

Abstract
Many colorectal cancers (CRCs) that exhibit microsatellite instability (MSI) are not explained by MLH1 promoter methylation or germline mutations in mismatch repair (MMR) genes, which cause Lynch syndrome (LS). Instead, these Lynch-like syndrome (LLS) patients have somatic mutations in MMR genes. However, many of these patients are young and have relatives with cancer, suggesting a hereditary entity. We performed germline sequence analysis in LLS patients and determined their tumor's mutational profiles using FFPE DNA. Six hundred fifty-four consecutive CRC patients were screened for suspected Lynch syndrome using MSI and absence of MLH1 methylation. Suspected LS cases were exome sequenced to identify germline and somatic mutations. Single nucleotide variants were used to characterize mutational signatures. We identified 23 suspected LS cases. Germline sequence analysis of 16 available samples identified 5 cases with LS mutations and 11 cases without LS mutations, LLS. Most LLS tumors had a combination of somatic MMR gene mutation and loss of heterozygosity. LLS patients were relatively young and had excess first-degree relatives with cancer. Four of the 11 LLS patients had rare likely pathogenic variants in genes that maintain genome integrity. Moreover, tumors from this group had a distinct mutational signature compared to tumors from LLS patients lacking germline mutations in these genes. In summary, more than a third of the LLS patients studied had germline mutations in genes that maintain genome integrity and their tumors had a distinct mutational signature. The possibility of hereditary factors in LLS warrants further studies so counseling can be properly informed.
Keywords: Colorectal cancer, Lynch-like syndrome, Lynch syndrome, Lynch syndrome-like, DNA repair genes
INTRODUCTION
About ten percent of all colorectal cancers (CRCs) have a mismatch repair (MMR)-deficient phenotype, characterized by microsatellite instability (MSI) and lack of expression of MMR proteins[1]. A third of these have germline mutations in a MMR gene that causes Lynch syndrome, the most common form of hereditary CRC. The rest have traditionally been thought to be sporadic cancers due to biallelic inactivation of the MMR gene MLH1 through hypermethylation of its promoter region[2]. However, a substantial number of CRCs that exhibit MSI have neither MLH1 methylation nor a germline mutation in any of the MMR genes—a class of patients that is referred to as Lynch syndrome-like or Lynch-like syndrome (LLS), the term we will use hereafter.
Some studies have shown that several of these unexplained MSI tumors have two somatic hits that inactivate a single MMR gene[2, 3]. Other authors have shown that germline mutations in the exonuclease domains of DNA polymerases POLD1 and POLE, or biallelic germline mutations in MUTYH could result in a Lynch syndrome phenotype with MSI CRCs and somatic MMR gene mutations[4-6]. Even some somatic DNA polymerase mutations were found to be associated with MMR somatic mutations[4]. Altogether raises the consideration that other defective mechanism such as proofreading or base excision repair could result in somatic MMR gene mutations leading to MMR deficiency and subsequently to MSI tumors.
The presence of germline genetic defects, different from the Lynch syndrome-causing MMR genes, could at least in part explain the findings that LLS patients and their cancer-affected relatives are often younger at diagnosis than sporadic CRCs and the incidence of familial CRC in LLS families is higher than expected[3, 7].
To better understand the nature of the LLS phenotype, we studied the spectrum of individuals with MMR-deficient tumors and evaluated tumor and germline mutations using whole exome sequence analysis in patients from a multiethnic study of consecutive CRC cases.
MATERIALS AND METHODS
Ascertainment and Recruitment
We studied a series of 654 CRC patients, 60% African American (AA) and 40% White European Americans (WEA) from the Chicago Colorectal Cancer Consortium (CCCC)[8]. Briefly, the CCCC is a collaborative research study that ascertained CRC cases and controls at Jesse Brown West Side Veterans Medical Center, John H. Stroger Jr. Hospital of Cook County, Rush University Medical Center, University of Chicago Medicine, and University of Illinois Hospital & Health Sciences System. Consecutive incident CRC patients were recruited prospectively in 2011–2012 (161 cases) and unselected retrospective cases identified through Departments of Pathology records from 1997 to 2010 (473 cases). The mean age at diagnosis was 63 and 56.7% were male. The study was conducted according to the corresponding approved IRB protocol at each institution.
Microsatellite and Methylation Analysis
Microsatellite instability status was assessed in all tumors with paired DNA samples from tumor and uninvolved tissue. The panel of mononucleotide markers included NR21, NR22, NR24, NR27, BAT25, and BAT26. Multiplex PCR amplified all markers, and PCR products were analyzed by capillary electrophoresis, as previously described[9]. The BRAF V600E mutation was analyzed through direct DNA sequencing as previously described[10].
To distinguish sporadic MSI from suspected LS, MSI cases were further tested for MLH1 promoter methylation. Tumor DNA was modified using the Qiagen Epitect Plus DNA Bisulfite Kit (Qiagen, Hilden, Germany). Converted DNA was used to amplify the MLH1 promoter region D by PCR using the Qiagen Pyromark kit (Qiagen, Hilden, Germany) with described primers[11]. The PCR product was cleaned with Biotage reagents (Biotage, Uppsala, Sweden) and streptavidin sepharose high-performance beads (GE Healthcare, Buckinghamshire, England). Pyrosequencing reactions were performed using PyroMark Gold Q96 reagents (Qiagen, Hilden, Germany) as recommended. Methylation data was analyzed using pyro Q-CpG Software (Biotage, Uppsala, Sweden). To assess the total methylation status for each sequence we calculated the mean percentage of methylation for all analyzed CpG sites. We classified each sequence as methylated when the mean methylation percentage was above 20%.
Immunohistochemistry analysis
Immunohistochemistry staining was performed to assess MMR (MLH1, MSH2, MSH6 and PMS2) protein expression as described[12]. Tumor cells were judged to be negative for expression only if they lacked staining in a sample in which normal colonocytes and stroma cells were positively stained.
Exome sequencing
Exome sequencing was performed in suspected LS patients at the Yale Center for Genome Analysis (YCGA). Fourteen cases with matching germline and tumor samples and one germline only case were available for exome analysis. Results from one patient who underwent MMR gene testing through the cancer genetics clinic were also included.
