

Summary
For the human pathogen Clostridioides (also known as Clostridium) difficile, the ability to adapt to nutrient availability is critical for its proliferation and production of toxins during infection. Synthesis of the toxins is regulated by the availability of certain carbon sources, fermentation products and amino acids (e.g., proline, cysteine, isoleucine, leucine and valine). The effect of proline is attributable at least in part to its role as an inducer and substrate of D-proline reductase (PR), a Stickland reaction that regenerates NAD+ from NADH. Many Clostridium spp. use Stickland metabolism (co-fermentation of pairs of amino acids) to generate ATP and NAD+. Synthesis of PR is activated by PrdR, a proline-responsive regulatory protein. Here we report that PrdR, in the presence of proline, represses other NAD+-generating pathways, such as the glycine reductase and succinate-acetyl CoA utilization pathways leading to butyrate production, but does so indirectly by affecting the activity of Rex, a global redox-sensing regulator that responds to the NAD+/NADH ratio. Our results indicate that PR activity is the favored mechanism for NAD+ regeneration and that both Rex and PrdR influence toxin production. Using the hamster model of C. difficile infection, we revealed the importance of PrdR-regulated Stickland metabolism in the virulence of C. difficile.
Keywords: C. difficile, Rex, NAD+/NADH ratio, Stickland metabolism, PrdR
Graphical Abstract
Clostridioides difficile is a major pathogenic bacterium that is responsible for large numbers of human infections that cause diarrhea and pseudomembranous colitis. Two proteins, PrdR and Rex, were found to control the expression of a large number of genes involved in metabolism, including those whose activities lead to production of butyrate, a factor that stimulates the expression of the major C. difficile toxin genes.
Introduction
Clostridioides difficile, a Gram-positive, anaerobic, spore-forming bacterium, is the leading cause of antibiotic-associated diarrhea. In recent years, morbidity and mortality rates due to C. difficile have been on the rise (Loo et al., 2005, Ricciardi et al., 2007, Gerding, 2010, Ong et al., 2017), leading to the designation of C. difficile as one of the most important emerging pathogens. The onset of C. difficile infection (CDI) is usually preceded by exposure to antibiotics that disrupt the normal gut microbiota, thereby allowing C. difficile to colonize the colon (Kyne & Kelly, 2001). For the vast majority of pathogenic C. difficile strains, the primary virulence factors are two large cytotoxins, TcdA and TcdB (Kuehne et al., 2010, Lyras et al., 2009, Voth & Ballard, 2005).
Upon colonization, C. difficile cells encounter changes in environmental conditions that may trigger expression of virulence factor genes (Deneve et al., 2009). In the laboratory, toxin production increases as cells enter stationary phase and is modulated in response to a range of environmental signals, such as temperature, biotin limitation, the presence of antibiotics, butyric acid, butanol, rapidly metabolized carbohydrates (e.g., glucose) and certain amino acids, such as proline, cysteine and the branched-chain amino acids (BCAAs) isoleucine, leucine and valine (Antunes et al., 2011, Bouillaut et al., 2013, Karlsson et al., 1999, Karlsson et al., 2000, Yamakawa et al., 1996). Some of the molecular mechanisms regulating C. difficile toxin gene expression have been elucidated. Transcription of the tcdA and tcdB genes depends on TcdR, an alternative sigma factor that directs RNA polymerase to the toxin gene promoters (Mani & Dupuy, 2001). The anti-sigma factor TcdC and the global regulators CodY and CcpA reduce toxin gene expression by blocking TcdR activity or expression (Antunes et al., 2011, Dineen et al., 2007, Dupuy & Sonenshein, 1998, Matamouros et al., 2007); the motility sigma factor, SigD, also contributes to tcdR expression (El Meouche et al., 2013). We previously demonstrated that PrdR, a sigma-54-activating positive regulator of the D-proline reductase (PR) operon, is required for the proline-dependent repression of C. difficile toxin production (Bouillaut et al., 2013). PR catalyzes one of the two major pathways of Stickland metabolism in C. difficile. Stickland reactions are pathways of coupled oxidation and reduction of pairs of amino acids (Stickland, 1935a, Stickland, 1935b). The oxidative pathway typically generates ATP and reducing power (NADH); the reductive pathway regenerates NAD+ from NADH. NAD+ is an essential co-factor for critical metabolic pathways, including metabolism of sugars; if the pool of NAD+ becomes exhausted, cells will be unable to grow. Several amino acids, including alanine, isoleucine, leucine and valine, can serve as energy-generating subjects of Stickland oxidation, but proline and glycine are the only amino acids that are substrates for reduction carried out by PR and glycine reductase (GR), respectively (Stadtman, 1956, Stadtman & Elliott, 1957, Stickland, 1935a, Stickland, 1935b). Whereas PrdR activates transcription of the PR pathway, it represses, directly or indirectly, expression of the GR gene cluster, suggesting that PrdR causes preferential utilization of proline for NAD+ regeneration (Bouillaut et al., 2013). However, the potential impact of PrdR and proline on global gene expression in C. difficile and the mechanisms by which PrdR controls expression of GR and the toxin genes are unknown.
A recent review summarized our current understanding of the complex mechanisms that control C. difficile metabolism (Neumann-Schaal et al., 2019). In the study presented here, the impact on global transcription of addition of exogenous proline and interruption of prdR was characterized using microarray analysis. Interestingly, most of the proline-dependent effects on gene expression in the wild-type strain turned out to be lost when prdR was mutated, implying that these genes are controlled directly or indirectly through PrdR. Moreover, most of the genes whose expression was strongly altered are involved in fermentation and reductive pathways that regenerate NAD+. We hypothesized that the effects of PrdR and proline on these pathways might be mediated by a regulatory protein that senses the relative concentrations of NAD+ and NADH. Rex, a protein found in many bacterial species, including Clostridium species, and whose activity is controlled by the intracellular NAD+/NADH ratio (Brekasis & Paget, 2003, Gyan et al., 2006, Nguyen et al., 2016, McLaughlin et al., 2010, Wang et al., 2013, Schwarz et al., 2017, Hu et al., 2016, Panitz et al., 2014, Christensen et al., 2015, Yang et al., 2018), was a candidate for such a regulator. We demonstrate here that C. difficile Rex is indeed a direct repressor of several reductive pathways involved in the production of butyrate and that its ability to bind to DNA is stimulated by NAD+ and inhibited by NADH. In addition, we show that Rex plays a role in toxin gene expression. A prdR mutation increased virulence in hamsters considerably, but, surprisingly, a rex mutation did not.
Results
Effects of proline and a prdR null mutation on global gene expression
We have previously shown that proline and PrdR regulate the expression of the genes that encode PR, GR and TcdA (Bouillaut et al., 2013). To place these regulatory phenomena in a broader context and to seek possible mechanisms for the observed regulation, we carried out microarray assays to compare gene expression in the parent strain JIR8094 grown to mid-exponential growth phase in the rich medium TY vs. TY supplemented with 30 mM L-proline (TYP) and JIR8094 vs. its prdR mutant both grown in TYP (note that TY contains an unknown amount of proline). All genes differentially expressed in response to excess proline and/or controlled by PrdR with a fold-change ≥2 are listed in Supplementary Tables S1 and S2. The 162 genes were differentially expressed (≥2-fold) when cells were grown with excess proline fell into 73 genetic loci, while the 182 genes differentially expressed (≥2-fold) in the prdR mutant when compared to the wild-type were mapped at 67 loci. Seventy genes that were underexpressed in excess proline medium were overexpressed in the prdR mutant and vice versa (Table S3)
The genes most highly affected (≥5-fold) by addition of proline or by a mutation in prdR are listed in Table 1; they are heavily biased toward reductive pathways that regenerate NAD+. In addition to the prd locus, which was overexpressed 30-to-54-fold when proline was added to the medium and underexpressed 25-to-30-fold in the prdR mutant, these highly regulated pathways include the genes that encode the GR pathway (CD2348-CD2358; Fig. 1 (pathway 2)) and the pathways leading to synthesis of butyryl-CoA from acetyl-CoA (CD1054–CD1059; Fig. 1 (pathway 4)) and succinate (CD2344–CD2338, Fig. 1 (pathway 3)). All of these latter genes were repressed when proline was added to the medium, but overexpressed when the prdR gene was inactivated. In addition, cobalamin biosynthesis genes, cobalt transport genes (CD0324-CD0327) and ethanolamine utilization genes (CD1906-CD1925; Fig. 1 (pathway 5)) were overexpressed in the prdR mutant (Table 1). Co-regulation of these genes is consistent with the fact that ethanolamine degradation to acetyl-CoA requires the coenzyme B12 (cobalamin) (Garsin, 2010). To confirm the results obtained with the microarrays, we selected a subset of three genes representative of different fermentation pathways (CD1054 (bcd2, encoding butyryl-CoA dehydrogenase, Fig. 1(pathway 4)), CD2344 (encoding a membrane protein in the succinate metabolism gene cluster, (Fig. 1(pathway 3)) and adhE (encoding acetaldehyde CoA-alcohol dehydrogenase, Fig. 1(pathway 6)) for quantitative reverse-transcription (qRT)-PCR analysis. The results were consistent with the microarray results for all the genes tested (Table 2), i.e., CD2344, bcd2 and adhE were reduced in expression in the presence of excess proline and overexpressed in the prdR mutant strain compared to the wild-type grown in excess proline. Consistent with the microarray results, we previously showed by qRT-PCR that prdA is induced by addition of proline to the medium and underexpressed in the prdR mutant, whereas grdE (encoding a component of glycine reductase) responds to proline and PrdR in the opposite manner (Bouillaut et al., 2013).
Table 1.
