

SUMMARY:
The accurate completion of DNA replication on the chromosome requires RecBCD and structure specific SbcCD ExoI nucleases. However, the substrates and mechanism by which this reaction occurs remains unknown. Here we show that these completion enzymes operate on plasmid substrates containing two replisomes, but are not required for plasmids containing one replisome. Completion on the two-replisome plasmids requires RecBCD, but does not require RecA and no broken intermediates accumulate in its absence, indicating that the completion reaction occurs normally in the absence of any double strand breaks. Further, similar to the chromosome, we show that when the normal completion reaction is prevented, an aberrant RecA-mediated recombination process leads to amplifications that drives most of the instabilities associated with the two-replisome substrates. The observations imply that the substrate SbcCD, ExoI, and RecBCD act upon in vivo is created specifically by two convergent replisomes, and demonstrate that the function of RecBCD in completing replication is independent of double strand break repair, and likely arises from a failure to join the strands of the convergent replication forks.
Keywords: Plasmid, DNA replication, RecBCD, SbcCD, ExoI, Completion of DNA replication
The accurate completion of DNA replication on the chromosome requires RecBCD and structure specific SbcCD ExoI nucleases. Here we show that the substrate(s) SbcCD, ExoI, and RecBCD act upon in vivo is created specifically by two convergent replisomes, and demonstrate that the function of RecBCD in completing replication is independent of double strand break repair, and likely arises from a failure to join the strands of the convergent replication forks.
INTRODUCTION
Cells tightly regulate DNA replication initiation, elongation, and completion to ensure that each daughter cell inherits an identical copy of the genetic information. While the mechanisms regulating initiation and elongation have been well characterized (reviewed in (Costa et al., 2013)), the process of how cells recognize replicated regions and complete replication at the precise point where all sequences have doubled has until recently, remained unknown. To complete replication accurately, cells must encode an enzymatic system that is capable of recognizing or counting in pairs, and joins the strands of converging replication forks at the point where all sequences have precisely doubled. The failure to complete a single replication event would be expected to result in a loss of genomic stability, mutation, or cell lethality. Yet, this reaction occurs thousands of times per generation along the chromosomes of human cells, and therefore must occur with remarkable efficiency. Given this critical role, and considering the large number of proteins that cells devote to ensure fidelity during replication initiation and elongation, it is not surprising that this final step is also tightly regulated and controlled enzymatically (Wendel et al., 2014; Courcelle et al., 2015; Wendel et al., 2018; et al., 2013; Dimude et al., 2015; Midgley-Smith et al., 2018; Dimude et al., 2018b; Dimude et al., 2018a).
A number of studies demonstrate that an ability to sense when all sequences in the genome have doubled is critical to genomic replication. In vitro, converging replisomes continue through their meeting point as one replisome displaces the other, resulting in over-replication, or a third copy, of the region where the forks meet (Hiasa and Marians, 1994). Over-replication also occurs in vivo and is prominently observed on the chromosome of cells lacking the helicases and exonucleases required to disrupt and degrade these events (Wendel et al., 2014; Wendel et al., 2018; Asai et al., 1994; de Massy et al., 1984b; de Massy et al., 1984a; Rudolph et al., 2009b; et al., 2013; Dimude et al., 2015; Dimude et al., 2016; Midgley-Smith et al., 2018; Dimude et al., 2018a). Other studies suggest that illegitimate initiations of replication associated with repair events or transcriptional processes occur frequently at single strand nicks, gaps, D-loops, and R-loops throughout the genomes of both prokaryotes and eukaryotes (de Massy et al., 1984a; Magee et al., 1992; Asai and Kogoma, 1994; Bhatia et al., 2014; Hamperl and Cimprich, 2014; Donnianni and Symington, 2013; Brochu et al., 2018). Each of these events would generate a third copy of the chromosomal region where the replication initiates. Thus, over-replication appears to be an inherent and promiscuous problem during the duplication of genomes. These considerations make it clear that enzymes catalyzing the accurate and efficient completion of replication is essential to genomic stability and are likely required for cellular life overall.
The completion step of DNA replication has been challenging to study in eukaryotic cells, in part because multiple origins are utilized with varying efficiencies and timing, making the regions where forks meet highly variable between cells and cell cycles (Heichinger et al., 2006; Wu and Nurse, 2009). By comparison, Escherichia coli is uniquely suited to dissect this fundamental aspect of cellular metabolism since the event can be localized to a single ~400 kb region of the chromosome, opposite to its bidirectional origin of replication (reviewed in (Hill, 1992)). This region is flanked by ter sequences which bind the protein Tus, blocking replication forks in an orientation specific manner (Kobayashi et al., 1989). Although ter ensures that completion occur within this region, it is not involved in the reaction, as chromosomes lacking ter replicate normally and are stably maintained (Duggin et al., 2008; Duggin and Bell, 2009; Roecklein et al., 1991).
Current models of completion in E.coli suggest that converging replisomes transiently bypass each other at the point where they converge, creating an over-replicated region that contains three copies of the genetic information (Figure 1). The completion reaction is thought to initiate through the action of SbcCD and ExoI structure specific nucleases which act on a structural intermediate created at the point where forks converge (Dimude et al., 2018a; Wendel et al., 2018). In the absence of the SbcCD and ExoI nucleases, the over-replicated region persists, leading to genomic instabilities and amplifications at these loci (Wendel et al., 2014; Wendel et al., 2018; et al., 2013; Dimude et al., 2018a). Following incisions by these enzymes, the RecBCD helicase-nuclease complex processes the over-replicated intermediate and is required to catalyze or recruit enzymes that promote joining of the convergent strands. In vitro, RecB and RecC interact with RecD to form a dual helicase–nuclease complex that unwinds and degrades double-strand DNA ends (Taylor and Smith, 2003; Taylor and Smith, 1985; Amundsen et al., 1986). Loss of RecB or -C inactivates the enzyme complex, whereas loss of RecD inactivates the nuclease but retains helicase activity and remains recombination proficient (Taylor and Smith, 2003; Taylor and Smith, 1985; Amundsen et al., 1986). On the chromosome, in the absence of RecB or C, the nascent ends of convergent replication forks are not joined, leading to excessive degradation and rendering cells unable to maintain the chromosome region where forks converge (Wendel et al., 2014; Wendel et al., 2018; et al., 2013). The inability to complete replication or maintain these regions of the genome severely compromises the viability and growth of recBCD cultures (Wendel et al., 2014; Courcelle et al., 2015; Wendel et al., 2018). In the absence of recD, degradation of the excess sequence is impaired, however joining appears to occur normally and viability is not compromised (Wendel et al., 2014; Courcelle et al., 2015; Wendel et al., 2018).
Figure 1. Current model for completing DNA replication.
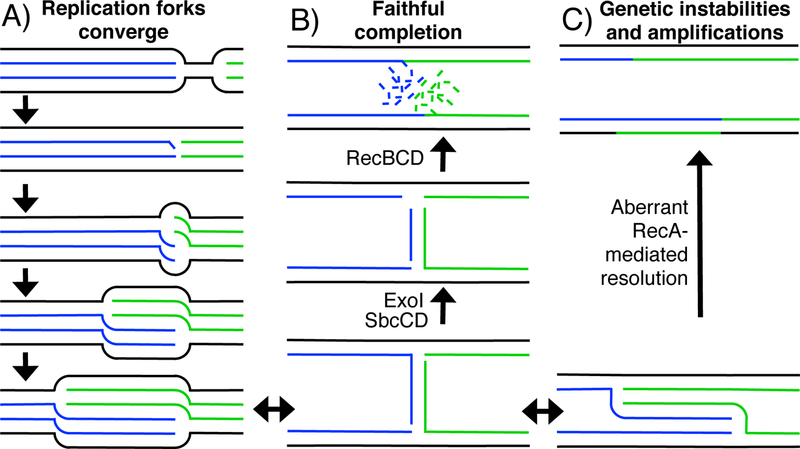
A) Convergent replication forks continue past their meeting point, creating a partially over-replicated substrate that contains three copies of the genetic information. B) SbcCD-ExoI recognize and cleave this branched over-replicated substrate which creates a DNA that can be resect and processed by RecBCD. Following resection, RecBCD promotes or recruit enzymes that join the convergent strands at the doubling point. C) When processing by SbcCD-ExoI is prevented or impaired, the over-replicated region persists, and an aberrant form of recombination then resolves the over-replicated region, leading to amplifications and genomic instabilities.