For exome sequence analysis, specimens from formalin-fixed, paraffin-embedded (FFPE) tissues were used. A 2 mm core of tissue was taken for DNA extraction. Non-involved tissue was used for germline DNA extraction. Due to its FFPE origin, DNA went through a repair step prior to being sheared to a mean fragment length of about 100 bp. Exome capture libraries were constructed from 500 ng of DNA using SeqCap EZ version 2 by Nimblegen (Roche NimbleGen, Madison, WI). The captured fragments were PCR amplified and purified. Capture efficiency was evaluated by quantitative PCR to confirm successful exome enrichment. Samples with a yield of ≥0.5 ng/ul DNA were used for sequencing. Sample concentrations were normalized to DNA content and loaded into Illumina version 3 flow cells at a concentration that yields 170-200 million passing filter clusters per lane. Samples were sequenced using 76 bp paired-end sequencing in an Illumina HiSeq 2000 according to Illumina protocols. First and last base were trimmed from each read. Tumor samples were sequenced to greater depth of coverage to permit detection of somatic mutations in tumors on account of admixture of normal and tumor cells in these samples. Sequences were aligned to the reference genome, and variants were called using the best-practices BWA-MEM, Picard, GATK 3 pipeline. We used the ANNOVAR tool[13] to annotate (hg19) single nucleotide variants (SNVs) and insertions/deletions, examine functional consequences of mutations on genes, report functional importance scores, find variants in conserved regions, and to identify variants reported in public databases[13]. Exome statistics for germline and tumor are provided in Supplementary Table 1.
Germline Mutational analysis
To identify LS patients, we performed an analysis including only MMR genes. To identify variants that were likely to change the function of the encoded proteins, we selected variants that were: (i) mutations previously documented in the ClinVar database as disease-causing and associated with LS, or (ii) loss-of-function mutations (premature termination, frameshift, and splice site). All germline samples were also investigated to identify exonic deletions or duplications. For the exons in the MMR genes, we calculated the highest read count per exon and we normalized that value by the median coverage of each sample. The normalization allowed us to compare data between samples and to identify any exonic deletion or duplication as a significant loss or gain of coverage between samples. In addition, copy number variation (large deletions or duplications) was assessed using ConIFER[14]. A case was classified as LS when a disease-causing germline mutation was identified in an MMR gene according the criteria given above or as LLS when a disease-causing germline mutation in an MMR gene was not present. Our analysis only included variants with high-quality calls (single nucleotide variants quality score >100 and indel variants quality score >1000).
Using the germline exome data generated we confirmed that patients under study did not have any pathogenic mutations in other known CRC predisposing genes including APC, MUTYH, POLE, POLD1, CHEK2, BMPR1A, GREM1, PTEN, STK11, TP53.
To test potentially cancer-predisposing DNA repair genes, we compared the mutational gene burden of DNA repair genes in LLS patients to healthy, cancer-free controls. The controls included 3,275 WEAs and 394 AAs from the private Yale Exome Database. Cases and controls were all processed at the YCGA using the same sequencing platform and bioinformatics pipelines. The DNA repair genes were selected as having been mutated in at least one cancer cell line of NCI-60 panel. The NCI-60 panel is derived from nine tissues, including breast, colon, skin, blood, central nervous system, lung, prostate, ovaries, and kidney[15]. There were 162 DNA repair genes after exclusion of the MMR genes (Supplementary Table 2). Exonic and splicing variants were identified in the 162 genes. We excluded missense and synonymous variants from the analysis. The rest of exonic and splicing variants with a population frequency ≤2×10−5 in the ExAC database were included. Inframe indels were included only if the variant was affecting a highly conserved amino acid. Once variants were identified in the 162 genes set, we compared the overall mutational burden (number of mutated alleles versus reference alleles) between cases and controls using a Fisher exact test.
Lastly, we analyzed germline mutations in CRC cases from The Cancer Genome Atlas (TCGA) and our LS patients to investigate whether candidate genes identified in LLS patients were also mutated in other types of CRCs. The TCGA Network[16] includes a non-selected series of CRC cases. The present analysis included germline exome sequence data from 297 CRC cases, 45 of them with MSI. In this analysis, we only included variants that were supported by more than 5% of the sequence reads with a total of at least 8 alternate sequence reads.
To evaluate the frequency of specific variants or variants in specific genes in the general population we accessed the vcf file from ExAC that excludes the 7,601 cancer patients from the TCGA. Variants with dubious annotation were not included in the analysis.
Somatic Mutational analysis
We used MuTect to identify somatic single-nucleotide variants (SNVs)[17]. To exclude potential false positives, we only included variants with a total coverage of at least 8 reads in both, normal and tumor tissue. Somatic SNVs were identified as present if the frequency of the non-reference allele was >5% in the tumor. We required that non-reference calls were covered by at least 3 sequence reads. Variants were annotated, and we excluded any variant present in a germline exome database (NHLBI, 1000G, and Yale’s).
Loss of heterozygosity (LOH) regions were identified by comparing the allele frequencies of tumor and matched normal samples. Genomic regions with allele frequencies in the tumor deviating from the frequencies in the normal tissue were marked as LOH. Tumor purity was estimated by averaged absolute b-allele frequency change in LOH regions multiplied by 2. Samples were excluded if tumor purity was lower than 20% (<0.20). Tumor purity is shown in Supplementary Table 3.
Identification of Mutational signatures
Different mutational processes (that may include one or more DNA damage and/or DNA maintenance mechanisms), commonly result in different combinations of mutations or signatures[18, 19]. To identify mutational signatures that characterize tumor phenotypes, we used the R package “SomaticSignatures”[20]. We followed the analytical pipeline described in the package’s vignette from Bioconductor.org. Each somatic SNV was categorized in relation to the base pair 5’ and 3’ to the SNV. To identify the characteristic signatures from the motif matrix, we used a non-negative matrix factorization (NMF) with the Brunet algorithm[21], implemented in a second R package[22]. We computed the residual sum of squares and the explained variance between the observed and fitted mutational spectrum for different numbers of signatures. Doing so we selected 3 signatures for the analysis. This number of signatures corresponded to the first inflection point of the residual sum of squares curve. After that point, increasing the number of signatures did not yield a significantly better approximation of the data[23]. For comparison purposes, we accessed somatic mutation data from the TCGA. Thirty-nine MSI cases were available for analysis. Moreover, to explore the potential differences between MSI cases and LLS tumors we used the package “pvclust” and performed hierarchical clustering of tumor phenotypes based on proportions of each motif. pvclust[24] is an R package to assess the uncertainty in hierarchical cluster analysis. For each cluster in hierarchical clustering, quantities called p-values are calculated via multiscale bootstrap resampling. P-value of a cluster is a value between 0 and 1, which indicates how strong the cluster is supported by data. pvclust provides two types of p-values: AU (Approximately Unbiased) p-value and BP (Bootstrap Probability) value. AU p-value, which is computed by multiscale bootstrap resampling, is a better approximation to unbiased p-value than BP value computed by normal bootstrap resampling. For a cluster with AU p-value > 0.95, the hypothesis that “the cluster does not exist” is rejected with significance level of 0.05. We used a correlation distance method and a mean (or average) linkage clustering, which finds all possible pairwise distances for points belonging to two different clusters and then calculates the average. 1,000 bootstrap replications were used.
