

Abstract
Imines formed in situ from 2-aminopyridines and aldehydes undergo Rh(III)-catalyzed imidoyl C–H activation and coupling with diazo esters to give pyrido[l,2-a]pyrimidin-4-ones. Aromatic and enolizable aliphatic aldehydes were both effective substrates, and a broad range of functional groups were incorporated at different sites on the heterocyclic products. In addition, methoxy and dimethylamino functionalities could be directly installed on the pyrimidine ring by employing trimethyl orthoformate or N,N-dimethylformamide dimethyl acetal in place of the aldehyde, respectively.
Graphical Abstract
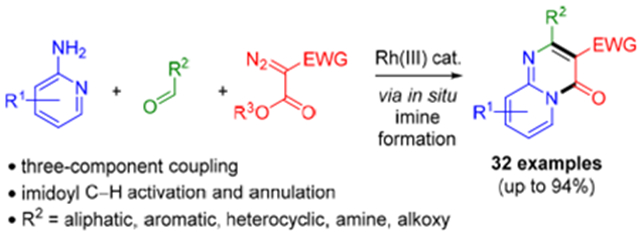
Nitrogen heterocycles are found in a large percentage of drugs and clinical candidates.1 To enable efficient syntheses of a range of different classes of nitrogen heterocycles from readily available inputs, we have focused on annulations that proceed through imidoyl C–H activation.2 A central tenet of this approach is the vast number of aldehydes and amines that theoretically could be used to generate imines for annulations. We initially implemented this approach for N-azolo imines 1, which can readily be prepared from amino azoles and aldehydes (Scheme 1A). Specifically, Rh(III)-catalyzed annulations of imines 1 with alkynes, diazo ketones, sulfoxonium ylides, and dioxazolones provide rapid access to a wide range of N-fused [5,6]-bicyclic heterocycles 2 and 3 (Scheme 1A).3,4 To further enhance reaction efficiency, we also recently reported a three-component coupling of aminopyrazoles 4, aldehydes 5, and sulfoxonium ylides 6 that proceeds via in situ imine formation and imidoyl C–H activation to provide privileged pyrazolopyrimidines 7 (Scheme 1B).5
Scheme 1.

