

Abstract
The oncofetal mRNA binding protein, IMP1 or insulin-like growth factor-2 mRNA-binding protein 1 [IGF2BP1], promotes the overexpression of several oncogenic proteins by binding to and stabilizing their mRNAs. IMP1 is frequently overexpressed in melanoma and is associated with a poor prognosis, but the full spectrum of IMP1 target transcripts remains unknown. Here we report identification of Protein Kinase C alpha (PKCα), as a novel molecular target of IMP1. Overexpression of IMP1 resulted in increased levels of PKCα, while RNAi knockdown of IMP1 resulted in decreased PKCα mRNA stability, PKCα protein levels, and MAPK/ERK activation. In addition to IMP1 acting as a positive regulator of PKCα mRNA, we also report the identification of miR-340 as a negative regulator of PKCα mRNA. In melanoma cancer cells, inhibition of miR-340 led to increased PKCα protein levels. PKCα plays important roles in numerous signaling pathways including the MAPK/ERK signaling pathway. PKCα activates RAF1, which in turn activates MEK1, and activates downstream transcriptional targets of MAPK through activation of JNK signaling. Together, these pathways provide a way to activate MAPK signaling downstream of BRAF and MEK1 inhibitors, which are commonly used to treat melanoma. Analysis of 117 melanoma tumors samples showed that overexpression of PKCα is associated with poorer overall survival. In patients harboring BRAFV600E or NRAS mutations, PKCα overexpression is associated with a 11-fold increased risk of death. Thus, PKCα mRNA is a novel target of IMP1, which is commonly overexpressed in melanoma and is linked to poorer overall survival.
Keywords: IMP1, IGF2BP1, protein kinase C alpha, mRNA-binding protein, melanoma
Introduction
MicroRNAs (miRNAs) and RNA-binding proteins (RBPs) regulate gene expression and are increasingly linked to melanoma development and progression. miRNAs are small ~22-nucleotide non-coding RNAs that negatively regulate mRNAs by complementary base pairing in the 3’UTR, 5’UTR, and protein coding regions of target mRNAs. miR-340 is a well characterized tumor suppressor in melanoma that negatively regulates the expression of microphthalmia-associated transcription factor (MITF), which is a master regulator of melanocyte development and commonly overexpressed in melanoma [1,2]. In contrast to miRNAs, interaction with RNA-binding proteins typically enhances the stability of mRNA targets. The RNA-binding protein IMP1, also known as insulin-like growth factor-2 mRNA-binding protein 1(IGF2BP1), is commonly upregulated in melanomas and linked to poor survival [3]. As a largely oncofetal protein, IMP1 binds to and stabilizes transcripts that are important in oncogenesis including c-Myc (MYC), MDR1 (ABCB1), and βTrCP1 (BTRC) [4-6]. IMP1 also attenuates miRNA-dependent degradation of several mRNAs, such as miR-340-mediated degradation of MITF [1,7]. In a recent study, depleting levels of IMP1 restored sensitivity to BRAF/MEK inhibitors in melanoma cells containing BRAFV600E mutations [8]. While efforts continue to identify all the mRNA targets of IMP1, a comprehensive list of IMP1 targets is lacking.
Here we report the identification of a previously undescribed IMP1 and miR-340 target, Protein Kinase C alpha (PKCα/PRKCA). PKCα belongs to the PKC protein family of serine/threonine kinases that includes classical (α, βI, βII, and γ), novel (δ, ε, η, and θ), and atypical PKC isozymes (ζ, ι, and λ). Classical PKCs are calcium-dependent for activation, whereas novel and atypical PKCs are not and often have differential roles in cancer cell proliferation and survival [9]. PKCα is an important target in cancer that promotes cell proliferation, apoptosis and migration in melanoma, breast, lung, and ovarian cancer [10–12]. PKCα induces phosphorylation of RAF1 (CRAF), which activates the extracellular signal-regulated kinase/mitogen-activated protein kinase (ERK/MAPK) cascade. The ERK/MAPK signaling pathway serves as a key signaling pathway in melanocytes, and the pathway is constitutively activated through BRAFV600E and NRAS mutations in the majority of melanomas [13]. BRAF inhibitors, such as dabrafenib and vemurafenib, promote rapid regression of tumors expressing the BRAFV600E mutant and improve overall survival in patients with metastatic melanoma [14,15]. However, tumors rapidly become resistant to these therapies, most commonly through reactivation of the MAPK signaling pathway secondary to alterations in MEK1, BRAF, and NRAS [16]. Thus, melanoma treatment has evolved to include combining BRAF inhibitors with downstream MEK inhibitors, which has further enhanced melanoma survival compared to BRAF monotherapy. Unfortunately, most melanomas acquire resistance to combined therapies through activation of PAKs and RAF1. PAKs and RAF1 activate JNK signaling, which in turn activates downstream transcriptional targets of the MAPK pathway (ELK1, FOS, JUN) [17].
