

Abstract
Circulating tumor DNA (ctDNA) is a component of cell-free DNA that is shed by malignant tumors into the bloodstream and other bodily fluids. Levels of ctDNA are typically low, particularly in patients with localized disease, requiring highly sophisticated methods for detection and quantification. Multiple liquid biopsy methods have been developed for ctDNA analysis in solid tumor malignancies and are now enabling detection and assessment of earlier stages of disease, post-treatment molecular residual disease (MRD), resistance to targeted systemic therapy, and tumor mutational burden. Understanding ctDNA biology, mechanisms of release, and clearance and size characteristics, in conjunction with the application of molecular barcoding and targeted error correction, have increased the sensitivity and specificity of ctDNA detection techniques. Combinatorial approaches including integration of ctDNA data with circulating protein biomarkers may further improve assay sensitivity and broaden the scope of ctDNA applications. Circulating viral DNA may be utilized to monitor disease in some virally induced malignancies. In spite of increasingly accurate methods of ctDNA detection, results need to be interpreted with caution given that somatic mosaicisms such as clonal hematopoiesis of indeterminate potential (CHIP) may give rise to genetic variants in the bloodstream unrelated to solid tumors, and the limited concordance observed between different commercial platforms. Overall, highly precise ctDNA detection and quantification methods have the potential to transform clinical practice via non-invasive monitoring of solid tumor malignancies, residual disease detection at earlier timepoints than standard clinical and/or imaging surveillance, and treatment personalization based on real-time assessment of the tumor genomic landscape.
1. Cell-Free DNA
Mandel and Métais [1] first described cell-free DNA (cfDNA) in 1948, referring to extracellular DNA found in the blood plasma. Present in various forms, cfDNA can be encapsulated in lipid membrane microvesicles, trapped by leukocytes, or bound to nucleosomes, serum and/or lipoproteins [2]. cfDNA circulates mostly in blood plasma, but can also be found in various bodily fluids, including urine [3, 4, 5, 6, 7, 8], cerebrospinal fluid [9, 10, 11], pleural fluid [12, 13, 14], ascites [14], and saliva [15, 16]. Passive release via apoptosis, necrosis, and phagocytosis account for the primary mechanisms of cfDNA release [17, 18, 19] (Fig. 1). Active secretion via extracellular vesicles or protein complexes is also thought to contribute to cfDNA [20, 21, 22, 23], although the exact mechanisms have not been elucidated (Fig. 1).
Fig. 1:
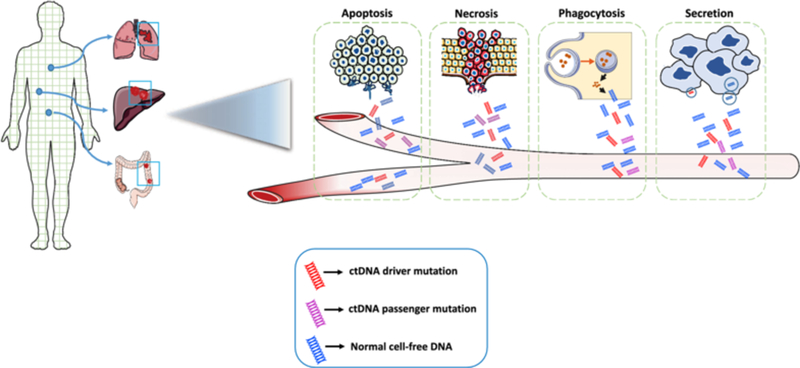
ctDNA release into the bloodstream from solid tumors.
Mechanisms of release into the bloodstream include cellular apoptosis, necrosis, phagocytosis, and active secretion. ctDNA driver and passenger mutations can be released in these ways, as can normal germline DNA, which typically comprises the majority of cell-free DNA even in cancer patients. ctDNA circulating tumor DNA
The clearance of cfDNA is also not fully understood. In 1963, Tsumita and Iwanaga [24] studied the kinetics of foreign DNA clearance by injecting radioactive DNA into mice. They showed that 99% of the radioactivity was cleared from the bloodstream in 30 min, and the highest increase of radioactivity was in the kidneys, followed by liver and spleen [24]. However, the plasma levels of cfDNA did not seem to be dramatically different in patients with chronic kidney disease or patients on peritoneal dialysis or hemodialysis, suggesting that renal elimination may not be the main mechanism of cfDNA clearance [25]. Other studies indicated that cfDNA is cleared from the circulation via nuclease action [26, 27]. As a whole, cfDNA has a short half-life ranging between 16 min and 2.5 h [26, 28]. The half-life of cfDNA may be longer when it is bound to protein complexes or inside membrane vesicles since the cfDNA is less vulnerable to degradation by phagocytes [2]. Accumulation of cfDNA can be attributed to an excessive release of DNA caused by massive cell death, inefficient removal of dead cells, or a combination of the two [29].