Heterocycle Synthesis by Imidoyl C–H Activation
To expand upon the types of nitrogen heterocycles accessible by imidoyl C–H activation we have begun to explore alternative imine coupling partners. Herein, we report the rapid three-component synthesis of drug relevant [6,6]-bicyclic heterocycles 10 from imines, formed in situ from 2-aminopyridines 8 and aldehydes 5, and diazo esters 9 (Scheme 1C).6,7 The use of a commercially available and air stable Rh(III) precatalyst, convenient benchtop set up, and short reaction times with microwave (MW) heating further facilitates the preparation of substituted heterocycles 10. Broad scope is observed for both aromatic and enolizable aliphatic aldehydes. Additionally, methoxy and dimethylamino functionalities can be directly installed on the pyrimidine ring by employing trimethyl orthoformate or N,N-dimethylformamide dimethyl acetal in place of the aldehyde, respectively. Finally, good functional group compatibility is observed for this three-component reaction, with amide, ester, ether, carbamate, halide, secondary anilide, sulfone and phosphonate functionalities all successfully incorporated.
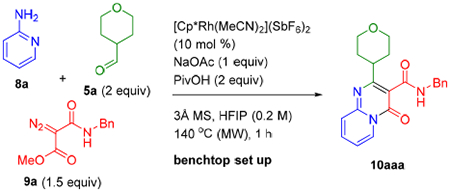
We began our investigation by examining a large variety of reaction parameters for coupling of 2-aminopyridine (8a), aldehyde 5a, and diazo ester 9a to give pyridopyrimidinone 10aaa (Table 1). Good yields of 10aaa could be obtained by straightforward benchtop set up using the commercially available and air stable cationic catalyst [Cp*Rh(MeCN)3](SbF6)2 with NaOAc, pivalic acid (PivOH) and 3 Å molecular sieves as additives in hexafluoroisopropanol (HFIP) (0.2 M) under microwave (MW) conditions at 140 °C for 1 h (entry 1, Table 1). Switching the stoichiometry to limiting amounts of aldehyde and 2 equiv of the aminopyridine led to a lower yield (entry 2). When the non-cationic rhodium precatalyst [Cp*RhCl2]2 was used, only a slight reduction in yield was observed (entry 3). As expected, when a Rh(III) catalyst was not added, no product was obtained (entry 4). We observed 22% of competitive esterification of PivOH with diazo ester 9a, which was used in excess (1.5 equiv). The stoichiometry of the PivOH additive was therefore reduced to 1 equiv (entry 5) and 0.5 equiv (entry 6), which resulted in a reduction in the ester byproduct to 13% and 6%, respectively. However, the yield of the desired product 10aaa also decreased slightly. Moreover, removing PivOH led to a significant drop in the yield of 10aaa (entry 7). Acetic acid was less effective than PivOH (entry 8), and the removal of NaOAc resulted in a moderately lower yield (entry 9).
Table 1.
Reaction Parameters for Annulation to 10aaaa
 | ||
|---|---|---|
| entry | variation | Yield %b |
| 1 | none | 73 (74)c |
| 2 | 0.1 mmol 5a + 2 equiv 8a +1.5 equiv 9a | 55 |
| 3 | [Cp*RliCl2]2 (5%) | 65 |
| 4 | no Rh | 0 |
| 5 | PivOH (1 equiv) | 71 |
| 6 | PivOH (0.5 equiv) | 68 |
| 7 | no PivOH | 48 |
| 8 | AcOH instead of PivOH | 57 |
| 9 | no NaOAc | 60 |
| 10 | 150 °C | 73 |
| 11 | TFE as solvent | 70 |
| 12 | EtOH as solvent | 10 |
| 13 | dioxane as solvent | 4 |
| 14 | 0.4 M | 61 |
| 15 | 0.1 M | 74 |
| 16 | [Cp*Rh(MeCN)3](SbF6)2 (5%) | 72 |
| 17 | [Cp*Rh(MeCN)3](SbF6)2(2%), 150 °C | 65 |
| 18 | no sieves | 68 |
| 19 | conventional heating, 100 °C, 20 h | 70 (72%)c |
| 20 | [Cp*Co(MeCN)3](SbF6)2 (10%) | 0 |
| 21 | [Cp*IrCl2]2 (5%) and AgSbF6 (20%) | 72 |
Conditions: 8a (0.10 mmol), 5a (0.20 mmol), 9a (0.15 mmol).
Yields determined by 1H NMR integration relative to 1,3,5-trimethoxybenzene as external standard.
Isolated yield at 0.30 mmol scale of limiting reagent 8a.
Complete consumption of the limiting aminopyridine 8a was observed at 140 °C, but this does not establish whether or not complete conversion of stable intermediates to 10aaa had occurred (see Scheme 8 for proposed mechanism). The reaction was therefore performed at a higher temperature; however, an identical yield was obtained indicating that more forcing conditions are not necessary (entry 10). An acidic protic solvent was crucial for this transformation as trifluoroethanol (TFE) was found to be equally effective as HFIP (entry 11), but ethanol and dioxane only resulted in formation of small amounts of the desired product (entries 12 and 13). Concentration has the potential to be an important parameter for three-component coupling, but increasing the concentration to 0.4 M led to only a slight reduction in yield (entry 14), while lowering the concentration to 0.1 M had no effect (entry 15). Consistent with our goal to provide access to diverse pyridopyrimidinones 10 at short reaction times, we chose 10 mol % catalyst loading for evaluating the substrate scope. However, 5 and 2.5 mol % catalyst loading did afford good yields of product (entries 16 and 17). Lower catalyst loadings are therefore likely to be applicable to many starting material combinations (see Scheme 6). It is also worth noting that acceptable reaction yields were obtained without molecular sieves (entry 18), and conventional heating at 100 °C for 20 h gave a comparable isolated yield to that observed under microwave conditions (entry 19). Finally, we found that Cp*Co(III) was not an effective catalyst (entry 20), whereas Cp*Ir(III) was comparable to Cp*Rh(III) (entry 21).
Scheme 8.

Proposed Catalytic Cycle for Annulation
Scheme 6.

A 2 mmol Scale Reaction at 5 mol % Catalyst
Using the optimal reaction conditions (i.e., entry 1, Table 1), we first explored the scope of aliphatic aldehydes for three-component coupling with 2-aminopyridine (8a) and diazo ester 9a (Scheme 2). A series of cyclic, α-branched carboxaldehydes gave 10aaa to 10daa in good yields (64–74%). Acetaldehyde and a β-branched aldehyde were also effective coupling partners affording 10eaa (47%) and 10faa (67%), respectively. The incorporation of ether (10gaa) and ester (10haa) functionalities by employing the correspondingly functionalized linear aldehydes was also demonstrated. Methoxy (10iaa) or dimethylamino (10jaa) functionalities were also installed directly onto the pyrimidine ring by employing trimethyl orthoformate and N,N-dimethylformamide dimethyl acetal in place of the aldehyde, respectively. Under the standard conditions, only a trace amount of 10kaa was obtained from sterically hindered pivaldehyde.
Scheme 2. Scope of Aliphatic Aldehydes.a.

