordering of antitoxin regions that are intrinsically disordered (Kamada and Hanaoka 2005; Li et al. 2008; De Jonge et al. 2009; Garcia-Pino et al. 2010). This disordered-to-ordered transition has been shown to be important for stabilization of the antitoxin for transcriptional repression (Loris and Garcia-Pino 2014). In the heterotetrameric HigBA complex, the last eleven C-terminal residues of HigA (amino acids 94–104) are disordered in the presence of HigB but this region does not interact with HigB (Schureck et al. 2014). We next solved the X-ray crystal structure of antitoxin HigA in the absence of HigB to 1.9 Å (Fig. 2A; Table S1). HigA crystallizes in the C2 space group with one HigA dimer per asymmetric unit. The final HigA model was built for residues 3–94 and 6–99 (out of 104 total residues) for monomers A and B of the dimer, respectively. HigA is a single domain protein and adopts a compact, five α−helix bundle with α2 and α3 comprising the HTH motif that is exposed at its N-terminus (Fig. 2A). HigA dimerization is mediated via interactions between α5 and its C-termini that extend to interact with each HigA monomer. Recognition of the hig operator is likely mediated by α3 while α2 helps to position α3 in the major groove consistent with other classical bacteriophage transcriptional repressors that HigA structurally resembles (Shimon and Harrison 1993; Watkins et al. 2008). These bacteriophage repressors form dimers that separate the recognition helices, one from each monomer, by the same distance as successive major grooves of the operator DNA. Sedimentation velocity analytical ultracentrifugation (AUC) studies show that HigA is a 2.15 S particle with an estimated molecular mass of 25.9 kDa (the molecular mass of each HigA polypeptide is 11.5 kDa) providing additional support that HigA is a dimer (Fig. 2B).
Figure 2. HigA is an obligate dimer.
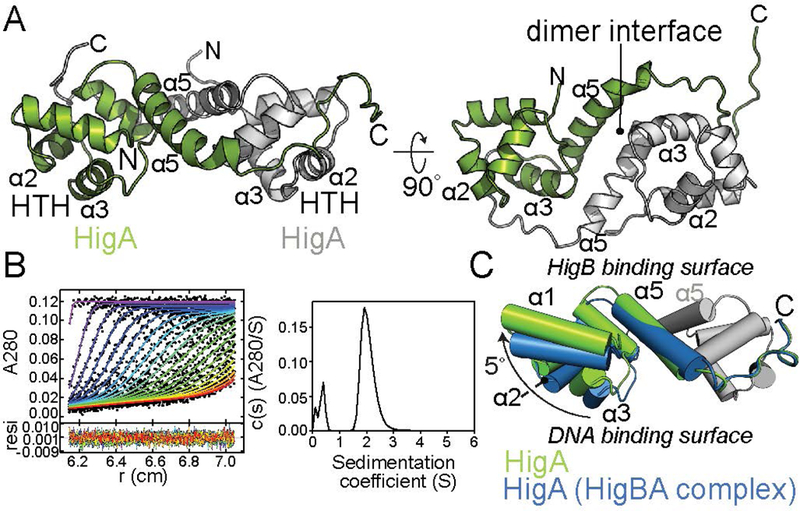
A. 1.9 Å X-ray crystal structure of HigA reveals the maintenance of the dimer interface in the absence of the HigB toxin. The DNA binding helix-turn-helix (HTH) motif and the dimer interface are indicated. B. Analytical ultracentrifugation of HigA produced a signal-average s20,w peak at 2.15 S corresponding to an estimated molecular weight of 25.9 kDa. C. Comparison of the HigA dimer (green) and HigA in the context of the HigBA complex (blue; PDB code 4MCT) reveals a 5° move away from the DNA binding surface involving α1, α2, and α3.
The overall structural architecture of the HigA dimer remains largely unchanged in the presence or absence of the toxin HigB (root mean square deviation (rmsd) of ~1.1 Å for 185–186 α carbon backbone atoms aligned). This low rmsd provides support that the toxin HigB does not induce large conformational rearrangements of HigA upon binding in contrast to other systems as mentioned above. However, one noticeable change is seen at the HigA dimer interface (Fig. 2C). Aligning a single protomer of the HigA dimer reveals a movement of HTH α-helices 1, 2, and 3 backbone residues of ~4.0 Å and a shift outward of 5.1° in the absence of toxin HigB (Fig. 2C). This movement increases the distance between each HTH motif by ~1.5 Å (to 29.4 Å from 27.9 Å as measured from Arg40 in each protomer) as compared to the HigA dimer in the HigBA complex (Schureck et al. 2014). If the spacing between α3 of the two HigA protomers is important for recognition of the hig operator, the remodeling of the HigA dimeric interface induced by HigB binding could modulate the strength of HigB binding to hig.
Structure of HigA bound to higO2.
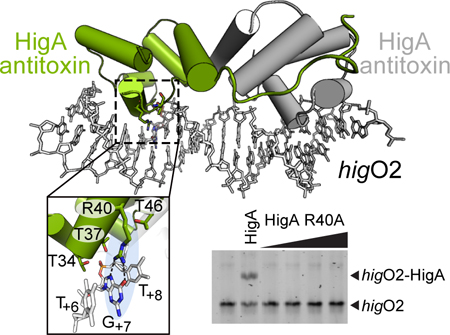
To understand how HigA recognizes its operator at the molecular level, we solved the X-ray crystal structure of HigA bound to the higO2 DNA operator to 2.9 Å (Fig. 3; Table S1). The HigA-DNA complex crystallizes in the space group P65 with three HigA dimers and three 21 nucleotide, double-stranded DNA in the asymmetric unit. The final HigA model was built for residues 4–92 and 4–91 (out of 104 total residues) in dimer A, 5–92 and 5–95 for dimer B, and 6–96 and 7–92 for dimer C. The three HigA dimers in the asymmetric unit are similar with rmsd values ranging 0.424 – 0.537 Å. A DALI search reveals that HigA-DNA is similar to the Shewanella oneidensis HipA-HipB-DNA toxin-antitoxin complex (PDB code 4PU4), Pseudomonas aeruginosa transcriptional repressor RsaL-DNA complex (PDB code 5J2Y), and Escherichia coli HipB antitoxin-DNA complex (PDB code 4YG1) with Z-scores of 6.2, 5.8, and 5.3, and r.m.s.d. values of 5.5, 2.1, and 2.9 Å, respectively (using 66, 55 and 59 aligned Cα atoms, respectively) (Holm and Rosenstrom 2010).
Figure 3. Structural basis of HigA-DNA operator recognition.
