

Summary
The Enterobacter cloacae complex (ECC) consists of closely-related bacteria commonly associated with the human microbiota. ECC are increasingly isolated from healthcare-associated infections, demonstrating that these Enterobacteriaceae are emerging nosocomial pathogens. ECC can rapidly acquire multidrug resistance to conventional antibiotics. Cationic antimicrobial peptides (CAMPs) have served as therapeutic alternatives because they target the highly conserved lipid A component of the Gram-negative outer membrane. Many Enterobacteriaceae fortify their outer membrane with cationic amine-containing moieties to prevent CAMP binding, which can lead to cell lysis. The PmrAB two-component system (TCS) directly activates 4-amino-4-deoxy-l-arabinose (l-Ara4N) biosynthesis to result in cationic amine moiety addition to lipid A in many Enterobacteriaceae such as E. coli and Salmonella. In contrast, PmrAB is dispensable for CAMP resistance in E. cloacae. Interestingly, some ECC clusters exhibit colistin heteroresistance, where a subpopulation of cells exhibit clinically significant resistance levels compared to the majority population. We demonstrate that E. cloacae lipid A is modified with l-Ara4N to induce CAMP heteroresistance and the regulatory mechanism is independent of the PmrABEcl TCS. Instead, PhoPEcl binds to the arnBEcl promoter to induce l-Ara4N biosynthesis and PmrAB-independent addition to the lipid A disaccharolipid. Therefore, PhoPQEcl contributes to regulation of CAMP heteroresistance in some ECC clusters.
Keywords: heteroresistance, antimicrobial resistance, lipid A, outer membrane, colistin, Enterobacteriaceae
Abbreviated Summary

Enterobacter cloacae acquires resistance to conventional antibiotic treatments and has demonstrated heteroresistance to colistin, which is a cationic antimicrobial peptide used to supplement our dwindling antibiotic arsenal. The PhoPQ two-component system contributes to colistin heteroresistance in E. cloacae. Here we show that PhoP directly binds to the arn promoter to induce transcription, which culminates in modification of lipid A and colistin heteroresistance.
Introduction
Gram-negative bacteria assemble a highly conserved outer membrane (OM) barrier. Glycerophospholipids comprise the periplasmic monolayer of the asymmetric lipid bilayer, while the surface-exposed monolayer is enriched with lipopolysaccharide (LPS). The LPS glycolipid is organized into three domains; an O-antigen carbohydrate repeat, core oligosaccharide, and the membrane anchor, lipid A (1). The lipid A domain is initially synthesized as a β−1’,6-linked glucosamine disaccharide that is both phosphorylated and fatty acylated. Lipid A is the bioactive portion of LPS and robustly activates the human Toll-like receptor 4 (TLR-4) and myeloid differentiation factor 2 (MD-2) immune complex to induce immune reactivity (1–4). Gram-negative pathogens encode highly conserved regulatory mechanisms that modify lipid A to prevent TLR-4/MD-2 recognition and to fortify the OM against immune effectors and antimicrobials, which promotes survival in the host (5).
Lipid A modification enzymes are transcriptionally regulated by two-component systems (TCS) (6, 7). The PmrAB and PhoPQ TCSs are well-studied phosphorelay signaling systems that regulate lipid A modifications in response to specific environmental signals (8–10). PmrAB and PhoPQ are highly conserved among pathogenic Enterobacteriaceae (11). PmrAB responds to high Fe3+ concentrations, cationic antimicrobial peptides (CAMPs), and slightly acidic pH to directly activate eptA (also known as pmrC) and arn operon expression (12–14), which encode phosphoethanolamine (pEtN) and 4-amino-4-deoxy-l-arabinose (l-Ara4N) transferases, respectively (15–18). Cationic amine addition to the lipid A domain of LPS neutralizes the surface charge to protect the cell from CAMP-mediated lysis (15, 17).
PhoPQ is activated in response to depletion of divalent cations such as Mg2+ and Ca2+ and the presence of CAMPs (8, 10, 19). PhoPQ phosphotransfer directly activates transcription of genes encoding PagL (only in Salmonella (5)) and PagP, which add or remove acyl chains from lipid A, respectively (8, 20–22). Additionally, it directly activates arn expression in Klebsiella and Yersinia spcs. (23, 24). While the PmrAB and PhoPQ TCSs each regulate distinct subsets of genes, the independent signaling pathways also converge through the connector protein, PmrD (25–27). PmrD binds phospho-PmrA, which prevents PmrB-mediated dephosphorylation (25, 28–30). Constitutive PmrA-dependent gene expression increases pEtN and l-Ara4N lipid A modifications.
The Enterobacter cloacae complex (ECC) is composed of thirteen closely-related Gram-negative bacterial clusters (designated C-I to C-XIII) (31). ECC are typically associated with the host microbiota. However, many clusters have been associated with hospital-acquired infections, especially in immunocompromised patients (32). Infections manifest in a wide range of host tissues with symptoms including skin, respiratory tract, urinary tract, wound and blood infections (33). ECC have increasingly emerged in nosocomial settings and are problematic because they harbor multidrug resistance (MDR) mechanisms, which limit treatment options (32, 34–36). Alternative last-line therapeutics used to treat MDR Gram-negative infections include the CAMP, colistin (polymyxin E), which binds the lipid A portion of LPS to perturb the outer membrane and lyse the bacterial cell. Despite success as a last-line therapeutic (37, 38), many ECC clusters demonstrate heteroresistance, where a subset of the clonal population is colistin resistant (34, 39–41). We do not fully understand the underlying molecular mechanism(s) that regulate colistin heteroresistance in ECC; further characterization will advance our understanding of antimicrobial resistance and could help inform new treatment strategies.
A previous report showed that colistin heteroresistance naturally occurs within clonal ECC clusters (34). Moreover, colistin heteroresistance in E. cloacae was induced by innate immune defenses within a murine infection model, which led to treatment failure (39). Transcriptional analysis of susceptible and resistant populations suggested that pEtN and l-Ara4N lipid A modifications contribute to heteroresistance (39) and PhoPQ contributed to regulation (34, 39), as described in other Enterobacteriaceae (11). However, it was not established that the lipid A modifications actually occur, nor has PhoPQ-dependent, PmrAB-independent regulation of colistin heteroresistance been fully described in E. cloacae or other ECC isolates.
Herein, we demonstrate that E. cloacae colistin heteroresistance involves PhoPQEcl, which regulates l-Ara4N modification of lipid A. The PhoPEcl response regulator directly binds to the promoter region of arnBEcl, which is the first gene of a seven-gene operon (arnBCADTEFEcl). Transcriptomics analysis supports a model of PhoPQ-dependent, PmrAB-independent arnEcl regulation. Furthermore, l-Ara4N modification of lipid A increased in response to growth in limiting Mg2+, which amplified colistin resistance in a PhoPQEcl-dependent manner. Lastly, sequencing of twelve ECC colistin-susceptible isolates pinpointed mutations within the PhoPQ-dependent lipid A modification pathway that promoted colistin susceptibility. This study advances our understanding of the molecular mechanisms that mediate colistin heteroresistance in ECC.