Selected genes whose expression was altered in the prdR mutant and/or in excess of proline.
| Class a | Gene number | Gene Name | Putative Product | prdR/WTb | TYP/TYb |
|---|---|---|---|---|---|
| Cell Wall | CD1413 | Putative membrane protein | 6.11 | ||
| Cell Wall | CD0830 | Putative membrane protein | 0.17 | ||
| Fermentation | CD1054–1059 | bcd2, etfB, etfA, crt2, hdb, thlA1 | Butyrate production | 5.92–9.26 | 0.03–0.05 |
| Fermentation | CD2338–2344 | 4hbD*,cat2*, abfD*, sucD, cat1 | Succinate utilization | 3.33 – 10.44 | 0.04.0.17 |
| Fermentation | CD2966 | adhE | Aldehyde-alcohol dehydrogenase | 12.77 | 0.05 |
| Membrane Transport | CD0324–0327 | cbiM, cbiN, cbiQ, cbiO | Cobalamin biosynthesis protein, cobalt ABC-type transport system | 8.04–12.07 | 0.25–0.27 |
| Membrane Transport | CD2666–2667 | ptsG-A, ptsG-BC | PTS system, glucose-specific transporter | 0.04 | |
| Membrane Transport | CD3136–3138 | bglA, bglF, bglG | PTS system, b-glucoside transporter | 0.11 – 0.29 | |
| Membrane Transport | CD2954–2961 | atpDBAFCEKI | ATP synthase | 2.90–3.49 | 0.13–0.19 |
| Membrane Transport | CD3036 | Transporter, Major Facilitator Superfamily | 6.03 | ||
| Metabolism-Amino Acid | CD2348–2358 | grdDCBAE, trxA2, trxB3, grdX | Glycine reductase | 6.61 – 64.16 | 0.04–0.1 |
| Metabolism-Amino Acid | CD3236–3247 | prdC, prdR, prdABDEE2F | Proline reductase | 0.03–0.05 | 3.44–64.29 |
| Metabolism-Amino Acid | CD0994–0996 | ser | Serine biosynthesis | 0.19–0.21 | |
| Metabolism-Carbon | CD1767 | gapB | Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) | 0.07 | |
| Metabolism-Carbon | CD3248 | Polysaccharide deacetylase | 0.17 | 7 | |
| Metabolism-Lipids | CD1906–1925 | eutG, eutS, eutPVWABCLMEKTDNHQ | Ethanolamine utilization | 5.55 – 61.95 | |
| Regulation | CD1811 | Putative two-component sensor histidine kinase (N-terminal region) | 5.25 | ||
| Regulation | CD2668 | Transcription antiterminator, LicT family | 0.19 |
A selection of genes whose expression was affected more than 5-fold by a prdR mutation or by growth in excess proline. Not all genes that were affected >2-fold by the prdR mutation were affected >2-fold by the presence of proline and vice versa. (All genes whose expression was affected ≥2-fold by a prdR mutation or by proline excess are listed in Tables S1 and S2.)
The cutoff for significant change was set at 5-fold. Three genes, indicated by asterisks (*), had a fold-change less than 5-fold but were included because they are apparently in the same transcription unit as genes regulated ≥5-fold.
Figure 1. Metabolic map of prdR regulated genes.
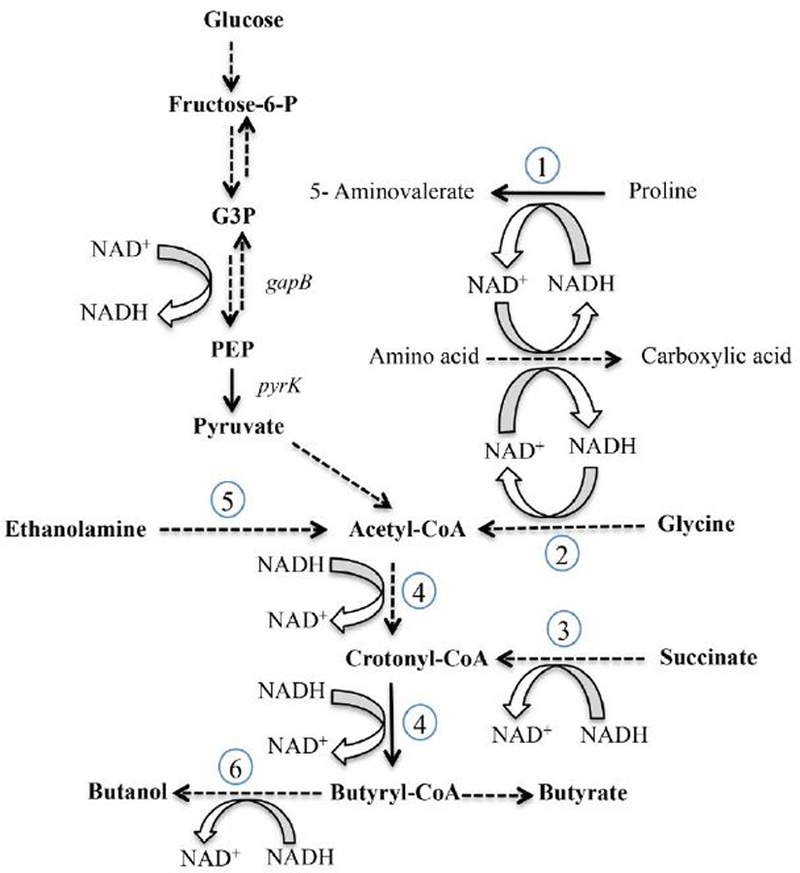
Conversion of glucose to pyruvate (glycolysis) and oxidation of amino acids to carboxylic acids (Stickland metabolism) generate NADH. Proline reductase (1), glycine reductase (2) and the succinate-acetyl CoA pathway to butyrate (3, 4) generate NAD+. Solid lines indicate single enzyme steps, whereas dotted lines indicate multienzyme steps in the pathways. Specific pathways and genes that encode the relevant enzymes are denoted as follows: (1) D-proline reductase (CD3236–3244, prd); (2) glycine reductase (CD2348-CD2358, grd); (3) succinate reduction (CD2344–CD2338); (4) conversion of acetyl CoA to butyryl CoA (CD1054–CD1059); (5) ethanolamine utilization (CD1906-CD1925, eut); and (6) alcohol dehydrogenase (CD2966, adhE). In the prdR mutant, the prd genes were underexpressed, but the other indicated pathways were all overexpressed at least 5-fold compared to the wild-type.
Table 2.
Validation of microarray data.
| CD2344 | bcd2 | adhE | |
|---|---|---|---|
| WT-TYP/WT-TY | 0.046 (± 0.029) | 0.022 (± 0.024) | 0.002 (± 0.0003) |
| prdR-TYP/WT-TYP | 374.5 (± 103.9) | 21.27 (± 5.33) | 251.7 (± 126.4) |
Expression of the CD2344, bcd2, and adhE genes was measured by qRT-PCR in JIR8094 (WT) and prdR mutant strains. Cells were grown in TY medium with or without supplementation with 30 mM L-proline (TYP) to mid-exponential phase as described under Experimental procedures. The means and standard error of the means of at least three biological replicates are shown.
Activation of the PR gene cluster by PrdR in response to excess proline availability (Bouillaut et al., 2013) leads to production of 5-aminovalerate and to regeneration of NAD+ from NADH (Fig 1). Interestingly, PrdR also appears to activate expression of genes for carbohydrate transport (CD2666–2667, CD3136–3138, CD2566–2569) and glycolysis (CD3394–3395, CD3171–3174) (Tables 2, S1 and S2), a process that converts NAD+ to NADH. Thus, PrdR seems to co-regulate two sets of genes whose functions are antithetical. On the other hand, PrdR negatively regulates alternative reductive pathways [CD1054–CD1059 (Fig. 1 (4)) and CD2344–CD2338 (Fig. 1 (3)] that regenerate NAD+, implying that PrdR somehow controls which pathways take precedence. Two possible mechanisms by which proline and PrdR could repress the reductive pathways are: (i) that upon activation by proline, PrdR acts as a direct repressor of all the reductive pathways; or, (ii) that the direct activation of PR expression by PrdR affects the NAD+/NADH ratio in the cell, thereby influencing the activity of another protein that is the direct regulator of the alternative reductive pathways. Because we observed that expression of CD2344, bcd2 and grdE was up-regulated in a prdB mutant lacking PR activity as much as in a prdR mutant (data not shown and (Bouillaut et al., 2013), we favored the second hypothesis.
Rex controls expression of NAD+- generating pathways
Several Gram-positive bacteria (e.g., Clostridium acetobutylicum (Wietzke & Bahl, 2012), Streptomyces coelicolor (Brekasis & Paget, 2003), Bacillus subtilis (Gyan et al., 2006, Schau et al., 2004), and Staphylococcus aureus (Pagels et al., 2010)) have a redox-dependent transcriptional repressor (Rex) that plays a key role in regulating important physiological processes related to energy and carbohydrate metabolism, fermentation pathways, nitrate/nitrite and sulfate/sulfite reduction pathways and the NAD(P)H biogenesis pathways (Ravcheev et al., 2012). Rex is a DNA-binding protein containing a Rossmann-fold dinucleotide binding site and a DNA-binding domain. Structural studies of Thermus aquaticus Rex-NAD+/NADH interactions have shown that binding of NADH induces a conformational change as a result of which binding of Rex to its DNA target sites (TTGTGAA[a/t6]TTCACAA) is blocked (Sickmier et al., 2005).
In C. acetobutylicum, rex lies upstream of a cluster of genes involved in butyrate synthesis and has been shown to control production of butyrate (Wietzke & Bahl, 2012). Although the genetic context is different in C. difficile, BLAST analysis indicated that a homolog of Rex is also present in C. difficile. In addition, a search for the C. difficile Rex regulon, based on the presence of an apparent consensus sequence for Rex-binding sites (defined by comparative genomics) using the RegPrecise database (http://regprecise.lbl.gov/RegPrecise/regulon.jsp?regulon_id=29872, (Novichkov et al., 2010)), suggested that Rex may play a role in regulation of several pathways, including the succinate and butyrate reductive pathways (CD2344–CD2338, Fig. 1 (pathway 3) and CD1054–CD1059, Fig. 1 (pathway 4), respectively), the alcohol-aldehyde dehydrogenase (adhE) gene, an NADH oxidase gene (fprA), and the rex gene.
Although rex gene expression was affected less than 2-fold by addition of proline to the medium or by the prdR null mutation (hence its absence from Supplementary Tables S1 and S2), we tested whether the Rex protein plays a role in the proline-dependent regulation of the genes required to regenerate NAD+. To do so, we created an insertion mutation in rex using TargeTron technology (see Experimental procedures and Fig. S1 for verification of the mutation). Unlike the prdR mutation, the rex mutation did not have a negative impact on cell culture density and, in fact, led to increased culture density in the presence of proline (Fig. S2). Using qRT-PCR, we measured the relative amounts of prdA (CD3244), grdE (CD2354), CD2344 and bcd2 (CD1054) mRNAs in the wild-type and rex mutant (LB-CD24) strains. We found that in the absence or in the presence of excess proline, a rex mutation had only a weak effect (< 2-fold) on prdA expression compared to the wild-type (Fig. 2A). However, in the absence of added proline, transcripts of CD2344, bcd2 and grdE in the rex mutant were increased 100-fold, 12-fold and 5-fold, respectively, compared to the wild-type (Fig 2B–D). Moreover, when the cells were grown in the presence of excess proline, mRNA levels for these genes were much higher in the rex mutant than in wild-type cells grown under the same conditions (Fig. 2B–D). These results indicate that Rex contributes to the negative regulation of these alternative reductive pathways in both the presence and absence of excess proline.