aStandard conditions: 8a (0.30 mmol), 5 (0.60 mmol), and 9a (0.45 mmol). b10 equiv (3.0 mmol) trimethyl orthoformate was used in place of 5; no molecular sieves. c1.1 equiv (0.33 mmol) N,N-dimethylformamide dimethyl acetal in place of 5. Isolated yields are reported.
We next evaluated the scope of aromatic aldehydes (Scheme 3). Benzaldehyde is a good coupling partner giving bicyclic product 10laa in high yield. Para-substituted electron-rich and deficient benzaldehydes provided good yields of products 10maa and 10naa, respectively, establishing that the reaction is effective regardless of the electronic properties of the aldehyde input. Benzaldehydes substituted at the meta- and ortho-positions were also effective coupling partners as exemplified by the halo substituted products 10oaa and 10paa, respectively, although a lower yield was observed for the ortho-substituted derivative. An acidic secondary anilide could also be incorporated under standard conditions to give 10qaa. The five-membered pyrazole-, furan-, and thiophene carboxaldehydes also provided 10raa to 10taa in good yields (63–77%).
Scheme 3. Scope of Aromatic Aldehydes.a.

aStandard conditions: 8a (0.30 mmol), 5 (0.60 mmol), and 9a (0.45 mmol). Isolated yields are reported.
The influence of the substitution pattern of the employed 2-aminopyridines is provided in Scheme 4. A series of methyl-substituted 2-aminopyridines at the 3-, 4-, and 5-positions were effective coupling partners affording 10aba to 10ada in good to excellent yields (73–94%). However, 6-methyl-2-aminopyridine was not effective; only trace amount of bicyclic product 10aea was observed. Methoxy (10afa), bromo (10aga) and fluoro (10aha) substituents were also incorporated using the correspondingly substituted aminopyridines 8.
Scheme 4. Influence of the Substitution Pattern of the Employed 2-Aminopyridinesa.

aStandard conditions: 8 (0.30 mmol), 5a (0.60 mmol), and 9a (0.45 mmol). Isolated yields are reported.
Finally, the scope of diazo esters was examined (Scheme 5). In addition to employing the benzylamide diazo ester 9a, which provided products with an unbranched (N-benzyl amide substituent, (N-α-branched secondary amides can also be obtained in good yield as exemplified by the isopropyl amide product 10aab. Tertiary amides could also be prepared in reasonable yield as demonstrated by 10aac. Symmetrical diazo malonates were also effective partners and provided the corresponding ester-containing bicyclic products 10aad (76%) and 10aae (62%). Moreover, the reaction is not limited to carboxylic acid derivatives as exemplified by the preparation of phosphonate 10aaf and sulfone 10aag.
Scheme 5. Scope of Diazo Estersa.

aStandard conditions: 8a (0.30 mmol), 5a (0.60 mmol), and 9 (0.45 mmol). bThree equiv of diazophosphonate 9f (0.90 mmol) were used. Isolated yields are reported.
To demonstrate the scalability of this method, a 2 mmol scale reaction was carried out for the preparation of 10aid at a lower catalyst loading of 5 mol % (Scheme 6). Pyridopyrimidinone 10aid was then employed for further transformations (Scheme 7). The chlorine substituent provides a versatile site for diversification. For example, Suzuki cross-coupling provided 11 in 95%,9 while Buchwald-Hartwig amination with aniline and morpholine also proceeded in very high yields to give the secondary aniline 12 and tertiary amine 13, respectively.10 Saponification of the ester in 10aid also cleanly provided 14 with the carboxylic acid functionality amenable further synthetic elaboration.
Scheme 7.