Results
Colistin heteroresistance in E. cloacae is regulated by PhoPQEcl, but not PmrABEcl.
To elucidate the underlying mechanisms that regulate colistin heteroresistance in ECC, we analyzed a collection of E. cloacae subsp. cloacae strain ATCC 13047 genetic mutants by calculating the colony forming units (CFUs) during exponential growth in the absence and presence of colistin (Fig 1A). While wild type and all mutant E. cloacae strains grew in standard growth media, ∆phoPQEcl was not viable when 10 µg/ml of colistin was added to the media. Clinical resistance to colistin is defined as >4 µg/ml (42). The decrease in ∆phoPQEcl cell viability suggested that PhoPQEcl contributes to colistin heteroresistance. Furthermore, wild type E. cloacae grown in colistin demonstrated approximately ten-fold less CFUs at hour two (P <0.05), suggesting a survival defect in early logarithmic growth phase. However, the fitness defect was no longer significant at hour three. By hour four, CFUs were equivalent to growth without colistin (Fig 1A).
Figure 1: Survival of colistin heteroresistant E. cloacae is dependent on PhoPQEcl, but not PmrABEcl-regulated lipid A modifications.
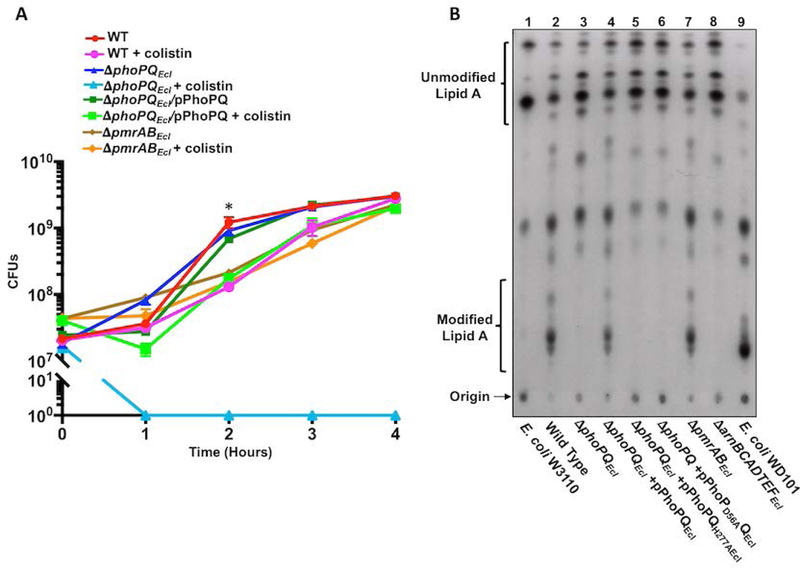
(A) E. cloacae logarithmic phase growth over time as measured by colony forming units (CFUs). At two hours, the growth rate between wild type grown in LB was significantly (*) different from cells grown in LB + colistin (P value <0.05). (B) 32P-radiolabeled lipid A was isolated from wild type and mutant E. cloacae strains and separated based on hydrophobicity using thin layer chromatography. Lipid A species are labeled as unmodified or modified as determined by E. coli W3110 (lane 1) and WD101 (lane 9) lipid A, respectively.
Due to reports of colistin heteroresistance in E. cloacae and other ECC strains (34, 39), we subjected wild type, ∆phoPQEcl, ∆phoPQEcl/pPhoPQEcl, and ∆pmrABEcl E. cloacae to colistin E-test strip analysis, which provides a convenient method to observe heteroresistance (Fig S1). Squatter colonies within the zone of inhibition indicated colistin heteroresistance in wild type, ∆phoPQEcl/pPhoPQEcl, and ∆pmrABEcl strains, but not ∆phoPQEcl. We confirmed colistin heteroresistance by population analysis profiling (PAP) (Table 1) (43). Minimal inhibitory concentration (MIC) values were calculated using the broth microdilution (BMD) method (Table 1). Wild type, ∆phoPQEcl/pPhoPQEcl, and ∆pmrABEcl E. cloacae all demonstrated MICs >256 µg/ml, while the ∆phoPQEcl, ∆phoPQEcl/pPhoPQH277A, ∆phoPQEcl/pPhoPD56AQ and ∆arnEcl (arnBCADTEFEcl) MIC was 0.5 µg/ml. Together, these studies confirm that PhoPQEcl signal transduction and the arnEcl biosynthetic operon (l-Ara4N) contribute to colistin heteroresistance in E. cloacae.
Table 1.
MICs of colistin and PAP analysis for each E. cloacae mutant
| Frequency of appearance of subpopulations (PAPsa) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Concentration of colistin | |||||||||
| Isolate | MIC of colistin (µg/ml) by BMDb | MIC of colistin (µg/ml) by Etest | 1 µg/ml | 2 µg/ml | 4 µg/ml | 8 µg/ml | 16 µg/ml | 32 µg/ml | 64 µg/ml |
| Wild type | ≥256* | 0.125** | 9.5 X 10−3 | 6.3 X 10−2 | 5.2 X 10−3 | 7.5 X 10−3 | 7.6 X 10−2 | 6.3 X 10−3 | 9.3 10−3 |
| ∆phoPQEcl | 0.5 | 0.125 | 2.6 X 10−3 | 0 | 0 | 0 | 0 | 0 | 0 |
| ∆phoPQEcl +pPhoPQEcl | ≥256* | 0.125** | 3.6 X 10−2 | 1.3 X 10−2 | 4.3 X 10−3 | 1.6 X 10−2 | 4.1 X 10−3 | 1.7 X 10−3 | 4.5 10−4 |
| ∆phoPQEcl +pPhoPQH277AEcl | 0.5 | 0.125 | 3.1 X 10−2 | 0 | 0 | 0 | 0 | 0 | 0 |
| ∆phoPQ +pPhoPD56AQEcl | 0.5 | 0.125 | 7.4 X 10−2 | 0 | 0 | 0 | 0 | 0 | 0 |
| ∆pmrABEcl | ≥256* | 0.125** | 2.9 X 10−2 | 1.8 X 10−3 | 7.7 X 10−2 | 9.2 X 10−3 | 5.5 X 10−2 | 1.3 X 10−3 | 2.3 X 10−4 |
| ∆arnTEcl | 0.5 | 0.125 | 4.0 X 10−2 | 0 | 0 | 0 | 0 | 0 | 0 |
PAP : Population Analysis Profile using an initial culture of 1010 CFU/mL
BMD : Broth microdilution method
Presence of skip wells
Presence of squatter colonies inside the zone of inhibition
Since lipid A modifications induce colistin resistance in pathogenic Enterobacteriaceae (11), we analyzed wild type and mutant E. cloacae lipid A for modifications. 32P-radiolabelled lipid A was isolated and chromatographically separated based on hydrophobicity. As controls, we also analyzed lipid A from E. coli strain W3110 (Fig 1B, lane 1), which does not significantly modify its lipid A, and E. coli strain WD101 (Fig 1B, lane 9), which constitutively expresses pmrA to produce modified lipid A (15). Thin layer chromatography (TLC) analysis indicated that wild type E. cloacae produced a mixture of lipid A consistent with modified and unmodified species (Fig 1B, lane 2). ∆phoPQEcl and the ∆arnEcl strains did not produce modified lipid A (Fig 1B, lanes 3 and 8). PhoPQ complementation fully restored production of modified lipid A in the phoPQ mutant (Fig 1B, lane 4). Furthermore, site-directed mutagenesis to substitute H277 in PhoQEcl or D57 in PhoPEcl with alanine limited lipid A assembly to only unmodified species (Fig 1B lanes 5 and 6). These results confirm that PhoPQEcl phosphotransfer and l-Ara4N biosynthesis are essential for lipid A modification in E. cloacae. Interestingly, the pmrABEcl mutant assembled a modified lipid A, similar to wild type (Fig 1B, lane 7), and exhibited colistin heteroresistance (Fig 1A, Table 1, Fig S1A), suggesting that PmrABEcl does not regulate colistin heteroresistance in E. cloacae.