The completion reaction can occur normally in the absence of RecA or recombination (Wendel et al., 2014; Courcelle et al., 2015; Wendel et al., 2018). However, when completion is impaired or prevented from processing the over-replicated intermediates, viability and growth become dependent on an aberrant form of RecA-mediated recombination that leads to genetic instabilities and amplifications of these loci (Wendel et al., 2018). Similar instabilities and amplifications are observed in eukaryotes lacking the homologs Mre11-Rad50 and Sae2, suggesting that the completion reaction is one that is conserved throughout evolutionarily diverged organisms (Deng et al., 2015; Bruhn et al., 2014; Lengsfeld et al., 2007).
Here, we used plasmid mini-chromosomes to further characterize the completion reaction. We show that SbcCD ExoI and RecBCD are required to propagate substrates containing two replisomes but not one replisome, suggesting that the substrates acted upon by these enzymes is specific to a structure created when two replisomes converge. We further show that similar to completion sites on the chromosome, genetic instability on plasmids containing two replisomes are driven by amplifications associated with an aberrant, RecA-mediated recombinational reaction.
RESULTS
Construction and characterization of plasmids replicated by two replisomes.
To further characterize the completion of replication reaction, we utilized a plasmid containing a bidirectional origin of replication that would allow us to characterize the genes and sequences associated with maintaining a minichromosome that contains convergent replication forks (Fig 2A). Plasmid pCL01 contains a lambda origin of replication, which loads dual helicases and utilizes the host’s replication proteins (reviewed in (Stillman, 1994)). For the purposes of comparison and control, we compared the replication and stability of these two-replisome plasmids to pBR322, a well-characterized plasmid that maintains a moderate copy number and also utilizes the host’s replication machinery, but has a unidirectional ColE1 origin of replication and propagates using a single replisome (Martin-Parras et al., 1991).
Figure 2. Plasmids replicated by two-replisomes are less stable than those replicated by one replisome.
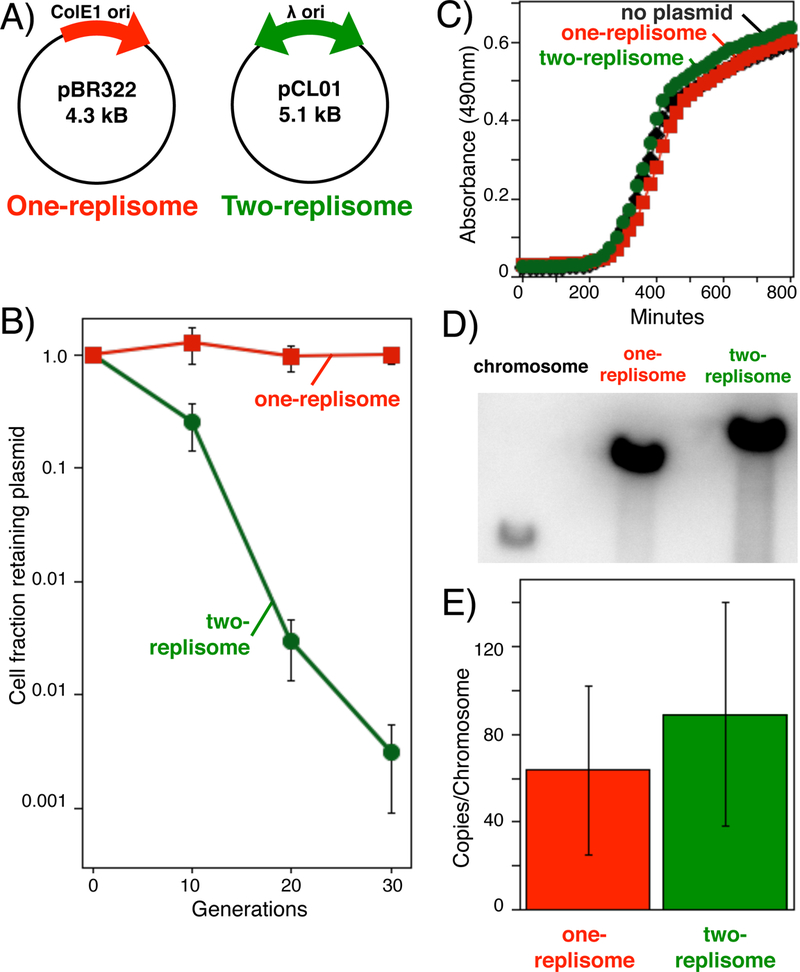
A) Diagram of the one-replisome and two replisome plasmids, containing a ColE1 and lambda origin or replication, respectively. B) In the absence of selection, the two-replisome plasmid is lost more rapidly than the one-replisome plasmid. Cultures containing the one-replisome (pBR322) or two-replisome (pCL01) plasmid were grown without selection. Serial dilutions of the culture were then plated with and without ampicillin selection to determine the fraction of cells that retained the plasmid over time. Error bars represent the standard error of 4 or more independent experiments. C) The instability of the two-replisome plasmid, relative to the one-replisome plasmid is not due a reduced growth rate of cells containing the two-replisome plasmid. The absorbance at 630 nm of cultures containing no plasmid, pBR322, or pCL01 grown at 37 °C is plotted over time. D) The instability of the two-replisome plasmid, relative to the one-replisome plasmid is not due to a reduced copy number during growth. A representative Southern analysis of the ampicillin resistance gene on the chromosome, on the plasmid pBR322, and on the plasmid pCL01 is shown. Total genomic DNA was purified from HL946 (Courcelle et al., 1999), containing a chromosomal copy of the ampicillin resistance gene, SR108 containing pBR322, and SR108 containing pCL01. Purified DNA was digested with EcoRV to linearize the plasmids and equal cell equivalents were then loaded and analyzed by Southern analysis using a P32-labeled ampicillin resistance gene as a probe. D) The copy number of each plasmid, relative to the chromosome is plotted. Error bars represent the standard error from five independent experiments. No plasmid (diamonds); pBR322 (squares); pCL01 (circles).
The two-replisome plasmid was stably propagated in the presence of selection. However, when grown without selection, the two-replisome plasmids was far less stable than the one-replisome plasmid (Figure 2B). Whereas the unidirectionally-replicating pBR322 was stably maintained without loss over 30 generations, only 0.1–1.0% of cells maintained the bidirectionally-replicating plasmids over the same duration.
The reduced stability of the two-replisome plasmid, relative to the one replisome plasmid, was not due to an inhibition on growth rate or a lower overall copy number. Cultures containing no plasmid, one-replisome plasmids, or two replisome plasmids all grew at similar rates (Fig 2C). To examine plasmid copy number, total genomic DNA was purified from cultures containing the ampicillin resistance gene integrated into the chromosome, as well as cultures containing the one-replisome and two-replisome plasmids which also contain this gene. The purified DNA was then digested with a restriction enzyme that linearizes the plasmid before DNA from equal cell equivalents was analyzed by southern analysis following agarose-gel electrophoresis using a 32P-labeled ampicillin resistance gene as a probe. As shown in Fig 2D, both the one-replisome and two-replisome plasmids were maintained at similar copy numbers and ranged between 50–80 copies per chromosome. These numbers are similar to those previously reported for pBR322 and plasmids containing a phage lambda origin (Atlung et al., 1999; Boros et al., 1984; Lupski et al., 1986; Boyd and Sherratt, 1995).
These initial observations indicate that the two-replisome plasmid, pCL01 can replicate and propagate in cells, but that it is less stable than the one-replisome plasmid, pBR322. However, the reduced stability is not due to an effect on the growth rate of cells containing the plasmid or a lower copy number, relative to pBR322. Further, neither plasmid contain partitioning genes or sequences that would account for the difference in stability.
Plasmids with two-replisomes, but not one-replisome, require enzymes needed to complete replication on the chromosome.
We next asked whether the genes required to complete replication on the chromosome were also needed to maintain the two-replisome plasmids. Completion of replication on the chromosome requires RecB and -C to resect and join the over-replicated region where forks converge. In their absence, joining does not occur and cells are unable to maintain these regions of their genome (Wendel et al., 2014; Courcelle et al., 2015; Wendel et al., 2018) (Fig S1). Similarly, we found that the ability of cells to propagate plasmids containing two-replisomes was severely impaired in the absence of RecBC. Transformation of the two-replisome plasmid was reduced by more than two orders to magnitude in recBC mutants (Figure 3). By comparison, the absence of RecD does not impair the cell’s ability to maintain regions where completion occurs, and transformation of the two replisome plasmid was not reduced in recD mutants. The impaired ability to transform recBC mutants was specific to the plasmid with two replisomes, as one replisome plasmids, pBR322 and pREP4, were transformed in recBC mutants at frequencies similar to wildtype cultures (Fig 3 & S2). The observation suggests that RecBC is specifically required to process a structure created when two replisomes converge.