In healthy individuals, most of the cfDNA originates from hemopoietic cells such as erythrocytes, leukocytes, and endothelial cells, because hemopoietic cells are abundant, turn over rapidly, and have ready access to the vasculature [30, 31]. Normal tissues that undergo damage by ischemia, trauma, infection, or inflammation can also release cfDNA [32, 33]. Specifically, cfDNA has been shown to be elevated in patients with myocardial infarction [34], cerebral infarction [35, 36], lung transplant [37], trauma [38], parasitic infections [39], urinary tract infections [5], inflammatory conditions such as systemic inflammatory response syndrome [40], rheumatoid arthritis [41], systemic lupus erythematosus [42], as well as in patients who are pregnant [43] or undergo intense exercise [44, 45, 46]. For each specific tissue type, factors such as the rates of proliferation and apoptosis (cell turnover), total mass/volume, and vascularity are primary determinants in the degree of contribution from that tissue to the cfDNA pool [2].
cfDNA is typically comprised of fragments of double-stranded DNA ranging between 150 and 200 base pairs in length [47]. The commonly observed length of 166 bp corresponds to the length of DNA wrapped around a nucleosome (~ 147 bp) plus linker DNA associated with histone H1 [17, 48, 49, 50]. Compared to cfDNA derived from non-cancer cells, circulating tumor DNA (ctDNA) has been shown to be shorter [17, 51, 52, 53]. In fact, Mouliere et al. [54] demonstrated that selecting for fragments between 90 and 150 bp improved the detection sensitivity of ctDNA, with more than twofold median enrichment in > 95% of cases and more than fourfold enrichment in > 10% of cases. Similarly, the size difference between circulating fetal and maternal DNA has been used to improve the sensitivity of non-invasive prenatal testing [48, 55, 56, 57]. These studies suggest that the cell of origin of the cfDNA can affect its specific fragmentation patterns, which may offer clues regarding cell type and biology and enhance the sensitivity and specificity of ctDNA detection [23, 31, 51, 54, 58].
2. Circulating Tumor DNA (ctDNA) as a Tumor Biomarker
In 1987, Stroun et al. [59] first reported a potential correlation between cfDNA and cancer in a report where ten of 37 patients with advanced cancer had quantifiable cfDNA in their plasma, compared with zero of 50 normal controls. More recently, tumor-derived cfDNA, commonly referred to as ctDNA, has shown incredible potential as a highly sensitive and specific cancer biomarker [47, 60, 61]. Quantitative characterization of ctDNA via liquid biopsy has been associated with clinical and pathologic features of cancer, including stage, tumor burden, vascularization, and response to therapy [47, 60, 61, 62]. ctDNA is reflective of the mutations present within the tumor from which it arose and thus includes both driver and passenger mutations [63, 64, 65] (Fig. 1). The short half-life of ctDNA ensures that its detection captures tumor burden in real-time [66, 67].
As a whole, ctDNA has emerged as a dynamic plasma-based biomarker with the translational potential to facilitate molecular profiling at diagnosis [6, 63, 64, 65, 68, 69, 70, 71, 72, 73, 74], targeted therapy selection [6, 63, 68, 74, 75, 76, 77, 78, 79, 80], molecular residual disease (MRD) detection [6, 63, 64, 81, 82, 83, 84], and post-treatment tumor surveillance [6, 63, 64, 66, 68, 69, 74, 76, 85, 86, 87] (Fig. 2). The molecular precision of longitudinal tumor surveillance via serial ctDNA measurement enables the identification of mutations that drive cancer progression and treatment resistance [63, 64, 66, 68, 74, 75, 76, 77, 78, 79, 80] (Fig. 2). Crucial to its potential role in guiding therapeutic decision-making, ctDNA may better capture intra-tumoral heterogeneity than invasive biopsy, although highly sensitive methods are required to detect subclonal mutations [64, 65, 74, 75, 88, 89, 90].
Fig. 2:
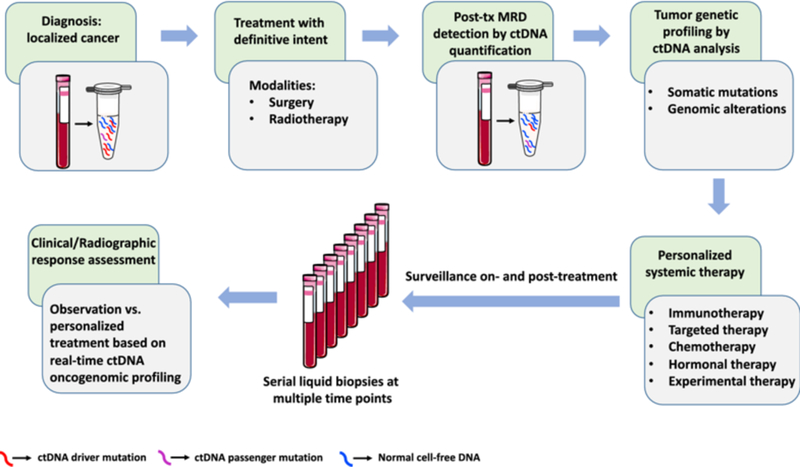
Potential diagnostic and therapeutic approaches for solid tumors based on ctDNA detection and oncogenomic profiling.