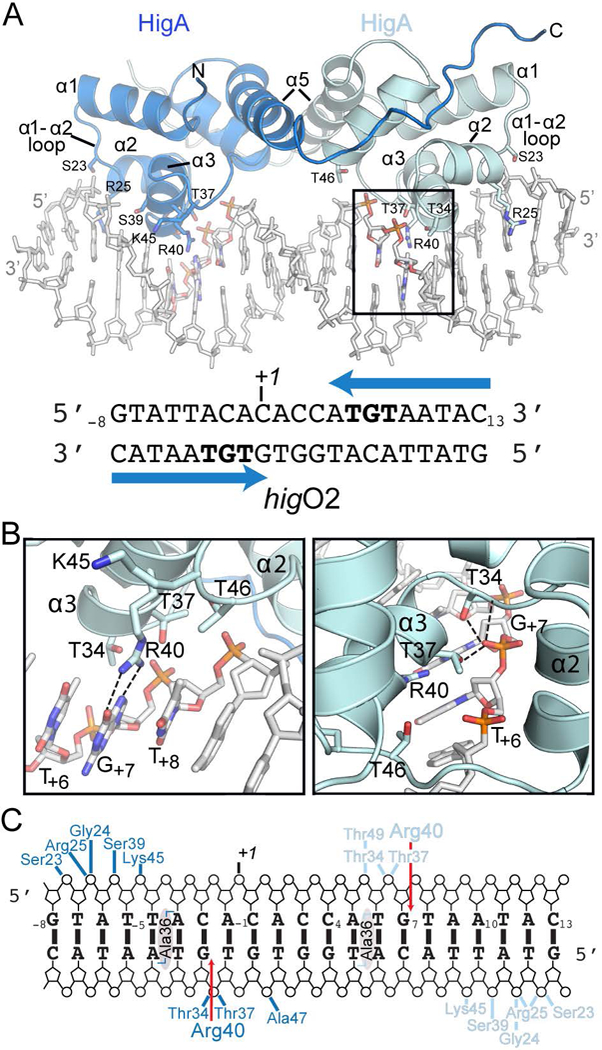
A. 2.9 Å X-ray crystal structure of HigA bound to higO2. One helix-turn-helix (HTH) motif of a HigA monomer is boxed. The higO2 is shown with the blue arrows indicating the inverted repeats HigA recognizes and the specific nucleotides contacted are shown in bold. B. Zoomed in view of HigA α2 and α3 of the HTH motif that interact directly with nucleotides T+6, G+7 and T+8. HigA residue Arg40 hydrogen bonds to G+7. The phosphate of G+7 is contacted by Thr34 and Thr37 that may serve to stabilize the interaction between Arg40 and the nucleobase of G+7 (right panel). C. Schematic representation of interactions between HigA residues with higO2. Van der waals interactions between Ala36 in both monomers is shaded grey.
Each HigA dimer recognizes a single inverted repeat of the operator with no apparent crosstalk between HigA dimers (Fig. 3A). Sequence specific interactions are mediated by α3 recognition of the nucleobases in the major groove while sequence-independent interactions with the phosphate backbone on the opposite strand are mediated by the α1-α2, α2-α3 and α3-α4 loops. As is often the case with HTH motifs (Shimon and Harrison 1993), three residues (Thr34, Thr36 and Arg40) are displayed on the surface of the α3 recognition helix facing the major groove of the inverted operator repeats (Fig. 3B). The hydroxyl groups of Thr34 and Thr37, and the backbone amine of Thr34, interact with the G+7 phosphate while the side chain methyl groups of Ala36 and Thr34 form van der Waals interactions with the nucleobase C5 methyl of T+6 (Fig. 3C). HigA also makes hydrophobic interactions in recognition of the operator via the side chain methyl group of Thr46 with the C5 methyl of T+8. Sequence-specific interactions include hydrogen bonds between the hydroxyl groups of Ser23 and Ser39 with the G+12 and T+10 phosphate oxygens, respectively, and an ionic interaction between the Lys45 amino and the T+9 phosphate oxygen. Non-sequence specific interactions between HigA and its operator are between the backbone amines of both Gly24 and Arg25 and the phosphate oxygens of A+11.
HigA α3 residue Arg40 specifically recognizes the DNA major groove. The Arg40 side chain guanidino moiety makes bifurcated hydrogen bonding interactions with the Hoogsteen face of G+7 (via the O6 and N7 atoms) (Fig. 3B). Additionally, the guanidino moiety stabilizes interactions with the C5 methyl group of T+8 via cation-pi interactions. The interaction between Arg40 and the T+8G+7 dinucleotide is similar to an evolutionary conserved mode of recognition of TG and methylated CG dinucleotides by prokaryotic and eukaryotic transcription factors termed YpG interactions (where Y indicates a pyrimidine) (Lamoureux et al. 2004; Liu et al. 2013). Together, these interactions likely facilitate HigA specific recognition of higO2.
To identify how HigA binding might alter higO2, we analyzed the backbone and nucleobase geometry of higO2 DNA using the program Curves+ (Blanchet et al. 2011). The most prominent finding of this analysis was that there are local deformations of the major and minor grooves as compared to B-form DNA. In the structure of HigA bound to higO2, the major groove was narrowed at the site where the HTH inserts (~9.9–10 Å) and widens (~11.8 Å) between both HTH motifs on the opposite side of the DNA (Fig. S1) (the major groove of B-form DNA is 11.4 Å). Likewise, the minor groove expands (~7.4–7.7 Å) on the opposite side of where the HTH motifs contact DNA and contracts in the center of higO2 between the HigA monomers (~6.9 Å). These backbone deformations from B-form DNA may be required to facilitate productive HigA-DNA interactions (Fig. S1).
Arginine 40 governs hig recognition and transcriptional repression by HigA.
To investigate the importance of Arg40 in operator recognition, we constructed a HigA variant with an Arg40 to Ala substitution and analyzed its function both in vivo and in vitro. To confirm that Arg40 is important for the transcriptional repression function of HigA and not its toxin neutralization function, we performed the spot-dilution assay used described in Fig. 1C to evaluate the ability of the point mutant to inhibit HigB toxicity. Normal cell growth was observed when HigB and the HigA R40A variants were co-expressed, indicating that HigA Arg40 was dispensable for its toxin neutralization function in vivo (Fig. 4A). To examine the importance of Arg40 in the ability of HigA to control higB-higA transcription, a pBAD33 plasmid with the HigA R40A variant was introduced into E. coli BW25113 bearing the Phig-lacZ transcriptional reporter plasmid. When the strain was grown to mid-exponential phase in minimal medium with 0.2% arabinose, it produced nearly the same amount of β-gal activity as the strain with pBAD33 without higA (Fig. 4B). This result demonstrates that Arg40 is important for HigA to repress transcription from the hig promoter. We next determined that HigA R40A is unable to bind to its DNA operator in vitro, even up to a concentration of 1 μM HigA R40A protein (Fig. 4C). Further, we confirmed that HigA R40A retained its ability to form a dimer (molecular weight of ~30 kDa), an oligomeric state known to be required for DNA binding in vitro (Schureck et al. 2014) (Fig. 4D). Lastly, we mutated both guanosines in each inverted repeat to adenosines (G+7 as shown in Fig. 4B) and observed no wild-type HigA binding (again to a concentration of 1 mM) (Fig. 4E). These data demonstrate that HigA Arg40 is important for operator recognition and interacts specifically with nucleotide G+7 but is not critical for HigA dimerization or HigB neutralization.
Figure 4. HigA Arg40 and G+7 are necessary for HigA recognition of higO2.