Materials and Methods
Cell Culture
Cells were maintained in the following culture media: SK-MEL2: RPMI-1640, supplemented with 10% FBS; HEK 293T: DMEM-F12 supplemented with 10% FBS.
Plasmids and Oligonucleotides
The IMP1 mammalian expression plasmid was created as follows: The IMP1 coding region was amplified by PCR with a pair of primers, 5’-CAGCTGGTACCATGAACAAGCTTTACATCGGCAAC and 5’-GTATTCTAGACCTCACTTCCTTCGTGCCTGGGCCTG. The PCR product was digested with Kpn I and Xba I, and then cloned into pCMV-hERα to generate pCMV-IMP1 [4]. The empty plasmid pTZ18U served as a negative control. Locked nucleic acid (LNA) anti-sense oligonucleotides for miR-340 and a miR-control were purchased from Exiqon, Inc.
Transient Transfections
SK-MEL2 cells were plated at 300,000 cells/well in 6-well plates and transfected the following day with Dharmafect 1 transfection reagent and either a non-coding siRNA SmartPool (Dharmacon) or IMP1 specific siRNA SmartPool. HEK 293T cells were plated at 150,000 cells/well in 24-well plates. The next day, 1 μg total DNA (PTZ18U or pCMV-IMP1) was transfected using a ratio of 1:3 for DNA to Lipofectamine 2000 (Invitrogen). For transient transfections using the LNA miR inhibitors, SK-MEL2 cells were plated at 100,000 cells/well in 12-well plates. The next day, LNA inhibitors were added to cells using Lipofectamine 2000 according to the manufacturer’s instructions.
Endogenous Gene Expression and Western Blots
Cell extraction of RNA and protein, qRT-PCR, and Western blotting was performed, as previously described [4]. The following antibodies were used: IMP1 antibody (sc-21026, Santa Cruz, CA), PKCα antibody (sc-208, Santa Cruz), ERK (4695, Cell Signaling), p-ERK (sc-208, Cell Signaling), and ɑ-tubulin (A1978, Sigma).
Tumor Microarray Data Analysis
Analysis of histologically normal skin (GSE15605) was compared to melanomas (GSE15605, GSE7553, GSE19234). PRKCA expression was evaluated for its ability to predict survival in 71 melanoma patients (GSE53118) and 46 cutaneous melanoma patients (GSE15605). Microarray data analysis was performed using the BRB ArrayTools (version 4.2.1) and R software version 3.2.5. Gene expression values from CEL files were normalized by use of the standard quantile normalization method. HRs were estimated using Cox’s regression analysis. Risk prediction using the supervised principle components method was visualized using Kaplan–Meier plots and compared using log-rank tests.
Statistical Analysis
Results are expressed as the mean ± S.E.M. of at least three independent experiments. Significance was established when p < 0.05. Student’s t-test was used for comparison of the means between two groups.
Results
Knockdown of IMP1 Decreases PKCα mRNA and Protein and Overexpression of IMP1 Increases PKCα Protein in HEK 293T Cells
To identify novel molecular targets of IMP1, we performed in silico analysis using publically available microarrays in which IMP1 had been depleted [18]. We were especially interested in genes known to play a role in cancer, specifically in cell proliferation and migration. We focused on a previously undescribed IMP1 target, PKCα, which plays important roles in cell proliferation, survival, invasion, migration, and anticancer drug resistance through interaction with several signal transduction pathways [19]. To confirm and extend these results, RNAi knockdown of IMP1 was performed in HEK 293T cells. In cells treated with IMP1 siRNA, we observed a 4-fold decrease in PKCα mRNA and a decrease in PKCα protein levels (Fig. 1a and 1b). If IMP1 enhances the stability of PKCα mRNA, then overexpression of IMP1 should lead to increased PKCα mRNA and protein levels. We tested this by transiently transfecting a control plasmid or an IMP1 expression plasmid into HEK 293T cells. In cells transfected with the IMP1 expression plasmid, PKCα protein levels were substantially increased (Fig. 1c). Together, these results demonstrate that IMP1 regulates PKCα levels.