ctDNA may be measured pre-and post-definitive treatment of localized cancer using highly sensitive techniques. Assessment of both driver and passenger mutations can maximize the clinical sensitivity of detection. Based on post-treatment ctDNA MRD detection and oncogenomic profiling, personalized systemic therapy could potentially be offered, and patients could potentially be monitored by ctDNA surveillance with subsequent treatment based on real-time ctDNA-based oncogenomic profiling. ctDNA circulating tumor DNA, MRD molecular residual disease, tx treatment
Here we explore the key ctDNA detection methods, highlight ctDNA applications in various solid tumors, and discuss the potential clinical utility of ctDNA in oncology.
3. Challenges of ctDNA Detection
To utilize ctDNA effectively as a cancer biomarker in clinical settings, its measurement methods must be reliable and accurate. For most solid tumor malignancies, ctDNA levels in the blood plasma are low, with mutant allele fractions typically less than 10% in advanced metastatic disease [73, 75], and less than 1% in locally advanced non-metastatic disease [19, 63, 73]. ctDNA levels decrease further in early-stage cancers [63, 64, 73, 91] and after curative-intent treatment [63, 64, 83], where mutant allele fractions are often less than 0.1% [63, 64, 82, 84, 91]. Thus, highly sensitive methods are necessary to measure ctDNA both pre-treatment and post-treatment. In cancer patients with comorbidities such as anemia and poor performance status, clinical samples can be limited, thus decreasing the number of cfDNA molecules available for assessment and further complicating ctDNA detection [60, 91].
ctDNA detection can also be confounded by clonal hematopoiesis of indeterminate potential (CHIP) [92, 93]. CHIP arises when age-dependent acquired mutations accumulate in hematopoietic progenitor cells, leading to the formation of a genetically distinct subpopulation that contributes disproportionately to the population of mature blood cells [94, 95]. These distinct subclones have driver mutations and have been implicated in hematologic diseases, such as myelodysplastic syndrome (MDS) [96], acute myeloid leukemia [97], chronic lymphocytic leukemia (CLL) [98], and hairy cell leukemia (HCL) [99]. However, CHIP typically occurs in elderly healthy individuals in the absence of clinically apparent hematologic disease [100, 101, 102, 103]. Next-generation sequencing (NGS) studies have shown that blood cells derived from CHIP are rarely found in individuals younger than 60 years old, but can be as high as 5–18% for people older than 70 years old [95, 104]. Recent studies applying ultra-sensitive targeted sequencing suggest that rates of CHIP mutations are much higher, as high as 92–95%, with the majority of CHIP mutations being present at low mutant allele fractions [92, 103]. This highlights the prevalence of CHIP, and underscores the specificity challenge of detecting solid tumor-derived ctDNA.
In the measurement of ctDNA, CHIP can result in false-positive results due to detection of non-reference variants in the blood plasma, which is especially problematic when the ctDNA mutant allele fraction is low in the setting of MRD detection [92, 93]. Focusing on clonal mutations may lead to more specific ctDNA detection by avoiding low allele fraction CHIP variants. Assays that involve sequencing of paired peripheral blood mononuclear cells (PBMCs) may also lead to more specific ctDNA detection by enabling filtration of variants present in both cfDNA and PBMCs [63, 69, 84, 92]. Reassuringly, the majority of CHIP mutations involve DNMT3A, TET2, and ASXL1, genes implicated in hematologic cancer but not commonly involved in solid tumor malignancy [93, 94, 95, 101, 102, 103, 104, 105, 106]. However, TP53 is among the next most commonly mutated gene in CHIP [92, 93, 95, 101, 102, 103, 106], accounting for up to ~10% of all impacted genes [92], which is challenging given its high prevalence as a driver mutation in solid tumors [107] that is commonly tracked in ctDNA [108]. Thus, CHIP must be properly accounted for in order to specifically measure ctDNA, such as by sequencing matched PBMCs to similar depth [92, 93], especially when using ultra-sensitive assays that are capable of achieving detection of low mutant allele fraction variants.
In addition to CHIP, other sources of somatic mosaicism may serve as potential confounders in ctDNA analysis. Somatic mosaicism can arise from limitations in the fidelity of genetic material inheritance, such as the intrinsic error rate of DNA replication, which generates base substitutions occurring at an estimated frequency of 10–9 per replicative cycle in vivo [109, 110]. Environmental factors can also contribute to somaticism, leading to chromosome losses, deletions, duplications, and inversions [110]. Since these variants may not represent malignant transformation, it is important to refine ctDNA detection techniques to identify somatic mosaicism, such as by sequencing matched PBMCs and healthy donor plasma and using these results to filter variants detected in the cell-free compartment [63, 69, 84, 92, 93].
Given the multitude of factors affecting efficiency of ctDNA detection, sensitive and specific methods are crucial [60, 84, 91, 111]. Currently available ctDNA detection techniques are summarized in Table 1.