Figure 2. Effects of a rex mutation on expression of NAD+-regeneration genes.
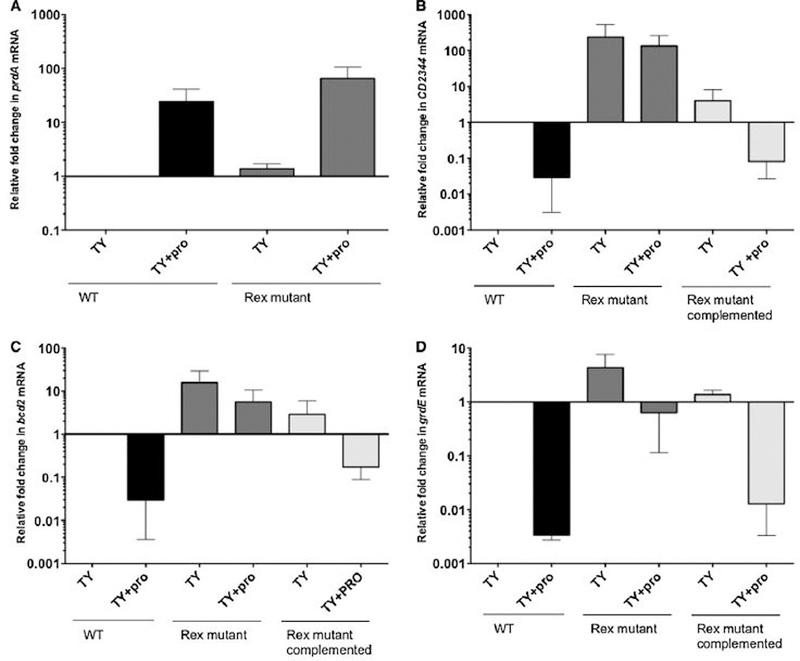
Expression of the prdA (A), CD2344 (B), bcd2 (C), and grdE (D) genes was measured by qRT-PCR in wild- type, rex mutant and complemented rex mutant strains. Cells were grown in TY medium with or without supplementation with 30 mM L-proline to mid-exponential phase as described under Experimental procedures. The means and standard error of the means of at least three biological replicates are shown.
To confirm that proline-responsive regulation was due to the disruption of rex, we complemented strain LB-CD24 by integrating the wild-type rex gene into the LB-CD24 chromosome, creating strain ND-CD23 (see Experimental procedures). As shown in Fig. 2B–D, complementation of LB-CD24 with the wild-type rex gene restored fully or at least partially to wild-type levels the amounts of the CD2344, bcd2 and grdE mRNAs. The results support a role for Rex in regulation of these genes, but do not rule out the possibility that PrdR, the known proline-responsive regulator, further contributes to this regulation either directly or by controlling the activities of regulators other than Rex.
Binding of Rex to promoter regions
To determine whether Rex controls expression of these pathways directly, the C. difficile Rex protein containing a six-histidine-tag at the C-terminus was expressed in and purified from Escherichia coli, as described in Experimental procedures. Purified Rex was used in DNA-binding assays with DNA fragments corresponding to the upstream regions of the aldehyde CoA-alcohol dehydrogenase gene (adhE) and of the first genes in the proline reductase (prdA), succinate (CD2344), butyrate (bcd2) and glycine reductase (grdE) operons. Radiolabeled PCR-generated probes were prepared for each of these sites (Table 3). As shown in Fig. 3, Rex bound to the sites upstream of grdE, CD2344, adhE and bcd2, but not prdA. Surprisingly, the addition of NAD+, which is known to increase the binding affinity of Rex (Pagels et al., 2010), had a relatively small impact on the apparent affinity for Rex binding (Fig. 3). To assess the role of NADH, we mixed NAD+ and NADH at varying ratios. When Rex was present at 100 nM and NAD+ at 10 mM, binding was nearly complete for the adhE, grdE, and CD2344 samples (Fig. 4). The addition of 1 mM NADH, however, substantially inhibited binding; addition of NADH at 5 mM blocked binding almost completely. When NADH was present at 1 mM, increasing the concentration of NAD+ overcame the inhibitory effect of NADH (Fig. 5). These results support a model in which Rex binds upstream of four genes whose expression is derepressed in a rex mutant strain and the extent of Rex binding is determined by the relative concentrations of NAD+ and NADH. Although not tested here, the relatively small impact of NAD+ on Rex binding in the absence of NADH may mean that at least a fraction of the purified Rex protein population had NAD+ bound to it.
Table 3.
DNA samples used for in vitro Rex binding assays
| Target gene | Gene product | Oligonucleotides used for creating DNAs as targets of Rex binding | Location of DNA sequence used for in vitro binding relative to start codon |
|---|---|---|---|
| grdE | subunit of glycine reductase | oLB185 & oLB186, 321-bp product | −303 to +17 |
| adhE | aldehyde-alcohol dehydrogenase | oLB403 & oLB402, 236-bp product | −224 to +12 |
| CD2344 | putative membrane protein of the succinate metabolism cluster | oLB400 & oLB401, 446-bp product | −411 to +24 |
| bcd2 | butyryl-CoA dehydrogenase | oLB404 & oLB369, 179-bp product | −277 to −99 |
| prdA | subunit of proline reductase | oLB198 & oLB184, 246-bp product | −294 to −49 |
Figure 3. Gel mobility shift assays of Rex binding to the upstream regions of potential target genes.
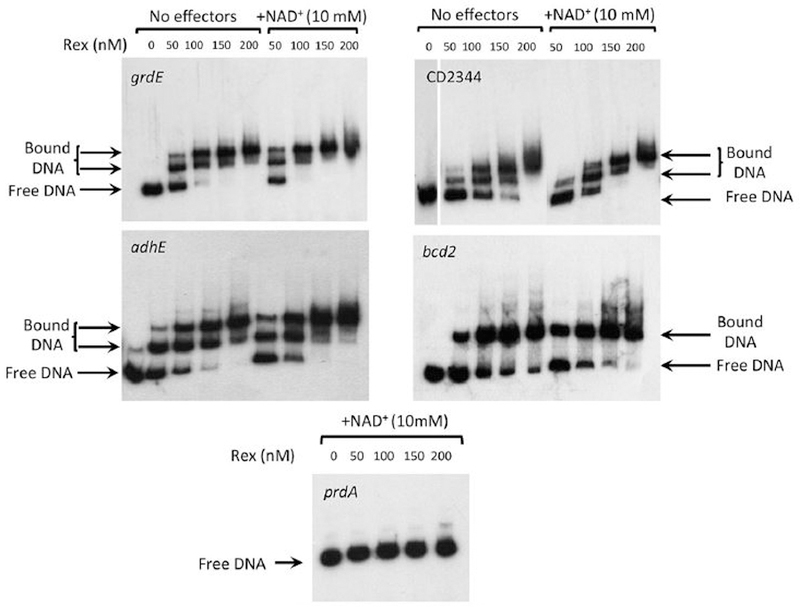
DNA samples corresponding to upstream regions of potential Rex target genes were created by PCR using radioactive oligonucleotides listed in Table S4. For each potential target, the radioactive DNA was incubated with varying concentrations of purified, His-tagged Rex protein with or without the addition of NAD+. The samples were then subjected to electrophoresis in non-denaturing polyacrylamide gels and analyzed by autoradiography.
Figure 4. Impact of NADH/NAD+ ratios on binding of Rex to target sites for the adhE. grdE and CD2344 genes.
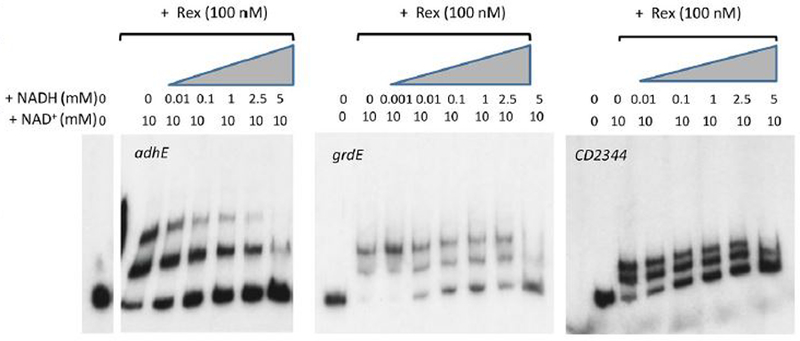
As described in Figure 3, radioactive DNAs carrying the adhE, grdE and CD2344 Rex-binding sites were incubated with Rex protein in the absence or presence of 10 mM NAD+ and at varying concentrations of NADH.
Figure 5. Impact of varying concentrations of NAD+ on binding of Rex to target sites for the adhE, grdE, bcd2 and CD2344 genes.
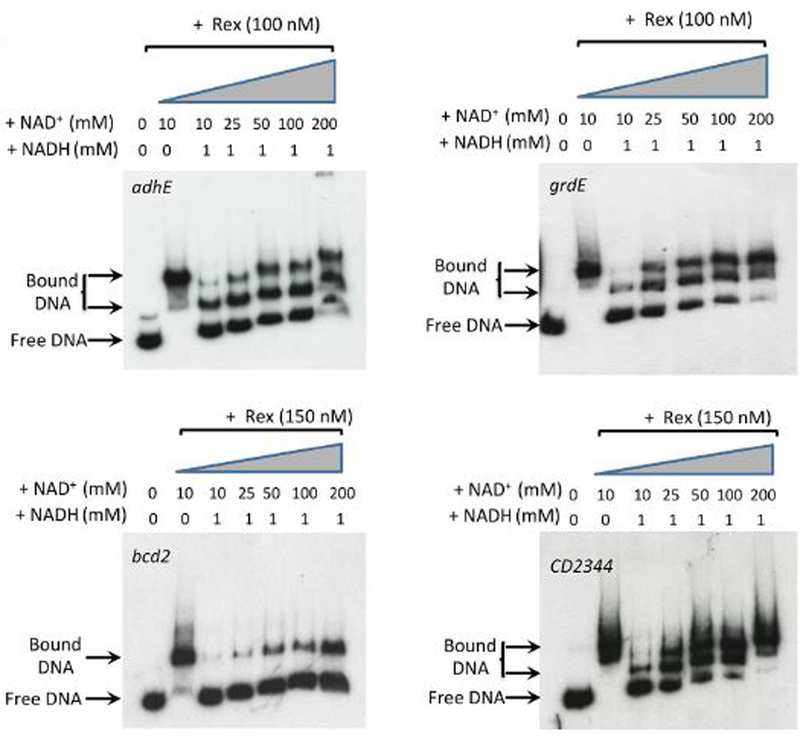
As described in Figure 3, radioactive DNAs carrying the adhE, grdE, bcd2 and CD2344 Rex-binding sites were incubated with Rex protein in the presence of varying concentrations of NAD+ with and without 1 mM NADH.