Figure 3. Transformation of plasmids with two replisomes, but not one replisome, depends on the enzymes required to complete replication on the chromosome.
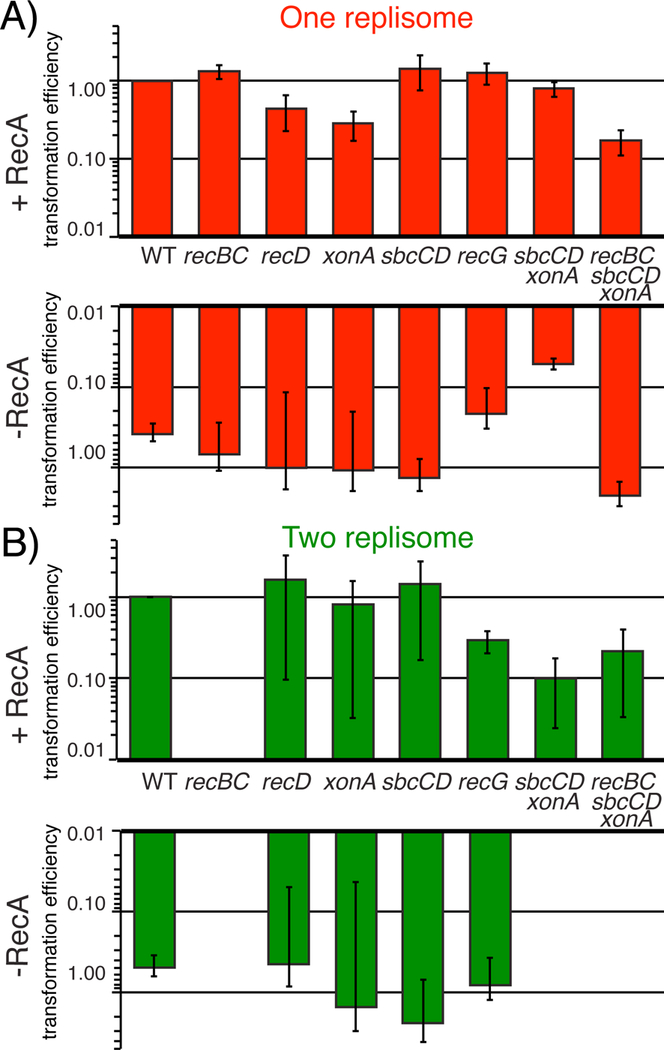
A) The transformation efficiency relative to wild type cells is shown for the strains indicated following electroporation of competent cells with 50ng of pBR322 (one-replisome). B) The transformation efficiency relative to wild type cells is shown for the strains indicated following electroporation of competent cells with 50ng of pCL01 (two-replisomes). Error bars represent the standard error of at least two independent experiments.
If the impaired ability of two-replisome plasmids to transform recBC mutants were due to defective repair of double strand breaks that arise on these plasmid substrates, then one would expect that mutants lacking RecA, which is essential for double strand break repair, to exhibit a similar deficiency. However, unlike recBC mutants, the two-replisome plasmid transformed recA mutants at frequencies comparable to wild type cells. Thus, similar to what is observed on the chromosome, the RecBC-mediated completion reaction on the plasmid does not require recombination or RecA, and is unlikely to be associated with a double strand break-intermediate.
On the chromosome, the completion reaction is initiated by the SbcCD and ExoI nucleases, encoded by sbcC, sbcD, and xonA (also called sbcB (Kushner et al., 1972)). Together, these enzymes are required to incise and/or resect the DNA structure created at sites where convergent replication forks meet (Wendel et al., 2018). When these nucleases are inactivated, the over-replicated region where replication forks converge persists, preventing replication from completing normally. Under these conditions, the ability to maintain these regions and continue to grow becomes dependent on an aberrant RecA-mediated recombinational process that results in genomic instabilities and amplifications at sites where forks converge (Wendel et al., 2018) (Fig S1). To determine if the two-replisome plasmids also rely upon these enzymes, we compared the ability of the two-replisome plasmid to transform sbcCD exoI mutants, both in the presence and absence of RecA. We found that strains lacking the SbcCD ExoI nucleases could be transformed by the two-replisome plasmid. However, in the absence of these gene products, transformation depended on the presence of RecA. The observations are consistent with the idea that, similar to the chromosome, when the normal mechanism of completion is impaired or inactivated, the reaction is shunted through a recombinational mechanism that depends on RecA (Wendel et al., 2018). Additionally, RecA-dependence of sbcCD xonA mutants was specific to plasmids containing two replisomes, as the single replisome, pBR322, could successfully transform sbcCD xonA mutants in the presence or absence of RecA (Fig 3A).
For comparison, we also examined mutants lacking RecG. On the chromosome, over-replicated regions persist in recG mutants, similar to sbcCD xonA mutants. However, the over-replication in recG mutants is thought to arise from a failure to disrupt illegitimate replication initiation events from R-and D-loops (Rudolph et al., 2009a; Rudolph et al., 2009b; Rudolph et al., 2010; et al., 2013; Lloyd and Rudolph, 2016; Dimude et al., 2016; Midgley-Smith et al., 2018), rather than a direct involvement in the completion reaction (Wendel et al., 2014). We found that the two-replisome plasmid transformed recG mutants both in the presence or absence of RecA, arguing that the RecA dependence does not extend to all mutants exhibiting an over-replication phenotype.
Similar to the chromosome, the effect of the sbcCD and xonA mutations were additive (Wendel et al., 2018), as the absence of either gene product alone did not prevent the ability to maintain the two-replisome plasmid in the absence of RecA. Additionally, inactivation of both SbcCD and ExoI restored the ability of recBC mutants to transform and maintain the bidirectional plasmid through a mechanism that depended upon RecA (Fig 3B), consistent with the idea that without initiation of the completion reaction by SbcCD ExoI, the downstream function of RecBCD becomes unnecessary, as the reaction is shunted to the aberrant RecA-mediated pathway.
These observations are all consistent with the mechanisms that complete replication on the chromosome and suggest that the two-replisome plasmid may serve as a useful model to dissect the intermediates involved in this reaction. Additionally, requirement for the completion enzymes on the two-replisome plasmid, but not the one-replisome plasmid argues that the substrate RecBCD, SbcCD, and ExoI act upon in vivo is specifically created when two replisomes converge.
The aberrant recombinational pathway is responsible for the instability of plasmids with two replisomes.
The results above show that transformation of two-replisome, but not one-replisome plasmids depends on the factors needed to complete chromosome replication and that plasmids containing two-replisomes exhibit instability relative to those containing one replisome, On the chromosome, when the normal mechanism of completion is impaired or prevented, the reaction occurs through an aberrant RecA-mediated pathway that is associated with genomic instabilities and amplifications in this region (Wendel et al., 2018).
To examine if recombination played a role in instability associated with the two-replisome plasmid, we examined the ability of recA mutants to maintain these plasmids in the absence of selection. We found that most of the instability on the two-replisome plasmid was due to RecA and was associated with the appearance of amplifications and aberrant plasmid species. Inactivation of RecA markedly increased the stability of the two-replisome plasmid (Fig 4A). The increase in stability or the recA mutant containing the two replisome plasmids approached that seen for the one-replisome plasmid, indicating that most of the instability on the two-replisome plasmid was driven by recombinational mechanisms. We next examined the form of these plasmids in cells. To this end, total genomic DNA was purified from cultures containing the plasmid and examined by southern analysis using a 32P-labelled plasmid as the probe. We observed that, relative to the one-replisome plasmid, the two-replisome plasmid contained a higher proportion of amplifications and aberrant multimeric species (Figure 4B&C). However, in the absence of recA, the proportion of aberrant multimeric plasmid species noticeably diminished and a higher proportion of the molecules were maintained as monomeric circles (Fig 4B&C).
Figure 4. Similar to completion on the chromosome, amplifications and instability on two-replisome plasmids are driven by an aberrant RecA-mediated form of recombination.