Table 1:
Summary of circulating tumor DNA detection techniques.
| Technique | Example technologies |
Scale of analysis | Method | Detection limit (% of Cost cfDNA) |
Assay personalization |
Advantages | Limitations | |
|---|---|---|---|---|---|---|---|---|
| AS-PCR | ARMS [112] | Single-locus or multiplexed assays | Preferentially amplifies rare mutant DNA molecules | ~ 0.1–1 | $ | Some required | Ease of use; ideal for detecting recurrent ‘hotspot’ point mutations | Can only query small number of variants
concurrently; cannot detect mutations not known a priori |
| dPCR | dPCR [117] ddPCR [119,125,126,127] BEAMing [128, 129] |
Single-locus or multiplexed assays | Partitions target DNA into different reactions for massively parallel qPCR |
~0.01 | $$ | Some required | High sensitivity | Can only query small number of variants
concurrently; cannot detect mutations not known a priori |
| WGS | WGS [143, 144] Plasma-Seq [139, 140] PARE [145] |
Genome-wide | NGS of whole genome |
~10 | $$– $$$ | Not required | Entire genome is interrogated | Low sensitivity; mostly limited to SCNA detection |
| FAST-SeqS [141] | ||||||||
| Retrotransposon-based amplicon NGS | mFAST-SeqS [152] WALDO [142] |
Genome-wide retrotransposon sites | PCR amplification of retrotransposon insertion sites prior to NGS analysis | ~ 5 | $$ | Not required | Rapid aneuploidy assessment with lower cost
than WGS |
Limited to aneuploidy detection |
| WES | WES [79] | Exome-wide | NGS of whole exome | ~ 5 | $$$ | Not required | Entire exome is interrogated | Low sensitivity |
| Multiplex PCR-based NGS | TAm-Seq [155] Enhanced TAm-Seq [156] Safe-SeqS [111] Natera® [64] |
Targeted sequencing | PCR amplification enriches targets prior to NGS analysis | ~ 0.01–2.0 | $$ | Some required | High sensitivity (modern methods) | Less comprehensive than other NGS
methods; unable to detect SCNAs; unable to detect fusions without assay personalization |
| Hybrid Capture-based NGS | CAPP-Seq [70,84] TEC-Seq [73] Guardant360® [161, 162] FoundationOne® Liquid [163] |
Targeted sequencing | Subset of exome is hybridized to biotinylated probes and captured for NGS analysis | ~ 0.001–0.5 | $$ | Not required | High sensitivity; detects multiple mutation types; broadly applicable without personalization |
Less comprehensive than WGS or WES |
| Combination approaches | CAPP-Seq + GRP [149] CancerSEEK [169] UroSEEK [7] |
Single-locus to genome-wide | Combines different ctDNA detection methods, sometimes including protein biomarkers | Variable | $$–$$$ | Variable | Improved detection compared to standard ctDNA analysis alone in certain settings | Potentially more time and resource intensive |
ARMS Amplification Refractory Mutation System, AS-PCR allele-specific PCR, BEAMing beads, emulsion, amplification and magnetics, CAPP-Seq Cancer Personalized Profiling by Deep Sequencing, cfDNA cell-free DNA, ctDNA circulating tumor DNA, ddPCR digital droplet PCR, dPCR digital PCR, FAST-SeqS Fast Aneuploidy Screening Test-Sequencing System, GRP Genome Representation Profiling, mFAST-SeqS modified FAST-SeqS, NGS next-generation sequencing, PARE Personalized Analysis of Rearranged Ends, Plasma-Seq plasma sequencing, Safe-SeqS Safe Sequencing System, SCNA somatic copy number alternation, TAm-Seq Tagged-Amplicon Sequencing, TEC-Seq Targeted Error Correction Sequencing, WALDO Within Sample Aneuploidy Detection, WES whole exome sequencing, WGS whole genome sequencing
4. ctDNA Detection Methods
4.1. PCR Assays
Initial efforts to characterize ctDNA relied on allele-specific PCR (AS-PCR) [112], which demonstrated clinical utility [113, 114] but had limited detection sensitivity [115, 116]. Digital PCR (dPCR) was subsequently developed [117], which enabled highly sensitive detection of specific ctDNA mutant alleles [83, 118, 119]. While PCR-based techniques have been implemented clinically, including the US Food and Drug Administration (FDA)-approved Cobas® test to detect EGFR (epidermal growth factor receptor) mutations in lung cancer patients [120], they are limited by their ability to assess only a small number of pre-defined mutations at a time [19].