Figure 1.
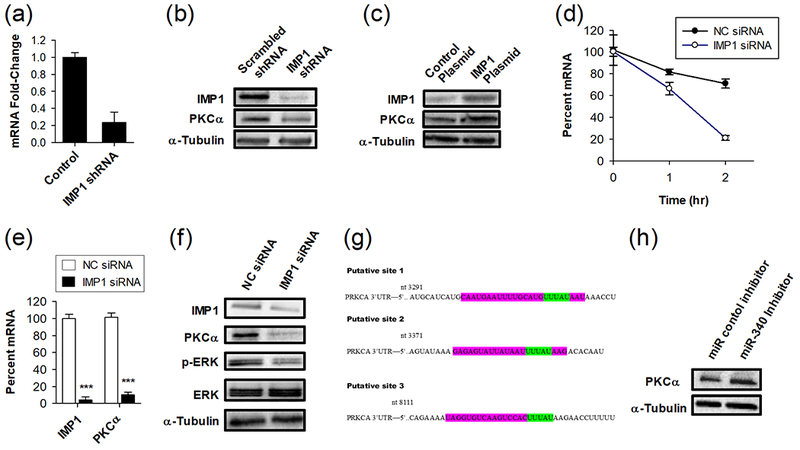
IMP1 and miR-340 regulate PKCα mRNA and protein levels, and downstream activation of ERK signaling. (a) qRT-PCR analysis of PKCα mRNA levels and (b) Western blot analysis of IMP1 and PKCα protein levels following shRNA knockdown of IMP1 in HEK 293T cells. HEK 293T cells were treated with scrambled shRNA or IMP1 shRNA for 72 hours (n = 4). (c) Western blot analysis of IMP1 and PKCα protein levels following overexpression of IMP1 in HEK 293T cells. (d) IMP1 knockdown enhances the degradation rate of PKCα mRNA in SK-MEL2 cells. Cell pretreated with 100 nM non-coding control (NC) or IMP1 siRNA SmartPool for 72 hours, followed by treatment with actinomycin D for the indicated times (n = 4). (e) qRT-PCR analysis showing IMP1 and PKCα mRNA levels and (f) Western blot showing protein levels of IMP1, PKCα, ERK and p-ERK in SK-MEL2 melanoma cancer cells after treatment with either non-coding (NC) control siRNA or IMP1 siRNA. Cells were treated with 100 nM non-coding control (NC) or IMP1 siRNA SmartPool for 72 hours (n = 4). (g) Predicted miRNA sites in the PKCα mRNA 3′-UTR region produced by miRWalk and other programs (purple) and IMP1 binding sites (green). (h) Transient transfection of a miR-340 inhibitor increases PKCα protein levels in SK-MEL2 melanoma cells. Cells were transiently transfected with an inactive control microRNA inhibitor or a miR-340 inhibitor, and PKCα protein levels were analyzed by Western blot.
Knockdown of IMP1 Enhances Degradation of PKCα mRNA, Decreases PKCα mRNA and Protein Levels, and Decreases ERK Pathway Activation in Melanoma Cells
We next wanted to assess whether IMP1 can regulate PKCα levels in melanoma cells. If IMP1 enhances the stability of PKCα mRNA, then IMP1 knockdown should increase the degradation of PKCα mRNA and reduce the total mRNA levels. To test this, we applied the widely used technique of blocking new mRNA synthesis using actinomycin D to follow the decay of PKCα mRNA in cells pre-treated with either non-coding or IMP1 siRNA. IMP1 knockdown significantly increased PKCα mRNA degradation (Fig 1d), led to a 10-fold reduction in PKCα mRNA (Fig. 1e), and produced a robust decline in PKCα protein levels (Fig. 1f). Since PKCα stimulates survival and proliferation in part through activation of the Ras/Raf/MEK/ERK [20], we assessed whether knockdown of IMP1 reduced ERK signaling. IMP1 knockdown in SK-MEL2 cells led to a decrease in Thr-202/Thr-204 phosphorylation of ERK, which is a common readout of MAPK activation (Fig. 1f).