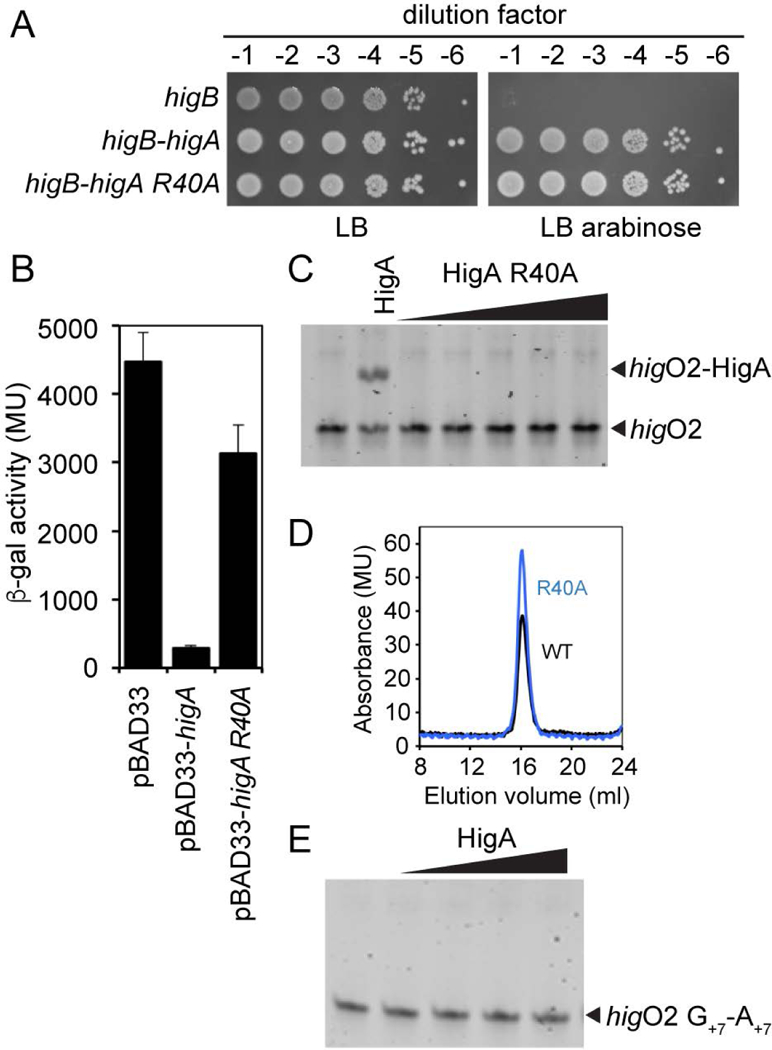
A. Spot dilution assay of E. coli BW25113 transformed with indicated plasmids, overexpressed with the indicated amounts were plated on LB and LB in the presence of 0.2% arabinose. B. β-gal assays of E. coli BW25113 transformed with indicated plasmids. C. Electrophoretic mobility shift assay of HigA R40A and higO2. D. Size exclusion chromatography analysis of HigA wild-type and the R40A variant. E. Electrophoretic mobility shift assay of the wild-type HigA and higO2 containing a G+7 to A+7 mutation (along with a C+7 to U+7 to maintain Watson-Crick base-pairing).
Comparison of HigBA, HigA and HigA-higO2 structures.
Although the HigB toxin is not required for the formation of the HigA dimer (Schureck et al. 2014), binding of HigB does influence the overall architecture of HigA as evidenced by superimposition (Fig. 5A). The distance between the HigA HTH motifs located in each protomer when bound to HigB is 27.2 Å (as measured by the distance between Arg40 located in the middle of each operator recognition helix α3 of each promoter). HigA binding to higO2 increases the distance between the HTH motifs to by ~4 Å to 31.0 Å and moves a HigA promoter 12° relative to the other promoter. This movement is necessary to expand the distance between the two recognition helices of the HigA dimer to bind to the two inverted repeat sequences and two major grooves of the operator. In the presence of HigB, the distance between the HTH motifs decreases to a distance that would cause a steric clash between HigA helix α3 and higO2 (Fig. 5A).
Figure 5. Structural rearrangements of HigA during operator recognition.
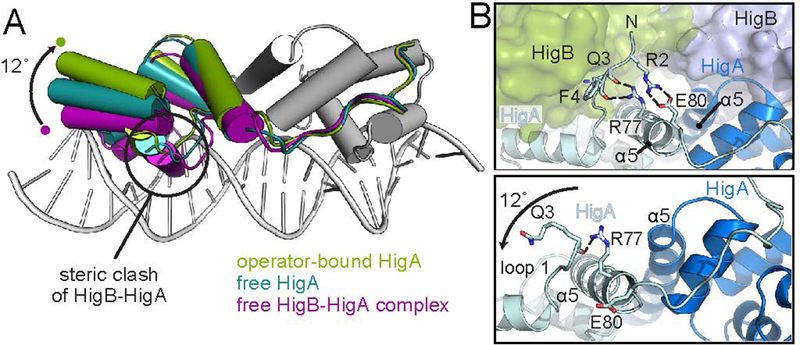
A. Superposition of free HigA (this study), the HigB-HigA complex (PDB code 4MCT), and DNA operator-bound HigA (this study) using the second HigA monomer (grey) as an anchor point. The HigA dimer hinges ~12° away from the DNA surface upon DNA recognition as compared to HigA bound to HigB. In the context of both apo HigA and in the HigB-HigA complex, clashes between α2 and α3 with DNA would occur. Rearrangement of the one monomer of HigA is required to allow the HTH motif to fully engage DNA. B. The HigA N terminus packs against the toxin HigB and forms interactions with α5 that may restrict its conformation (top panel). In the absence of HigB, the interactions between loop 1 and α5 are disrupted (bottom panel).
These structural comparisons also suggest other changes to HigA that may occur upon HigB binding (Fig. 5B). For example, in the presence of the toxin HigB but the absence of higO2, the N-terminal loop of HigA packs against HigB and forms a hydrogen bonding and salt bridge network between HigA residues Arg2 and Gln3 with Glu80 and Arg77 of HigA α5, respectively (Fig. 5B, top). In the HigA dimer structure solved in this study, these interactions between HigA loop 1 and α5 are weakened by rotation of α5 residues that ablate and remodel these interactions (Fig. 5B, bottom). This disruption between Arg2 and Glu80 is also seen in the HigA-higO2 operator structure. Considering that the HigA dimerization occurs at the α5-α5 interface and remodeling of the HigA protomers relative to the other in the absence of HigB appears to be mediated by the α5-α5 interface (Fig. 2A, 2C), the consequence of disrupting interactions between HigA loop 1 and α5 may contribute to the increase in distance between HigA HTH recognition helices (Fig. 5A). These structural comparisons suggest that HigB may help to rigidify HigA’s conformational freedom of rotation.
The HigBA complex binds two-fold less tight than HigA to higO1 or higO2.
Previously, we and others have found that the HigBA complex binds DNA (Tian et al. 2001; Schureck et al. 2014). However, it is unclear how the conformational changes observed in HigA alone versus in complex with HigB will affect operator binding. Therefore, we next performed electromobility shift assays (EMSAs) to assess the ability of purified HigA or HigBA to bind to a 61 base-pair DNA fragments that contained higO1 with higO2 sequence scrambled or higO2 with higO1 sequence scrambled (Fig. 6 and Fig. S2). HigA binds to each of operator with approximately the same affinity (Kd values of 140 ± 0.03 and 125 ± 0.03 nM for higO1 with higO2, respectively). Interestingly, HigBA binds to each operator with a ~2-fold lower affinity (Kd values of 364 ± 0.10 and 243 ± 0.04 nM for higO1 with higO2, respectively) (Fig. 6B and Fig. S2).