The lipid A anchor of LPS is a pathogen associated molecular pattern (PAMP) that is bound with high affinity by the mammalian host TLR-4/MD-2 complex (44), which activates a proinflammatory response to clear the bacterial infection (45). Structural alterations to lipid A can dramatically alter TLR-4/MD-2-dependent host immune activation (2) and a previous report nicely demonstrated that E. cloacae colistin heteroresistance was induced by innate immune effectors (39). Therefore, we examined if E. cloacae containing modified or unmodified lipid A would differentially activate TLR-4/MD-2 in a human embryonic kidney reporter cell line (HEK-blue) (2). Wild type and phoPQEcl mutant strains stimulated TLR-4/MD-2-dependent activation equally (Fig S1B), suggesting that lipid A modifications do not significantly alter host immune recognition. Reporter activation by E. cloacae lipid A was attenuated compared to E. coli lipid A at higher cell densities, suggesting differential recognition by the human TLR-4/MD-2 complex. The Gram-positive Staphylococcus aureus, which does not produce lipid A, did not stimulate the TLR-4/MD-2 complex (Fig S1B). Thus, while PhoPQEcl-dependent lipid A modifications contribute to CAMP resistance in E. cloacae, they do not significantly affect innate immune recognition and reactivity.
Determination of E. cloacae lipid A modifications.
In order to define outer membrane modifications, we isolated lipid A from wild type E. cloacae grown in media supplemented with 10 µg/ml of colistin and from ∆phoPQEcl, which was grown without colistin. Purified lipid A was analyzed by direct infusion nanoESI. The MS1 spectra with a range of m/z 750–2000 are shown in Figure S2. The expanded MS1 spectrum (m/z 850–1200) of lipid A isolated from wild type E. cloacae demonstrated three distinct modifications: (i) addition of either one or two l-Ara4N moieties (red), (ii) palmitate (C16:0) addition (green), and (iii) hydroxylation (Fig 2A). The MS1 spectrum of lipid A isolated from ∆phoPQEcl did not produce l-Ara4N modified lipid A (Fig 2B). Hydroxyl addition was not labeled for simplicity, but correlates with a m/z shift of 8 of the doubly-charged molecular ions. Higher-energy collisional dissociation (HCD) and ultraviolet photodissociation (UVPD) MS/MS spectra were obtained for the ions of m/z 1042.68 and 1161.79 from wild type and the ions of m/z 911.62 and 1030.73 from ∆phoPQ E. cloacae (Fig S3, S4, S5 and S6). Analysis of the MS/MS spectra from wild type (m/z 1042.68) indicated PhoPQEcl-dependent addition of l-Ara4N at both the 1- and 4’-phosphates (Fig S3). The MS/MS spectra for the ion of m/z 1161.79 (wild type E. cloacae) showed addition of l-Ara4N at both the 1- and 4’-phosphates and palmitate addition to the R-2-hydroxymyristate (Fig S4). Analysis of lipid A from the phoPQEcl mutant (m/z 911.62) completely lacked l-Ara4N modified lipid A (Fig S5) and analysis of the m/z 1030.73 ion from the phoPQEcl mutant demonstrated that palmitate addition at the R-2-hydroxymyristate position of lipid A occurred independent of PhoPQEcl (Fig S6).
Figure 2: Expanded MS1 spectra of lipid A isolated from.

(A) wild type E. cloacae grown in media supplemented with 10 µg/ml colistin and (B) …phoPQEcl, which was grown in media without antibiotics. The chemical structures associated with the MS1 spectra are illustrated on the right. The presence of aminoarabinose groups are denoted by L-Ara4N (red), while addition of palmitoyl groups are denoted by +C16:0 (green). Hydroxylation is not illustrated, but is indicated by an m/z shift of 8 relative to doubly-charged lipid A ions in the spectra.
Based on transcriptomics studies, a previous report suggested that E. cloacae adds pEtN and l-Ara4N to lipid A to develop colistin heteroresistance (39). However, our genetic and high resolution mass spectrometry analysis demonstrate that only l-Ara4N modifies the 1- and 4’-phosphates of lipid A in a PhoPQEcl-dependent manner (Fig 2A and B) and this amine-containing modification correlates with colistin heteroresistance (Fig 1A and Table 1).
l-Ara4N lipid A modifications are dependent on PhoPQEcl, but not PmrABEcl.
To further characterize lipid A modifications in the ∆pmrABEcl mutant, we analyzed purified lipid A from wild type and mutant E. cloacae using MALDI-TOF mass spectrometry. Wild type produced a lipid A mixture, which included l-Ara4N modified lipids (Fig S7A and B). In contrast, analysis of ∆phoPQEcl and ∆arnEcl lipid A indicated that l-Ara4N modified lipids were not present. Expression of PhoPQEcl in trans from an IPTG-inducible promoter restored l-Ara4N modified lipid A in the phoPQEcl mutant. Furthermore, ∆pmrABEcl produced the l-Ara4N modification, similar to wild type (Fig S7A). The m/z of each prominent peak in our MALDI-MS analysis corresponded with the exact mass of an expected structure with only the l-Ara4N-containing structures demonstrating colistin resistance (Fig S7B). Here, we confirmed that l-Ara4N modification of lipid A in E. cloacae is not dependent on PmrABEcl.
PhoPEcl directly binds to the arnBEcl promoter.
The arn operon is composed of seven genes and expression is driven by a promoter upstream of arnB (16). This genetic organization is conserved in E. cloacae as illustrated in Fig 3A. phoP expression is autoregulated in Enterobacteriaceae, where PhoP binds to the PhoP box to interact with RNA polymerase, which induces transcription (46). The putative PhoP box in the phoP promoter region (PphoP) is conserved in E. coli, Salmonella, and E. cloacae (Fig 3B). Alignment of the E. cloacae arnB promoter region (ParnB) with E. coli, Salmonella, and E. cloacae PphoP suggested a putative PhoP box region. Importantly, E. cloacae ParnB, which contains a putative PhoP box, is highly conserved among ECC. However, this feature was not conserved within E. coli ParnB, suggesting regulatory mechanisms that control promoter activation are different (Fig 3B).
Figure 3: PhoPEcl binds to the arnB promoter of E. cloacae (Ecl), but not E. coli (Ec).