miR-340 Regulates PKCα Protein Levels
MicroRNAs (miRs) inhibit gene expression by binding to target mRNAs and enhancing their degradation. To identify miRs that target PKCα, and might exhibit interplay with IMP1 in regulating PKCα mRNA levels, we used four cited algorithms, miRanda – mirSVR, miRDB, miRWalk, and Targetscan, and identified miR-340 as a top predicted microRNA. Three putative binding sites of miR-340 were identified in the 3′-UTR of PKCα and their overlap with IMP1 binding sequences is shown (Fig. 1g). Given these in silico results, we then performed a transient transfection using a miR-340 antagonist and found that treatment with the miR-340 inhibitor resulted in an increase in PKCα protein levels (Fig.1h). This suggests a role for miR-340 in regulation of PKCα levels.
PKCα is Upregulated in Melanomas and is Associated with Reduced Survival in Patients with Melanoma
Prior studies have shown that IMP1 is often upregulated in melanomas, and higher expression is associated with a poor prognosis and resistance to chemotherapy. Since IMP1 upregulates PKCα levels, we assessed whether PKCα levels were increased in melanomas compared to normal skin tissue using publically available tissue samples. Melanoma samples demonstrated significantly higher IMP1 and PKCα expression compared to normal skin (Fig. 2a). We next explored if PKCα expression was predictive of survival from melanoma in several publically available datasets. Using data from 71 melanoma patients, we used median PKCα mRNA expression to divide patients into low- or high-PKCα groups, and observed reduced melanoma survival for patients overexpressing PKCα (hazard ratio (HR) = 1.9, 95% confidence interval (CI): 0.96-11.21), which trended toward significance (p = 0.055) (Fig. 2b). A second cohort of 46 melanoma tumors was evaluated, which included information on the presence or absence of NRAS or BRAF mutations. In the 46 melanoma patients, patients with high PKCα expression had a significantly higher risk of death (HR 4.4, 95% CI: 1.2-15.9) (Fig. 2c). In patients with a BRAF or NRAS mutation, elevated expression of PKCα was associated with a 11.2-fold higher risk of death from melanoma (Fig. 2d and 2e). In contrast, patients with wild-type BRAF or NRAS showed no significant increase in risk of death from melanoma (HR 0.63, 95% CI: 0.54-7.3) (Fig. 2e). Thus, the presence of BRAF or NRAS mutations in melanomas serves as an effect modifier for predicting survival in patients with elevated expression of PKCα.
Figure 2.
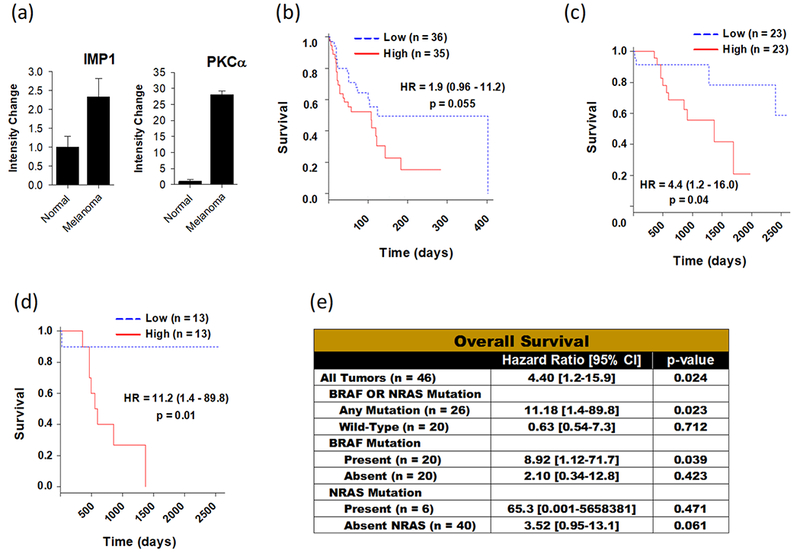
PKCα is upregulated in melanoma and overexpression of PKCα is associated with reduced survival in melanoma. (a) mRNA expression levels of IMP1 and PKCα in samples of normal skin (n = 4) and cutaneous melanoma (n = 14). P-values represent comparison to histologically normal tissue. Kaplan-Meier plot showing overall survival as a function of the mRNA expression of PKCα in different patient cohorts with cutaneous melanoma including (b) GSE53118 (n = 71) and (c) GSE15605 (n = 46), with hazard ratios calculated between low and high PKCα expression groups. (d) Overall survival in patients with BRAF or NRAS mutations, as a function of PKCα expression (n = 26), with hazard ratios calculated between low and high PKCα expression groups. (e) PKCα overexpression predicts poor overall survival in patients with BRAF or NRAS mutations. Cox regression analysis of PKCα expression in cutaneous melanoma patients (n = 46) and subgroups (Any Mutation, BRAF Mutation, and NRAS mutation). Median gene expression value was used to classify tumors into high- and low risk groups.