4.1.1. Allele-Specific PCR (AS-PCR): Amplification Refractory Mutation System (ARMS) or Amplification of Specific Alleles (PASA)-PCR
AS-PCR, also known as amplification refractory mutation system (ARMS) or amplification of specific alleles (PASA), is a modified form of PCR designed to detect recurrent hot-spot point mutations or single nucleotide polymorphisms (SNPs) in plasma and serum [112]. Invented by Newton et al. [112] in the late 1980s, AS-PCR [113] gained popularity in the mid-2000s. Instead of selecting primers from an invariant region of the genome to amplify a polymorphic area between them as in traditional PCR, AS-PCR selects primers from at least one polymorphic region, with the mutations of interest located at (or near) its 3′-end [121, 122]. DNA polymerase only amplifies DNA when the nucleotide at the 3′-end of the primer perfectly complements the base at the variant, thereby preferentially amplifying mutant DNA molecules [123]. While AS-PCR is relatively affordable, has a limit of detection of ~0.1–1% [115, 116], and has successfully detected key mutations in non-small cell lung cancer (NSCLC) [114], it can only query one genetic locus per assay [112]. This drawback has limited the widescale application of AS-PCR for ctDNA detection.
4.1.2. Digital PCR (dPCR) and Beads, Emulsions, Amplification, and Magnetics (BEAM)-ing PCR
In an effort to improve ctDNA detection sensitivity using PCR, Vogelstein and Kinzler [117] introduced a novel technique in 1999 called digital PCR (dPCR) [117]. Compared to conventional real-time quantitative PCR (qPCR), dPCR offers improved identification, absolute quantification, and rare allele detection by partitioning samples into multiple parallel quantitative PCR reactions [117, 124]. These parallel reactions can take place in a single reaction tube, with PCR performed within aqueous microdroplets in an oil emulsion, thus earning the name digital droplet PCR (ddPCR) [119, 125, 126, 127]. ddPCR maximizes sensitivity by having a single molecule per compartment, where some portions of these reactions contain the target molecule (positive) while others do not (negative) [118, 119]. Variations of ddPCR, such as Beads, Emulsions, Amplification, and Magnetics (BEAM)-ing PCR, have also been devised [128, 129]. Indeed, dPCR and ddPCR have been applied to detect ctDNA in various cancers, such as NSCLC [80, 130, 131, 132], colorectal cancer [129, 133], breast cancer [83, 134], melanoma [135], and bladder cancer [136], with a high sensitivity detection threshold of ~ 0.01% [83]. While ddPCR is highly sensitive, it requires assay customization, has very limited multiplexing capacity, and cannot detect mutations not known a priori, thus limiting its ability to detect tumor heterogeneity and emergent mutations [19, 137, 138].
4.2. Genome-Wide Next-Generation Sequencing (NGS) Approaches
NGS approaches have become prevalent for tumor sequencing and have also been applied to cfDNA for ctDNA detection [19, 60]. Whole genome sequencing (WGS), retrotransposon-based amplicon sequencing, and whole exome sequencing (WES) enable mutation assessment across broad areas of the genome, and are not limited to querying variants known a priori [19, 60]. This is a clear advantage over PCR-based methods. However, WGS and WES are costly, which limits sequencing depth and assay sensitivity [19, 60]. Typically, WGS assays applied to cfDNA achieve a sequencing depth of ~ 0.1× [60, 139, 140] and WES achieves a sequencing depth of ~ 100× [74, 79]. Additionally, retrotransposon-based amplicon sequencing is limited to aneuploidy assessment [141, 142]. These disadvantages make these methods less practical for routine clinical use than more targeted methods (Fig. 3).
Fig. 3:
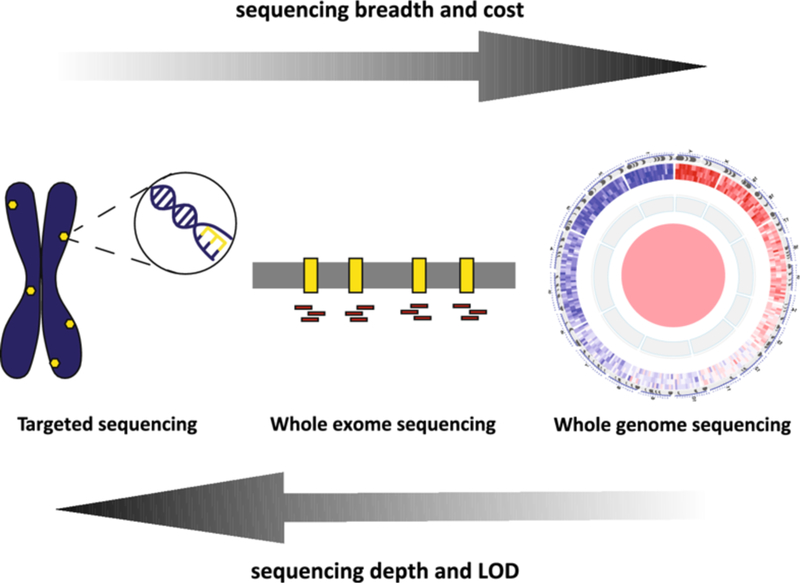
Trade-offs between sequencing breadth, depth, cost, and limit of detection.