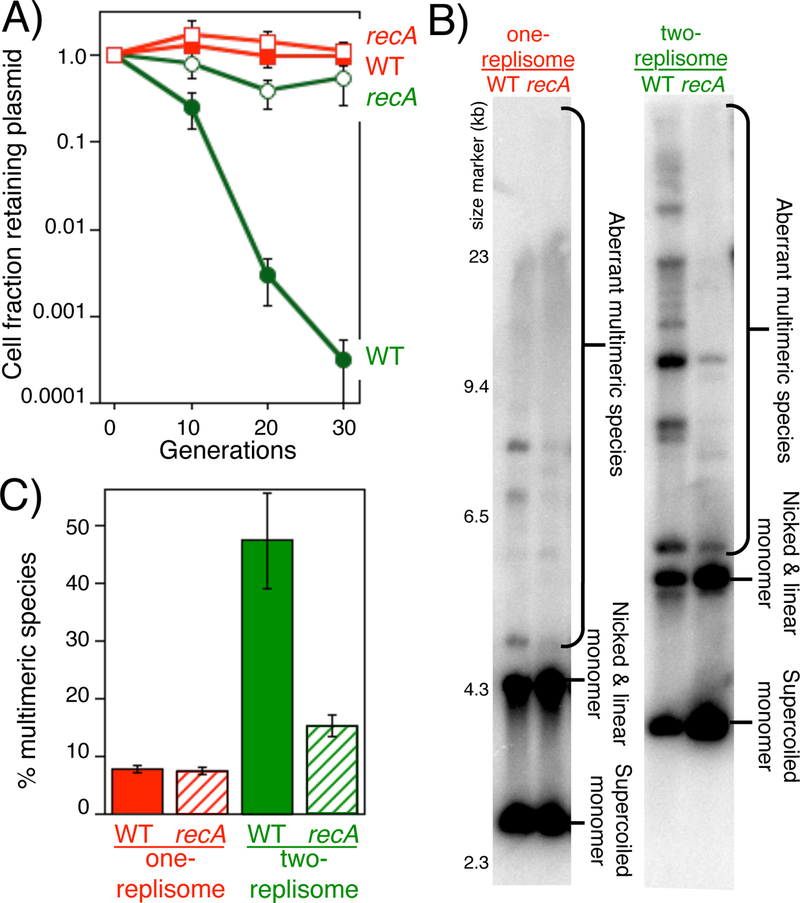
A) Inactivation of RecA restores the stability of the two-replisome plasmid to levels that approach that of the one-replisome plasmid. Cultures were treated as in Fig 2B. The fraction of cells retaining the one-replisome and two-replisome plasmid in wild type and recA mutants is plotted over time. Error bars represent the standard error of 4 independent experiments. WT pBR322 (filled squares); recA pBR322 (open squares); WT pCL01 (filled circles); recA pCL01 (open circles) B) Plasmid instability promoted by RecA correlates with an increased level of amplifications and aberrant multimeric species. Total genomic DNA from cells containing the one-replisome or two-replisome plasmid was purified and analyzed by southern analysis following agarose gel electrophoresis using P32-labeled pBR322 or pCL01 as a probe. (C) The fraction of unit-length monomeric plasmid is plotted for the one-replisome and two-replisome plasmid in the presence and absence of RecA. Error bars represent the standard error of four independent experiments.
We additionally examined several other mutants, including recD, recG and sbcCDxonA, to determine if the absence of other recombination mutants would increase the stability of the two-replisome plasmid similar to recA. None of the mutants examined improved the stability to the two-replisome plasmid relative to wild type cells (Fig5A). We were unable to examine recBC mutants due to their impaired ability to transform or stably maintain the two-replisome plasmid. We also examined the form of the plasmid in these mutants and found that the mutations also did not reduce the overall proportion of aberrant multimeric plasmid species that were observed (Fig 5B). In some preparations, a large fraction of the plasmid DNA in recD and the recBCsbcCDxonA strains migrate as high molecular weight multimers (Fig S4). This also occurs on one-replisome plasmids in these strains and reduces the stability of these plasmids overall (Wendel et al., 2014; Wendel et al., 2018; Seelke et al., 1987; Silberstein and Cohen, 1987; Niki et al., 1990). On the two-replisome plasmid, the high proportion of mutlimeric species did not appear to further reduce the plasmid loss rate beyond the relatively high rate already observed in wild type cells. However, we noted that in all strains where the plasmid were unstable, there was significant variation observed between the total and proportion of plasmid observed in strains during analysis (Fig S4).
Figure 5. Stability of the two-replisome plasmid in various mutants.
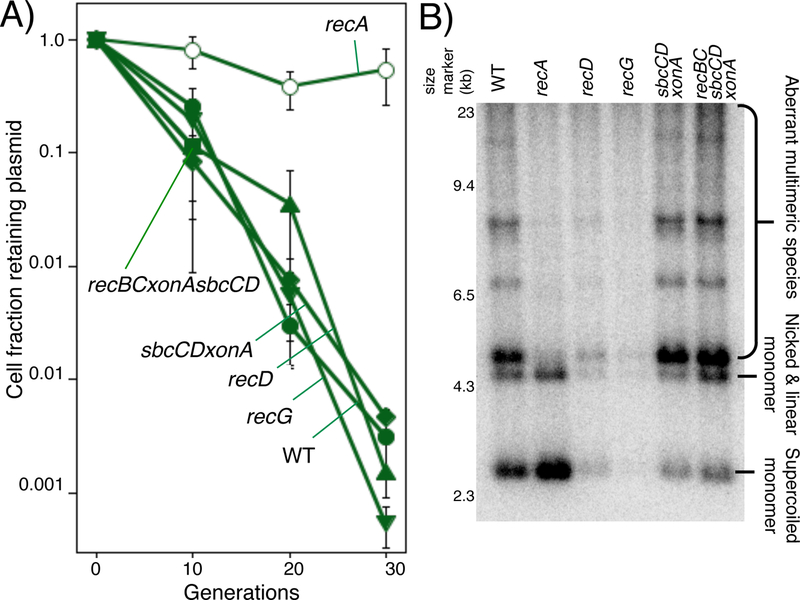
A) The fraction of cells retaining the two-replisome plasmid for each strain is plotted over time. Cultures were treated as in Fig 2B. WT (filled circles); recA (open circles); recD (filled triangles); recG (filled inverted triangles); sbcCDxonA (diamonds); recBCsbcCDxonA (filled squares); Error bars represent the standard error of at least three independent experiments. B) A representative Southern analysis of plasmid pCL01 grown in each strain is shown. DNA was purified and analyzed as in Fig 4B.
Taken together, Fig 4 and 5 argue that a RecA-mediated recombinational reaction is responsible for the amplifications and aberrant species on the two-replisome plasmid, and that these products are driving the instability on these plasmids, similar to that seen at sites where replication completes on the chromosome.
DISCUSSION
Here, we show that maintaining plasmids containing two replisomes depends on the enzymes needed to complete replication on the chromosome. On the chromosome, the completion of replication requires RecBCD to join the strands of convergent replication forks on the chromosome. In its absence, DNA ends persist, are extensively degraded, and cells fail to maintain these regions of the chromosome ( et al., 2013; Dimude et al., 2018a; Wendel et al., 2014; Courcelle et al., 2015; Wendel et al., 2018). Similarly, we show that transformation of two-replisome plasmids in recBC mutants is severely impaired and the plasmids fail to propagate in cells under selection. On the chromosome, the SbcCD ExoI structure-specific nucleases are required to initiate the faithful completion reaction. In the absence of these proteins, normal completion cannot occur, excess over-replicated regions persist, and the reaction is shunted through a RecA-mediated pathway that is associated with amplifications and genetic instabilities ( et al., 2013; Dimude et al., 2018a; Wendel et al., 2014; Courcelle et al., 2015; Wendel et al., 2018). Here, we show that transformation of two-replisome plasmids in sbcCD xonA mutants similarly depends on the RecA-mediated pathway. Thus, both the normal recBCD-mediated reaction, and the aberrant recombinational process appear to operate on the two-replisome plasmids. Additionally, we show that the requirement for these completion enzymes is specific to plasmids with two-replisomes, as transformation was not impaired in the one-replisome plasmid, pBR322. The observation implies that the substrate acted upon by SbcCD ExoI and RecBCD during the completion reaction is specific to a structure created when two replisomes converge.