Discussion
We show that IMP1, which itself is overexpressed and linked to BRAF inhibitor resistance and poor overall survival in melanoma [7], positively regulates the levels of PKCα mRNA and protein in melanoma cells and has downstream effects on the RAS-RAF-MAPK pathway by increasing phosphoactivation of ERK1/2. IMP1 upregulates PKCα protein levels by increasing PKCα mRNA levels. IMP1 likely stabilizes PKCα mRNA through binding to the consensus sequence (UUUAY) in the 3’-UTR of PKCα. In contrast, miR-340 binds to the 3’-UTR of PKCα mRNA to downregulate PKCα levels.
Since IMP1 upregulates PKCα, IMP1 likely promotes resistance in part through upregulation of PKCα. Overexpression of PKCα leads to activation of several pathways that promote melanoma progression and resistance to subsequent therapy. First, PKCα activates RAF1, which leads to activation of MAPK signaling independent of upstream activation of BRAF and NRAS. In patients treated with BRAF inhibitors, PKCα-dependent activation of RAF1 provides a means for reactivation of ERK signaling. Second, PKCα induces the phosphoactivation of JNK and subsequent downstream activation of ELK1, JUN, and FOS. These targets are common downstream targets of both MAPK and JNK signaling that promote cell proliferation. In patients treated with BRAF and MEK1 inhibitors, JNK activation provides a way to activate downstream transcription factors in the MAPK pathway, despite blocking upstream BRAF and MEK1 activation [17].
We explored whether the relationship between IMP1 and PKCα was predictive of survival in melanoma, and showed that PKCα overexpression portends a poor prognosis, especially in patients with primary melanomas containing BRAFV600E or NRAS mutations. In patients expressing either BRAFV600E or NRAS mutations, increased expression of PKCα is associated with an 11-fold higher risk of death. Patients with BRAFV600E mutations are commonly treated with the BRAF inhibitors, vermurafenib and abrafenib. While these inhibitors block BRAF activation of MAPK signaling, high aberrant expression of PKCα leads to activation of RAF1, which in turn activates MAPK signaling. Similarly, in patients with NRAS mutations, downstream activation of MAPK signaling by PKCα circumvents attempts to block upstream signaling. As evidenced by our survival data, these patients don’t respond well to treatment and often die.
Although there is much interest in elucidating the role of IMP1 in cancer through identification of its regulatory targets, a comprehensive list has proved challenging, given the complexity of regulatory networks that operate in cancer subtype-specific contexts. Our data highlights PKCα as a novel molecular target of IMP1 in cancer and explores the effect of IMP1 knockdown on the RAS–RAF–MAPK signal transduction pathway in melanoma. Future studies that explore regulatory mechanisms relating IMP1 and PKCα expression, including miRNA regulatory networks will provide greater understanding of IMP1 in cancer biology and may represent new avenues for therapeutic interventions to improve the prognosis of IMP1 positive cancers.
Acknowledgements
Source of Funding: This research was funded by NIH R21CA173527 and RO1 DK071909 (to DS), and NIH F30CA192648 (to LM).