In general, there is an inverse correlation between sequencing breadth versus assay cost, and sequencing depth versus assay limit of detection. Since ctDNA mutant allele fractions are typically low, especially in the settings of early cancer detection and MRD detection, targeted approaches including hybrid capture-based NGS, PCR-based NGS, and digital PCR are favored over broader sequencing methods such as whole exome or whole genome sequencing. ctDNA circulating tumor DNA, LOD limit of detection, MRD molecular residual disease, NGS next-generation sequencing
4.2.1. Whole Genome Sequencing (WGS): Plasma-Seq and Personalized Analysis of Rearranged Ends (PARE)
WGS has demonstrated the ability to detect chromosomal alterations in the plasma of patients with various cancer types [143, 144]. Although such studies suggest feasible clinical application to some patients, the cost and time required to perform WGS and the associated analysis have proven prohibitive for routine clinical implementation [143, 144]. To mitigate these limitations, Heitzer et al. [139] devised a shallow whole genome-wide NGS approach called Plasma-Seq [140]. This method uses Illumina MiSeq, a benchtop high-throughput sequencing instrument to expedite runtime, with the goal of detecting somatic copy number alterations (SCNAs) in plasma-derived cfDNA with a sequencing depth of~ 0.1× [139, 140]. Using Plasma-Seq, investigators detected multiple SCNAs in chromosome arms 8q, 8p, and the androgen receptor locus in patients with metastatic prostate cancer, reporting a sensitivity and specificity of > 80% when plasma ctDNA concentrations were ≥ 10% [139]. However, Plasma-Seq’s shallow sequencing depth (~ 0.1x ) made it difficult to detect structural chromosomal rearrangements with high confidence, and the sequencing resolution was too low to identify single nucleotide variants (SNVs) [139, 140]. Leary et al. [145] thus implemented a technique called Personalized Analysis of Rearranged Ends (PARE), utilizing ligation-based WGS [146] of sheared tumor DNA to identify patient-specific genomic rearrangements, followed by the design of ddPCR assays targeting rearranged loci to quantify their levels within cfDNA for tumor monitoring. Other investigators have also used WGS-based techniques to successfully detect cancer-specific genomic rearrangements and SCNAs in blood plasma to quantify solid tumor disease burden [147, 148, 149, 150].
4.2.2. Retrotransposon-Based Amplicon Sequencing: Fast Aneuploidy Screening Test-Sequencing System (FAST-SeqS), modified FAST-SeqS (mFAST-SeqS), and Within Sample Aneuploidy Detection (WALDO)
Plasma-Seq performs best in tumors with high ctDNA mutant allele fractions (≥ 10%) because its sequencing depths are too low to reliably detect lower levels of ctDNA [139, 151]. Since plasma ctDNA concentration can vary widely [69, 150, 151], Belic et al. [152] devised a method to identify plasma samples with ≥ 10% ctDNA that are suitable for Plasma-Seq analysis. To estimate ctDNA percentages in plasma, Belic et al. [152] modified a method called Fast Aneuploidy Screening Test-Sequencing System (FAST-SeqS) described by Kinde et al. [141] that used specific primers to amplify L1 retrotransposon regions dispersed throughout the genome. Even with modified FAST-SeqS (mFast-SeqS) as a pre-screening technique, WGS remains limited by high costs and low detection sensitivity (Fig. 3), thus limiting its potential clinical utilization [19, 60, 153].
In samples with high aneuploidy, amplification based methods such as Fast-SeqS [141] or Within Sample Aneuploidy Detection (WALDO) [142] can provide genome-wide mutation information cost effectively by limiting the hypothesis space to long interspersed nuclear elements (LINEs). While FAST-SeqS relies on differences between read depths at specific genomic loci and the average read depth in those loci in other samples to identify aneuploidy [141], WALDO utilizes a matched normal or curated selection of control samples, eliminating batch effects that could otherwise skew data [142]. WALDO also controls for amplification bias derived from amplicon size and detects chromosomal aberrations with > 90% sensitivity and > 99% specificity when neoplastic cell fraction is > 5% [142]. The additional use of a Support Vector Machine allows for aneuploidy detection with 78% sensitivity at 99% specificity in samples with as little as 1% tumor-derived DNA [142]. Due to their mechanisms of detection, WALDO and Fast-SeqS have lower performance when applied to cancers with less aneuploidy.
4.2.3. Whole Exome Sequencing
To improve detection sensitivity and reduce cost while maintaining comprehensive coverage of likely mutated genomic regions, researchers turned to WES, which restricts the sequencing space to protein-coding exons. Although the exome only represents ~ 1.5% of the whole genome, it has been reported to be enriched for disease-causing somatic mutations [154]. Murtaza et al. [79] published the first application of this approach to cell-free DNA, using WES to analyze serial plasma samples containing high percentages of ctDNA (between 5% and 55%) to track genomic evolution and response to therapy in patients with metastatic cancer receiving systemic therapy. The average depth of sequencing coverage ranged from 31-to 160-fold across 19 plasma samples [79]. This study identified increased representation of mutant alleles associated with the emergence of therapeutic resistance, including an activating mutation in PIK3CA (phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha) after treatment with paclitaxel, a truncating mutation in RB1 (retinoblastoma 1) after treatment with cisplatin, and a truncating mutation in MED1 (mediator complex subunit 1) after treatment with tamoxifen and trastuzumab [79]. The authors recommended utilizing WES for advanced or metastatic cancers, where the median pre-treatment ctDNA mutant allele fraction in plasma is at least 5–10% [79], to identify acquired drug resistance and evaluate clonal evolution non-invasively.