Finally, we found that the two-replisome plasmid was less stable than the one replisome plasmid when propagated in the absence of selection, and showed that this instability was driven by amplifications arising from an RecA-mediated recombination reaction, similar to what is observed on the chromosome at sites where completion occurs (Wendel et al., 2018). Inactivation of RecA increased the stability of the two-replisome plasmid and reduced the proportion of abnormal amplification products that were observed (Fig 5). The high rate of RecA-driven instability on the two-replisome plasmid indicates that the aberrant pathway occurs at relatively high frequency on the plasmid, even when the normal, faithful pathway remains functional. We would speculate that this is likely due to the high copy number of the plasmid, and that the frequency of aberrant completion events on the chromosome is likely to be much less. Under normal conditions in E. coli, the completion enzymes are only required to catalyze a single reaction on the chromosome. Whereas the bi-directional plasmid, which is maintained at ~80 copies/chromosome (Fig 2), may exceed the reaction-capacity of the faithful pathway, whose genes are not highly expressed (Eichler and Lehman, 1977; Taylor and Smith, 1980), allowing the aberrant mechanism to operate more frequently than would normally occur.
To our knowledge, examples of other well-characterized plasmids with bi-directional origins have not been reported. Plasmid R6K was originally reported to have a bidirectional origin (Lovett et al., 1975), but subsequent work found the plasmid contained two unidirectional origins and employed a complex asymmetric mechanism of replication (Crosa et al., 1976). In our initial approach to this work, we initially utilized a plasmid containing the E. coli origin of replication, pOriC-2 (Sawitzke et al., 2012). However, consistent with previous studies on oriC plasmids, we found that the extensive homology with the chromosome led to integration events at high frequency (Yasuda and Hirota, 1977; Messer et al., 1978; Meijer et al., 1979). These frequent integration events prevented a direct analysis of plasmid stability, as cultures with integrated plasmids maintained resistance to ampicillin even after the extrachromosomal plasmid itself was lost (Fig S3).”
ExoI and SbcCD nuclease complexes are additive in their effect on the completion reaction and inactivation of the normal completion reaction required deletion of both genes (Wendel et al., 2014; Wendel et al., 2018; Dimude et al., 2018a). On the chromosome, in the absence of both nucleases, the over-replicated regions persist, and the ability to maintain growth and the chromosome region where forks converge becomes entirely dependent on RecA (Wendel et al., 2018). In this report, we show that transformation of two-replisome plasmids similarly becomes dependent on RecA only in the absence of both nucleases (Fig 3). Mechanistically, SbcCD and ExoI could functionally interact to cooperate and enhance their ability to degrade the substrate(s) created by convergent replication forks. This appears to occur in eukaryotes, where the homologous Mre11-Rad50 interacts with Sae2 and increase its exonuclease activity (Lengsfeld et al., 2007; Deng et al., 2015; Andres and Williams, 2017). Consistent with this, a recent study from Dimude et al. found that SbcCD alone prevented much of the degradation that occurs in recBC mutants (Dimude et al., 2018a), an observation we have confirmed in our lab. In vitro, SbcCD has been demonstrated to cleave a palindrome-like substrate similar to that predicted to occur when replication forks bypass each other (Lim et al., 2015; Saathoff et al., 2018). Alternatively, ExoI may act independently of SbcCD to suppress the aberrant recombinational pathway. Early studies suggested a strong physical interaction between Exo I exonuclease and RecA (Bedale et al., 1991; Bedale et al., 1993; Kowalczykowski et al., 1994). Association of this 3’ exonuclease with RecA would be expected to strongly degrade recombinagenic 3’ ends preventing RecA from initiating recombination at these sites.
In an alternative interpretation of RecBCD function, a recent study and review speculated that chromosome cleavage may frequently occur during septation at cell division, resulting in double strand breaks that require RecBCD for repair (Sinha et al., 2017; Michel et al., 2018). The authors based this argument primarily on the observation that the extensive degradation in recBC mutants centers upon the dif locus, where chromosomes ultimately separate as cells divide. However, several observations argue against this possibility. If the defects in recBC mutants were due to the presence of double strand breaks, then recA mutants, should be similarly, or more severely affected. Yet, in their initial study, the authors failed to examine or address recA mutants (Sinha et al., 2017). However, recA mutants grow at rates similar to wild type cultures, and, unlike recBC mutants, maintain this region of the chromosome normally (Courcelle et al., 2015; Wendel et al., 2018). Further, these authors and previous investigators all note that DNA breaks are not detected on the chromosome of recA mutants (Sinha et al., 2018; Wendel et al., 2014; Courcelle et al., 2015; Wendel et al., 2018; Pennington and Rosenberg, 2007). To explain the absence of DNA breaks in recA mutants, a subsequent study from this group proposed that the septation-induced breaks in recA genomes are degraded and therefore undetectable (Sinha et al., 2018). However, based on the viability of recBC mutants, ~90% of cells in culture would be experiencing these septation-induced breaks. Synthesis and subsequent degradation of genomes at this frequency would be expected to slow culture growth considerably and generate degradation intermediates that should be easily detectable, neither of which are observed (Sinha et al., 2018; Wendel et al., 2014; Courcelle et al., 2015; Wendel et al., 2018). Models proposing frequent double strand breaks also fail to address how the inactivation of exonucleases SbcCD and ExoI, which are not essential for double strand break repair, would restore recBCD mutant growth defects, or why maintaining the terminus region of the chromosome in the absence of exonucleases would depend on RecA (Templin et al., 1972; Lloyd and Buckman, 1985; Wendel et al., 2018). Finally, as shown here, plasmids containing two replisomes require RecBC to propagate but do not require RecA and are actually stabilized in its absence. These plasmids lack dif sequences, and segregate prior to and independent from cell division, and would therefore not be subject to septation-induced breaks. These observations are all inconsistent with the idea that the requirement for RecBCD is due to double strand breaks caused by cell division, and argue strongly its role in joining DNA ends of convergent forks can occur independently of recombination or RecA.
It is also worth considering that although current models for RecBCD in double-strand break repair propose that RecBCD acts prior to RecA, the early in vivo studies led several independent labs to conclude that RecBCD acted after RecA, at a late step in completing the recombination reaction (Wilkins, 1969; Willetts, 1975; Hall and Howard-Flanders, 1972; Birge and Low, 1974). Current models placing RecBCD as an initiator are heavily derived from biochemical studies in which linear double-stranded substrates were used to characterize enzyme binding, helicase, and exonuclease activities (reviewed in (Yeeles and Dillingham, 2010; Dillingham and Kowalczykowski, 2008; Smith, 2012)). The initial concept that RecBC acts late in recombination arose from the observation that although recA mutants receiving an F’ factor were unable to transfer chromosomal genes to another cell, recBC mutants could do so at frequencies that approached those of wild-type cells. However, over time (~1 generation), this ability rapidly declined (Hall and Howard-Flanders, 1972; Wilkins, 1969; Willetts, 1975). The authors inferred that recombination proceeds beyond the point at which the incoming DNA is joined to the chromosome in recBC mutants, but that recA mutants are blocked prior to this event. In recombinational crosses between Hfr and F− strains carrying noncomplementing mutations in lacZ, Birge and Low found that although recA mutants were entirely blocked, recBC mutants initially produced beta-galactosidase within two-fold of those seen in wild-type cells, indicating that recombination reactions progressed beyond the point where transcribable, mutation-free copies of LacZ+ were produced (Birge and Low, 1974). However, although these recombination intermediates were detectable, the completion of these recombination events was impaired in the absence of recBC and viable LacZ+ recombinant progeny were reduced 100–1000 fold. The authors inferred that “early steps in recombination can proceed efficiently in RecB-and RecC-strains, but that late steps, such as the degradation of excess DNA ‘tails’, might be defective.” Using combinations of single and double mutants, Willetts confirmed these previous studies and suggested that recombination proceeded past the first joining reaction of the two DNA molecules, but that RecBC was required for a second joining needed “to generate a circular unit” that could be inherited (Willetts, 1975). These interpretations are strikingly consistent with RecBCD’s apparent role in completing replication on the chromosome, and may suggest that RecBCD function during recombinational events is similar to its role in completing replication (Courcelle et al., 2015).
EXPERIMENTAL PROCEDURES
Strains and Plasmids.