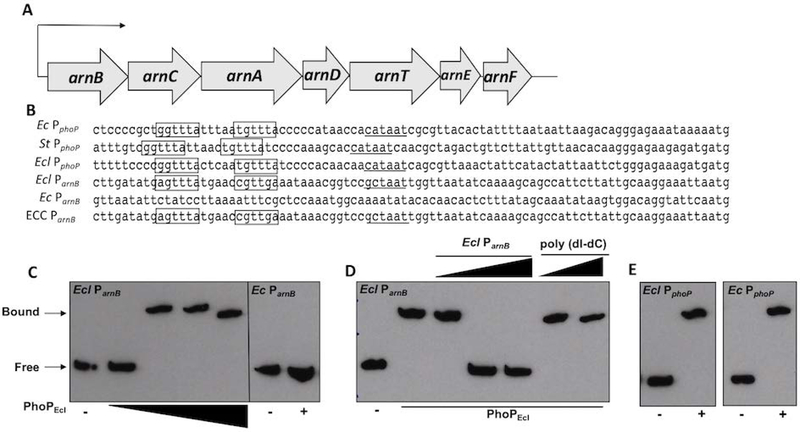
(A) Illustration of the arn operon organization. (B) Sequence alignment of the phoP promoter (PphoP) region in Ec, Salmonella (St), and Ecl, which each contain a PhoP box. The arnB promoter (ParnB) of Ecl contains a putative PhoP box binding site that is not present in Ec. The putative PhoP boxes have been boxed, while the −10 region is underlined. There were no putative PmrA boxes in the Ecl arnB or phoP promoter regions, so they were not included for simplicity. (C) Electrophoretic mobility shift assay (EMSA) of Ecl ParnB with increasing concentrations of PhoPEcl. PhoPEcl was used at concentrations of 0, 0.1, 1.0, 5.0 and 10.0 µM. EMSA using Ec ParnB in the absence or presence of PhoPEcl, respectively. (D) EMSA competition experiments where increasing concentrations (1:1, 2:1, 5:1) of unlabeled ParnB competes with biotin-labeled ParnB, but nonspecific unlabeled poly(dI-dC) (2:1, 5:1) does not. (E) PhoPEcl binds to both the Ecl and Ec phoP promoters.
We performed electrophoretic mobility shift assays (EMSAs) using E. cloacae ParnB to determine if PhoPEcl directly binds the promoter to activate arnEcl transcription. Increasing concentrations of purified PhoPEcl (Fig S8) induced a shift of the biotinylated arnBEcl promoter fragment, which contains the putative PhoP box binding motif (Fig 3C). Importantly, PhoPEcl does not bind to E. coli ParnB, which does not encode the PhoP box motif (Fig 3C). Furthermore, the PhoPEcl-arnBEcl promoter interaction was abrogated when unlabeled E. cloacae ParnB was added in increasing amounts, as a competitive inhibitor. We also show that the interaction is specific because addition of noncompetitive DNA (poly(dI-dC)) did not reduce the PhoPEcl and E. cloacae ParnB interaction (Fig 3D). Lastly, PhoPEcl bound E. cloacae and E. coli PphoP, which both encode the nucleotide sequence specific to the PhoP box (Fig 3E). Together, these findings suggest that E. cloacae encodes a mechanism that enables l-Ara4N biosynthesis to respond directly to PhoPQEcl.
RNA-sequencing analysis of the phoPQEcl and pmrABEcl mutants.
To better understand PhoPQEcl and PmrABEcl transcriptional regulation, we isolated and sequenced total RNA from wild type and mutant E. cloacae strains. A heat map illustrates the fold change of arnEcl, phoPQEcl, and pmrABEcl gene expression in the TCS mutants relative to wild type (Fig 4). Expression of the arnEcl genes were significantly down regulated in ∆phoPQEcl compared to wild type, suggesting that activation of the pathway is dependent on PhoPQEcl. In contrast, arnEcl gene expression was not significantly altered in the ∆pmrABEcl mutant relative to wild type. A complete list of the significant up- and down-regulated genes (P <0.05) is included in Table S4.
Figure 4: RNA-sequencing analysis of E. cloacae genes.

Heat map illustrating the altered expression of select operons in ∆phoPQEcl and ∆pmrABEcl mutants. Expression is shown as a ratio of mutant to wild type expression (P <0.05).
E. cloacae colistin resistance is amplified in response to limiting Mg2+.
Together, these analyses indicate colistin heteroresistance in wild type E. cloacae (Fig 1A and Table 1) is mediated by l-Ara4N modification of lipid A, which is regulated in a PhoPQEcl-dependent manner under standard growth conditions. In E. coli and Salmonella, PhoPQ is activated by various signals, including low Mg2+ and CAMPs (8, 19, 21). Here we analyzed if PhoPQEcl responds to similar physiological cues to induce colistin resistance in E. cloacae. Wild type and mutant E. cloacae were grown in N minimal medium with high (10 mM) or low (10 μM) Mg2+ levels. All cultures were exposed to colistin at mid-logarithmic growth. Wild type and complemented phoPQEcl mutant strains grown in high Mg2+ demonstrated some susceptibility to 5 and 10 μg/ml of colistin (Fig 5A, High Mg2+), suggesting colistin-susceptible and -resistant populations were present, which is indicative of heteroresistance. When grown under limiting Mg2+ conditions, E. cloacae cells were more resistant (Fig 5A, Low Mg2+). In contrast, phoPQEcl demonstrated a fitness defect in either Mg2+ concentration when exposed to colistin (Fig 5A). These data suggest that E. cloacae PhoPQEcl amplifies colistin resistance in response to limiting Mg2+ growth conditions.
Figure 5: PhoPQEcl-dependent activation of L-Ara4N addition induces colistin resistance in low Mg2+.

(A) Wild type and mutant E. cloacae strains were grown in N minimal medium with high (10mM, top) or low (10 μM, bottom) Mg2+. Strains were challenged with 0, 5, or 10 μg/ml of colistin for 1 h and plated for survival. Two biological replicates were each analyzed in triplicate with data from one representative set reported. P value <0.05. (B) 32P-radiolabeled lipid A was isolated from wild type and mutant E. cloacae strains and separated based on hydrophobicity using thin layer chromatography. The associated lipid A structures (right) are illustrated with black circles indicating L-Ara4N Addition. Lipid A species were labeled as determined by E. coli W3110 (unmodified) and WD101 (modified) lipid A.
PhoPQEcl responds to limiting Mg2+ conditions by inducing l-Ara4N lipid A modification.
To determine if increased colistin resistance was dependent on l-Ara4N modification of lipid A, we isolated lipid A after growth in either low or high Mg2+. TLC analysis demonstrated that wild type and the complemented phoPQEcl mutant primarily produced l-Ara4N-modified lipid A when Mg2+ concentrations were limiting (Fig 5B, Low Mg2+). In contrast, the same strains grown in excess Mg2+, produced a mixture of modified and unmodified lipid A, which is indicative of heteroresistance (Fig 5B, High Mg2+). Interestingly, growth in excess Mg2+ does not completely shut-off production of PhoPQEcl-dependent lipid A modification in E. cloacae, as was previously shown in E. coli (25). Together, these studies suggest that a subset of the clonal E. cloacae population activates PhoPQEcl-dependent l-Ara4N modification of lipid A under standard growth conditions to promote heteroresistance. However, depletion of Mg2+ amplifies l-Ara4N modification (Fig 5B) and colistin resistance (Fig 5A) throughout the population.