4.3. Targeted NGS Approaches
To improve sequencing depth and enable low-frequency mutant allele detection, NGS-based techniques have been tailored for targeted or ‘focused’ sequencing of specific gene panels [19, 60, 91], instead of untargeted or ‘broad’ WGS or WES (Fig. 3). These focused NGS-based approaches can be subdivided into amplicon-or hybrid capture-based sequencing.
4.3.1. Multiplex PCR-based NGS: Tagged-Amplicon Sequencing (TAm-Seq), Enhanced TAm-Seq, Safe Sequencing System (Safe-SeqS), and Natera®
In these approaches, PCR amplification of selected genes enriches for the target regions of interest prior to NGS analysis. Forshew et al. [155] pioneered Tagged-Amplicon Sequencing (TAm-Seq), demonstrating detection of cancer mutations with allele frequencies as low as 2%, with sensitivity and specificity over 97%, and successfully applied this technique to monitor cancer mutations in patients with advanced ovarian cancer [155]. This method was subsequently applied to monitor ctDNA in patients with metastatic breast cancer [85]. Enhanced TAm-Seq, which incorporates more efficient library preparation and statistical analysis, later demonstrated identification of > 97.66% point mutations at > 0.5% allele frequency [156].
To improve ctDNA detection, Kinde et al. [111] reported a method of unique molecular identifier (UMI)-based multiplex PCR followed by NGS called Safe-SeqS (Safe Sequencing System), where they tagged each template molecule with a UMI, amplified each tagged molecule using PCR to create UMI families, and performed NGS. This redundant sequencing approach enabled highly sensitive detection of rare variants. The UMIs were used to group sequencing results into PCR families, which robustly distinguished biologic variants from PCR errors, enabling reduction of the error rate to less than an average of 2.0 × 10–4 errors/bp [111]. Safe-SeqS was successfully applied in clinical cohorts where it demonstrated the ability to detect > 75% patients with multiple advanced cancer types [69] and highly specific detection of MRD after surgery for colorectal cancer [81, 82].
Abbosh et al. [64] applied a similar UMI-based multiplex PCR NGS approach that was patient-customized with primers designed to detect SNVs identified by tumor exome sequencing in conjunction with the company Natera® [64]. The authors reported results from TRACERx [Tracking Non-Small-Cell Lung Cancer (NSCLC) Evolution Through Therapy (Rx)], a multicenter prospective study that followed patients with stage IA through to IIIA NSCLC treated with curative-intent surgical resection [90, 157]. High-depth, multi-region tumor whole-exome sequencing (M-Seq) was performed to account for intra-tumoral heterogeneity and to construct phylogenetic trees [90, 157]. Personalized, patient-specific multiplex PCR assays targeting clonal and subclonal SNVs and incorporating UMIs were applied to cfDNA, then followed by NGS [64]. These assays prioritized coverage of SNVs in driver genes, but also included candidate passenger variants [64]. This technique yielded an analytical sensitivity of above 99% for detecting SNVs present at greater than 0.1% and a specificity of detecting a single SNV of 99.6% [64].
As a whole, amplicon-based targeted NGS methods are robust and highly sensitive and specific. However PCR amplification can potentially bias the observed mutant allele fraction [158], and these techniques are limited to mutation detection within the queried amplicon space [19, 60, 91].
4.3.2. Hybrid Capture-Based NGS: Cancer Personalized Profiling by Deep Sequencing (CAPP-Seq), Targeted Error Correction Sequencing (TEC-Seq), Guardant360®, and FoundationOne® Liquid
Hybrid capture-based NGS was recently developed to improve the detection of multiple cancer mutations with high sensitivity and without significant prior knowledge of the alterations [19, 60, 91]. In this ‘hybrid capture’ approach, relevant DNA sequences are hybridized to biotinylated probes [70]. Biotin is bound to streptavidin beads and then unbound DNA is washed away to ‘pull down’ the bait DNA for NGS analysis [70].