Figure 6. HigA and HigBA recognition of higO1 and higO2.
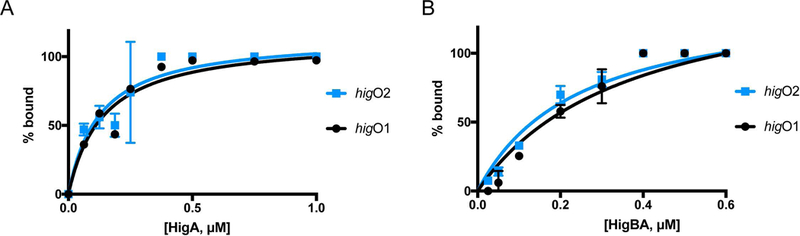
A. The band intensities from EMSAs as plotted as percent HigA bound versus HigA concentration for binding to Phig with scrambled higO1 and of HigA and Phig with scrambled higO2. In each assay, the HigA concentration increases from 0–1000 nM (gel shown in Fig. S2). B. The band intensities from EMSAs as plotted as percent HigBA bound versus HigBA concentration for binding to Phig with scrambled higO1 and of HigA and Phig with scrambled higO2. In each assay, the HigBA concentration increases from 0–600 nM (gel shown in Fig. S2). Curves represent the data from which binding affinities given in the main text were derived.
DISCUSSION
Much of what is known about the transcriptional regulatory mechanisms that establish and maintain appropriate levels of toxin-antitoxin complexes has been derived from the careful examination of a small set of model systems (reviewed in (Loris and Garcia-Pino 2014; Page and Peti 2016)). Studies of these model systems have led to significant advances in our understanding of how toxin-antitoxins are regulated but it is unclear whether this understanding can be broadly applied across diverse classes of toxin-antitoxins. One potential reason for the possibility of different repressor mechanisms is the rich diversity in the structure of antitoxins. Four predominant motifs are exploited by antitoxins for DNA binding: the RHH, Phd/YefM, SpoVT/AbrB or HTH motifs. There are only three known HTH-containing antitoxins and even among this group, there are clear differences in how they regulate transcriptional repression. For example, both antitoxins HipB and MqsA cause substantial DNA bending while we find in this study that HigA does not (Schumacher et al. 2009; Brown et al. 2013). Also, the direct and indirect readout of the operator sequences occurs by distinct mechanisms. We demonstrate that HigA recognizes its operator via a mechanism employed by both prokaryotic and eukaryotic transcription factors whereby the side chain of a conserved arginine residue makes both nucleobase-specific interactions with a guanosine and pi-cation stacking interactions with an adjacent thymine in recognition of a YpG dinucleotide (Lamoureux et al. 2004; Liu et al. 2013). In contrast, antitoxins HipB and MqsA do not recognize YpG dinucleotides. These data indicate that antitoxins containing HTH DNA-binding motifs do not necessarily use the same mechanism of transcriptional repression.
Previously it has been shown that regulation of transcription of toxin-antitoxin operons can be mediated by higher order toxin-antitoxin complexes, which are regulated by excess toxin functioning as a co-repressor (Afif et al. 2001; Overgaard et al. 2008; Garcia-Pino et al. 2010). For example, when CcdB, RelE or Doc toxins are present at levels below those of their cognate antitoxins, toxin binding to antitoxin-DNA operator sites increases the overall affinity and the level of repression at the toxin-antitoxin operon (Magnuson and Yarmolinsky 1998; Afif et al. 2001; Garcia-Pino et al. 2010). On a molecular level for the RelBE system which is the most similar to the HigBA system, this high affinity complex has been proposed to be a trimeric toxin-antitoxin complex (two antitoxins and one toxin molecule) when bound to its DNA operator (Overgaard et al. 2008; Boggild et al. 2012). At present, only a structure of the tetrameric RelBE complex has been observed in the absence of DNA (Boggild et al. 2012). As toxin levels increase to those similar to the antitoxin, the additional toxin disrupts the antitoxin-operator complexes and transcriptional repression is relieved (Overgaard et al. 2008). Additionally, in these systems, the antitoxin C terminus is disordered in the absence of its cognate toxin and gains structure upon toxin binding which also leads to an increase binding affinity for its DNA operator. These conditional co- and anti-repressor functions of toxins and their influence on antitoxin structure are the hallmarks of the current models describing toxin-antitoxin regulation.
Structural comparisons of apo HigA, HigA-higO2 complex and HigBA provide insights into how transcriptional repression could be mediated in this system. First, we find that the antitoxin HigA does not undergo a disorder-to-order transition at its C terminus in contrast to well-studied toxin-antitoxin systems (Magnuson and Yarmolinsky 1998; Afif et al. 2001; Overgaard et al. 2008; Garcia-Pino et al. 2010). Instead, we determine that the C terminus of HigA is required for its dimerization and this oligomeric state is, in turn, is necessary for DNA operator binding, despite each HigA monomer containing a single DNA operator. Toxin HigB binding does not order HigA providing further support that this system is not regulated by a disorder-to-order mechanism. Lastly, the HigBA complex, that we presume from our previous two crystal structures is a tetrameric complex (two HigBs and two HigAs) in the absence of DNA (Schureck et al. 2014), binds with a ~two-fold lower affinity than antitoxin HigA alone. These data demonstrate that toxin HigB does not function as a co-repressor in contrast to a number of other systems. Interestingly, in the case of the antitoxin MqsA, its cognate toxin MqsR also does not function as a co-repressor and shares a binding surface on MqsA with its operator DNA, effectively functioning as a competitor (Brown et al. 2013). One formal possibility is that the oligomeric state of HigBA changes in the context of binding the hig promoter, however, further detailed biophysical or structural insights are needed to unravel these molecular details.
These data together suggest a possible explanation for how toxin HigB may modulate the ability of the HigA antitoxin to repress transcription of the higBA genes. When HigB is present at levels below those of HigA, HigA binds tightly to its operator sites within the hig promoter and represses expression of higBA genes. When HigB reaches levels similar to those of HigA, the binding of two HigB monomers to the HigA dimer may restrict the complex to a conformation that is less favorable for operator binding than HigA alone. Indeed, structural superimpositions demonstrate that there are clashes of HigA’s DNA-binding motifs in the HigBA complex with higO2. Perhaps a single toxin HigB could bind to the HigA dimer which may not result in clashes with higO2, but at this time, there is no direct evidence that a trimeric HigBA exists. This possible model for how HigB could modulate the ability of HigA to repress gene expression is attractive in that it serves the same operating principles as those invoked to understand the well-studied toxin-antitoxins but does so through a distinct set of molecular mechanisms.
EXPERIMENTAL PROCEDURES
General methods.