All strains used in this work are derived from SR108, a thyA deoC derivative of W3110 (Mellon and Hanawalt, 1989) and listed in Table 1. pBR322 contains a ColE1 origin of replication (Bolivar et al., 1977). pCL01 is a derivative of pCB104 (Boyd and Sherratt, 1995) containing an ampicillin-resistant cassette from pBR322. Plasmid constructions were performed according to published protocols for in vivo recombineering (Sawitzke et al., 2012; Yu et al., 2000). Briefly, primers 5’gtcggttcagggcagggtcgtggatccactttagttacaacatacttattcgcggaacccctatttgttt and 5’ggcggtttgcgtattgggcgcatattagttacaacatcctatatggtctgacagttaccaatgc were used to amplify the ampR gene from pBR322. 0.2 µg gel purified PCR product was then combined with 0.5 µg BamHI-digested pCB104 and amplified for 25 cycles using Pfu Turbo Polymerase (Agilent). PCR products were examined by agarose gel electrophoresis and products running larger than 5kb were gel purified and transformed into recombineering strain DY329 (Yu et al., 2000) to generate the ampicillin resistant plasmid. A chi sequence was then removed from the plasmid using primer sets sets 5’attgctgataaatctgga / 5’ctttggaatccagtccctcttcctcctgctgatctgcgacttatcaac and 5’tccagatttatcagcaat / 5’gttgataagtcgcagatcagcaggaggaagagggactggattccaaag were used to amplify overlapping fragments of the plasmid template using Pfu Turbo Polymerase (Agilent). The fragments were then joined and transformed using Gibson assembly (New England Biolabs) to generate pCL01.
TABLE 1.
Strains and plasmids used in this study
| Strain/Plasmid | Relevant Genotype | Reference/Construction |
|---|---|---|
| SR108 parental | λ -thyA deo IN(rrnD-rrnE) | (Mellon and Hanawalt, 1989) |
| HL922 | SR108 recB21C22 argA81::Tn10 | (Courcelle et al., 1997) |
| CL851 | SR108 recB21C22 argA81::Tn10 recA::cam | (Chow and Courcelle, 2007) |
| HL923 | SR108 recD1011 argA81::Tn10 | (Courcelle et al., 1997) |
| CL726 | SR108 recD1011 argA81::Tn10 recA::cam | (Chow and Courcelle, 2007) |
| CL542 | SR108 recA::cam | (Chow and Courcelle, 2007) |
| CL039 | SR108 xonA::cam | (Courcelle and Hanawalt, 1999) |
| CL718 | SR108 xonA::Cat300 D(srlR-recA)306::Tn10 | (Wendel et al., 2018) |
| CL2344 | SR108 sbcCD::Gm | (Wendel et al., 2018) |
| CL3535 | SR108 sbcCD::Gm D(srlR-recA)306::Tn10 | (Wendel et al., 2018) |
| CL2357 | SR108 xonA::Cat300 sbcCD::Gm | (Wendel et al., 2014) |
| CL3539 | SR108 xonA::Cat300 sbcCD::Gm D(srlR-recA)306::Tn10 | (Wendel et al., 2018) |
| CL2542 | SR108 xonA::Cat300 sbcCD::Gm recB21C22 argA81::Tn10 | (Wendel et al., 2018) |
| CL2575 | SR108 xonA::Cat300 sbcCD::Gm recB21C22 argA81 D(srlR-recA)306::Tn10 | (Wendel et al., 2018) |
| CL2456 | SR108 recG6200::tet857 | P1 transduction of recG6200::tet857 from TP538 (Murphy et al., 2000) into SR108 |
| CL2579 | SR108 recG6200::tet857 recA::cam | P1 transduction of recA::cam from CL542 (Courcelle et al., 1997) into CL2456 |
Growth Rates.
Cultures containing 0.1-mL cultures were grown in LB medium supplemented with 10µg mL−1 thymine (LBthy) at 37°C with agitation in a 96-well microtiter dish. Absorbance at 630nm was measured over time using a BIO-Whittaker ELx808 plate reader. The number of viable colonies per ml in each culture was determined at the start of every experiment. Equal numbers of viable cells were compared in each case (Courcelle et al., 2012).
Transformation Efficiency.
Electro-competent cells were prepared by growing a 100-fold dilution of a fresh overnight culture in 10 ml LBthy to an OD600 of 0.4. Cells were then pelleted, and serially washed with 30 mL water, 30mL 10% glycerol, and then resuspended in 200 µL of 10% glycerol and stored at −80ºC. To determine transformation efficiency, 40 µL of competent cells were mixed with 50ng of plasmid and electroporated at 1.8kV 25µFD 200Ohms and allowed to recover at 37°C for 30 minutes in 1 mL SOC media. The transformation reactions were then diluted and aliquots were spread on LB thy plates with and without 50 µg mL−1 ampicillin to determine the number of transformants and viable cells, respectively. Colonies were counted following overnight incubation at 37°C. The same preparations of competent cells were used for all strains when comparing pBR322 and pCL01. The relative transformation efficiency of each strain was calculated as the ratio of transformants per viable cells in the mutant cultures to the transformants per viable cells in wild type cultures.
Plasmid Stability.
Cells from overnight cultures of strains containing the plasmid grown in LBthy medium with 50 µg mL−1 ampicillin were pelleted and used to inoculate 10ml cultures of LBthy medium at 1:1000 dilution. Cultures were grown without ampicillin selection at 37°C with aeration overnight. The resulting cultures were then sampled to determine the ratio of cells retaining the plasmid and used to reinoculate 10ml LBthy medium at 1:1000 dilution. This was repeated for iterations. To determine plasmid retention, 10-µl aliquots of serial 10-fold dilutions were spotted on LBthy plates in the presence or absence of 50 µg mL−1 ampicillin. Colonies were counted following overnight incubation at 37°C and compared to establish the percent of plasmid-containing cells (Wendel et al., 2014).
Southern Analysis of Plasmid Forms.
To purify total genomic DNA, 0.75 mL aliquots of culture containing the plasmid grown in LBthy medium with 100 µg mL−1 ampicillin were placed into 0.75 ml ice cold 2× NET (100 mM NaCl, 10 mM Tris, pH 8.0, 10 mM EDTA). Each sample was pelleted, resuspended in 140 µl of 1 mg mL−1 lysozyme and 0.2 mg mL−1 RNaseA in TE (10 mM Tris, pH 8.0, 1 mM EDTA), and lysed at 37 °C for 30 min. Proteinase K (10 µl, 10 mg mL−1) and Sarkosyl [10 µl, 20% (wt/wt)] were then added and incubated continued for 30 min. Samples were then serially extracted with 4 volumes phenol/chloroform (1/1) and 4 volumes chloroform followed by dialysis for 1 h on 47 mm Whatman 0.05-µm pore disks (Whatman #VMWP04700) floating on a 250-mL beaker of TE (1 mM Tris, pH 8.0, 1 mM EDTA).
Total genomic DNA from each strain was then partially digested with SacII (for strains containing pBR322) or ApaI (for strains containing pCL01) (Thermo Fisher Scientific). Restriction sites for these enzymes are absent on each plasmid. Samples were then extracted with 1 volume of chloroform. Equal cell equivalents were loaded on a 0.5% or 1.0% agarose gel as indicated, and electrophoresed in 1× TBE (220 mM Tris, 180 mM Borate, 5 mM EDTA, pH 8.3). Gels were transferred to Hybond N+ nylon membranes (Amersham-GE Healthcare) and probed with either 32P-labeled pBR322 or pCL01. Probes were prepared by nick translation according to the protocol supplied by Roche using [α−32P]dCTP (PerkinElmer). Radioactivity was visualized using a Storm 840 and its associated ImageQuant Software (Molecular Dynamics) (Wendel et al., 2014).
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the National Science Foundation (Grant MCB0130486) and National Institute of Environmental Health Sciences (Grant R15ES025953). We are grateful to Drs. G. Węgrzyn and S. Austin for providing plasmids.