To determine more precisely the locations of the Rex-binding sites, DNase I footprinting assays were performed, using a probe carrying the CD2344 promoter region. Figure 6A shows that Rex, in the presence of 10 mM NAD+, protected with different affinities three regions corresponding to positions −48 bp to −66 bp (BS-1, highest affinity), −111 bp to −120 bp (BS-2, intermediate affinity) and approximately −150 to −175 bp (BS-3, lowest affinity) with respect to the CD2344 start codon (Fig. 6B). Furthermore, we observed no protection when no NAD+ was added or when a mixture of 1 mM NADH and 10 mM NAD+ was added to the reaction (Fig. 6A), indicating that NAD+ stimulates Rex binding and NADH inhibits Rex binding. Consistent with repression by Rex, we found that the protected region BS-2 overlaps with the initiation site of transcription determined by 5’RACE-PCR (Fig. 6B) and that Rex BS-1 is located downstream of this transcription start. Dineen et al. (2010) reported the presence of a CodY-controlled promoter upstream of Rex BS-3 (Fig. 6B), suggesting that both CodY and Rex contribute to repression of the upstream promoter.
Figure 6. DNase I footprinting of the Rex binding site upstream of the CD2344 coding region.
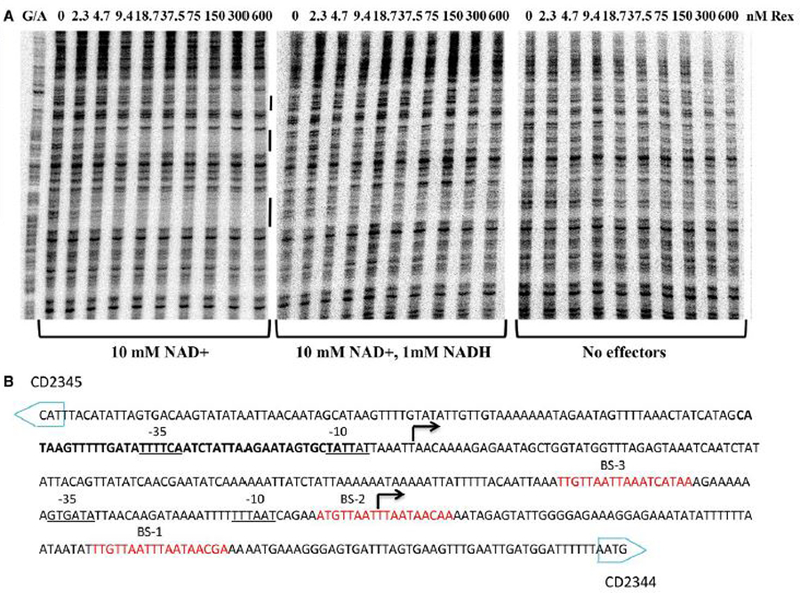
(A) Binding of Rex to the CD2344 regulatory region as detected by a footprinting assay. Radiolabelled DNA fragments were incubated with increasing amounts of purified Rex with 10 mM NAD+ (left panel), or with a mixture of 10 mM NAD+ and 1 mM NADH (middle panel) or no effectors (right panel). Rex concentrations used (nM) are indicated above each lane. The black bars delineate the regions of protection corresponding to Rex binding sites (BS-1, BS-2 and BS-3). (B) Sequence of the CD2344 regulatory region. Two sites of transcription initiation as determined by 5’-RACE are indicated in bold below the broken arrow. The CodY-protected area (Dineen et al., 2010) is in bold. Potential −10 and −35 regions of the promoter are indicated. The sequences protected by Rex in DNase I footprinting experiments located using a G/A ladder (see Experimental procedures) are underlined and predicted binding sites are italicized.
To verify the proposed binding site for Rex upstream of bcd2, we introduced a deletion mutation in the apparent site (Fig. 7). The deletion completely blocked binding of Rex to the bcd2 region.
Figure 7. Impact of deletion of the apparent Rex binding site on Rex binding in vitro.
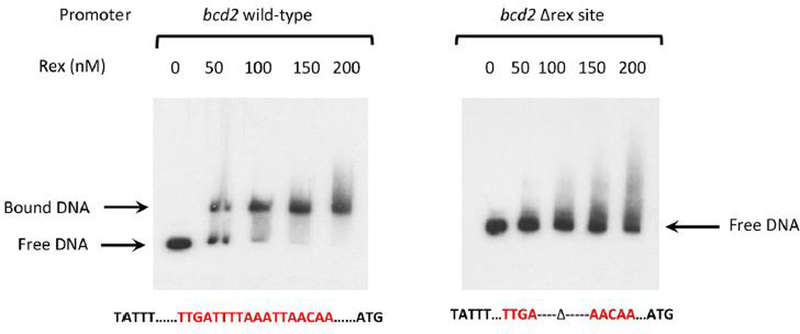
Radioactive DNA samples carrying the apparent wild-type Rex-binding site upstream of the bcd2 coding region or a deletion in the apparent site were tested for binding by purified Rex protein in vitro.
The grd gene region is composed of eight genes that appear to be arranged in two clusters (grdX-trxB3-trxA2 and grdEABCD). We observed weak binding (approximate KD >150 nM) of Rex to a DNA fragment containing the grdX promoter region by a gel mobility shift assay; DNase I footprinting did not reveal any protected region (data not shown), suggesting that the main impact of Rex might be elsewhere. Graentzdoerffer et al. (2001) reported the mapping of four different initiation sites of transcription for glycine reductase genes in Clostridium sticklandii, all of them lying upstream of grdE. We mapped, by 5’RACE-PCR, a site of initiation of transcription at position −58 relative to the start codon of grdE. This site is within the region bound by Rex in gel shift experiments (Fig. 3–5).
Analysis of the sequences of the protected regions detected here showed that a binding motif similar to the proposed consensus TTGTTAANNNNTTAACAA (Ravcheev et al., 2012) can be found (Fig. 8). Based on these sequences, we built a consensus sequence using MEME (Bailey & Elkan, 1994) (Fig. 8). This consensus sequence differs slightly from the previously proposed consensus by showing less variability in the four central positions.
Figure 8. Alignment of Rex binding motifs.
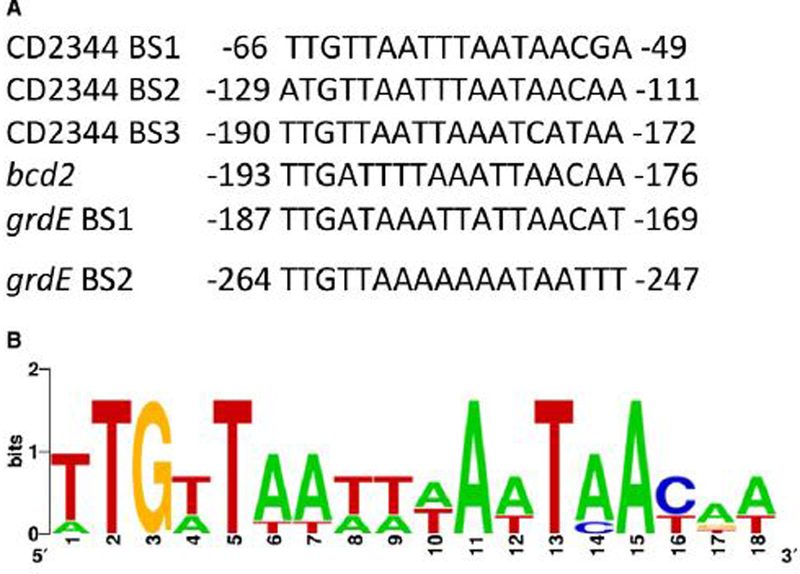
(A) To search for a consensus Rex binding site, the protected regions of the CD2344, grd and bcd2 genes were aligned and compared to the consensus proposed by Ravcheev et al. (Ravcheev et al., 2012). The location of the apparent binding motif is presented with respect to the start codon of the gene in question. (B) A motif logo based on the six C. difficile Rex-binding sites was generated using the MEME algorithm (http://meme.nbcr.net/meme/).
Taken together, these results indicate that Rex binds directly to the bcd2, CD2344, aldE and grdE upstream regions and its binding is affected by the relative concentrations of NAD+ and NADH.
A null mutation in prdR but not in rex increases the severity of C. difficile infection in hamsters
We previously reported that PrdR mediates proline-dependent negative regulation of tcdA expression in early stationary growth phase (Bouillaut et al., 2013). Given that Rex controls expression of pathways involved in butyrate production and that butyrate has been shown to activate toxin synthesis by a mechanism not yet known (Karlsson et al., 2000), we tested the role of Rex in toxin gene expression by qRT-PCR. We observed that relative amounts of tcdA mRNA were about 6.2-fold and 4.5-fold higher in a rex mutant and in a prdR mutant, respectively, compared to that in wild-type cells after 24 hours of growth in the defined medium CDMM (Fig. 9A). These results suggest that alterations in PrdR or Rex activities could influence the outcome of C. difficile infection. (The impact of PrdR and Rex may have been underestimated in these experiments, since CDMM contains 2.6 mM proline, a concentration considerably lower than that used to increase PrdR activity in TY medium (30 mM)). We therefore tested the roles of PrdR and Rex in C. difficile infection using the Syrian hamster model; this animal model recapitulates the most severe form of human CDI, pseudomembranous colitis (Chang et al., 1978). Hamsters were treated with clindamycin to induce sensitivity to C. difficile colonization and infection and were gavaged with 1,000 spores of C. difficile strain JIR8094 (wild-type) or the prdR mutant or the rex mutant. None of the uninfected animals died. Animals infected with either wild-type or rex mutant spores showed relatively mild signs of disease and virulence. (In fact, the rex mutant was somewhat less virulent than the wild-type.) However, the prdR mutant was able to cause fulminant CDI and exhibited increased virulence (log rank p-value < 0.05) and decreased time to death (hazard ratio 2.5, 95% CI 1.339 to 10.87) when compared to strain JIR8094 (Fig. 9B). These results imply that, in the absence of PrdR, virulence of C. difficile increases because of increased expression of toxin and/or other virulence genes. Since the rex mutant also overexpresses the tcdA gene, the lack of effect of a rex mutation on virulence may be attributable to one or more of its broad effects on NAD+-regenerating pathways (see Discussion). An additional possibility would be that the rex mutation reduces the ability of C. difficile cells to form spores or germinate their spores, two steps essential for virulence. However, a test of sporulation (and, indirectly, germination) showed no significant decrease in the abilities of the rex and prdR mutants to sporulate or for their spores to form colonies (Table S4).