RESULTS
Identification of Lynch-like syndrome cases
The characterization of CRCs as suspected LS cases is summarized in Figure 1. We first tested 654 tumors available through the CCCC for MSI and genotyped them for the BRAF mutation V600E because it is known to be associated with sporadic MSI. Fifty-six of the 654 tumors (8.6%) exhibited MSI. We were able to test 53 of the 56 MSI tumors for MLH1 promoter methylation and found that 30 (57%) had MLH1 promoter methylation. Twenty-three of the 30 tumors with MLH1 promoter methylation (77%) had the BRAF V600E mutation. No V600E BRAF mutations were found in tumors without MLH1 promoter methylation. From this analysis, 23 of the 654 cases (3.5%) tested were suspected as having a LS mutation.
Figure 1. Flow chart of the different tests performed to identify suspected Lynch syndrome cases.

Immunohistochemical analysis showed that all available tumors with MLH1 promoter methylation exhibited loss of MLH1 and PMS2 protein expression, as expected. Of the 23 suspected LS cases, 10 had loss of MLH1/PMS2 and 12 had loss of MSH2/MSH6. In one case, we were unable to perform IHC.
In 15 of the 23 suspected LS cases DNA was available for germline analysis. In these 15 patients, we identified 4 individuals (2 AA, 2 WEA) with LS mutations. An additional LS patient from this cohort was identified through the cancer genetics clinic and had a MLH1 mutation. In 11 cases, no disease-causing, germline MMR mutation was identified (7 AA and 4 WEA) (Table 1). In the 11 MMR mutation-negative cases, none had a likely pathogenic mutation in other genes known to cause gastrointestinal hereditary cancer syndromes, such as MUTYH or APC.
Table 1.
Genetic and protein expression alterations of mismatch repair genes in Lynch syndrome (LS) and Lynch-like syndrome (LLS)
| ID | Germline MMR gene mutation |
Somatic mutation of MMR genes |
LOH at MMR genes |
MMR protein expressionb |
Race |
|---|---|---|---|---|---|
| LS: MMR germline mutations | |||||
| 1 | Del MSH2/EPCAM |
MLH1 rs267607751c MSH2 c.376delG (G126fs) |
None identified | MSH2 | WEA |
| 2 | MLH1 rs63750781c | NA | NA | MLH1/PMS2 | WEA |
| 3 | MSH6 c.1701delTT (K567fs) |
MSH2 c.G2272A (D758N), MSH6 rs587779246c |
None identified | MSH2/MSH6 | AA |
| 4 | PMS2 rs201451115c | MLH1 c.A560C (N187T) | PMS2 | PMS2 | WEA |
| 5a | MLH1 c.C1219T (Q407X) | NA | NA | MLH1/PMS2 | AA |
| LLS: No MMR germline mutations | |||||
| 6 | none identified | None identified | MLH1 | MSH2/MSH6 | AA |
| 7 | none identified | MLH1 rs63750781d | MLH1 | MLH1/PMS2 | WEA |
| 8 | none identified | MSH2 c.211-2T>G (2:47630543T>G) | MSH2, MSH6 | MSH2/MSH6 | AA |
| 9 | none identified | NA | NA | MSH2/MSH6 | AA |
| 10 | none identified | None identified | None identified | MSH2 | AA |
| 11 | none identified |
MSH2 rs587779143c, MSH2 c.A193T(K65X) |
None identified | MSH2/MSH6 | AA |
| 12 | none identified | MLH1 rs63750303c | MLH1 | MLH1/PMS2 | AA |
| 13 | none identified | MLH1 rs267607765c | MLH1 | MLH1/PMS2 | WEA |
| 14 | none identified | MSH2 c.1387+2A>T (2:47690168T>A) | None identified | MSH2/MSH6 | AA |
| 15 | none identified |
MSH2 rs28929484c MSH2 rs63750508c |
None identified | MSH6 | WEA |
| 16 | none identified | NA | NA | MSH2/MSH6 | WEA |
LLS, Lynch-like syndrome; LOH, loss of heterozygosity; LS, Lynch syndrome; MMR, mismatch repair; NA, tumor sample not available for study. AA: African American; WEA: West European American;
Patient was identified as a mutation carrier through the genetics clinic.
Loss of tumor MMR protein expression as assessed by immunohistochemistry.
Variant described as likely/pathogenic in ClinVar.
Variant described as uncertain significance in ClinVar
All three available tumors from the LS patients had somatic alterations in the corresponding MMR gene (Table 1). IHC analysis in the LLS cases indicated loss of expression of one or more MMR proteins in all 11 cases. We had tumor DNA available for analysis in 9 of the 11 LLS cases. Four cases had a somatic point mutation in one MMR gene and loss of heterozygosity (LOH) of the corresponding normal copy of the MMR gene (cases 7, 8, 12, and 13). Two cases had two different pathogenic mutations in an MMR gene (possibly one in each allele) (cases 11 and 15). One tumor had only one mutation in an MMR gene (case 14). One case had LOH in MLH1 but loss of MSH2/MSH6 protein expression (case 6). One case lacked somatic alterations at MMR genes (case 10) (Table 1). Two tumors (cases 9 and 16) had a tumor purity <20% and were not analyzed for somatic mutations or LOH (Supplementary Table 3). In summary, our sequence analysis of the MMR genes in LLS patients revealed two independent somatic mutations in six out of the nine cases we were able to characterize. These data are consistent with previous reports[2, 3]. Finally, in all cases but one (case 6), the results of MMR expression were consistent with germline and somatic mutations.