Inactivation of colistin heteroresistance in ECC clinical isolates.
A previous report showed that while many ECC clinical isolates were colistin heteroresistant, some were susceptible to colistin–mediated lysis. Interestingly, ECC colistin heteroresistance was cluster dependent (34). To determine the genetic basis for colistin sensitivity in ECC clusters, we examined twelve colistin susceptible clinical isolates (CI) and compared them to heteroresistant wild type ATCC 13047. We confirmed colistin susceptibility in all isolates using the BMD method to determine MICs (Table S3) and sequenced each genome. We obtained ~70 X 106 reads for each susceptible isolate and the wild type strain, which were mapped to the annotated ATCC 13047 genome (47). The coverage region of each isolate varied between 82% to 99%, where most coverage variation occurred in plasmid DNA regions. Initial analysis of our wild type strain confirmed it did not encode mutations in the phoPQEcl, pmrABEcl, or the arnEcl coding sequences (Table S3), consistent with the published annotation. In contrast, multiple mutations were found in the phoPQEcl and arnEcl operons of the colistin-susceptible isolates. All of the amino acid changes in these coding regions are listed in Table S3. Interestingly, ten of the twelve isolates encoded single nucleotide polymorphisms (SNPs) in the phoPQEcl promoter sequence immediately upstream of the phoPEcl translational start codon (within 8 nucleotides), which indicated the SNPs likely disrupted the ribosome binding site (Fig 6A). To determine if the SNPs attenuated PhoPQ-dependent colistin resistance, clinical isolate-3 (CI-3) (ECC cluster III), which did not encode amino acid changes in the phoPQEcl or arnEcl genetic coding regions, was transformed with pPhoPQEcl. Complementation restored colistin resistance, where the MIC increased from 0.5 in CI-3 to >256 μg/ml after PhoPQEcl signaling was restored (Fig 6B). Complementation suggested that SNPs in the phoPQEcl ribosome binding sites contributed to colistin susceptibility. SNP mutations, which abrogate translation of PhoPQEcl, support a model where colistin heteroresistance is dependent on PhoPQEcl in diverse ECC isolates.
Figure 6: PhoPEcl single nucleotide polymorphisms that attenuate colistin heteroresistance in ECC clinical isolates.

(A) Sequence alignment of E. cloacae and other ECC phoP promoter (PphoP) regions. The region includes 25 nucleotides upstream of the start codon (boxed). Grey boxes indicate single nucleotide replacement mutations, while grey triangles indicate single nucleotide insertion mutations. (B) E-test strips of CI-3 and CI-3/pPhoPQEcl to visualize heteroresistant colonies. The black arrow indicates colistin resistant colonies.
Discussion
E. cloacae and other ECC members encode PmrABEcl and PhoPQEcl homologs. We hypothesized these TCS regulatory systems functioned together in a pathway to control l-Ara4N and pEtN modification of lipid A, based on previous transcriptomics analysis of resistant and susceptible populations (39) and because these lipid A modifications are highly conserved among Enterobacteriaceae (11). However, our genetic and high-resolution mass spectrometry analysis of E. cloacae lipid A determined that colistin heteroresistance in E. cloacae was mediated by PhoPQEcl-dependent, PmrABEcl-independent l-Ara4N lipid A modification. Therefore, we identified a mechanism of ECC colistin heteroresistance that involves the PhoPQ system.
E. cloacae and other ECC members do not encode a PmrD homolog, which couples PhoPQ signal transduction to regulation of PmrA-dependent genes in many Enterobacteriaceae (11). Moreover, PmrAEcl shares only 52% identity with E. coli PmrA and PmrBEcl shares only 57% identity with E. coli PmrB, suggesting the l-Ara4N lipid A modification pathway in E. cloacae diverged from E. coli and Salmonella. We confirmed direct binding of PhoPEcl to the arnBEcl promoter, which supports a model where l-Ara4N addition to lipid A and colistin heteroresistance in E. cloacae is dependent on PhoPQEcl, but not PmrABEcl.
Research from other groups has outlined a complex regulatory network in E. coli and Salmonella that tightly regulates lipid A l-Ara4N and EptA modifications (15–18, 25). We hypothesize that uncoupling PmrABEcl regulation from l-Ara4N modification bypasses an important regulatory checkpoint, which likely promotes misregulated PhoPQEcl-dependent arnEcl expression. Furthermore, colistin heteroresistance has also been associated with Klebsiella pneumoniae (48), another Enterobacteriaceae family member that activates arn expression independently of PmrAB (23). Since selection has driven ECC and other opportunistic pathogens to maintain an altered lipid A modification signaling network, we predict that it is advantageous to maintain a CAMP resistant subpopulation in some environments. Presumably, the alternative regulatory mechanism promotes bacterial fitness in environments specific to their commensal and pathogenic niches.
Colistin heteroresistance is not well-understood at the molecular level in Enterobacteriaceae. Our study indicates that PhoPQEcl signal transduction contributes to heteroresistance in ECC. However, additional studies are necessary to understand if there is a genetic determinant within the heteroresistant subpopulation that promotes resistance. Alternatively, colistin heteroresistance could be a byproduct of promoter noise-induced bimodality, which has been linked to heteroresistance in other bacteria (49, 50). Despite these two possibilities, our studies demonstrated that PhoPQEcl phosphotransfer (PhoQH227 and PhoPD56) is required for colistin heteroresistance (Table 1), suggesting the phenotype is regulated by PhoPQEcl.
Experimental Procedures
Bacterial Strains and Growth
E. cloacae subsp. cloacae ATCC 13047 and ECC strains were initially grown from freezer stocks on Luria-Bertani (LB) agar. Isolated colonies were used to inoculate LB broth or N minimal medium (0.1M Bis-Tris, pH 7.5 or 5.8, 5 mM KCl, 7.5 mM (NH4)2SO4, 0.5 M K2SO4, 1 mM KH2PO4, 0.10% casamino acids 0.2% glucose, 0.0002% thiamine, 15 μM FeSO4, 10 μM or 10 mM MgSO4) at 37° C. Strains were grown into mid-logarithmic growth (OD600 = 0.6) before analysis. Kanamycin was used at 25 μg/ml for selection and colistin was used at 5 μg/ml or 10 μg/ml where indicated.
All strains and plasmids used in this study are listed in Table S1. Briefly, E. cloacae subsp. cloacae 13047 mutant strains were constructed as previously described using recombineering with the plasmid pKOBEG (51). Linear PCR products were introduced in to the E. cloacae ATCC 13047/pKOBEG strain by electroporation and plated on selective media. Selected clones were transformed with pCP20 to cure the antibiotic resistance cassette.
To complement E. cloacae mutants, the coding sequence from phoPQEcl was cloned into the SalI and KpnI sites in pMMBKn (3). To generate point mutants in PhoQH277A and PhoQD56A, site directed mutagenesis was performed using Pfu Turbo using primers that incorporated the associated alanine-encoded nucleotide replacements. All constructs were validated using Sanger sequencing. IPTG inducible constructs were transformed into the phoPQ mutant and grown in 2.0 mM IPTG to induce expression.