Product Diversification
A plausible mechanism for this three-component coupling reaction is depicted in Scheme 8. We propose that imine A, formed in situ from 2-aminopyridine 8 and aldehyde 5, undergoes concerted metalation-deprotonation to give rhodacycle B. Evidence for rhodacycle B is suggested by a previous report of Rh(III)-catalyzed C-H oxidation of imine A in the presence of Cu(OAc)2.11,12 Moreover, we have reported the X-ray structural characterization of an analogous neutral rhodacycle derived from [Cp*RhCl2]2 and N-2-imidazo benzaldimine and further showed that it was catalytically competent for other types of annulation reactions.3 Carbene insertion of diazo ester 9 with release of N2 then provides the six-membered rhodacycle C.8 Protonolysis affords ester D and regenerates the Rh(III) catalyst. Under the reaction conditions, lactamization of D then provides the (V-fused [6,6]-bicyclic heterocycle product 10.13
In conclusion, imines formed in situ from 2-aminopyridines and aldehydes undergo Rh(III)-catalyzed imidoyl C–H activation and coupling with diazo esters to give pyrido[1,2-a]pyrimidin-4-ones. Both aromatic and enolizable aliphatic aldehydes are effective substrates. Methoxy and dimethylamino functionalities could also be directly installed on the pyrimidine ring by employing trimethyl orthoformate and N,N-dimethylformamide dimethyl acetal in place of the aldehyde, respectively, and a broad range of functional groups are compatible with the reaction conditions. The synthesis of additional classes of nitrogen heterocycles by annulations proceeding via imidoyl C–H activation of different types of imines will be reported in due course.
Supplementary Material
ACKNOWLEDGMENTS
The NIH (R35GM122473) is gratefully acknowledged for supporting this work.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Procedure details and NMR spectra (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).For an analysis of nitrogen heterocycles in pharmaceuticals, see:; Vitaku E; Smith DT; Njardarson JT. J. Med. Chem 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- (2).For recent reviews on heterocycle synthesis by C–H functionalization, see:; (a) Song G; Wang F; Li X. Chem. Soc. Rev 2012, 41, 3651–3678. [DOI] [PubMed] [Google Scholar]; (b) Gulías M; Mascareñas JL Angew. Chem. Int. Ed 2016. 55, 11000–11019. [DOI] [PubMed] [Google Scholar]; (c) Hummel JR; Boerth JA; Ellman JA Chem. Rev 2017. 117, 9163–9227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Halskov KS; Witten MR; Hoang GL; Mercado BQ; Ellman JA. Org. Lett 2018. 20 2464–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Hoang GL; Halskov KS; Ellman JA J. Org. Chem 2018, 83 9522–9529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Hoang GL; Streit AD; Ellman JA J. Org. Chem 2018, 83 15347–15360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).For leading references on transition metal-catalyzed syntheses of pyrido[1,2-a]pyrimidin-4-ones 10, see:; (a) Gulevich AV; Helan V; Wink DJ; Gevorgyan V Org. Lett. 2013, 15, 956–959. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liang Y-F; Steinbock R; Münch A; Stalke D; Ackermann L. Angew. Chem. Int. Ed. 2018, 57, 5384–5388. [DOI] [PubMed] [Google Scholar]
- (7).For biological activities of pyrido[1,2-a]pyrimidin-4-ones 10, see:; (a) Van der Mey M; Windhorst AD; Klok RP; Herscheid JDM; Kennis LE; Bischoff F; Bakker M; Langlois X; Heylen L; Jurzyk M; Leysen JE Biooig. Med. Chem 2006,14,4526–4534. [DOI] [PubMed] [Google Scholar]; (b) La Motta C; Sartini S; Mugnaini L; Simorini F; Taliani S; Salerno S; Marini AM; Da Settimo F; Lavecchia A; Novellino E; Cantore M; Failli P; Ciuffi M; J. Med. Chem 2007, 50, 4917–4927. [DOI] [PubMed] [Google Scholar]; (c) Peng L; Gao X; Duan L; Ren X; Wu D; Ding KJ Med. Chem 2011, 54, 7729–7733. [DOI] [PubMed] [Google Scholar]
- (8).For leading references on C–H functionalization with diazo compounds, see:; (a) Xia Y; Qiu D; Wang J Chem. Rev. 2017, 117, 13810–13889. [DOI] [PubMed] [Google Scholar]; (b) Chan W-W; Lo S-F; Zhou Z; Yu W-YJ Am. Chem. Soc 2012, 134, 13565–13568. [DOI] [PubMed] [Google Scholar]; (c) Hyster TK; Ruhl KE; Rovis TJ Am. Chem. Soc 2013, 135, 5364–5367. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Shi Z; Koester DC; Boultadakis-Arapinis M; Glorius FJ Am. Chem. Soc 2013, 135, 12204–12207. [DOI] [PubMed] [Google Scholar]
- (9).Aounzou M; Campos JF; Loubidi M; Berteina-Raboin S Molecules 2018, 23, 1159–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Bruno NC; Tudge MT; Buchwald SL Chem. Sci 2013, 4, 916–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).For a seminal report on Rh(I)-catalyzed ligand assisted hydroacylation via C-H alkylation of imines formed in situ from aldehydes and a catalytic amount of a 2-aminopyridine, see:; Jun C-H; Lee H; Hong J-B J. Org. Chem 1997, 62, 1200–1201. [Google Scholar]
- (12).For multicomponent synthesis of isoindolinones from 2-aminopyridines, aromatic aldehydes, and electron deficient alkenes in the presence of 2 equiv of Cu(OAc)2 by Rh(III)-catalyzed C–H oxidation of in situ formed imine to the corresponding amide followed by further aromatic C–H functionalization, see:; Zhang Y; Zhu H; Huang Y; Hu Q; He Y; Wen Y; Zhu G. Org. Lett 2019, 21, 1273–1277. [DOI] [PubMed] [Google Scholar]
- (13).For heterocycle synthesis by a related lactamization following Co(III)-catalyzed directed coupling of aryl and alkenyl C-H bonds with diazo esters, see:; Zhao D; Kim JH; Stegemann L; Strassert CA; Glorius F. Angew. Chem. Int. Ed 2015, 54, 4508–4511. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.