The strains, plasmids, and oligonucleotide primers used in this study are listed in Tables S2, S3, and S4, respectively. Strains were grown in lysogeny broth (LB; 1% tryptone, 0.5% yeast extract, 1% sodium chloride) and M9 minimal medium (48 mM sodium phosphate dibasic, 22 mM potassium phosphate monobasic, 8.6 mM sodium chloride, 19 mM ammonium chloride, 2.0 mM magnesium sulfate, 0.1 mM calcium chloride) with 0.4% maltose. When necessary, strains were grown in media supplemented with 100 μg ml−1 ampicillin, 20 μg ml−1 chloramphenicol, or 30 μg ml−1 kanamycin.
Plasmid construction.
To build a replicating plasmid with a lacZ transcriptional reporter for the Rts1 hig promoter, oligonucleotides oJM835 and oJM836 were annealed and then ligated to pQF50 that had been cut with PstI and HindIII to make pQF50 Phig-lacZ (pJM359) (Table S3). The pQF50 plasmid contains a multiple cloning site immediately upstream of a lacZ reporter gene with the strong ribosome-binding site from the E. coli lpp gene. To build a similar replicating plasmid with the higBA and lacZ genes under the control of the hig promoter, a DNA fragment containing the hig promoter, higB, and higA was chemically synthesized (IDT). The fragment was cut with PstI and HindIII and ligated to pQF50 cut with the same restriction enzymes to make pQF50-Phig-higBA-lacZ.
To construct a replicating plasmid with higB under the control of an inducible promoter, the higB sequence was amplified by PCR from pQF50-Rts1 Phig-higBA-lacZ with oligonucleotide primers oJM821 and oRJD2. The higB PCR product, as well as the pBAD33 plasmid (Guzman et al. 1995), were cut with PstI and HindIII, and then ligated to make pBAD33-higB (pJM346). A series of plasmids were also built containing both higB and higA under the control of the arabinose-inducible promoter in pBAD33. The higB-higA sequence was amplified by PCR from pQF50-Rts1 Phig-higBA-lacZ with oligonucleotide primers oJM821 and oJM837, cut with PstI and HindIII, and then ligated to pBAD33 cut with the same restriction enzymes. To build a derivative of this plasmid with the C-terminally truncated HigA, higAΔ84–104 was amplified from pQF50-Rts1 Phig-higBA-lacZ with oJM821 and oJM862, cut with PstI and HindIII, and then ligated to pBAD33. To make a derivative with the HigA R40A mutant, pBAD33-higB-higA was subjected to site-directed mutagenesis (according to the Strategene QuikChange protocol) with oJM870 and oJM871. All pBAD33 plasmids were confirmed by sequencing (Genewiz).
To build a transcriptional reporter plasmid with the higA and lacZ genes under the control of the hig promoter, oligonucleotides oJM863 and oJM864 were annealed and then ligated to pQF50 that had been cut with SphI and BamHI to make pQF50 Phig-lacZ (pJM379). The higA sequence was amplified by PCR from pQF50-Rts1 Phig-higBA-lacZ with oligonucleotide primers oJM865 and oJM837. The PCR product was cut with BamHI and HindIII, and then ligated to pJM379 cut with the same restriction enzymes to make pQF50 Phig-higA-lacZ (pJM384). The C-terminally truncated higA, higAΔ84–104, was also amplified with oligonucleotide primers oJM865 and oJM837, cut with BamHI and HindIII, and then ligated to pJM379 to make pQF50 Phig-higAΔ84–104-lacZ (pJM385).
To construct a plasmid with higA under the control of the arabinose inducible promoter, the higA sequence was amplified by PCR from pQF50-Rts1 Phig-higBA-lacZ with oligonucleotide primers oJM823 and oJM837. The PCR product was cut with PstI and HindIII, and then ligated with pBAD33 cut with the same restriction enzymes. To build a version of this plasmid containing the C-terminally truncated higA, higAΔ84–104 was amplified from pQF50-Rts1 Phig-higBA-lacZ with oJM823 and oJM862, cut with PstI and HindIII, and then ligated with pBAD33. To make a derivative with the higAR40A mutant, pBAD33-higA was subjected to site-directed mutagenesis with oJM870 and oJM871.
The expression plasmid used in this study, pET28a-his6-Rts1 higA, was a generous gift from the Woychik laboratory (Hurley and Woychik 2009). To make a derivative of this expression plasmid with the HigA R40A variant, pET28a-his6-Rts1 higA was subjected to site-directed mutagenesis with oJM870 and oJM871. The plasmids were confirmed by sequencing (Genewiz) using oJM437 and oJM438.
Spot dilution assays.
Strains were grown in 3 mL LB supplemented with chloramphenicol at 37°C with rolling to an OD600 of 0.5–1.0. The cultures were then serially diluted 10-fold in LB, 5 μl of each dilution (10−1 to 10−6 dilutions) was spotted on LB agar with chloramphenicol or LB agar with chloramphenicol and 0.2% arabinose, and cells were grown at 37°C overnight.
β-galactosidase assays.
Strains were grown in 3 mL medium (LB or M9 maltose) at 37°C with rolling until cell density reached until an OD600 of 0.5. Cultures were diluted 100-fold into 20 ml medium with or without 0.2% arabinose and grown at 37°C with rolling to an OD600 of ~0.5 (mid-exponential growth phase). 1 mL of each culture was centrifuged at 10,000 x g in a microcentrifuge tube for 1 min and cell pellets were stored at −20°C. Cell pellets were thawed on ice and resuspended in 500 μl cold buffer (60 mM sodium phosphate dibasic, 40 mM sodium phosphate monobasic, 10 mM potassium chloride, 1 mM magnesium sulfate, pH 7.0 with 50 mM β-mercaptoethanol). 10–200 μl of each cell suspension was added to microcentrifuge tubes containing 800–990 μl buffer (to a final combined volume of 1 mL, 100 μl chloroform, and 50 μL 0.1% SDS. Reactions were vortexed and incubated at 30°C for 10 min. 200 μl 4 mg ml−1ortho-nitrophenyl-β-galactosidase (in 0.1 M phosphate buffer, 60 mM sodium phosphate dibasic, 40 mM sodium phosphate monobasic, pH 7.0) was added to each sample. Reactions were vortexed briefly and incubated at 30°C for 10–30 min. Each reaction was terminated by the addition of 400 μL of 1 M sodium carbonate, vortexed and centrifuged to remove cell debris. 1 mL of each reaction supernatant was transferred to disposable cuvettes. Absorbance of each reaction was measured at 420 nm. β-galactosidase activity (in Miller units) was calculated as follows: (1000 x A420)/(reaction time in minutes x cell suspension volume in mL x OD600).
HigA and HigA variant expression and purification.
To overexpress wild-type Rts1 HigA or the HigA R40A variant, the expression strain E. coli BL21(DE3) was transformed with pET28a-his6-higA or pET28a-his6-higAR40A (pJM382) to make strains BL21(DE3) pET28a-his6-higA (ECJM901) and BL21(DE3) pET28a-his6-higA R40A (ECJM902), respectively. These expression strains were grown in 20 ml LB supplemented with kanamycin at 37°C with shaking. When the cultures reached mid-exponential phase (optical density (OD) at 600 nm of approximately 0.5), they were transferred to a 2.8 L baffled Fernbach flasks containing 1 L LB with kanamycin and incubated at 37°C with shaking. Once the cultures reached an OD600 of 0.6–0.7, they were cooled at 4°C for 15 min. Then 50 μl 1 M isopropyl-1-thio-ß-D-galactopyranoside (IPTG) was added to the 1 L cultures to give a final concentration of 50 μM, and the cultures were incubated at 18°C with shaking overnight (~18 hrs). The cultures were then centrifuged, the supernatants were removed, and the cell pellets were stored at −80°C.