Broth Microdilution assays
MICs of colistin were determined in triplicate by the broth microdilution (BMD) method. Briefly strains were inoculated from overnight cultures when OD600 = 0.1. Various concentrations (0 – 256 μg/ml) of colistin were added to each well and cultures were incubated overnight. Growth was measured by reading the OD600. The lowest concentration at which growth was inhibited was recorded as the MIC. E. coli W3110 and WD101 were used as control strains. In some cases, ‘skip wells’ were observed suggesting a heteroresistance phenomenon and the MIC was determined disregarding the clear wells (34).
Population Analysis
Population analysis profiling was performed by plating 1 X 1010 CFU onto LB agar containing 1 to 64 μg/ml colistin (in 2-fold increments). Plates were incubated overnight at 37° C and frequency of the subpopulation was determined by dividing by the total number of cells (43).
Isolation of Lipid A
Isolation of lipid A for TLC analysis involved 32P-radiolabeling of whole cells was performed as previously described with slight modifications (52). In brief, 12.5 ml of E. cloacae was grown at 37° C to OD600 = 1.0. Bacteria were harvested by centrifugation at 10,000 X g for 10 min. Lipid A extraction was carried out by mild-acid hydrolysis as previously described (53).
Mass Spectrometry
MS1 spectra of lipid A in Figure S7 were collected on a MALDI-TOF/TOF (Axima Performance, Shimadzu) mass spectrometer in the negative mode. All other spectra were collected in the negative mode on a Thermo Scientific Orbitrap Fusion Lumos mass spectrometer (San Jose, CA, USA) modified with a Coherent ExciStar XS ArF excimer laser (Santa Clara, CA), as previously described (54). HCD was performed with the normalized collision energy (NCE) of 25%. UVPD was performed with the laser emitting 193 nm photons at 5 mJ per laser pulse with 5 pulses per scan. The laser pulse repetition rate was 500 Hz. The instrument was operated at 120000 resolving power with a precursor isolation window of 3 m/z. All samples were dissolved in 50:50 MeOH:CHCl3 and directly infused into the mass spectrometer via a static nano-electrospray ionization source. The presented spectra are an average of 50 scans.
TLR-4 Signaling Assays
HEK-Blue hTLR4, cell line was maintained according to the manufacturer specifications (Invivogen). Overnight bacterial cultures in stationary phase were serial diluted for assays as previously described (2, 3). At least two biological replicates were each done in triplicate and one representative set was shown.
Colony Forming Unit Counts and Colistin Survival Assays
For colony forming unit counts (CFUs), E. cloacae subsp. cloacae 13047 and mutant strains were initially grown from freezer stocks on Luria-Bertani (LB) agar. Isolated colonies were resuspended and used to inoculate LB broth with 10 μg/ml or without colistin at an OD600 = 0.01. Cells were plated at designated time points on LB agar. Plates were grown overnight at 37° C and colony forming units (CFU) were counted and reported.
Colistin survival assays were performed as previously described with slight modifications (25). Briefly, wild type and mutant E. cloacae strains were grown overnight on LB agar. The following day, N minimal media pH = 7.5 containing either 10 µM MgSO4 (low Mg2+) or 10 mM MgSO4 (high Mg2+) were inoculated at OD600 = 0.05 with bacteria from overnight cultures after cells were washed with N minimal media without Mg2+. Cultures were grown until OD600 = 0.6, when they were split and treated with 0, 5 or 10 µg/ml of colistin (Polymyxin E). Cultures were incubated for 1 h at 37° C and then colony-forming units were plated on LB, grown, and calculated. Percent survival was calculated by dividing the number of bacteria after treatment with colistin relative to those incubated in the absence of colistin and then multiplied by 100.
Protein Purification
To purify the PhoPEcl protein, the coding sequence was cloned into pT7–7Kn, as previously described (55). Briefly, the phoPEcl coding sequence was amplified from E. cloacae cDNA with primers that added a C-terminal His8X tag. From an overnight starter culture, 1 Liter of LB broth containing 25 µg/ml of kanamycin was inoculated at 1:50 and grown at 37° C until the OD600 = 0.5. IPTG was added to a final concentration of 1mM, and the culture was incubated at 37° C for an additional 4 h. Bacteria were recovered by centrifugation at 10,000 x g for 10 min, and the bacteria were resuspended in lysis buffer. Bacteria were lysed using sonication and the soluble fraction was recovered by centrifugation at 10,000 x g for 30 min. PhoPEcl-His8X was purified on a Ni-nitrilotriacetic acid (NTA) beads according to the manufactures instructions (Qiagen).
Electrophoretic Mobility Shift Assay
PhoPEcl-His8X proteins were purified as described above. EMSAs were performed based on a modified protocol (56). 250-bp DNA fragments of phoPEcl and arnBEcl spanning −230 to +20 relative to the translational start site were amplified from E. cloacae or E. coli cDNA using 5’-biotinylated primers. PhoPEcl-His8X proteins were incubated with biotinylated DNA at 25° C for 20 min. For competition experiments, unlabeled E. cloacae ParnB and poly(dI-dC) were added at 1:1, 2:1, or 5:1 ratios relative to biotin-labeled ParnB DNA. 0.1 – 10 μM of PhoPEcl-His8X proteins were used. After electrophoresis at 4° C, protein/DNA was transferred onto a positively charged nylon membrane. Blots were blocked in 5% milk in TBS for 20 min and streptavidin conjugated HRP was used at a 1:300 dilution.
Nucleic Acid Extraction
Total RNA was extracted using the Direct-Zol RNA MiniPrep Kit (Zymo Research) from E. cloacae grown to a final OD600 = 0.6. Isolated RNA was treated with DNA-free DNA removal kit (Thermo-Fisher Scientific) to eliminate genomic DNA contamination. DNase-depleted RNA was used for qRT-PCR and RNA-seq.
RNA-sequencing
RNA-sequencing was performed as previously described (57). Briefly, DNA-depleted RNA was processed for Illumina sequencing using the NEB Next Ultra Directional RNA Library Prep kit for Illumina as described by the manufacturer (NEB). Sequencing was performed using Illumina HiSeq. Sequencing data was aligned to the E. cloacae subs. cloacae ATCC 13047 published genome annotations (47) using CLC genomic workbench software (Qiagen) and RPKM expression values were determined. The weighted proportions fold change of expression values between samples was determined and a Baggerley’s test on proportions was used to generate a false discovery rate corrected P-value. We then used a cut-off of 2-fold weighted proportions absolute change with a false-discovery rate corrected P-value of ≤ 0.05 to identify significantly differentially regulated genes between samples. The sequencing data for the clinical isolates has been deposited in the Nation Center for Biotechnology’s Gene Expression Omnibus (GSE127802).