Figure 9. Impact of Rex and PrdR on virulence.
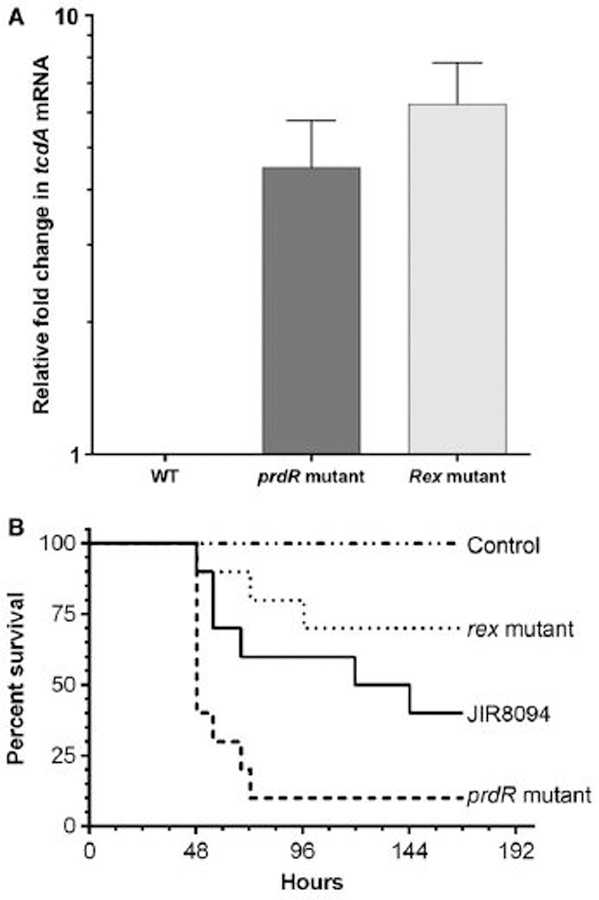
(A) Effects of a rex mutation on toxin gene expression. Expression of the tcdA gene was measured by qRT-PCR in strains JIR8094 (WT), LB-CD24 (rex mutant) and LB-CD8 (prdR mutant). Cells were grown in CDMM for 24 h. The amount of tcdA mRNA was normalized to that of the internal control transcript rpoA. The means and standard error of the means of at least three biological replicates are shown. (B) Virulence of rex and prdR mutants in the hamster model of CDI. Kaplan-Meier survival curves of clindamycin-treated Syrian hamsters inoculated with 1,000 spores of C. difficile JIR8094 (wild-type, black line) or the rex mutant (dotted line) or the prdR mutant (dashed line). Uninfected control animals were treated with clindamycin, but did not receive any C. difficile. Animals showing signs of C. difficile infection (wet tail, poor fur coat, lethargy) were euthanized.
Discussion
Stickland metabolism is an important source of energy for C. difficile and the proline reductive branch plays a role in toxin gene expression (Bouillaut et al., 2013, Jackson et al., 2006). Whereas several amino acids can feed the oxidative pathway to generate energy (ATP) and reducing power (NADH), only proline and glycine are reduced by PR and GR, respectively, to regenerate NAD+ from NADH. PrdR, an apparent sigma-54-dependent activator, controls both proline-dependent activation of the proline reductase gene cluster and proline-dependent repression of the glycine reductase operon (Bouillaut et al., 2013), suggesting the preferential use of proline for NAD+ regeneration in laboratory-grown cells. Interestingly, Janoir et al. observed that PR is up-regulated and GR is down-regulated during the course of infection of monoxenic mice (Janoir et al., 2013), suggesting that proline reduction may be a preferred reductive pathway in vivo as well.
In determining the impact of proline and PrdR on global gene expression, we observed that 4–5% of all genes are differentially regulated ≥2-fold in the presence of exogenous proline and in a prdR mutant (Tables 2, S1 and S2). Genes whose expression was altered fall roughly into five main categories: (i) energy metabolism, e.g., ATP synthase (CD2954–2961) and electron transport genes (CD1137–1142); (ii) carbohydrate metabolism, e.g., glycolysis (CD3394–3395, CD3171–3174) and PTS transport system genes (CD3136–3138); (iii) fermentation pathways, e.g., PR (CD3236–3244), GR (CD2348-CD2358) and the pathways producing butyryl-CoA from acetyl-CoA (CD1054–CD1059) and succinate (CD2344–CD2338); (iv) coenzyme and secondary metabolism pathways (CD0324-CD0327, CD1906-CD1925) and (v) cell wall components (CD1413, CD0830). Whereas the effects of proline and a prdR mutation on some of these genes may not be physiologically significant, the genes whose expression was most strongly affected by addition of proline or loss of PrdR function include the pathways that interconvert NAD+ and NADH (Table 1).
The extent to which PrdR regulates its target genes directly is unknown, but the results presented here support the hypothesis that many of the effects of PrdR are mediated directly by Rex. In several aerobic Gram-positive bacteria, the Rex protein acts as a repressor of genes important for fermentative growth. Rex directly senses changes in the redox status of the cell. When oxygen is available to support NAD+ regeneration by oxidative phosphorylation, Rex inhibits transcription of genes that code for alternative NAD+-regenerating pathways, but, when the NAD+/NADH ratio decreases, Rex dissociates from its operator sites, allowing the alternative pathways to be expressed (Brekasis & Paget, 2003). Rex also provides a redox-dependent regulatory function in controlling central fermentative pathways in C. acetobutylicum (Wietzke & Bahl, 2012). In C. difficile, apparent Rex boxes are found in the upstream regions of PrdR-regulated genes involved in the fermentation pathways producing butyryl-CoA from acetyl-CoA (CD1054–CD1059) or succinate (Ravcheev et al., 2012). As shown here, increased transcript levels for the respective genes (bcd2, CD2344 and grdE) were observed in a rex mutant (Fig. 2), indicating that Rex acts as a negative regulator of the genes tested. Moreover, Rex appears to be a direct repressor of at least four transcription units, since it binds to specific sites within the regulatory regions of CD2344, bcd2, adhE and grdE. In addition, its ability to bind to DNA is inhibited by NADH. C. difficile Rex binding to DNA also appears to be stimulated by NAD+, but it was previously reported that NADH co-purifies with Rex (Sickmier et al., 2005). Hence, a simple model is that NAD+ and NADH compete for binding to Rex and that binding of NAD+ removes the inhibitor of DNA binding (NADH) or that binding of NADH removes the activator of DNA binding (NAD+).
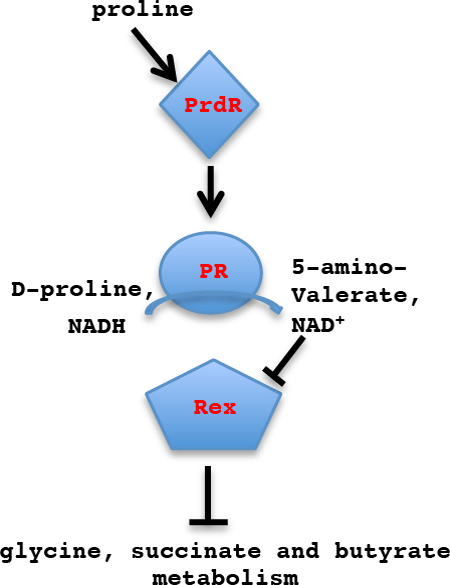
These findings support the following model for how PrdR and proline modulate Rex activity (Fig. 10). When proline is in excess, PrdR is active and stimulates PR expression; as a result, NADH is oxidized, the ratio of NADH to NAD+ is low, and Rex is active as a repressor of the alternative NAD+ regeneration pathways. When proline becomes limiting and the NADH/ NAD+ ratio increases, NADH prevents Rex from repressing expression of the alternative pathways. Unfortunately, we were unable to analyze reproducibly the relative concentrations of NAD+ and NADH in C. difficile, possibly because of instability of NAD+ or because the methods used do not reveal small differences.
Figure 10. Model for regulation of NAD+-regenerating pathways.
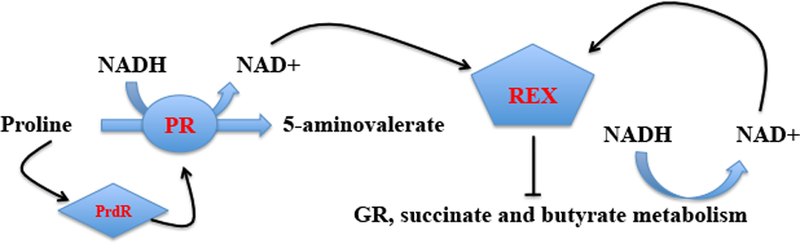
Proline reductase (PR) is proposed to be the preferred pathway for conversion of NADH to NAD+. When proline is present in an amount sufficient to activate PrdR, PR is expressed and produces enough NAD+ to activate Rex as a repressor of alternative NAD+-regenerating pathways. When proline is consumed, Rex loses DNA-binding activity and the alternative pathways are derepressed.
It has been previously reported that major global regulators, such as CodY and CcpA, play a role in controlling expression of these same pathways (Antunes et al., 2011, Dineen et al., 2007). In fact, CcpA, in response to fructose-1,6-bisphosphate, activates expression of the prd gene cluster and represses alternative NAD+-regenerating pathways, such as the glycine reductase gene cluster and the pathways from acetyl-CoA and succinate to butyrate (Antunes et al., 2011). CcpA binding sites were discovered upstream of prdA and grdX, but not for prdR or rex or the butyrate-production genes (Antunes et al., 2011). Thus, for some of these targets CcpA is likely to be a direct regulator, but an indirect regulator for others. CodY, in response to BCAAs and GTP, represses the pathways leading to butyryl-CoA, as well as the genes (ptb and buk) whose products interconvert butyryl CoA and butyrate (Dineen et al., 2010). A codY null mutation causes overexpression of the bcd2 gene cluster and the succinate metabolism operon; the same regions were shown to have CodY binding sites (Dineen et al., 2010). However, the prd and grd loci do not have CodY binding sites and their expression is not affected by a codY null mutation (Dineen et al., 2010). Together with the results presented here, these observations suggest (i) that C. difficile prioritizes NAD+ regeneration in favor of PR activity and (ii) butyrate production signals redox stress (high NADH/NAD+ ratio) and nutrient limitation (i.e., proline, BCAAs, carbohydrates). Regeneration of NAD+ using these alternative pathways leads to the production of butyrate, a stimulator of toxin synthesis (Karlsson et al., 2000), suggesting that integration of these environmental signals results in control of virulence. The molecular mechanism by which butyrate activates toxin synthesis remains unknown. In fact, a prdR mutant strain has increased virulence in a hamster model (Fig. 9B), implying that the functioning of the PR pathway normally keeps virulence under control. In addition to controlling tcdA expression, PrdR controls expression of genes involved in ethanolamine utilization (eut genes), cobalt transport and cobalamine biosynthesis (cbi genes) (Tables 2 and S2). Ethanolamine is a compound that can be readily derived from eukaryotic cell membranes; some intestinal bacteria use it as a source of carbon and/or nitrogen (Garsin, 2010). Interestingly, a recent report indicates that C. difficile eut genes are up-regulated during infection in monoxenic mice (Janoir et al., 2013).