We had family history information on six LLS patients, and all six had at least one first-degree relative (FDR) with a solid malignancy and, on average these individuals had 1.8 FDRs with malignant tumors (Table 2). By contrast, analyzing the 222 prospectively recruited CRC patients from whom reliable family history information was available, the average number of FDRs with solid organ cancers was 0.8, which was significantly different from the frequency in LLS patients (P=0.014; Mann-Whitney test).
Table 2:
Clinical information and family history of cancer
| ID | Age | Race | Sex | CRC Locationa |
Family History of Cancer |
|---|---|---|---|---|---|
| LS: MMR germline mutation | |||||
| 1 | 68 | WEA | F | R and L | Mother: breast Father: renal cell |
| 2 | 27 | WEA | M | R | Great grandmother: colon Grandmother: colon, uterus Great grand uncle: colon Aunt: colon, pancreas Cousin: colon |
| 3 | 78 | AA | M | L | n/a |
| 4 | 62 | WEA | F | L | n/a |
| 5b | 55 | AA | F | R | Aunt: breast |
| LLS: No MMR germline mutations | |||||
| 6 | 67 | AA | M | n/a | n/a |
| 7 | 53 | WEA | M | R | Mother: breast Father: prostate |
| 8 | 70 | AA | F | R | Brother: esophagus |
| 9 | 65 | AA | F | R | Father and brother: prostate Sister: breast Brother: renal cell Aunt: neck |
| 10 | 46 | AA | F | n/a | n/a |
| 11 | 86 | AA | M | R | Father: brain |
| 12 | 81 | AA | F | R | Mother: Uterus |
| 13 | 47 | WEA | M | R | n/a |
| 14 | 77 | AA | M | R | n/a |
| 15 | 58 | WEA | M | L | n/a |
| 16 | 50 | WEA | F | L | Mother: brain Brother: colon |
AA, African American; CRC, colorectal cancer; F, female; LLS, Lynch-like syndrome; LS, Lynch syndrome; M, male; n/a: not available, no information; WEA, White European American.
Tumor location is divided into tumors in the proximal colon, including the cecum, ascending colon, hepatic flexure, and transverse colon (R, right), and tumor in the distal colorectum, including the splenic flexure, descending colon, sigmoid colon, and rectum.
This case was seen in the University of Illinois at Chicago Familial Cancer Genetics Clinic, where she was found to have a LS mutation
In addition, the median age of the LLS patients’ group was significantly younger than the CCCC cases with a methylated MLH1 promoter (65 vs. 75, P value=0.04; Mann-Whitney U Test). The median age of LS patients was 62.
Identification of mutations in genes that maintain genome integrity in Lynch-like syndrome
We hypothesized that the somatic mutations in MMR genes resulted from a mutator genotype. The genomic instability that would result from mutations in these mutator genes would set the stage for mutations in MMR genes and the consequent acquisition of MSI. To test this hypothesis, we analyzed exome sequence data generated from the 11 LLS patients for rare and likely pathogenic variants in selected DNA repair genes. One hundred and sixty-two DNA repair genes were chosen based on the criterion that at least one mutation had been identified in the NCI-60 cell line panel. In the analysis of these 162 DNA repair genes, we identified one stop-gain mutation each in WRN, MCPH1, and BARD1, and a deletion of a conserved amino acid in REV3L (Table 3). None of the corresponding tumors harbored second hits in the same genes.
Table 3.
Genetic mutations found in genes that maintain genome integrity in Lynch-like patients and general population allele frequencies of the germline mutations present
| ID | Gene | Mutation | ExACa
(non-TCGA) |
dbSNP 146 | Race |
|---|---|---|---|---|---|
| 6 | BARD1 | K352X 2: 215645487 T>A |
NA | NA | AA |
| 7 | WRN | Q1242X 8: 31012176 C>T |
2/106,208 | rs762379051 | WEA |
| 8 | MCPH1 | L542X 8: 6302868 T>G |
2/106,208 | rs748011724 | AA |
| 11 | REV3L | L418del 6: 111701384 CAAG>C |
2/106,208 | rs758595911 | AA |
Data from ExAC excluding the 7,601 cancer patients from the TCGA.
NA: not available; AA: African American; WEA: West European American.
To determine whether these four genes have a high degree of genetic variability in the general population, we identified all likely pathogenic variants in the four genes from ExAC non-TCGA. Adding up the number of mutated alleles, these 4 genes were infrequently mutated and had an overall mutational burden of 0,26% (278/105,932) (Supplementary table 4). All individual variants were uncommon in the general population and had an allele frequency <5×10−4 suggesting that heterozygous loss of these genes is not a common event and may result in pathogenicity. In our limited series of LLS patients, the overall mutation burden for these 4 genes was approximately 22% (4 mutated alleles vs. 18 non-mutated alleles). The specific likely pathogenic variants identified in LLS cases were not identified in Yale controls.
To determine how likely it is that the observed number of mutations in the candidate DNA repair genes identified in LLS patients arose by chance, we compared the overall mutation burden from the 162 DNA repair genes between cases and controls. Including 3,275 WEAs and 394 AAs from the Yale control series, we identified 197 likely pathogenic variants in the 162 genes. The LLS patients exhibited a significant enrichment of likely pathogenic variants in DNA repair pathway genes (162 genes; mutated alleles 4/18 cases vs. 197/7,141 controls, P=0,003; Fisher exact test). We also searched for likely pathogenic variants among these 162 genes in LS patients. Only one patient had an indel variant in POLD1 (chr 19, 50917091, G>GGT). Lastly, we analyzed germline Illumina exome sequencing data of 297 sporadic CRC patients from the TCGA of which 45 exhibited MSI and 252 exhibited MSS. We identified 9 likely pathogenic variants (Supplementary Table 5). The LLS group had the highest frequency of mutated alleles in the non-MMR DNA repair genes compared to LS and sporadic CRC groups (Table 4).
Table 4:
Mutation burden of likely pathogenic variants in DNA repair candidate genes present in different colorectal cancer phenotypes and controls
| Mutated Alleles |
Reference Alleles |
Mutation Burden |
P-valuea | |
|---|---|---|---|---|
| LLS | 4 | 18 | 0,222 | |
| LS | 1 | 7 | 0,143 | ns |
| TCGA | 9 | 585 | 0.015 | 0.0007 |
| Controls | 197 | 7,141 | 0,027 | 0.003 |
LLS, Lynch-like syndrome; LS, Lynch syndrome; TCGA: The Cancer Genome Atlas database.