Genomic-sequencing
Genomic sequences were analyzed as previously done (3). Briefly, samples were processed for Illumina sequencing using the NEB Next Ultra DNA Library Prep kit (NEB). Sequencing was performed using Illumina HiSeq. Reads were aligned to E. cloacae strain ATCC 13047 published genome annotations using CLC genomic workbench software (Qiagen) with 90% length fraction and 90% similarities parameters. Mapped reads were locally realigned and fixed ploidy detection identified low and high frequency variants. E. cloacae variant tracks were compared to identify mutations. Mutations not present in strain ATCC 13047 were called if 95% of aligned reads contained the variant. The sequence data have been submitted to the GenBank under accession number SUB4176618.
Supplementary Material
Acknowledgements
This work was supported by NIH GM103655 (J.S.B), and Welch Foundation F-1155 (J.S.B.). Funding from the UT System for support of the UT System Proteomics Core Facility Network is gratefully acknowledged.
We would like to thank Cara Boutte and Mark Pellegrino for thoughtful review of the manuscript.
References
- 1.Whitfield C, Trent MS. (2014). Biosynthesis and export of bacterial lipopolysaccharides. Annu Rev Biochem 83:99–128. [DOI] [PubMed] [Google Scholar]
- 2.Needham BD, Carroll SM, Giles DK, Georgiou G, Whiteley M, Trent MS. (2013). Modulating the innate immune response by combinatorial engineering of endotoxin. Proc Natl Acad Sci U S A 110:1464–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boll JM, Crofts AA, Peters K, Cattoir V, Vollmer W, Davies BW, Trent MS. (2016). A penicillin-binding protein inhibits selection of colistin-resistant, lipooligosaccharide-deficient Acinetobacter baumannii. Proc Natl Acad Sci U S A 113(41):E6228–E6237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boll JM, Tucker AT, Klein DR, Beltran AM, Brodbelt JS, Davies BW, Trent MS. (2015). Reinforcing Lipid A Acylation on the Cell Surface of Acinetobacter baumannii Promotes Cationic Antimicrobial Peptide Resistance and Desiccation Survival. mBio 6:e00478–00415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Raetz CRH, Reynolds CM, Trent MS, Bishop RE. (2007). Lipid A modification systems in gram-negative bacteria. Annu Rev Biochem 76:295–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prost LR, Miller SI. (2008). The Salmonellae PhoQ sensor: mechanisms of detection of phagosome signals. Cell Microbiol 10:576–582. [DOI] [PubMed] [Google Scholar]
- 7.Gunn JS. (2008). The Salmonella PmrAB regulon: lipopolysaccharide modifications, antimicrobial peptide resistance and more. Trends Microbiol 16:284–290. [DOI] [PubMed] [Google Scholar]
- 8.GarcíaVéscovi E, Soncini FC, Groisman EA. (1996). Mg2+ as an extracellular signal: environmental regulation of Salmonella virulence. Cell 84:165–174. [DOI] [PubMed] [Google Scholar]
- 9.Bader MW, Sanowar S, Daley ME, Schneider AR, Cho U, Xu W, Klevit RE, Le Moual H, Miller SI. (2005). Recognition of antimicrobial peptides by a bacterial sensor kinase. Cell 122:461–472. [DOI] [PubMed] [Google Scholar]
- 10.Gunn JS, Richards SM. (2007). Recognition and integration of multiple environmental signals by the bacterial sensor kinase PhoQ. Cell Host Microbe 1:163–165. [DOI] [PubMed] [Google Scholar]
- 11.Needham BD, Trent MS. (2013). Fortifying the barrier: the impact of lipid A remodelling on bacterial pathogenesis. Nat Rev Microbiol 11:467–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herrera CM, Hankins JV, Trent MS. (2010). Activation of PmrA inhibits LpxT-dependent phosphorylation of lipid A promoting resistance to antimicrobial peptides. Mol Microbiol 76:1444–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wösten MM, Kox LF, Chamnongpol S, Soncini FC, Groisman EA. (2000). A signal transduction system that responds to extracellular iron. Cell 103:113–125. [DOI] [PubMed] [Google Scholar]
- 14.Perez JC, Groisman EA. 2007. Acid pH activation of the PmrA/PmrB two-component regulatory system of Salmonella enterica. Mol Microbiol 63:283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trent MS, Ribeiro AA, Lin S, Cotter RJ, Raetz CR. (2001). An inner membrane enzyme in Salmonella and Escherichia coli that transfers 4-amino-4-deoxy-L-arabinose to lipid A: induction on polymyxin-resistant mutants and role of a novel lipid-linked donor. J Biol Chem 276:43122–43131. [DOI] [PubMed] [Google Scholar]
- 16.Gunn JS, Lim KB, Krueger J, Kim K, Guo L, Hackett M, Miller SI. (1998). PmrA-PmrB-regulated genes necessary for 4-aminoarabinose lipid A modification and polymyxin resistance. Mol Microbiol 27:1171–1182. [DOI] [PubMed] [Google Scholar]
- 17.Zhou Z, Ribeiro AA, Lin S, Cotter RJ, Miller SI, Raetz CR. (2001). Lipid A modifications in polymyxin-resistant Salmonella typhimurium: PMRA-dependent 4-amino-4-deoxy-L-arabinose, and phosphoethanolamine incorporation. J Biol Chem 276:43111–43121. [DOI] [PubMed] [Google Scholar]
- 18.Lee H, Hsu F-F, Turk J, Groisman EA. (2004). The PmrA-regulated pmrC gene mediates phosphoethanolamine modification of lipid A and polymyxin resistance in Salmonella enterica. J Bacteriol 186:4124–4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bader MW, Sanowar S, Daley ME, Schneider AR, Cho U, Xu W, Klevit RE, Le Moual H, Miller SI. (2005). Recognition of antimicrobial peptides by a bacterial sensor kinase. Cell 122:461–472. [DOI] [PubMed] [Google Scholar]
- 20.Miller SI, Kukral AM, Mekalanos JJ. (1989). A two-component regulatory system (phoP phoQ) controls Salmonella typhimurium virulence. Proc Natl Acad Sci U S A 86:5054–5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Richards SM, Strandberg KL, Conroy M, Gunn JS. (2012). Cationic antimicrobial peptides serve as activation signals for the Salmonella Typhimurium PhoPQ and PmrAB regulons in vitro and in vivo. Front Cell Infect Microbiol 2:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kawasaki K, Ernst RK, Miller SI. (2005). Inhibition of Salmonella enterica serovar Typhimurium lipopolysaccharide deacylation by aminoarabinose membrane modification. J Bacteriol 187:2448–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mitrophanov AY, Jewett MW, Hadley TJ, Groisman EA. (2008). Evolution and dynamics of regulatory architectures controlling polymyxin B resistance in enteric bacteria. PLoS Genet 4:e1000233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Winfield MD, Latifi T, Groisman EA. (2005). Transcriptional regulation of the 4-amino-4-deoxy-L-arabinose biosynthetic genes in Yersinia pestis. J Biol Chem 280:14765–14772. [DOI] [PubMed] [Google Scholar]