The apparent relationship between the activities of PrdR and Rex are in contrast to the results obtained during infection of hamsters. That is, when PrdR is inactive, the increased NADH/NAD+ ratio should favor inactivation of Rex and, as a result, the pathways to production of butryrate, an activator of toxin production, should increase in expression. Hence, the fact that a prdR mutant is more virulent than the wild-type fits with this prediction. However, the reduced virulence of a rex mutant is at odds with this explanation of the effect of a prdR mutation. Moreover, a recent publication (Battaglioli et al., 2018) reported that proline is required for growth of C. difficile strain 630 in vitro and that proline and the wild-type prdA gene are required for strain 630 to survive and grow in mice. Moreover, expression of the C. difficile prdA gene was detectable in infected dysbiotic mice (i.e., mice with reduced intestinal microbiota), but not in infected healthy mice. Furthermore, after C. difficile infection, a prdB mutant was undetectable in the stool of healthy mice and was only capable of reduced colonization and toxin B production in infected dysbiotic mice. Since mutants defective in prdA or prdB are expected to have a reduced ability to convert NADH to NAD+ (in a proline-containing medium), the Rex protein should be relatively inactive in such strains and, as a result, the conversion of succinate and acetyl CoA to butyrate, an inducer of toxin synthesis, should be higher than in wild-type cells.
One possible explanation would be that PrdR controls virulence factor gene expression in part via regulation of Rex activity and in part by a mechanism that is independent of Rex. Alternatively, a rex mutant may accumulate unacceptably high levels of NAD+ and consequently be more sensitive to oxidative stress in vivo, as has been reported for C. acetobutylicum (Zhang et al., 2014).
Experimental Procedures
Bacterial strains and growth conditions
The bacterial strains and plasmids used in this study are listed in Table 4. C. difficile strains were grown in TY complex medium (Dupuy & Sonenshein, 1998) or in defined CDMM (Cartman & Minton, 2010), supplemented with 250 µg D-cycloserine per ml, 40 µg kanamycin per ml, 20 µg thiamphenicol per ml, 20 µg lincomycin per ml or 5 µg erythromycin per ml, as needed. C. difficile strains were maintained at 37°C in an anaerobic chamber (Coy Laboratory Products) with an atmosphere of 10 % H2, 5 % CO2 and 85 % N2. E. coli strains were grown at 37°C in L medium supplemented with 20 µg chloramphenicol per ml or 100 µg ampicillin per ml, as needed.
Table 4.
Rex-binding sites
| Gene | Rex-binding sites | Transcription start sites | Start codon |
|---|---|---|---|
| grdE | 2723649-2723632 2723726-2723709 |
2723519 | 2723461 |
| bcd2 | 1246727-1246744 | 1246818 | 1246920 |
| CD2334 | 2713745-2713728 2713808-2713791 2713869-2713851 |
2713799 2713982 |
2713679 |
The Rex-binding sites were determined by gel mobility shift and DNase I footprinting assays; the transcription start points were determined by 5’-RACE. The numbers refer to sites within the whole genome of strain JIR8094.
Strain and plasmid constructions.
Genomic DNA (gDNA) of C. difficile strain JIR8094, an erythromycin-sensitive derivative of strain 630 (GenBank accession number AM180355) was used as a template for PCR amplification. Sequencing of cloned DNA fragments was performed by the Tufts University Core Facility. Oligonucleotides used in this study are listed in Supplementary Table S4.
To create a rex insertional mutation by TargeTron mutagenesis, we first generated a rex-targeted intron fragment using primers oLB329, oLB330, oLB331 and EBS universal and a mixture of two templates, pBL64 and pBL65. The PCR fragment was cloned between the HindIII and BsrGI restriction sites of pBL100 (Bouillaut et al., 2013). The resulting plasmid, pBL130, was introduced by electroporation into E. coli strain HB101 (pRK24) and the recombinant strain obtained was subsequently mated with C. difficile JIR8094 as described (Bouillaut et al., 2011). Insertional disruption of rex was verified by PCR using primers oLB332, oLB333 and EBSu (see Fig. S1).
The plasmid pBL26 used for complementation experiments was created as follow: pSMB47 (Manganelli et al., 1998) was digested with EcoRI and NcoI, blunted and self-ligated, creating pBL18. The catP gene including its own promoter (950 bp) from pJIR1456 (amplified using primers 78 and 83) was inserted at the SmaI site of pMMOrf (Lampe et al., 1996) creating pMMOrf-Cat. The latter and ITR primer (Liu et al., 2013) were used to amplify the catP gene while adding a SphI site at each end; the PCR-generated fragment was digested and inserted at the SphI site of pBL18, resulting in pBL26. To complement the rex gene disruption, a 785-bp fragment containing the rex gene and its upstream region was amplified using primers oLB345 and oLB368 and cloned between the BamHI and BspHI sites of pBL26, generating pND1. This plasmid was introduced into B. subtilis strain BS49; homologous recombination within the transposon Tn916 was selected on chloramphenicol plates. The transfer of Tn916 containing the rex gene into strain LB-CD24 was performed as previously described (Bouillaut et al., 2011), resulting in strain ND-CD23.
Microarray design, DNA-array hybridization and transcriptome analysis
A microarray of the C. difficile 630 genome (GEO database accession number GPL10556) was designed as previously described (Saujet et al., 2011). Transcriptome analysis was performed using four different RNA preparations for each condition (JIR8094 in TY medium with and without added proline (30 mM); JIR8094 vs. the prdR mutant in proline-supplemented medium). After reverse transcription of RNA extracted from each culture, hybridization of labeled cDNA to microarrays and array scanning were done as previously described (Saujet et al., 2011). The slides were analyzed using R and limma software (Linear Model for Microarray Data) from Bioconductor (www.bioconductor.org). We corrected background with the ‘normexp’ method (Breitling et al., 2004), resulting in strictly positive values and reducing variability in the log ratios for genes with low levels of hybridization signal. Then, we normalized each slide with the ‘loess’ method (Smyth & Speed, 2003). To test for differential expression, we used Bayesian adjusted t-statistics and performed the multiple testing correction of Benjamini & Hochberg based on the false discovery rate (FDR) (Benjamini & Hochberg, 1995). A gene was considered as differentially expressed when the p-value was < 0.05. The complete data set was deposited in the GEO database with a series record accession number GSE42472.
Purification of His6-tagged Rex.
The rex gene was amplified from JIR8094 genomic DNA using Phusion polymerase (New England Biolabs) and the primers oLB334/oLB335 (Table S3). The PCR product was digested with NdeI and BamHI and cloned in pET16b (Novagen, San Diego, CA), resulting in pBL131, which encodes the Rex protein with a six-histidine extension at the C-terminus. Rex-His6 expression in E. coli Rosetta BL21(DE3) (Novagen) was induced by exposure to 0.5 mM isopropyl-β-D-thiogalactopyranoside (IPTG) for 3 hrs at 37ºC and the protein was purified using cobalt-charged resin according to the manufacturer’s protocol (Novagen). As analyzed by SDS-PAGE electrophoresis, the Rex protein was the principal protein in the samples used for in vitro assays (data not shown).
Labeling of DNA fragments.
Fragments containing the regulatory regions of the CD2344, bcd2, grdE,grdX and prdA genes were radioactively labeled by PCR from genomic DNA of C. difficile strain JIR8094 with primers listed in Table S4. For each primer set used for PCR amplification, the left-hand primer was end-labelled with T4 polynucleotide kinase (Fermentas) and γ−32P-adenosine triphosphate (3000 Ci.mM−1; Perkin-Elmer) as recommended by the manufacturer. After PCR, the amplified, labeled fragment was then purified using the QIAquick Nucleotide Removal kit (Qiagen™).
Gel mobility shift and DNase I footprinting experiments.
Purified Rex-His6 was incubated with γ32-P-labeled promoter fragments in a GS buffer (20 mM Tris-Cl (pH 8.0), 50 mM KCl, 2 mM MgCl2, 5% glycerol, 0.5 mM EDTA, 1 mM dithiothreitol (DTT), 0.05% Nonidet P-40, and 25 μg salmon sperm DNA per ml). Reactions (10 μl) containing various amounts of Rex and less than 1 fmole DNA were incubated for 30 min at room temperature. In some experiments, NAD+ (15 mM in GS buffer), NADH (1 mM) or various concentrations of NAD+ and NADH, were added to the incubation mixture. For gel mobility shift assays, samples were separated on 8% non-denaturing polyacrylamide gels in Tris-glycine buffer (50 mM Tris, 384 mM glycine, 1 mM EDTA) at 200 v. After electrophoresis (2 h at 13 V/cm), the gel was dried, transferred to filter paper, and analyzed by autoradiography.
For DNase I footprinting experiments, radiolabeled DNA (20–40 fmoles) was incubated with various amounts of purified Rex-His6 in GS buffer for 20 min at room temperature. Incubation was continued for 1 min after addition of 6 mM MgCl2, 6 mM CaCl2, and 0.15 or 0.25 U of RQ1 DNase I (Promega). The reactions were stopped by the addition of 4 μl of sequencing gel loading buffer and heated for 5 min at 80ºC. Samples were then subjected to electrophoresis in an 8 M urea-6% polyacrylamide gel. For each probe, a G/A ladder was generated by incubating the radiolabelled DNA probe with formic acid (Liu & Hong, 1998) and used to locate the position of the protected area.