Fisher exact test performed between LLS and each other group. ns: non-statistical
Mutational signatures of Lynch-like syndrome tumors
To determine whether LLS patients can be distinguished by the types of mutational profiles that occur in the resulting tumors, we compared the mutational signatures with the ones from other CRC MSI phenotypes. All tumors, regardless of MSI status, showed a high frequency of mutations in N[C>T]G (where N is any base). MSI tumors exhibited increased frequencies of mutations in A[C>T]G, C[C>T]G, G[C>T]A, G[C>T]G, and C[C>A]T motifs. The most remarkable difference among LS, LLS and MSI sporadic tumors was the frequency of mutations in T>C motifs, which progressively decreased from LS > LLS > MSI sporadic (Figure 2A). Using the NMF algorithm, we identified two different mutational signatures (Figure 2B) with a distinct contribution to each tumor phenotype (Figure 2C). Signature 1 is the main contributor to the sporadic MSI phenotype; signature 2 is the main contributor to LS tumor. In these series, we saw that signature 2 contributes importantly to LLS cases with germline mutations in DNA repair genes (LLS-1), similarly to what we saw in LS. On the other hand, signature 1 was the main contributor to LLS without germline mutations in DNA repair genes (LLS-2). The proportion of the contributions of signatures 1 and 2 to the LLS-2 group was similar to the one seen in sporadic MSI cases. These results were further confirmed through the evaluation of the uncertainty of the hierarchical cluster analysis using the 4 phenotypes based on the frequency of each motif. The analysis significantly separated LLS-1 cases on one side, from LLS-2 and sporadic MSI tumors on the other side (Figure 3).
Figure 2: Somatic signatures that characterize colorectal tumors.
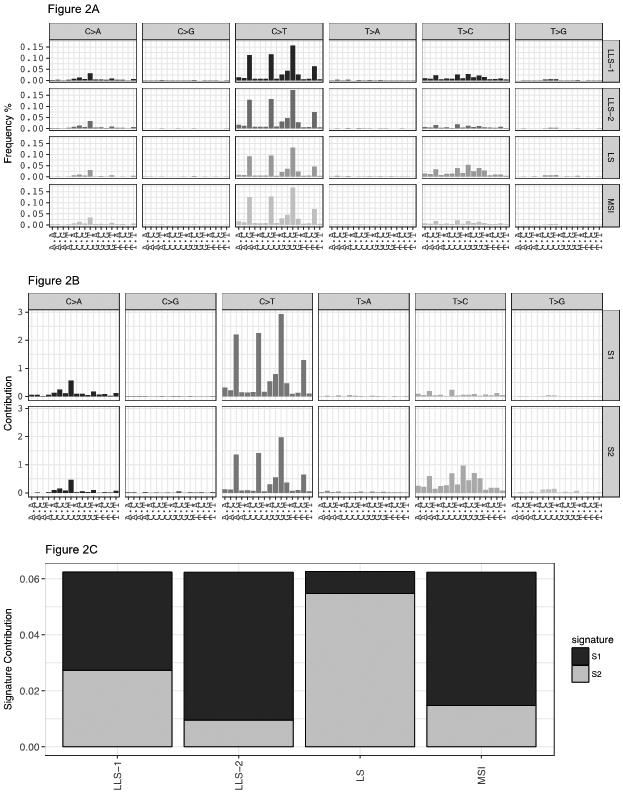
A: Mutation spectrum of the different CRC phenotypes. In the X axis there is a representation of all the 96 possible nucleotide triplets, 16 combinations per each type of mutation. B: Composition of somatic signatures estimated with the non-negative matrix factorization (NMF) represented as a bar chart. C: Contribution of the different signatures to the distinct CRC phenotypes estimated with the NMF represented as a bar chart. LLS-1, cases of Lynch-like syndrome with DNA repair gene mutations; LLS-2, cases of Lynch-like syndrome without DNA repair gene mutations; LS, Lynch syndrome; MSI, MSI colorectal cancers from The Cancer Genome Atlas.
Figure 3: Hierarchical cluster analysis of colorectal tumor phenotypes.
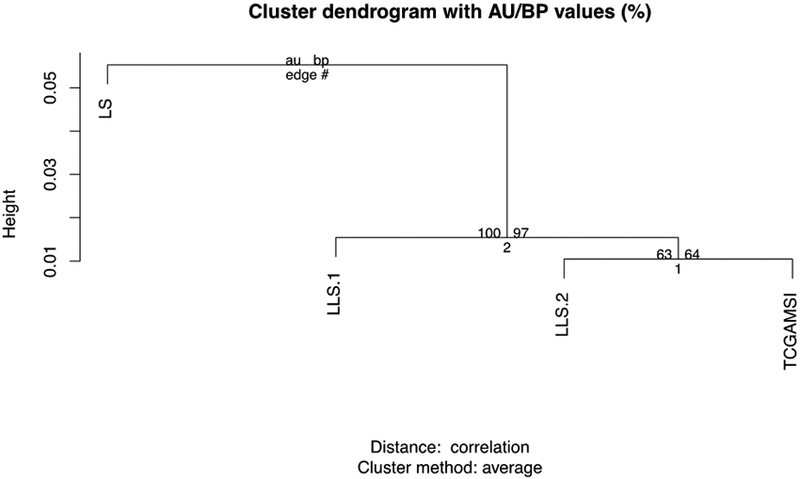
AU (Approximately Unbiased) p-value (on the left) and BP (Bootstrap Probability) value (on the right). LLS-1: Lynch-like syndrome with DNA repair gene variants; LLS-2: Lynch-like syndrome without DNA repair gene variants; LS: Lynch Syndrome; MSI: MSI colorectal cancers from The Cancer Genome Atlas.
DISCUSSION
In the present study, we found rare likely pathogenic variants in genes that promote genomic integrity in some cases of CRCs with MSI and lack of MLH1 promoter methylation; these cases traditionally have been thought to have LS with unidentified germline MMR repair gene mutations. The data suggest that the LLS patients with DNA repair gene mutations have a constitutional genomic instability that predisposes the mutation carriers to somatic mutations, which promote cancer development. The excess family history of cancer in LLS patients is consistent with a genetic predisposition. In addition, the mutation signatures associated with carriers of DNA repair gene mutations significantly separated from the other types of cases, implying that the route to MMR gene inactivation is associated with a genomic instability specific to these mutation carriers. If these data can be corroborated in follow-up studies, they would identify carriers of certain pathogenic DNA repair gene mutations as a cancer predisposed group.