- 25.Rubin EJ, Herrera CM, Crofts AA, Trent MS. (2015). PmrD is required for modifications to Escherichia coli endotoxin that promote antimicrobial resistance. Antimicrob Agents Chemother 59:2051–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gunn JS, Miller SI. (1996). PhoP-PhoQ activates transcription of pmrAB, encoding a two-component regulatory system involved in Salmonella typhimurium antimicrobial peptide resistance. J Bacteriol 178:6857–6864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soncini FC, Groisman EA. (1996). Two-component regulatory systems can interact to process multiple environmental signals. J Bacteriol 178:6796–6801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kato A, Groisman EA. (2004). Connecting two-component regulatory systems by a protein that protects a response regulator from dephosphorylation by its cognate sensor. Genes Dev 18:2302–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luo S-C, Lou Y-C, Rajasekaran M, Chang Y-W, Hsiao C-D, Chen C. (2013). Structural basis of a physical blockage mechanism for the interaction of response regulator PmrA with connector protein PmrD from Klebsiella pneumoniae. J Biol Chem 288:25551–25561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kox LF, Wösten MM, Groisman EA. (2000). A small protein that mediates the activation of a two-component system by another two-component system. EMBO. J 19:1861–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morand PC, Billoet A, Rottman M, Sivadon-Tardy V, Eyrolle L, Jeanne L, Tazi A, Anract P, Courpied J-P, Poyart C, Dumaine V. (2009). Specific distribution within the Enterobacter cloacae complex of strains isolated from infected orthopedic implants. J Clin Microbiol 47:2489–2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sanders WE, Sanders CC. (1997). Enterobacter spp.: pathogens poised to flourish at the turn of the century. Clin Microbiol Rev 10:220–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.John JF, Sharbaugh RJ, Bannister ER. (1982). Enterobacter cloacae: bacteremia, epidemiology, and antibiotic resistance. Rev Infect Dis 4:13–28. [DOI] [PubMed] [Google Scholar]
- 34.Guérin F, Isnard C, Sinel C, Morand P, Dhalluin A, Cattoir V, Giard J-C. (2016). Cluster-dependent colistin hetero-resistance in Enterobacter cloacae complex. J Antimicrob Chemother 71:3058–3061. [DOI] [PubMed] [Google Scholar]
- 35.Mezzatesta ML, Gona F, Stefani S. (2012). Enterobacter cloacae complex: clinical impact and emerging antibiotic resistance. Future Microbiol 7:887–902. [DOI] [PubMed] [Google Scholar]
- 36.Davin-Regli A, Pagès J-M. (2015). Enterobacter aerogenes and Enterobacter cloacae; versatile bacterial pathogens confronting antibiotic treatment. Front Microbiol 6:392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carlet J, Mainardi J-L. (2012). Antibacterial agents: back to the future? Can we live with only colistin, co-trimoxazole and fosfomycin? Clin Microbiol Infect Off Publ Eur Soc Clin Microbiol Infect Dis 18:1–3. [DOI] [PubMed] [Google Scholar]
- 38.Nation RL, Li J. (2009). Colistin in the 21st century. Curr Opin Infect Dis 22:535–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Band VI, Crispell EK, Napier BA, Herrera CM, Tharp GK, Vavikolanu K, Pohl J, Read TD, Bosinger SE, Trent MS, Burd EM, Weiss DS. (2016). Antibiotic failure mediated by a resistant subpopulation in Enterobacter cloacae. Nat Microbiol 1:16053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Napier BA, Band V, Burd EM, Weiss DS. (2014). Colistin Heteroresistance in Enterobacter cloacae Is Associated with Cross-Resistance to the Host Antimicrobial Lysozyme. Antimicrob Agents Chemother 58:5594–5597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuper KM, Boles DM, Mohr JF, Wanger A. (2009). Antimicrobial susceptibility testing: a primer for clinicians. Pharmacotherapy 29:1326–1343. [DOI] [PubMed] [Google Scholar]
- 42.Reis AO, Luz DAM, Tognim MCB, Sader HS, Gales AC. (2003). Polymyxin-resistant Acinetobacter spp. isolates: what is next? Emerg Infect Dis 9:1025–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.El-Halfawy OM, Valvano MA. (2015). Antimicrobial heteroresistance: an emerging field in need of clarity. Clin Microbiol Rev 28:191–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park BS, Song DH, Kim HM, Choi B-S, Lee H, Lee J-O. (2009). The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 458:1191–1195. [DOI] [PubMed] [Google Scholar]
- 45.Kawai T, Akira S. (2005). Pathogen recognition with Toll-like receptors. Curr Opin Immunol 17:338–344. [DOI] [PubMed] [Google Scholar]
- 46.Soncini FC, Véscovi EG, Groisman EA. (1995). Transcriptional autoregulation of the Salmonella typhimurium phoPQ operon. J Bacteriol 177:4364–4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ren Y, Ren Y, Zhou Z, Guo X, Li Y, Feng L, Wang L. (2010). Complete genome sequence of Enterobacter cloacae subsp. cloacae type strain ATCC 13047. J Bacteriol 192:2463–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jayol A, Nordmann P, Brink A, Poirel L. (2015). Heteroresistance to colistin in Klebsiella pneumoniae associated with alterations in the PhoPQ regulatory system. Antimicrob Agents Chemother 59:2780–2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hung K-H, Wang M-C, Huang A-H, Yan J-J, Wu J-J. (2012). Heteroresistance to cephalosporins and penicillins in Acinetobacter baumannii. J Clin Microbiol 50:721–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van Merode AEJ, van der Mei HC, Busscher HJ, Krom BP. (2006). Influence of culture heterogeneity in cell surface charge on adhesion and biofilm formation by Enterococcus faecalis. J Bacteriol 188:2421–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guérin F, Isnard C, Cattoir V, Giard JC. (2015). Complex Regulation Pathways of AmpC-Mediated β-Lactam Resistance in Enterobacter cloacae Complex. Antimicrob Agents Chemother 59:7753–7761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hankins JV, Madsen JA, Giles DK, Childers BM, Klose KE, Brodbelt JS, Trent MS. (2011). Elucidation of a novel Vibrio cholerae lipid A secondary hydroxy-acyltransferase and its role in innate immune recognition. Mol Microbiol 81:1313–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou Z, Lin S, Cotter RJ, Raetz CR. (1999). Lipid A modifications characteristic of Salmonella typhimurium are induced by NH4VO3 in Escherichia coli K12. Detection of 4-amino-4-deoxy-L-arabinose, phosphoethanolamine and palmitate. J Biol Chem 274:18503–18514. [DOI] [PubMed] [Google Scholar]
- 54.Klein DR, Holden DD, Brodbelt JS. (2016). Shotgun Analysis of Rough-Type Lipopolysaccharides Using Ultraviolet Photodissociation Mass Spectrometry. Anal Chem 88:1044–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boll JM, Hendrixson DR. (2013). A regulatory checkpoint during flagellar biogenesis in Campylobacter jejuni initiates signal transduction to activate transcription of flagellar genes. mBio 4:e00432–00413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boll JM, Hendrixson DR. (2011). A specificity determinant for phosphorylation in a response regulator prevents in vivo cross-talk and modification by acetyl phosphate. Proc Natl Acad Sci U S A 108:20160–20165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Davies BW, Bogard RW, Young TS, Mekalanos JJ. 2012. Coordinated regulation of accessory genetic elements produces cyclic di-nucleotides for V. cholerae virulence. Cell 149:358–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.