To purify the wild-type and protein variants, the frozen cell pellets were thawed and resuspended in 25 ml HisTrap buffer A (50 mM Tris, 250 mM KCl, 5 mM MgCl2, 5 mM imidazole, 5 mM β-mercaptoethanol, 10% glycerol, pH 7.4) with ProBlock Gold protease inhibitor (GoldBio) and 50 μl 10 mg/ml DNase I. The cells were lysed with French press at 15,000 psi. The resulting cell lysates were centrifuged at 10,000 x g at 4°C for 20 min. Cleared lysates were filtered and loaded onto 5 ml HisTrap HP column in an ÅKTApurifier chromatography system (GE Healthcare). His6-tagged proteins were eluted from the column with an increasing linear gradient of HisTrap buffer B (50 mM Tris, 250 mM KCl, 5 mM MgCl2, 500 mM imidazole, 5 mM β-mercaptoethanol, 10% glycerol, pH 7.4). To remove the hexahistidine tag, HisTrap elution fractions were pooled and incubated with thrombin (1 unit per mg of protein) at room temperature for 2 hrs and then at 4°C overnight. The thrombin proteolysis reactions were subjected to size exclusion chromatography on a HiLoad 16/60 Superdex 200 column and buffer exchanged into storage buffer (40 mM Tris-HCl, pH 7.5, 250 mM KCl, 5 mM MgCl2, and 5 mM β-mercaptoethanol). To remove any remaining thrombin or His-tagged HigA protein, the gel filtration fractions were pooled and loaded onto 1 ml HiTrap Benzamidine FF and 1 mL HisTrap FF columns arranged in tandem. Finally, HigA was dialyzed back into storage, concentrated and stored in aliquots at −80°C. The HigA protein lacking any tags is 11.5 kDa.
HigA crystallization, data collection, and structure determination.
HigA crystals (10 mg ml−1) were grown by sitting drop vapor diffusion in 0.2 M NaSCN and 20% w/v polyethylene glycol (PEG) 3350 at 20°C. Crystals were cryoprotected by gradually increasing the ethylene glycol concentration to 30% w/v while decreasing the PEG 3,350 concentration to 10% w/v followed by freezing in liquid nitrogen. Three hundred and sixty degrees of diffraction data were collected at the Northeastern Collaborative Access Team (NE-CAT) beamline 24-IDC at the Advanced Photon Source (APS) (Chicago, IL, USA). The x-ray diffraction data were indexed, integrated and scaled in XDS (Kabsch 2010). The structure was solved by molecular replacement using a HigA monomer from the HigB-HigA complex (PDB code 4MCT) as a search model in PHENIX AutoMR to 1.9 Å (Adams et al. 2010). Two HigA proteins per asymmetric unit were identified and form a dimer within the asymmetric unit. The model was built in Coot (Emsley et al. 2010) for residues 3–94 and 6–99 followed by refinement of xyz coordinates, occupancies and B-factors in PHENIX (Adams et al. 2010) to a final Rwork/Rfree of 17.5/22.2%.
Analytical ultracentrifugation studies of HigA.
Purified HigA was dialyzed against 20 mM Tris, 100 mM NaCl, 10 mM MgCl2, pH 7.5. Sedimentation velocity experiments were performed on a 0.11 mg/mL sample (0.4 mL) at 182,000 x g (50,000 rpm) at 20°C in a Beckman Coulter ProteomeLab XLI analytical ultracentrifuge using standard procedures (Zhao et al. 2013). Absorbance scans were taken at 280 nm in continuous mode using a radial spacing of 0.003 cm at approximately 3.6 min intervals in an An-60 rotor equipped with 12 mm path length double sector cells and sapphire windows. Absorbance values were fit to the Lamm equation along with the meniscus position, baseline and time-invariant noise using the continuous c(s) distribution model of SEDFIT, version 15.01c (http://analyticalultracentrifugation.com) integrating between 0 and 10 S at 0.1 S increments (Schuck 2000; Dam and Schuck 2004). Fitting was done using maximum entropy regularization with a confidence interval of 0.68. Both the simplex and Marquardt-Levenberg nonlinear least squares fitting algorithms were tested and produced equivalent results. Buffer density, viscosity and the partial specific volume of HigA (0.730 mL g−1) were estimated using SEDNTERP (http://sednterp.unh.edu) (Cole et al. 2008). The sedimentation coefficient was corrected to s20,w using the buffer density and viscosity. The molecular weight of HigA was obtained from the c(s) peak by deconvolution of the contribution of diffusion to the observed signal as described (Schuck 2000; Dam and Schuck 2004). AUC results were plotted using GUSSI version 1.2.1 (Schuck P 2016).
HigA-DNA operator complex crystallization, data collection, and structure determination.
The hig operator 2 DNA was formed by incubating two 21 nt (pHigCryst3 and pHigCryst4), single-stranded DNA at 95°C for 2 mins and then cooled to room temperature for two hrs in DNA buffer (100 mM NaCl, 1 mM EDTA and 10 mM Tris, pH 8.0). HigA (3.2 mg ml−1) was complexed with 1.6 mg ml−1 of the higO2 DNA in 1X binding buffer (20 mM Tris, pH 8, 100 mM NaCl, 10 mM MgCl2) to form a complex consisting of 1 HigA dimer per 1 duplex DNA molar ratio. Crystals were grown by sitting drop vapor diffusion in 0.2 M CaCl2 and 10–25% w/v PEG 3,350 at 20°C producing rod-shaped crystals after two days. Crystals were cryoprotected by three serially increases of ethylene glycol to final concentration of 30% w/v followed by freezing in liquid nitrogen. The HigA-DNA crystals are of the P65 space group and diffracted anisotropically. A total of 120° of diffraction data was collected on the SER-CAT 22ID beamline. The data were indexed, integrated and scaled to 2.9 Å in XDS (Kabsch 2010). Structure determination was performed by molecular replacement using the structure of HigA in isolation as a search model in PHENIX AutoMR (Adams et al. 2010). Three copies of a HigA dimer bound to double-stranded DNA are found per asymmetric unit. Manual modification of the model was performed in Coot (Emsley et al. 2010) followed by refinement of XYZ coordinates with secondary structure restraints and B-factors in PHENIX. The final model has an Rwork/Rfree of 22.5/26.0%. All figures were created in PyMOL (Schrodinger 2010). Alignments between HigBA, HigA, and HigA-DNA were constructed using either the secondary structure matching or least-squares fit functions in Coot (Emsley et al. 2010).
Electromobility shift assay (EMSA).