REFERENCES
- Rudolph CJ, Upton AL, Stockum A, Nieduszynski CA, and Lloyd RG (2013) Avoiding chromosome pathology when replication forks collide. Nature 500: 608–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amundsen SK, Taylor AF, Chaudhury AM, and Smith GR (1986) recD: the gene for an essential third subunit of exonuclease V. Proc Natl Acad Sci U S A 83: 5558–5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres SN, and Williams RS (2017) CtIP/Ctp1/Sae2, molecular form fit for function. DNA Repair (Amst) [DOI] [PMC free article] [PubMed]
- Asai T, Bates DB, and Kogoma T (1994) DNA replication triggered by double-stranded breaks in E. coli: dependence on homologous recombination functions. Cell 78: 1051–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai T, and Kogoma T (1994) D-loops and R-loops: alternative mechanisms for the initiation of chromosome replication in Escherichia coli. J Bacteriol 176: 1807–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atlung T, Christensen BB, and Hansen FG (1999) Role of the rom protein in copy number control of plasmid pBR322 at different growth rates in Escherichia coli K-12. Plasmid 41: 110–119. [DOI] [PubMed] [Google Scholar]
- Bedale WA, Inman RB, and Cox MM (1993) A reverse DNA strand exchange mediated by recA protein and exonuclease I. The generation of apparent DNA strand breaks by recA protein is explained. J Biol Chem 268: 15004–16. [PubMed] [Google Scholar]
- Bedale WA, Inman RB, and Cox MM (1991) RecA protein-facilitated DNA strand breaks. A mechanism for bypassing DNA structural barriers during strand exchange. J Biol Chem 266: 6499–6510. [PubMed] [Google Scholar]
- Bhatia V, Barroso SI, Garcia-Rubio ML, Tumini E, Herrera-Moyano E, and Aguilera A (2014) BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 511: 362–365. [DOI] [PubMed] [Google Scholar]
- Birge EA, and Low KB (1974) Detection of transcribable recombination products following conjugation in rec+, reCB-and recC-strains of Escherichia coli K12. J Mol Biol 83: 447–457. [DOI] [PubMed] [Google Scholar]
- Bolivar F, Rodriguez RL, Greene PJ, Betlach MC, Heyneker HL, Boyer HW, Crosa JH, and Falkow S (1977) Construction and characterization of new cloning vehicles. II. A multipurpose cloning system. Gene 2: 95–113. [PubMed] [Google Scholar]
- Boros I, Posfai G, and Venetianer P (1984) High-copy-number derivatives of the plasmid cloning vector pBR322. Gene 30: 257–260. [DOI] [PubMed] [Google Scholar]
- Boyd AC, and Sherratt DJ (1995) The pCLIP plasmids: versatile cloning vectors based on the bacteriophage lambda origin of replication. Gene 153: 57–62. [DOI] [PubMed] [Google Scholar]
- Brochu J, Vlachos-Breton É, Sutherland S, Martel M, and Drolet M (2018) Topoisomerases I and III inhibit R-loop formation to prevent unregulated replication in the chromosomal Ter region of Escherichia coli. PLoS Genet 14: e1007668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruhn C, Zhou ZW, Ai H, and Wang ZQ (2014) The essential function of the MRN complex in the resolution of endogenous replication intermediates. Cell Rep 6: 182–195. [DOI] [PubMed] [Google Scholar]
- Chow KH, and Courcelle J (2007) RecBCD and RecJ/RecQ initiate DNA degradation on distinct substrates in UV-irradiated Escherichia coli. Radiat Res 168: 499–506. [DOI] [PubMed] [Google Scholar]
- Costa A, Hood IV, and Berger JM (2013) Mechanisms for initiating cellular DNA replication. Annu Rev Biochem 82: 25–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courcelle CT, Landstrom AJ, Anderson B, and Courcelle J (2012) Cellular Characterization of the Primosome and Rep Helicase in Processing and Restoration of Replication following Arrest by UV-Induced DNA Damage in Escherichia coli. J Bacteriol 194: 3977–3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courcelle J, Crowley DJ, and Hanawalt PC (1999) Recovery of DNA replication in UV-irradiated Escherichia coli requires both excision repair and recF protein function. J Bacteriol 181: 916–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courcelle J, and Hanawalt PC (1999) RecQ and RecJ process blocked replication forks prior to the resumption of replication in UV-irradiated Escherichia coli. Mol Gen Genet 262: 543–51. [DOI] [PubMed] [Google Scholar]
- Courcelle J, Wendel BM, Livingstone DD, and Courcelle CT (2015) RecBCD is required to complete chromosomal replication: Implications for double-strand break frequencies and repair mechanisms. DNA Repair (Amst) 32: 86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courcelle J, Carswell-Crumpton C, and Hanawalt PC (1997) recF and recR are required for the resumption of replication at DNA replication forks in Escherichia coli. Proc Natl Acad Sci U S A 94: 3714–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosa JH, Luttropp LK, and Falkow S (1976) Mode of replication of the conjugative R-plasmid RSF1040 in Escherichia coli. J Bacteriol 126: 454–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Massy B, Fayet O, and Kogoma T (1984a) Multiple origin usage for DNA replication in sdrA(rnh) mutants of Escherichia coli K-12. Initiation in the absence of oriC. J Mol Biol 178: 227–36. [DOI] [PubMed] [Google Scholar]
- de Massy B, Patte J, Louarn JM, and Bouche JP (1984b) oriX: a new replication origin in E. coli. Cell 36: 221–7. [DOI] [PubMed] [Google Scholar]
- Deng SK, Yin Y, Petes TD, and Symington LS (2015) Mre11-Sae2 and RPA Collaborate to Prevent Palindromic Gene Amplification. Mol Cell 60: 500–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillingham MS, and Kowalczykowski SC (2008) RecBCD enzyme and the repair of double-stranded DNA breaks. Microbiol Mol Biol Rev 72: 642–71, Table of Contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimude JU, Midgley-Smith SL, and Rudolph CJ (2018a) Replication-transcription conflicts trigger extensive DNA degradation in Escherichia coli cells lacking RecBCD. DNA Repair (Amst) 70: 37–48. [DOI] [PubMed] [Google Scholar]
- Dimude JU, Midgley-Smith SL, Stein M, and Rudolph CJ (2016) Replication Termination: Containing Fork Fusion-Mediated Pathologies in Escherichia coli. Genes (Basel) 7: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimude JU, Stein M, Andrzejewska EE, Khalifa MS, Gajdosova A, Retkute R, Skovgaard O, and Rudolph CJ (2018b) Origins Left, Right, and Centre: Increasing the Number of Initiation Sites in the. Genes (Basel) 9: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimude JU, Stockum A, Midgley-Smith SL, Upton AL, Foster HA, Khan A, Saunders NJ, Retkute R, and Rudolph CJ (2015) The Consequences of Replicating in the Wrong Orientation: Bacterial Chromosome Duplication without an Active Replication Origin. MBio 6: e01294–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnianni RA, and Symington LS (2013) Break-induced replication occurs by conservative DNA synthesis. Proc Natl Acad Sci U S A 110: 13475–13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggin IG, and Bell SD (2009) Termination structures in the Escherichia coli chromosome replication fork trap. J Mol Biol 387: 532–539. [DOI] [PubMed] [Google Scholar]
- Duggin IG, Wake RG, Bell SD, and Hill TM (2008) The replication fork trap and termination of chromosome replication. Mol Microbiol 70: 1323–1333. [DOI] [PubMed] [Google Scholar]
- Eichler DC, and Lehman IR (1977) On the role of ATP in phosphodiester bond hydrolysis catalyzed by the recBC deoxyribonuclease of Escherichia coli. J Biol Chem 252: 499–503. [PubMed] [Google Scholar]
- Hall JD, and Howard-Flanders P (1972) Recombinant F’ factors from Escherichia coli K-12 strains carrying recB or recC. J Bacteriol 110: 578–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamperl S, and Cimprich KA (2014) The contribution of co-transcriptional RNA:DNA hybrid structures to DNA damage and genome instability. DNA Repair (Amst) 19: 84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heichinger C, Penkett CJ, Bahler J, and Nurse P (2006) Genome-wide characterization of fission yeast DNA replication origins. EMBO J 25: 5171–5179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiasa H, and Marians KJ (1994) Tus prevents overreplication of oriC plasmid DNA. J Biol Chem 269: 26959–26968. [PubMed] [Google Scholar]