Determination of transcription start points using rapid amplification of cDNA ends (5’-RACE)
cDNA samples were synthesized from 1 μg of RNA in 20 μl reactions using 2 pmol of CD2344- or bcd2-specific primers, oMC155 or oLB282, respectively, and SuperScript II reverse transcriptase (Invitrogen) per the manufacturer’s instructions. Poly(A) tails were added to the 3’ end using terminal transferase (New England Biolabs), and cDNA was purified using the PCR Purification kit (Qiagen) and eluted in a 50 μl volume. First-round PCR products were generated using 2 μl of purified cDNA as template, universal anchor primer oKZ69, and primers oMC155 or oLB282 specific for the CD2344 and bcd2 genes, respectively. The products were PCR-amplified again using the universal amplification primer oKZ70 and the specific primer oLB337 or oLB338 and then sequenced. The nucleotide at the junction between the gene-specific sequence and the stretch of A nucleotides, generated from sequencing the cDNA corresponding to the poly(A) tail, was taken to represent the apparent 5′ end of mRNA (Frohman, 1994).
Quantitative reverse-transcription PCR (qRT-PCR) analysis.
Cultures of C. difficile grown in TY or CDMM medium were harvested at OD600 = 0.4–0.7 (mid-exponential phase) or after 24 hours of growth (stationary phase) and DNA-free RNA was prepared as previously described (Dineen et al., 2007, McBride & Sonenshein, 2011). RNA was quantified by absorbance (A260 and A260/A280 ratio) using a NanoDrop ND-1000 spectrophotometer (Thermo Scientific). Primers for qRT-PCR were designed using the online PrimerQuest tool from Integrated DNA Technologies (http://www.idtdna.com/Scitools/Applications/Primerquest), and amplification efficiencies for each primer set were determined prior to use. To control for chromosomal DNA contamination, mock cDNA synthesis reactions containing no reverse transcriptase were used as negative controls in subsequent amplifications. cDNA samples (500 ng or 1000 ng) were used as templates for quantitative RT-PCR of rpoC (primers oLB122/oLB123), rpoA (oLB273/oLB274), prdA (primers oLB170/oLB171), grdE (primers oLB176/oLB177), CD2344 (oMC154/oMC155) (Dineen et al., 2010), tcdA (oLB131/oLB132), adhE (oLB341/oLB342) and bcd2 (oLB281/oLB282) using Roche SYBR Green I PCR mix and a Roche LightCycler 480 II thermocycler. Reactions were performed in a final volume of 20 µl. Reactions were performed in triplicate using RNA extracted from each of a minimum of three biological replicates, and results are presented as the means and standard deviations of the data obtained. Amplification included 45 cycles of the following steps: 10 s at 95°C, 10 s at 53°C, 15 s at 72°C. Results were calculated using the comparative cycle threshold method (Schmittgen & Livak, 2008), in which the amount of target mRNA is normalized to that of an internal control transcript (rpoC or rpoA).
Animal experimental conditions
Female Syrian hamsters (80 g – 120 g) were purchased from Charles River Laboratories and individually housed in sterile cages. During the experiment, animals had ad libitum access to food and water. Five days before inoculation with C. difficile spores, hamsters were sensitized to infection with a single gavage of 30 mg of clindamycin per kg. Hamsters (10 per strain) were infected with 1,000 spores of wild-type C. difficile JIR8094 or prdR or rex mutant strains. Five hamsters served as antibiotic-treated, non-infected controls. Animals were monitored for signs of disease (wet tail, lethargy, poor fur coat) and moribund animals were humanely euthanized by CO2 asphyxia followed by thoracotomy as a secondary means of death in accordance with Panel on Euthanasia of the American Veterinary Medical Association. All animal studies were performed with prior approval from the Texas A&M University Institutional Animal Care and Use Committee. Differences in survival of the animals were determined using the Mantel-Cox (log-rank) test and differences in the time to death were determined using the Hazard Ratio (logrank) using the GraphPad Prism software.
Supplementary Material
Table 5.
Strains and plasmids used in this study
| Characteristics | Reference or source | |
|---|---|---|
| Strain | ||
| E. coli | ||
| HB101 | F–supE44 hsdS20(r-Bm-B) recA13 ara-14 proA2 lacY1 galK2 rpsL20 xyl-5 mtl-1 | |
| DH5α | F– Φ80lacZΔM15 Δ(lacZYA-argF) U169 recA1 endA1 hsdR17 (rK–, mK+) phoA supE44 λ– thi-1 gyrA96 relA1 | |
| C. difficile | ||
| JIR8094 | EmS derivative of strain 630 | (O’Connor et al., 2006) |
| LB-CD8 | JIR8094 prdR::ermB | (Bouillaut et al., 2013) |
| LB-CD24 | JIR8094 rex::ermB | This study |
| ND-CD23 | JIR8094 rex::ermB Tn916::rex | This study |
| B. subtilis | ||
| BS49 | (Manganelli et al., 1998) | |
|
Plasmid | ||
| pBL26 | pSMB47 containing catP | This study |
| pBL64 | pCR2.1-intron template part A | (Bouillaut et al., 2013) |
| pBL65 | pCR2.1-intron template part B | (Bouillaut et al., 2013) |
| pBL100 | pBL68 containing un-targeted groupII intron – Targetron vector | (Bouillaut et al., 2013) |
| pBL130 | pBL100 targeted to rex | This study |
| pCR2.1 | Invitrogen® | |
| pND1 | pBL26 containing rex gene with its own promoter | This study |
| pRK24 | Tra+, Mob+, AmpR, TcR | (Trieu-Cuot et al., 1991) |
| pSMB47 | (Manganelli et al., 1998) | |
Acknowledgments
We thank Boris Belitsky for helpful suggestions and discussions during the course of this work and Audrey Hamiot for verifying the location of the rex mutation and providing the growth curves of the various strains. This work was supported by research grants from the US National Institute of General Medical Sciences (R01GM042219) to ALS and from the US National Institute of Allergy and Infectious Diseases (R56AI108987) to JAS, and was funded by the Institut Pasteur and by Integrative Biology of Emerging Infectious Diseases (LabEX IBEID) as part of the framework of the French Government’s “Programme Investissements d’Avenir”. This work was also supported by an American Heart Association National Scientist Development grant to JAS (No. 11SDG7160013) and by a Roux Fellowship (Institut Pasteur) to TD. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or of the NIGMS or NIAID. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- Antunes A, Martin-Verstraete I & Dupuy B, (2011) CcpA-mediated repression of Clostridium difficile toxin gene expression. Mol Microbiol 79: 882–899. [DOI] [PubMed] [Google Scholar]
- Bailey TL & Elkan C, (1994) Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2: 28–36. [PubMed] [Google Scholar]
- Battaglioli EJ, Hale VL, Chen J, Jeraldo P, Ruiz-Mojica C, Schmidt BA, Rekdal VM, Till LM, Huq L, Smits SA, Moor WJ, Jones-Hall Y, Smyrk T, Khanna S, Pardi DS, Grover M, Patel R, Chia N, Nelson H, Sonnenburg JL, Farrugia G & Kashyap PC, (2018) Clostridioides difficile uses amino acids associated with gut microbial dysbiosis in a subset of patients with diarrhea. Science translational medicine 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y & Hochberg Y, (1995) Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B-Methodological 57: 289–300. [Google Scholar]
- Bouillaut L, McBride SM & Sorg JA, (2011) Genetic manipulation of Clostridium difficile. Curr Protoc Microbiol Chapter 9: Unit 9A 2. [DOI] [PMC free article] [PubMed]
- Bouillaut L, Self WT & Sonenshein AL, (2013) Proline-dependent regulation of Clostridium difficile Stickland metabolism. Journal of bacteriology 195: 844–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitling R, Armengaud P, Amtmann A & Herzyk P, (2004) Rank products: a simple, yet powerful, new method to detect differentially regulated genes in replicated microarray experiments. FEBS Lett 573: 83–92. [DOI] [PubMed] [Google Scholar]
- Brekasis D & Paget MS, (2003) A novel sensor of NADH/NAD+ redox poise in Streptomyces coelicolor A3(2). EMBO J 22: 4856–4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartman ST & Minton NP, (2010) A mariner-based transposon system for in vivo random mutagenesis of Clostridium difficile. Applied and environmental microbiology 76: 1103–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TW, Bartlett JG, Gorbach SL & Onderdonk AB, (1978) Clindamycin-induced enterocolitis in hamsters as a model of pseudomembranous colitis in patients. Infect Immun 20: 526–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen GA, Zane GM, Kazakov AE, Li X, Rodionov DA, Novichkov PS, Dubchak I, Arkin AP & Wall JD, (2015) Rex (encoded by DVU_0916) in Desulfovibrio vulgaris Hildenborough is a repressor of sulfate adenylyl transferase and is regulated by NADH. Journal of bacteriology 197: 29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deneve C, Janoir C, Poilane I, Fantinato C & Collignon A, (2009) New trends in Clostridium difficile virulence and pathogenesis. Int J Antimicrob Agents 33 Suppl 1: S24–28. [DOI] [PubMed] [Google Scholar]
- Dineen SS, McBride SM & Sonenshein AL, (2010) Integration of metabolism and virulence by Clostridium difficile CodY. Journal of bacteriology 192: 5350–5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dineen SS, Villapakkam AC, Nordman JT & Sonenshein AL, (2007) Repression of Clostridium difficile toxin gene expression by CodY. Mol Microbiol 66: 206–219. [DOI] [PubMed] [Google Scholar]
- Dupuy B & Sonenshein AL, (1998) Regulated transcription of Clostridium difficile toxin genes. Mol Microbiol 27: 107–120. [DOI] [PubMed] [Google Scholar]
- El Meouche I, Peltier J, Monot M, Soutourina O, Pestel-Caron M, Dupuy B & Pons JL, (2013) Characterization of the SigD regulon of C. difficile and its positive control of toxin production through the regulation of tcdR. PLoS One 8: e83748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohman MA, (1994) On beyond classic RACE (rapid amplification of cDNA ends). PCR Methods Appl 4: S40–58. [DOI] [PubMed] [Google Scholar]
- Garsin DA, (2010) Ethanolamine utilization in bacterial pathogens: roles and regulation. Nat Rev Microbiol 8: 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerding DN, (2010) Global epidemiology of Clostridium difficile infection in 2010. Infect Control Hosp Epidemiol 31 Suppl 1: S32–34. [DOI] [PubMed] [Google Scholar]