Footnotes
Conflicts of Interest: None declared
References
- 1.Goswami S, Tarapore RS, Poenitzsch Strong AM, TeSlaa JJ, Grinblat Y, Setaluri V et al. : MicroRNA-340-mediated degradation of microphthalmia-associated transcription factor (MITF) mRNA is inhibited by coding region determinant-binding protein (CRD-BP). The Journal of biological chemistry 2015; 290: 384–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Poenitzsch Strong AM, Setaluri V, Spiegelman VS: MicroRNA-340 as a modulator of RAS-RAF-MAPK signaling in melanoma. Arch Biochem Biophys 2014; 563: 118–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Craig EA, Weber JD, Spiegelman VS: Involvement of the mRNA binding protein CRD-BP in the regulation of metastatic melanoma cell proliferation and invasion by hypoxia. J Cell Sci 2012; 125: 5950–5954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mahapatra L, Andruska N, Mao C, Le J, Shapiro DJ: A Novel IMP1 Inhibitor, BTYNB, Targets c-Myc and Inhibits Melanoma and Ovarian Cancer Cell Proliferation. Transl Oncol 2017; 10: 818–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Noubissi FK, Elcheva I, Bhatia N, Shakoori A, Ougolkov A, Liu J et al. : CRD-BP mediates stabilization of betaTrCP1 and c-myc mRNA in response to beta-catenin signalling. Nature 2006; 441: 898–901. [DOI] [PubMed] [Google Scholar]
- 6.Sparanese D, Lee CH: CRD-BP shields c-myc and MDR-1 RNA from endonucleolytic attack by a mammalian endoribonuclease. Nucleic Acids Res 2007; 35: 1209–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elcheva I, Goswami S, Noubissi FK, Spiegelman VS: CRD-BP protects the coding region of betaTrCP1 mRNA from miR-183-mediated degradation. Mol Cell 2009; 35: 240–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim T, Havighurst T, Kim K, Albertini M, Xu YG, Spiegelman VS: Targeting insulin-like growth factor 2 mRNA-binding protein 1 (IGF2BP1) in metastatic melanoma to increase efficacy of BRAF(V600E) inhibitors. Mol Carcinog 2018; 57:678–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Halder K, Banerjee S, Bose A, Majumder S, Majumdar S: Overexpressed PKCdelta downregulates the expression of PKCalpha in B16F10 melanoma: induction of apoptosis by PKCdelta via ceramide generation. PloS one 2014; 9: e91656. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 10.Byers HR, Boissel SJ, Tu C, Park HY: RNAi-mediated knockdown of protein kinase C-alpha inhibits cell migration in MM-RU human metastatic melanoma cell line. Melanoma Res 2010; 20: 171–178. [DOI] [PubMed] [Google Scholar]
- 11.Lahn M, Kohler G, Sundell K, Su C, Li S, Paterson BM et al. : Protein kinase C alpha expression in breast and ovarian cancer. Oncology 2004; 67: 1–10. [DOI] [PubMed] [Google Scholar]
- 12.Smith SD, Enge M, Bao W, Thullberg M, Costa TD, Olofsson H et al. : Protein kinase Calpha (PKCalpha) regulates p53 localization and melanoma cell survival downstream of integrin alphav in three-dimensional collagen and in vivo. The Journal of biological chemistry 2012; 287: 29336–29347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S et al. : Mutations of the BRAF gene in human cancer. Nature 2002; 417: 949–954. [DOI] [PubMed] [Google Scholar]
- 14.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J et al. : Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011; 364: 2507–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M et al. : Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012; 380: 358–365. [DOI] [PubMed] [Google Scholar]
- 16.Fedorenko IV, Gibney GT, Sondak VK, Smalley KS: Beyond BRAF: where next for melanoma therapy? Br J Cancer 2015; 112: 217–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu H, Liu S, Zhang G, Bin W, Zhu Y, Frederick DT et al. : PAK signalling drives acquired drug resistance to MAPK inhibitors in BRAF-mutant melanomas. Nature 2017; 550: 133–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P et al. : Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 2010; 141: 129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakashima S: Protein kinase C alpha (PKC alpha): regulation and biological function. J Biochem 2002; 132: 669–675. [DOI] [PubMed] [Google Scholar]
- 20.Wen-Sheng W, Jun-Ming H: Activation of protein kinase C alpha is required for TPA-triggered ERK (MAPK) signaling and growth inhibition of human hepatoma cell HepG2. J Biomed Sci 2005; 12: 289–296. [DOI] [PubMed] [Google Scholar]