The four genes found mutated in LLS patients have not previously been linked to CRC; however, the association of colorectal tumor development with their alteration is plausible given their function in maintenance of genomic integrity. Moreover, statistical inference indicates that the association is significant as the overall mutational burden of putatively pathogenic variants in our LLS patients was high whereas in the general population is low. Of note, several recently published studies have identified likely pathogenic variants in other genes that maintain DNA integrity, including NTHL1, FAN1, and MCM9 in cases with familial CRC with unknown genetic basis[25-28] and mutations in BLM in cases with early-onset CRC[29].
The genomic instability genes we found mutated here encode factors that influence homologous recombination, particularly in response to DNA replication-associated damage. The gene mutated in Werner syndrome WRN is an ATP-dependent DNA helicase of the RecQ family. One role of WRN is to remove secondary DNA structures, such a G4 DNA, that can impede replication fork progression, particularly at telomeres[30]. Microcephalin 1 (MCPH1) is a chromatin remodeling protein and bona fide tumor suppressor gene involved in replication-associated DNA damage responses. Its three BRCA1 C-terminal (BRCT) domains interact with the N-terminal region of BRCA2 during recruitment of RAD51 to the repair site[31]. It also functions to stabilize p53 during DNA damage response [32]. REV3L (REV3-like) is the catalytic subunit of polymerase polζ. It functions in lesion bypass by effecting translesion DNA synthesis. Mutations in REV3L have been identified in tumors resistant to platinum-based chemotherapies and it functions as a tumor suppressor in animal models[33]. Lastly, BARD1 (BRCA1 Associated RING Domain 1) forms a complex with BRCA1 that has important functions in regulating homologous recombination at replication forks and double strand breaks. Germline mutations in BARD1 have also been associated with hereditary breast and ovarian cancer syndrome[34-36].
The genomic instability of carriers of rare DNA repair gene mutations could result from inactivation of the normal copy of the gene in a somatic cell, which is the predominant mechanism in LS, or it could result from haploinsufficiency. Although we did not identify a second mutation or LOH that could account for the inactivation of the second allele in the tumors of our select LLS patients, we cannot rule out an inactivation through a different mechanism such as epigenetic silencing. For example, WRN has been found to be methylated in some CRCs[37]. Alternatively, haploinsufficiency of certain genomic integrity genes could cause a modest decrease in fidelity of the transmission of the genetic material from cell generation to cell generation. As an example, haploinsufficiency of TP53 contributes to tumor development as some tumors arising in Li-Fraumeni patients (due to TP53 germline mutations) present with a retained normal allele; normal allele retention may be associated with later-onset disease in Li-Fraumeni syndome[38]. Later onset and less severe disease phenotypes are a common feature of tumors generated in a haploinsufficient context, and haploinsufficiency can be viewed as a form of partial penetrance for a classical two-hit tumor suppressor gene[38]. Evidence of haploinsufficiency has been shown in several DNA repair genes including WRN[39], BLM[40], MUTYH[41], and MSH2[42, 43].
In these series of LLS patients, we did not find any germline mutations in DNA polymerases POLD1/POLE, or biallelic germline mutations in MUTYH as previously reported[4-6]. This could be due to differences in the series as, while the present report included consecutive CRC patients, other studies reported on patients selected on the basis of familial clustering of CRC[4, 6]. As opposed to other series[4], we did not find any somatic DNA polymerase mutations in our LLS tumors. Finally, double somatic MMR mutations have been described in patients with Hodkin’s lymphoma suggesting an association with anti-cancer treatment[44]. In our series, none of our LLS tumors had had lymphoma. One patient had prior leukemia but also had a germline mutation in REV3L.
Our analysis of mutational signatures suggests that LLS individuals with mutations in genes that maintain genome integrity (LLS-1) comprise a group with a distinctive mutational profile. Somatic mutational signatures have been studied and characterized for a variety of different tumor types (http://cancer.sanger.ac.uk/cosmic/signatures). Four signatures have been observed in tumors with deficient DNA mismatch repair (signatures #6, 15, 20, and 26). Signature #6, characterized by N[C>T]G, was the common signature of all MSI tumors (Signature 1). Signature #6 is most common in colorectal and uterine cancers. On the other hand, we identified a signature that was a major contributor to both, LS and LLS-1 patients (Signature 2), and much less to the other cancer subgroups, including LLS-2. Signature 2 shows an important contribution of T>C motifs, which could represent a combination of signatures #20 and #26. Signatures #20 and #26 have been observed in stomach, breast, cervical and uterine cancers. These data suggest that the main contributor to germline predisposed tumors (LS and LLS-1) might be different from sporadic MSI or LLS-2 with a significant contribution of signature #6. Interestingly, the presence of signature #20 and #26 in other tumor types suggests the potential presence of germline DNA repair alterations in other cancer types.
The data support the interpretation that LLS tumors are comprised of at least two different cancer phenotypes distinguished by the presence or absence of germline alterations in genes that maintain genome integrity. To our knowledge, this is the first study reporting mutational signatures in the different CRC phenotypes. Mutational signatures may be used in the future to differentiate genetically predisposed MSI cases from their somatic counterparts.
The hypothetical mutator phenotype of the DNA repair gene mutation carriers most likely increases the probability of mutations in MMR genes, which results in the somatic acquisition of a more hyper-mutability (i.e., MSI) and eventually CRC development. A similar mechanism could explain the previously described LLS cases with germline DNA polymerase mutations. Another possible scenario is that the mutator phenotype could increase the probability of mutations in other CRC genes, such as APC. Further analysis of LLS patients and other increased-risk states will be needed to determine the full spectrum of mutator genes that can give rise to this condition and the full spread of carcinogenetic mechanisms that are impacted by the mutator phenotype.
The clinical implications of these findings remain to be fully elucidated, but it seems premature to label the tumors that arise in LLS patients as sporadic cancers. Whether more intensive or earlier CRC screening should be employed needs to be investigated. It is also unclear if more intense screening for some other cancers would be advised. The LLS patients were younger than patients with sporadic MSI tumors. On the other hand, the median age of the entire CCCC cohort was lower than the population average, possibly due to the lower socioeconomic status of the CCCC catchment area[8]. Finally, it should not come as a surprise that the median age of the identified LS patients was 62, as this age is always higher when it is assessed through population-based cohorts (like this one) as opposed to high risk clinic-based studies that consistently report younger ages at diagnosis[7].