- Hill TM (1992) Arrest of bacterial DNA replication. Annu Rev Microbiol 46: 603–633. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Hidaka M, and Horiuchi T (1989) Evidence of a ter specific binding protein essential for the termination reaction of DNA replication in Escherichia coli. EMBO J 8: 2435–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczykowski SC, Dixon DA, Eggleston AK, Lauder SD, and Rehrauer WM (1994) Biochemistry of homologous recombination in Escherichia coli. Microbiol Rev 58: 401–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushner SR, Nagaishi H, and Clark AJ (1972) Indirect suppression of recB and recC mutations by exonuclease I deficiency. Proc Natl Acad Sci U S A 69: 1366–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengsfeld BM, Rattray AJ, Bhaskara V, Ghirlando R, and Paull TT (2007) Sae2 is an endonuclease that processes hairpin DNA cooperatively with the Mre11/Rad50/Xrs2 complex. Mol Cell 28: 638–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim CT, Lai PJ, Leach DR, Maki H, and Furukohri A (2015) A novel mode of nuclease action is revealed by the bacterial Mre11/Rad50 complex. Nucleic Acids Res 43: 9804–9816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd RG, and Buckman C (1985) Identification and genetic analysis of sbcC mutations in commonly used recBC sbcB strains of Escherichia coli K-12. J Bacteriol 164: 836–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd RG, and Rudolph CJ (2016) 25 years on and no end in sight: a perspective on the role of RecG protein. Curr Genet 62: 827–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovett MA, Sparks RB, and Helinski DR (1975) Bidirectional replication of plasmid R6K DNA in Escherichia coli; correspondence between origin of replication and position of single-strand break in relaxed complex. Proc Natl Acad Sci U S A 72: 2905–2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR, Projan SJ, Ozaki LS, and Godson GN (1986) A temperature-dependent pBR322 copy number mutant resulting from a Tn5 position effect. Proc Natl Acad Sci U S A 83: 7381–7385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee TR, Asai T, Malka D, and Kogoma T (1992) DNA damage-inducible origins of DNA replication in Escherichia coli. Embo J 11: 4219–4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Parras L, Hernandez P, Martinez-Robles ML, and Schvartzman JB (1991) Unidirectional replication as visualized by two-dimensional agarose gel electrophoresis. J Mol Biol 220: 843–53. [DOI] [PubMed] [Google Scholar]
- Meijer M, Beck E, Hansen FG, Bergmans HE, Messer W, von Meyenburg K, and Schaller H (1979) Nucleotide sequence of the origin of replication of the Escherichia coli K-12 chromosome. Proc Natl Acad Sci U S A 76: 580–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellon I, and Hanawalt PC (1989) Induction of the Escherichia coli lactose operon selectively increases repair of its transcribed DNA strand. Nature 342: 95–98. [DOI] [PubMed] [Google Scholar]
- Messer W, Bergmans HE, Meijer M, Womack JE, Hansen FG, and von Meyenburg K (1978) Mini-chromosomes: plasmids which carry the E. coli replication origin. Mol Gen Genet 162: 269–275. [DOI] [PubMed] [Google Scholar]
- Michel B, Sinha AK, and Leach DRF (2018) Replication Fork Breakage and Restart in Escherichia coli. Microbiol Mol Biol Rev 82: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midgley-Smith SL, Dimude JU, Taylor T, Forrester NM, Upton AL, Lloyd RG, and Rudolph CJ (2018) Chromosomal over-replication in Escherichia coli recG cells is triggered by replication fork fusion and amplified if replichore symmetry is disturbed. Nucleic Acids Res 46: 7701–7715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy KC, Campellone KG, and Poteete AR (2000) PCR-mediated gene replacement in Escherichia coli. Gene 246: 321–330. [DOI] [PubMed] [Google Scholar]
- Niki H, Ogura T, and Hiraga S (1990) Linear multimer formation of plasmid DNA in Escherichia coli hopE (recD) mutants. Mol Gen Genet 224: 1–9. [DOI] [PubMed] [Google Scholar]
- Pennington JM, and Rosenberg SM (2007) Spontaneous DNA breakage in single living Escherichia coli cells. Nat Genet 39: 797–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roecklein B, Pelletier A, and Kuempel P (1991) The tus gene of Escherichia coli: autoregulation, analysis of flanking sequences and identification of a complementary system in Salmonella typhimurium. Res Microbiol 142: 169–75. [DOI] [PubMed] [Google Scholar]
- Rudolph CJ, Mahdi AA, Upton AL, and Lloyd RG (2010) RecG protein and single-strand DNA exonucleases avoid cell lethality associated with PriA helicase activity in Escherichia coli. Genetics 186: 473–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph CJ, Upton AL, Harris L, and Lloyd RG (2009a) Pathological replication in cells lacking RecG DNA translocase. Mol Microbiol 73: 352–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph CJ, Upton AL, and Lloyd RG (2009b) Replication fork collisions cause pathological chromosomal amplification in cells lacking RecG DNA translocase. Mol Microbiol 74: 940–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saathoff JH, Käshammer L, Lammens K, Byrne RT, and Hopfner KP (2018) The bacterial Mre11-Rad50 homolog SbcCD cleaves opposing strands of DNA by two chemically distinct nuclease reactions. Nucleic Acids Res [DOI] [PMC free article] [PubMed]
- Sawitzke JA, Youngren B, Thomason LC, Baker T, Sengupta M, Court D, and Austin S (2012) The segregation of Escherichia coli minichromosomes constructed in vivo by recombineering. Plasmid 67: 148–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seelke R, Kline B, Aleff R, Porter RD, and Shields MS (1987) Mutations in the recD gene of Escherichia coli that raise the copy number of certain plasmids. J Bacteriol 169: 4841–4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberstein Z, and Cohen A (1987) Synthesis of linear multimers of OriC and pBR322 derivatives in Escherichia coli K-12: role of recombination and replication functions. J Bacteriol 169: 3131–3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha AK, Durand A, Desfontaines JM, Iurchenko I, Auger H, Leach DRF, Barre FX, and Michel B (2017) Division-induced DNA double strand breaks in the chromosome terminus region of Escherichia coli lacking RecBCD DNA repair enzyme. PLoS Genet 13: e1006895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha AK, Possoz C, Durand A, Desfontaines J-M, Barre F-X, Leach DRF, and Michel B (2018) Broken replication forks trigger heritable DNA breaks in the terminus of a circular chromosome. PLoS Genet 14: e1007256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GR (2012) How RecBCD enzyme and Chi promote DNA break repair and recombination: a molecular biologist’s view. Microbiol Mol Biol Rev 76: 217–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stillman B (1994) Initiation of chromosomal DNA replication in eukaryotes. Lessons from lambda. J Biol Chem 269: 7047–7050. [PubMed] [Google Scholar]
- Taylor A, and Smith GR (1980) Unwinding and rewinding of DNA by the RecBC enzyme. Cell 22: 447–57. [DOI] [PubMed] [Google Scholar]
- Taylor AF, and Smith GR (1985) Substrate specificity of the DNA unwinding activity of the RecBC enzyme of Escherichia coli. J Mol Biol 185: 431–443. [DOI] [PubMed] [Google Scholar]
- Taylor AF, and Smith GR (2003) RecBCD enzyme is a DNA helicase with fast and slow motors of opposite polarity. Nature 423: 889–893. [DOI] [PubMed] [Google Scholar]
- Templin A, Kushner SR, and Clark AJ (1972) Genetic analysis of mutations indirectly supressing recB and recC mutations. Genetics 72: 205–215. [PMC free article] [PubMed] [Google Scholar]
- Wendel BM, Cole JM, Courcelle CT, and Courcelle J (2018) SbcC-SbcD and ExoI process convergent forks to complete chromosome replication. Proc Natl Acad Sci U S A 115: 349–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendel BM, Courcelle CT, and Courcelle J (2014) Completion of DNA replication in Escherichia coli. Proc Natl Acad Sci U S A 111: 16454–16459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins BM (1969) Chromosome transfer from F-lac+ strains of Escherichia coli K-12 mutant at recA, recB, or recC. J Bacteriol 98: 599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willetts NS (1975) Recombination and the Escherichia coli K-12 sex factor F. J Bacteriol 121: 36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu PY, and Nurse P (2009) Establishing the program of origin firing during S phase in fission Yeast. Cell 136: 852–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda S, and Hirota Y (1977) Cloning and mapping of the replication origin of Escherichia coli. Proc Natl Acad Sci U S A 74: 5458–5462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeeles JT, and Dillingham MS (2010) The processing of double-stranded DNA breaks for recombinational repair by helicase-nuclease complexes. DNA Repair (Amst) 9: 276–285. [DOI] [PubMed] [Google Scholar]
- Yu D, Ellis HM, Lee EC, Jenkins NA, Copeland NG, and Court DL (2000) An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci U S A 97: 5978–5983. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.