- Gyan S, Shiohira Y, Sato I, Takeuchi M & Sato T, (2006) Regulatory loop between redox sensing of the NADH/NAD(+) ratio by Rex (YdiH) and oxidation of NADH by NADH dehydrogenase Ndh in Bacillus subtilis. Journal of bacteriology 188: 7062–7071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L, Huang H, Yuan H, Tao F, Xie H & Wang S, (2016) Rex in Clostridium kluyveri is a global redox-sensing transcriptional regulator. Journal of biotechnology 233: 17–25. [DOI] [PubMed] [Google Scholar]
- Jackson S, Calos M, Myers A & Self WT, (2006) Analysis of proline reduction in the nosocomial pathogen Clostridium difficile. Journal of bacteriology 188: 8487–8495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janoir C, Deneve C, Bouttier S, Barbut F, Hoys S, Caleechum L, Chapeton-Montes D, Pereira FC, Henriques AO, Collignon A, Monot M & Dupuy B, (2013) Adaptive strategies and pathogenesis of Clostridium difficile from in vivo transcriptomics. Infect Immun 81: 3757–3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson S, Burman LG & Akerlund T, (1999) Suppression of toxin production in Clostridium difficile VPI 10463 by amino acids. Microbiology 145 ( Pt 7): 1683–1693. [DOI] [PubMed] [Google Scholar]
- Karlsson S, Lindberg A, Norin E, Burman LG & Akerlund T, (2000) Toxins, butyric acid, and other short-chain fatty acids are coordinately expressed and down-regulated by cysteine in Clostridium difficile. Infect Immun 68: 5881–5888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehne SA, Cartman ST, Heap JT, Kelly ML, Cockayne A & Minton NP, (2010) The role of toxin A and toxin B in Clostridium difficile infection. Nature 467: 711–713. [DOI] [PubMed] [Google Scholar]
- Kyne L & Kelly CP, (2001) Recurrent Clostridium difficile diarrhoea. Gut 49: 152–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampe DJ, Churchill ME & Robertson HM, (1996) A purified mariner transposase is sufficient to mediate transposition in vitro. EMBO J 15: 5470–5479. [PMC free article] [PubMed] [Google Scholar]
- Liu H, Bouillaut L, Sonenshein AL & Melville SB, (2013) Use of a mariner-based transposon mutagenesis system to isolate Clostridium perfringens mutants deficient in gliding motility. Journal of bacteriology 195: 629–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ST & Hong GF, (1998) Three-minute G + A specific reaction for DNA sequencing. Anal Biochem 255: 158–159. [DOI] [PubMed] [Google Scholar]
- Loo VG, Poirier L, Miller MA, Oughton M, Libman MD, Michaud S, Bourgault AM, Nguyen T, Frenette C, Kelly M, Vibien A, Brassard P, Fenn S, Dewar K, Hudson TJ, Horn R, Rene P, Monczak Y & Dascal A, (2005) A predominantly clonal multi-institutional outbreak of Clostridium difficile-associated diarrhea with high morbidity and mortality. N Engl J Med 353: 2442–2449. [DOI] [PubMed] [Google Scholar]
- Lyras D, O’Connor JR, Howarth PM, Sambol SP, Carter GP, Phumoonna T, Poon R, Adams V, Vedantam G, Johnson S, Gerding DN & Rood JI, (2009) Toxin B is essential for virulence of Clostridium difficile. Nature 458: 1176–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manganelli R, Provvedi R, Berneri C, Oggioni MR & Pozzi G, (1998) Insertion vectors for construction of recombinant conjugative transposons in Bacillus subtilis and Enterococcus faecalis. FEMS Microbiol Lett 168: 259–268. [DOI] [PubMed] [Google Scholar]
- Mani N & Dupuy B, (2001) Regulation of toxin synthesis in Clostridium difficile by an alternative RNA polymerase sigma factor. Proc Natl Acad Sci U S A 98: 5844–5849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matamouros S, England P & Dupuy B, (2007) Clostridium difficile toxin expression is inhibited by the novel regulator TcdC. Mol Microbiol 64: 1274–1288. [DOI] [PubMed] [Google Scholar]
- McBride SM & Sonenshein AL, (2011) Identification of a genetic locus responsible for antimicrobial peptide resistance in Clostridium difficile. Infect Immun 79: 167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin KJ, Strain-Damerell CM, Xie K, Brekasis D, Soares AS, Paget MS & Kielkopf CL, (2010) Structural basis for NADH/NAD+ redox sensing by a Rex family repressor. Molecular cell 38: 563–575. [DOI] [PubMed] [Google Scholar]
- Neumann-Schaal M, Jahn D & Schmidt-Hohagen K, (2019) Metabolism the Difficile Way: The Key to the Success of the Pathogen Clostridioides difficile. Frontiers in microbiology 10: 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen NP, Linder S, Flitsch SK, Schiel-Bengelsdorf B, Durre P & Soucaille P, (2016) Cap0037, a Novel Global Regulator of Clostridium acetobutylicum Metabolism. mBio 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novichkov PS, Rodionov DA, Stavrovskaya ED, Novichkova ES, Kazakov AE, Gelfand MS, Arkin AP, Mironov AA & Dubchak I, (2010) RegPredict: an integrated system for regulon inference in prokaryotes by comparative genomics approach. Nucleic Acids Res 38: W299–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor JR, Lyras D, Farrow KA, Adams V, Powell DR, Hinds J, Cheung JK & Rood JI, (2006) Construction and analysis of chromosomal Clostridium difficile mutants. Mol Microbiol 61: 1335–1351. [DOI] [PubMed] [Google Scholar]
- Ong GK, Reidy TJ, Huk MD & Lane FR, (2017) Clostridium difficile colitis: A clinical review. American journal of surgery 213: 565–571. [DOI] [PubMed] [Google Scholar]
- Pagels M, Fuchs S, Pane-Farre J, Kohler C, Menschner L, Hecker M, McNamarra PJ, Bauer MC, von Wachenfeldt C, Liebeke M, Lalk M, Sander G, von Eiff C, Proctor RA & Engelmann S, (2010) Redox sensing by a Rex-family repressor is involved in the regulation of anaerobic gene expression in Staphylococcus aureus. Mol Microbiol 76: 1142–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panitz JC, Zverlov VV, Pham VT, Sturzl S, Schieder D & Schwarz WH, (2014) Isolation of a solventogenic Clostridium sp. strain: fermentation of glycerol to n-butanol, analysis of the bcs operon region and its potential regulatory elements. Systematic and applied microbiology 37: 1–9. [DOI] [PubMed] [Google Scholar]
- Ravcheev DA, Li X, Latif H, Zengler K, Leyn SA, Korostelev YD, Kazakov AE, Novichkov PS, Osterman AL & Rodionov DA, (2012) Transcriptional regulation of central carbon and energy metabolism in bacteria by redox-responsive repressor Rex. Journal of bacteriology 194: 1145–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricciardi R, Rothenberger DA, Madoff RD & Baxter NN, (2007) Increasing prevalence and severity of Clostridium difficile colitis in hospitalized patients in the United States. Arch Surg 142: 624–631; discussion 631. [DOI] [PubMed] [Google Scholar]
- Saujet L, Monot M, Dupuy B, Soutourina O & Martin-Verstraete I, (2011) The key sigma factor of transition phase, SigH, controls sporulation, metabolism, and virulence factor expression in Clostridium difficile. Journal of Bacteriology 193: 3186–3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schau M, Chen Y & Hulett FM, (2004) Bacillus subtilis YdiH is a direct negative regulator of the cydABCD operon. Journal of Bacteriology 186: 4585–4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD & Livak KJ, (2008) Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3: 1101–1108. [DOI] [PubMed] [Google Scholar]
- Schwarz KM, Grosse-Honebrink A, Derecka K, Rotta C, Zhang Y & Minton NP, (2017) Towards improved butanol production through targeted genetic modification of Clostridium pasteurianum. Metabolic engineering 40: 124–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sickmier EA, Brekasis D, Paranawithana S, Bonanno JB, Paget MS, Burley SK & Kielkopf CL, (2005) X-ray structure of a Rex-family repressor/NADH complex insights into the mechanism of redox sensing. Structure 13: 43–54. [DOI] [PubMed] [Google Scholar]
- Smyth GK & Speed T, (2003) Normalization of cDNA microarray data. Methods 31: 265–273. [DOI] [PubMed] [Google Scholar]
- Stadtman TC, (1956) Studies on the enzymic reduction of amino acids: a proline reductase of an amino acid-fermenting Clostridium, strain HF. Biochem J 62: 614–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtman TC & Elliott P, (1957) Studies on the enzymic reduction of amino acids. II. Purification and properties of D-proline reductase and a proline racemase from Clostridium sticklandii. J Biol Chem 228: 983–997. [PubMed] [Google Scholar]
- Stickland LH, (1935a) Studies in the metabolism of the strict anaerobes (genus Clostridium): The oxidation of alanine by Cl. sporogenes. IV. The reduction of glycine by Cl. sporogenes. Biochem J 29: 889–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stickland LH, (1935b) Studies in the metabolism of the strict anaerobes (Genus Clostridium): The reduction of proline by Cl. sporogenes. Biochem J 29: 288–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trieu-Cuot P, Carlier C, Poyart-Salmeron C & Courvalin P, (1991) An integrative vector exploiting the transposition properties of Tn1545 for insertional mutagenesis and cloning of genes from gram-positive bacteria. Gene 106: 21–27. [DOI] [PubMed] [Google Scholar]
- Voth DE & Ballard JD, (2005) Clostridium difficile toxins: mechanism of action and role in disease. Clin Microbiol Rev 18: 247–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Venkataramanan KP, Huang H, Papoutsakis ET & Wu CH, (2013) Transcription factors and genetic circuits orchestrating the complex, multilayered response of Clostridium acetobutylicum to butanol and butyrate stress. BMC systems biology 7: 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wietzke M & Bahl H, (2012) The redox-sensing protein Rex, a transcriptional regulator of solventogenesis in Clostridium acetobutylicum. Appl Microbiol Biotechnol 96: 749–761. [DOI] [PubMed] [Google Scholar]
- Yamakawa K, Karasawa T, Ikoma S & Nakamura S, (1996) Enhancement of Clostridium difficile toxin production in biotin-limited conditions. J Med Microbiol 44: 111–114. [DOI] [PubMed] [Google Scholar]
- Yang X, Teng K, Su R, Li L, Zhang T, Fan K, Zhang J & Zhong J, (2018) AcrR and Rex control mannitol and sorbitol utilization through their cross-regulation of aldehyde-alcohol dehydrogenase (AdhE) in Lactobacillus plantarum. Applied and environmental microbiology [DOI] [PMC free article] [PubMed]
- Zhang L, Nie X, Ravcheev DA, Rodionov DA, Sheng J, Gu Y, Yang S, Jiang W & Yang C, (2014) Redox-responsive repressor Rex modulates alcohol production and oxidative stress tolerance in Clostridium acetobutylicum. Journal of Bacteriology [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.