This study has some important limitations. Even though we only included likely pathogenic variants, further studies in cell lines and animal models are needed to determine whether heterozygous mutations in these DNA repair genes truly confer a mutator phenotype. Another limitation is that we were unable to study disease co-segregation due to lack of genetic material from family members of LLS individuals. Thus, while there was a clear enrichment of cancers in families from LLS patients, we could not determine if affected individuals shared the mutations. Future studies should be carried out to address these limitations.
In summary, Lynch-like syndrome seems to be a heterogeneous phenotype with a significant number of cases harboring mutations in genes that maintain genome integrity. Tumors from these cases have distinct mutational signatures. The current study can set the basis for the definition of a new genetically predisposing multi-cancer condition that should allow for proper counseling of these patients.
Supplementary Material
Acknowledgments
Grant support:
This work was supported by grants from the American Cancer Society Illinois Division (223187, X.L.), the National Cancer Institute (U01 CA153060 and P30 CA023074, N.A.E. and 1K01CA204431-01A1 R.M.X) and the Prevent Cancer Foundation (RMX). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations:
- CRC
Colorectal cancer
- LS
Lynch syndrome
- LLS
Lynch-like syndrome
- MMR
mismatch repair
- AA
African American
- WEA
White European Americans
- MSI
microsatellite instability
- MSS
microsatellite stability
- IHC
immunohistochemistry
- YCGA
Yale Center for Genome Analysis
- SNVs
single-nucleotide variants
Footnotes
Disclosures of Potential Conflicts of Interest: None of the authors have any potential conflicts of interest to disclose that are relevant to the manuscript. The results published here are in whole or part based upon data generated by The Cancer Genome Atlas managed by the NCI and NHGRI. Information about TCGA can be found at http://cancergenome.nih.gov.
REFERENCES
- 1.Llor X (2012) When should we suspect hereditary colorectal cancer syndrome? Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association 10(4): 363–7 DOI 10.1016/j.cgh.2011.12.022 [DOI] [PubMed] [Google Scholar]
- 2.Geurts-Giele WR, Leenen CH, Dubbink HJ, et al. (2014) Somatic aberrations of mismatch repair genes as a cause of microsatellite-unstable cancers. J Pathol 234(4): 548–59 DOI 10.1002/path.4419 [DOI] [PubMed] [Google Scholar]
- 3.Mensenkamp AR, Vogelaar IP, van Zelst-Stams WA, et al. (2014) Somatic mutations in MLH1 and MSH2 are a frequent cause of mismatch-repair deficiency in Lynch syndrome-like tumors. Gastroenterology 146(3): 643–6.e8 DOI 10.1053/j.gastro.2013.12.002 [DOI] [PubMed] [Google Scholar]
- 4.Jansen AM, van Wezel T, van den Akker BE, et al. (2016) Combined mismatch repair and POLE/POLD1 defects explain unresolved suspected Lynch syndrome cancers. European journal of human genetics : EJHG 24(7): 1089–92 DOI 10.1038/ejhg.2015.252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morak M, Heidenreich B, Keller G, et al. (2014) Biallelic MUTYH mutations can mimic Lynch syndrome. European journal of human genetics : EJHG 22(11): 1334–7 DOI 10.1038/ejhg.2014.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elsayed FA, Kets CM, Ruano D, et al. (2015) Germline variants in POLE are associated with early onset mismatch repair deficient colorectal cancer. European journal of human genetics : EJHG 23(8): 1080–4 DOI 10.1038/ejhg.2014.242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rodriguez-Soler M, Perez-Carbonell L, Guarinos C, et al. (2013) Risk of cancer in cases of suspected lynch syndrome without germline mutation. Gastroenterology 144(5): 926–32 e1; quiz e13-4 DOI 10.1053/j.gastro.2013.01.044 [DOI] [PubMed] [Google Scholar]
- 8.Xicola RM, Gagnon M, Clark JR, et al. (2014) Excess of proximal microsatellite-stable colorectal cancer in African Americans from a multiethnic study. Clinical cancer research : an official journal of the American Association for Cancer Research 20(18): 4962–70 DOI 10.1158/1078-0432.ccr-14-0353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xicola RM, Llor X, Pons E, et al. (2007) Performance of different microsatellite marker panels for detection of mismatch repair-deficient colorectal tumors. J Natl Cancer Inst 99(3): 244–52. [DOI] [PubMed] [Google Scholar]
- 10.Goel A, Xicola RM, Nguyen TP, et al. (2010) Aberrant DNA methylation in hereditary nonpolyposis colorectal cancer without mismatch repair deficiency. Gastroenterology 138(5): 1854–62 DOI S0016-5085(10)00102-2 [pii] 10.1053/j.gastro.2010.01.035 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gausachs M, Mur P, Corral J, et al. (2012) MLH1 promoter hypermethylation in the analytical algorithm of Lynch syndrome: a cost-effectiveness study. European journal of human genetics : EJHG 20(7): 762–8 DOI 10.1038/ejhg.2011.277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Llor X, Pons E, Xicola RM, et al. (2005) Differential features of colorectal cancers fulfilling Amsterdam criteria without involvement of the mutator pathway. Clinical cancer research : an official journal of the American Association for Cancer Research 11(20): 7304–10. [DOI] [PubMed] [Google Scholar]
- 13.Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38(16): e164 DOI 10.1093/nar/gkq603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krumm N, Sudmant PH, Ko A, et al. (2012) Copy number variation detection and genotyping from exome sequence data. Genome Res 22(8): 1525–32 DOI 10.1101/gr.138115.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shoemaker RH (2006) The NCI60 human tumour cell line anticancer drug screen. Nature reviews Cancer 6(10): 813–23 DOI 10.1038/nrc1951 [DOI] [PubMed] [Google Scholar]