To construct the double-stranded DNA (dsDNA) for the EMSA, pairs of complementary single-stranded oligonucleotides were diluted to 2 μM each in 10 mM Tris, 1 mM EDTA, 100 mM NaCl, pH 8.0. The 61-nt oligonucleotide mixtures of the hig promoter fragment were incubated at 95°C for 2 min and then cooled at room temperature for 2 hr. The wild-type operator was generated from pHigA_F and pHigA_R, scrambled operator 1 with wild-type operator 2 from pHigA_F_Scra1 and pHigA_R_Scra1 and wild-type operator 1 and scrambled operator 2 from pHigA_F_Scra2 and pHigA_R_Scra2. For the EMSA, the dsDNA oligos were diluted to 150 nM in EMSA binding buffer (100 mM NaCl, 10 mM MgCl2, 5% glycerol, 0.01 mg/ml BSA). Purified wild-type HigA and HigA R40A proteins were diluted to 10 μM in EMSA binding buffer and serially diluted to give a series of final protein concentrations ranging from 62.5 nM to 1.0 μM in the EMSAs. HigBA was purified as previously described (Schureck et al. 2014) and diluted to 8 μM in EMSA binding buffer and serially diluted to give a series of final protein concentrations ranging from 25 nM to 600 nM in the EMSAs. The binding reactions were incubated on ice for 20 min and 5 μL of each reaction was loaded onto an 8% native, polyacrylamide-0.5X TBE-glycerol gels (50 mM Tris, 50 mM boric acid, 5 mM EDTA, 10% glycerol) and subjected to electrophoresis at 100 volts at 4°C. To visualize the DNA and DNA-protein complexes, the gels were stained with SYBR green nucleic acid gel stain (ThermoFisher Scientific) in 0.5X TBE glycerol for 30 mins with gentle agitation, and then the fluorescence was imaged with a Typhoon Trio phosphoimager (GE Healthcare; 488 nm excitation and 526 nm emission). Assays were performed in duplicate with representative gels shown. Band intensities for both free and bound hig DNA were quantified with ImageQuant 1D gel analysis using the rolling ball background subtraction. For HigA or HigBA bound to either higO1 or higO2, the hig DNA were fit using a one site specific binding equation in GraphPad .
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by in part by a National Science Foundation CAREER award MCB 0953714 (CMD), a National Institutes of Health (NIH) Biochemistry, Cellular and Developmental Biology Graduate Training Grant (5T32GM8367), and a NIH National Research Service Award Fellowship GM108351 (MAS). CMD is a Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Disease. We thank F. M. Murphy IV and staff members of the NE-CAT beamlines for assistance during data collection, Dr. G. Conn for critical reading of the manuscript, and Dunham lab member S. Miles for technical assistance. This work is based upon research conducted at the NE-CAT beamlines, which are funded by the NIGMS from the NIH (P41 GM103403), and at the SER-CAT beamline. The Pilatus 6M detector on 24-ID-C beam line is funded by a NIH-ORIP HEI grant (S10 RR029205). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02–06CH11357.
Footnotes
Data deposition: Crystallography, atomic coordinates, and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB codes 6CF1, 6CHV)
REFERENCES
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N et al. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta crystallographica 66: 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afif H, Allali N, Couturier M, Van Melderen L. 2001. The ratio between CcdA and CcdB modulates the transcriptional repression of the ccd poison-antidote system. Mol Microbiol 41: 73–82. [DOI] [PubMed] [Google Scholar]
- Blanchet C, Pasi M, Zakrzewska K, Lavery R. 2011. CURVES+ web server for analyzing and visualizing the helical, backbone and groove parameters of nucleic acid structures. Nucleic Acids Res 39: W68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boggild A, Sofos N, Andersen KR, Feddersen A, Easter AD, Passmore LA, Brodersen DE. 2012. The crystal structure of the intact E. coli RelBE toxin-antitoxin complex provides the structural basis for conditional cooperativity. Structure 20: 1641–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown BL, Grigoriu S, Kim Y, Arruda JM, Davenport A, Wood TK et al. 2009. Three dimensional structure of the MqsR:MqsA complex: a novel TA pair comprised of a toxin homologous to RelE and an antitoxin with unique properties. PLoS Pathog 5: e1000706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown BL, Lord DM, Grigoriu S, Peti W, Page R. 2013. The Escherichia coli toxin MqsR destabilizes the transcriptional repression complex formed between the antitoxin MqsA and the mqsRA operon promoter. J Biol Chem 288: 1286–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen SK, Maenhaut-Michel G, Mine N, Gottesman S, Gerdes K, Van Melderen L. 2004. Overproduction of the Lon protease triggers inhibition of translation in Escherichia coli: involvement of the yefM-yoeB toxin-antitoxin system. Mol Microbiol 51: 1705–1717. [DOI] [PubMed] [Google Scholar]
- Christensen-Dalsgaard M, Jorgensen MG, Gerdes K. 2010. Three new RelE-homologous mRNA interferases of Escherichia coli differentially induced by environmental stresses. Mol Microbiol 75: 333–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JL, Lary JW, T PM, Laue TM. 2008. Analytical ultracentrifugation: sedimentation velocity and sedimentation equilibrium. Methods Cell Biol 84: 143–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dam J, Schuck P. 2004. Calculating sedimentation coefficient distributions by direct modeling of sedimentation velocity concentration profiles. Methods Enzymol 384: 185–212. [DOI] [PubMed] [Google Scholar]
- De Jonge N, Garcia-Pino A, Buts L, Haesaerts S, Charlier D, Zangger K et al. 2009. Rejuvenation of CcdB-poisoned gyrase by an intrinsically disordered protein domain. Mol Cell 35: 154–163. [DOI] [PubMed] [Google Scholar]
- Dienemann C, Boggild A, Winther KS, Gerdes K, Brodersen DE. 2011. Crystal structure of the VapBC toxin-antitoxin complex from Shigella flexneri reveals a hetero-octameric DNA-binding assembly. Journal of molecular biology 414: 713–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta crystallographica 66: 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Pino A, Balasubramanian S, Wyns L, Gazit E, De Greve H, Magnuson RD et al. 2010. Allostery and intrinsic disorder mediate transcription regulation by conditional cooperativity. Cell 142: 101–111. [DOI] [PubMed] [Google Scholar]
- Garcia-Pino A, Christensen-Dalsgaard M, Wyns L, Yarmolinsky M, Magnuson RD, Gerdes K, Loris R. 2008. Doc of prophage P1 is inhibited by its antitoxin partner Phd through fold complementation. J Biol Chem 283: 30821–30827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes K, Christensen SK, Lobner-Olesen A. 2005. Prokaryotic toxin-antitoxin stress response loci. Nat Rev Microbiol 3: 371–382. [DOI] [PubMed] [Google Scholar]