- 16.Network TCGA (2012) Comprehensive molecular characterization of human colon and rectal cancer. Nature 487(7407): 330–7 DOI 10.1038/nature11252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cibulskis K, Lawrence MS, Carter SL, et al. (2013) Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 31(3): 213–9 DOI 10.1038/nbt.2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roberts SA, Gordenin DA (2014) Hypermutation in human cancer genomes: footprints and mechanisms. Nature reviews Cancer 14(12): 786–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. (2013) Signatures of mutational processes in human cancer. Nature 500(7463): 415–21 DOI 10.1038/nature12477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gehring JS, Fischer B, Lawrence M, Huber W (2015) SomaticSignatures: inferring mutational signatures from single-nucleotide variants. Bioinformatics: DOI 10.1093/bioinformatics/btv408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brunet JP, Tamayo P, Golub TR, Mesirov JP (2004) Metagenes and molecular pattern discovery using matrix factorization. Proc Natl Acad Sci U S A 101(12): 4164–9 DOI 10.1073/pnas.0308531101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nik-Zainal S, Alexandrov LB, Wedge DC, et al. (2012) Mutational processes molding the genomes of 21 breast cancers. Cell 149(5): 979–93 DOI 10.1016/j.cell.2012.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hutchins LN, Murphy SM, Singh P, Graber JH (2008) Position-dependent motif characterization using non-negative matrix factorization. Bioinformatics 24(23): 2684–90 DOI 10.1093/bioinformatics/btn526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suzuki R, Shimodaira H (2006) Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 22(12): 1540–2 DOI 10.1093/bioinformatics/btl117 [DOI] [PubMed] [Google Scholar]
- 25.Weren RD, Ligtenberg MJ, Kets CM, et al. (2015) A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nature genetics 47(6): 668–71 DOI 10.1038/ng.3287 [DOI] [PubMed] [Google Scholar]
- 26.Segui N, Mina LB, Lazaro C, et al. (2015) Germline Mutations in FAN1 Cause Hereditary Colorectal Cancer by Impairing DNA Repair. Gastroenterology 149(3): 563–6 DOI 10.1053/j.gastro.2015.05.056 [DOI] [PubMed] [Google Scholar]
- 27.Goldberg Y, Halpern N, Hubert A, et al. (2015) Mutated MCM9 is associated with predisposition to hereditary mixed polyposis and colorectal cancer in addition to primary ovarian failure. Cancer Genet 208(12): 621–4 DOI 10.1016/j.cancergen.2015.10.001 [DOI] [PubMed] [Google Scholar]
- 28.Arora S, Yan H, Cho I, et al. (2015) Genetic Variants That Predispose to DNA Double-Strand Breaks in Lymphocytes From a Subset of Patients With Familial Colorectal Carcinomas. Gastroenterology 149(7): 1872–83 e9 DOI 10.1053/j.gastro.2015.08.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Voer RM, Hahn MM, Mensenkamp AR, et al. (2015) Deleterious Germline BLM Mutations and the Risk for Early-onset Colorectal Cancer. Scientific reports 5: 14060 DOI 10.1038/srep14060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ozgenc A, Loeb LA (2006) Werner Syndrome, aging and cancer. Genome Dyn 1: 206–17 DOI 10.1159/000092509 [DOI] [PubMed] [Google Scholar]
- 31.Wu X, Mondal G, Wang X, et al. (2009) Microcephalin regulates BRCA2 and Rad51-associated DNA double-strand break repair. Cancer Res 69(13): 5531–6 DOI 10.1158/0008-5472.can-08-4834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang B, Wang E, Dai H, et al. (2013) BRIT1 regulates p53 stability and functions as a tumor suppressor in breast cancer. Carcinogenesis 34(10): 2271–80 DOI 10.1093/carcin/bgt190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wittschieben JP, Patil V, Glushets V, Robinson LJ, Kusewitt DF, Wood RD (2010) Loss of DNA polymerase zeta enhances spontaneous tumorigenesis. Cancer Res 70(7): 2770–8 DOI 10.1158/0008-5472.can-09-4267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De Brakeleer S, De Greve J, Loris R, et al. (2010) Cancer predisposing missense and protein truncating BARD1 mutations in non-BRCA1 or BRCA2 breast cancer families. Human mutation 31(3): E1175–85 DOI 10.1002/humu.21200 [DOI] [PubMed] [Google Scholar]
- 35.Ratajska M, Antoszewska E, Piskorz A, et al. (2012) Cancer predisposing BARD1 mutations in breast-ovarian cancer families. Breast cancer research and treatment 131(1): 89–97 DOI 10.1007/s10549-011-1403-8 [DOI] [PubMed] [Google Scholar]
- 36.Norquist BM, Harrell MI, Brady MF, et al. (2016) Inherited Mutations in Women With Ovarian Carcinoma. JAMA oncology 2(4): 482–90 DOI 10.1001/jamaoncol.2015.5495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kawasaki T, Ohnishi M, Suemoto Y, et al. (2008) WRN promoter methylation possibly connects mucinous differentiation, microsatellite instability and CpG island methylator phenotype in colorectal cancer. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc 21(2): 150–8 DOI 10.1038/modpathol.3800996 [DOI] [PubMed] [Google Scholar]
- 38.Santarosa M, Ashworth A (2004) Haploinsufficiency for tumour suppressor genes: when you don’t need to go all the way. Biochim Biophys Acta 1654(2): 105–22 DOI 10.1016/j.bbcan.2004.01.001 [DOI] [PubMed] [Google Scholar]
- 39.Cabelof DC (2012) Haploinsufficiency in mouse models of DNA repair deficiency: modifiers of penetrance. Cell Mol Life Sci 69(5): 727–40 DOI 10.1007/s00018-011-0839-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gruber SB, Ellis NA, Scott KK, et al. (2002) BLM heterozygosity and the risk of colorectal cancer. Science 297(5589): 2013. [DOI] [PubMed] [Google Scholar]
- 41.Theodoratou E, Campbell H, Tenesa A, et al. (2010) A large-scale meta-analysis to refine colorectal cancer risk estimates associated with MUTYH variants. British journal of cancer 103(12): 1875–84 DOI 10.1038/sj.bjc.6605966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bouffler SD, Hofland N, Cox R, Fodde R (2000) Evidence for Msh2 haploinsufficiency in mice revealed by MNU-induced sister-chromatid exchange analysis. British journal of cancer 83(10): 1291–4 DOI 10.1054/bjoc.2000.1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.DeWeese TL, Shipman JM, Larrier NA, et al. (1998) Mouse embryonic stem cells carrying one or two defective Msh2 alleles respond abnormally to oxidative stress inflicted by low-level radiation. Proc Natl Acad Sci U S A 95(20): 11915–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rigter LS, Snaebjornsson P, Rosenberg EH, et al. (2018) Double somatic mutations in mismatch repair genes are frequent in colorectal cancer after Hodgkin’s lymphoma treatment. Gut 67(3): 447–55 DOI 10.1136/gutjnl-2016-312608 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.