- Gonzalez Barrios AF, Zuo R, Hashimoto Y, Yang L, Bentley WE, Wood TK. 2006. Autoinducer 2 controls biofilm formation in Escherichia coli through a novel motility quorum-sensing regulator (MqsR, B3022). J Bacteriol 188: 305–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177: 4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadzi S, Garcia-Pino A, Haesaerts S, Jurenas D, Gerdes K, Lah J, Loris R. 2017. Ribosome-dependent Vibrio cholerae mRNAse HigB2 is regulated by a beta-strand sliding mechanism. Nucleic Acids Res 45: 4972–4983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison JJ, Wade WD, Akierman S, Vacchi-Suzzi C, Stremick CA, Turner RJ, Ceri H. 2009. The chromosomal toxin gene yafQ is a determinant of multidrug tolerance for Escherichia coli growing in a biofilm. Antimicrob Agents Chemother 53: 2253–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, Rosenstrom P. 2010. Dali server: conservation mapping in 3D. Nucleic Acids Res 38: W545–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley JM, Woychik NA. 2009. Bacterial toxin HigB associates with ribosomes and mediates translation-dependent mRNA cleavage at A-rich sites. J Biol Chem 284: 18605–18613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W 2010. Xds. Acta crystallographica 66: 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada K, Hanaoka F. 2005. Conformational change in the catalytic site of the ribonuclease YoeB toxin by YefM antitoxin. Mol Cell 19: 497–509. [DOI] [PubMed] [Google Scholar]
- Kamada K, Hanaoka F, Burley SK. 2003. Crystal structure of the MazE/MazF complex: molecular bases of antidote-toxin recognition. Mol Cell 11: 875–884. [DOI] [PubMed] [Google Scholar]
- Kedzierska B, Lian LY, Hayes F. 2007. Toxin-antitoxin regulation: bimodal interaction of YefM-YoeB with paired DNA palindromes exerts transcriptional autorepression. Nucleic Acids Res 35: 325–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick CL, Martins D, Redder P, Frandi A, Mignolet J, Chapalay JB et al. 2016. Growth control switch by a DNA-damage-inducible toxin-antitoxin system in Caulobacter crescentus. Nat Microbiol 1: 16008. [DOI] [PubMed] [Google Scholar]
- Lamoureux JS, Maynes JT, Glover JN. 2004. Recognition of 5’-YpG-3’ sequences by coupled stacking/hydrogen bonding interactions with amino acid residues. Journal of molecular biology 335: 399–408. [DOI] [PubMed] [Google Scholar]
- Li GW, Burkhardt D, Gross C, Weissman JS. 2014. Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell 157: 624–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GY, Zhang Y, Inouye M, Ikura M. 2008. Structural mechanism of transcriptional autorepression of the Escherichia coli RelB/RelE antitoxin/toxin module. Journal of molecular biology 380: 107–119. [DOI] [PubMed] [Google Scholar]
- Liu Y, Zhang X, Blumenthal RM, Cheng X. 2013. A common mode of recognition for methylated CpG. Trends Biochem Sci 38: 177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loris R, Garcia-Pino A. 2014. Disorder- and dynamics-based regulatory mechanisms in toxin-antitoxin modules. Chemical reviews 114: 6933–6947. [DOI] [PubMed] [Google Scholar]
- Magnuson R, Yarmolinsky MB. 1998. Corepression of the P1 addiction operon by Phd and Doc. J Bacteriol 180: 6342–6351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyed HS, Bertrand KP. 1983. hipA, a newly recognized gene of Escherichia coli K-12 that affects frequency of persistence after inhibition of murein synthesis. J Bacteriol 155: 768–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overgaard M, Borch J, Jorgensen MG, Gerdes K. 2008. Messenger RNA interferase RelE controls relBE transcription by conditional cooperativity. Mol Microbiol 69: 841–857. [DOI] [PubMed] [Google Scholar]
- Page R, Peti W. 2016. Toxin-antitoxin systems in bacterial growth arrest and persistence. Nat Chem Biol 12: 208–214. [DOI] [PubMed] [Google Scholar]
- Ruangprasert A, Maehigashi T, Miles SJ, Giridharan N, Liu JX, Dunham CM. 2014. Mechanisms of Toxin Inhibition and Transcriptional Repression by Escherichia coli DinJ-YafQ. J Biol Chem 289: 20559–20569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrodinger, LLC. 2010. The PyMOL Molecular Graphics System, Version 1.3r1.
- Schuck P 2000. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophysical journal 78: 1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck P ZH, Brautigam CA, Ghirlando R. . 2016. Basic Principles of Analytical Ultracentrifugation. CRC Press, Boca Raton. [Google Scholar]
- Schumacher MA, Piro KM, Xu W, Hansen S, Lewis K, Brennan RG. 2009. Molecular mechanisms of HipA-mediated multidrug tolerance and its neutralization by HipB. Science 323: 396–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schureck MA, Dunkle JA, Maehigashi T, Miles SJ, Dunham CM. 2015. Defining the mRNA recognition signature of a bacterial toxin protein. Proc Natl Acad Sci U S A 112: 13862–13867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schureck MA, Maehigashi T, Miles SJ, Marquez J, Cho SE, Erdman R, Dunham CM. 2014. Structure of the Proteus vulgaris HigB-(HigA)2-HigB toxin-antitoxin complex. J Biol Chem 289: 1060–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schureck MA, Maehigashi T, Miles SJ, Marquez J, Dunham CM. 2016a. mRNA bound to the 30S subunit is a HigB toxin substrate. RNA 22: 1261–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schureck MA, Repack A, Miles SJ, Marquez J, Dunham CM. 2016b. Mechanism of endonuclease cleavage by the HigB toxin. Nucleic Acids Res 44: 7944–7953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimon LJ, Harrison SC. 1993. The phage 434 OR2/R1–69 complex at 2.5 A resolution. Journal of molecular biology 232: 826–838. [DOI] [PubMed] [Google Scholar]
- Tian QB, Ohnishi M, Murata T, Nakayama K, Terawaki Y, Hayashi T. 2001. Specific protein-DNA and protein-protein interaction in the hig gene system, a plasmid-borne proteic killer gene system of plasmid Rts1. Plasmid 45: 63–74. [DOI] [PubMed] [Google Scholar]
- Tian QB, Ohnishi M, Tabuchi A, Terawaki Y. 1996. A new plasmid-encoded proteic killer gene system: cloning, sequencing, and analyzing hig locus of plasmid Rts1. Biochem Biophys Res Commun 220: 280–284. [DOI] [PubMed] [Google Scholar]
- Turnbull KJ, Gerdes K. 2017. HicA toxin of Escherichia coli derepresses hicAB transcription to selectively produce HicB antitoxin. Mol Microbiol 104: 781–792. [DOI] [PubMed] [Google Scholar]
- Van Melderen L, Bernard P, Couturier M. 1994. Lon-dependent proteolysis of CcdA is the key control for activation of CcdB in plasmid-free segregant bacteria. Mol Microbiol 11: 1151–1157. [DOI] [PubMed] [Google Scholar]
- Wang X, Kim Y, Hong SH, Ma Q, Brown BL, Pu M et al. 2011. Antitoxin MqsA helps mediate the bacterial general stress response. Nat Chem Biol 7: 359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins D, Hsiao C, Woods KK, Koudelka GB, Williams LD. 2008. P22 c2 repressor-operator complex: mechanisms of direct and indirect readout. Biochemistry 47: 2325–2338. [DOI] [PubMed] [Google Scholar]
- Zhao H, Lomash S, Glasser C, Mayer ML, Schuck P. 2013. Analysis of high affinity self-association by fluorescence optical sedimentation velocity analytical ultracentrifugation of labeled proteins: opportunities and limitations. PLoS One 8: e83439. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.