Using this principle, Newman et al. [70] implemented an ultrasensitive ctDNA detection technique called Cancer Personalized Profiling by Deep Sequencing (CAPP-Seq) [70]. Recurrently mutated genomic regions in the population of a given cancer type are identified through bioinformatic analysis of cancer WES and/or WGS data from databases such as The Cancer Genome Atlas (TCGA) and Catalog of Somatic Mutations in Cancer (COSMIC) [70]. Biotinylated probes are designed against these recurrently mutated regions, and are referred to collectively as the ‘Selector’ [70]. The ‘Selector’ is applied to cfDNA of cancer patients, which is followed by NGS in order to quantitate ctDNA with a detection limit of ~ 0.02% [19, 70]. The detection limit was significantly improved to ~ 0.001% when a NGS error-correction strategy called integrated digital error suppression (IDES) was implemented. This strategy utilized UMIs to reduce the effect of PCR errors and a bioinformatic error correction step called ‘polishing’ to reduce the effect of stereotypical background artifacts [84]. An analogous hybrid capture NGS method with error correction, targeted error correction sequencing (TEC-Seq), was developed by Phallen et al. [73] and demonstrated ability to detect early-stage cancers. A key difference, however, is that CAPP-Seq leverages duplex sequencing in its detection approach while TEC-Seq does not [73, 84], thus CAPP-Seq likely has a lower error rate per base and may achieve a lower theoretical limit of detection than TEC-Seq.
CAPP-Seq has some unique advantages compared to other methods. It has the capability to detect SNVs, insertions/deletions (indels), SCNAs, and genomic rearrangements without assay personalization [70]. It incorporates the Fusion And Chromosomal Translocation Enumeration and Recovery Algorithm (FACTERA), a highly sensitive and specific method for identifying gene rearrangements [159]. Additionally, CAPP-Seq is generalizable to nearly all tumor types and does not require patient-specific optimization [19, 70, 72, 86, 149, 160]. Compared to amplicon-based NGS, CAPP-Seq can more reliably detect copy number changes and allow for detection of fusion proteins [19, 70, 158, 159]. Finally, because the CAPP-Seq Selector is not customized per patient, but rather enables broad assessment within a cancer type [19, 70], results from sequencing can reveal new and complex mechanisms of carcinogenesis and treatment resistance [75, 76]. CAPP-Seq has been applied to the detection of MRD in localized lung cancer after treatment [63], early response assessment of diffuse large B cell lymphoma (DLBCL) after the first cycle of chemotherapy [86], as well as to bladder cancer post-treatment surveillance using a version of the assay adapted to urinary cfDNA [6].
In addition to CAPP-Seq and Tec-Seq, several other ctDNA sequencing techniques utilize hybrid capture-based NGS. For instance, Guardant360® (Guardant Health, Inc.) utilizes a 150 kb panel encompassing 73 cancer-related genes for hybrid capture, followed by NGS with an average sequencing depth of ~ 15,000x, noise filtering and molecular tracking, and variant calling for SNVs, indels, CNAs, and fusions [161, 162]. Validation of the Guardant360® assay in adult patients with advanced-stage solid tumors revealed high clinical sensitivity (85.9%), and SNV detection specificity reported to be 97% [162]. Another commercial assay that utilizes hybrid capture-based NGS technology, Foundation Medicine Inc.’s FoundationOne® Liquid, includes 70 genes implicated in cancer growth and genomic biomarkers for microsatellite instability in its capture panel [163]. It is reported to have > 99% analytical sensitivity for SNVs, indels, and gene rearrangements at mutant allele fractions > 0.5%, and 99% positive predictive value for all alterations [163]. While these commercially available platforms have high self-reported accuracy, specificity, and sensitivity to detect and quantify ctDNA, a comparative study of two hybrid capture-based NGS platforms demonstrated that 40% of patients had ctDNA alterations that were completely incongruent between the two assays [164]. Furthermore, by not incorporating sequencing information from matched PBMCs in these commercial assays, their reported results may be confounded by CHIP [92, 93]. Future comparative studies are needed to determine the reliability and potential utility of these platforms in the clinical management of patients with cancer.
4.4. Combinatorial Approaches
While some tumor types such as lung cancer and melanoma have high tumor mutational burden (TMB) with hotspot regions containing SNVs and indels that are amenable to ctDNA analysis using the aforementioned techniques [69, 160, 165], other cancer types may contain mutations at low prevalence and/or harbor a more heterogeneous spectrum of mutation types including genome-wide SCNAs [149, 165]. To optimize ctDNA analysis in these scenarios, investigators have incorporated approaches that combine multiple detection methods. For example, Przybyl et al. [149] combined CAPP-Seq and Genome Representation Profiling (GRP), a shallow whole-genome sequence technique for the assessment of genome-wide SCNAs [166], to detect tumor-derived alterations within the cell-free compartment of leiomyosarcoma patients. Springer et al. [7] developed a technique for detecting bladder cancer from urine sediment, called UroSEEK, that applied three separate tests to urine sediment: a ten-gene panel for Safe-SeqS (that included KRAS, TP53, and PIK3CA), a TERT promoter-specific Safe-SeqS test, and FAST-SeqS, which was used to evaluate genome-wide aneuploidy using an unpaired PCR primer targeting ~ 38,000 members of long interspersed nucleotide element-1 retrotransposons (LINEs) [7].
In another recent study by O’Leary et al. [68], ctDNA was assessed at baseline and end of treatment from 195 breast cancer patients in the PALOMA-3 (Palbociclib [PD-0332991] Combined with Fulvestrant in Hormone Receptor+ HER2-Negative Metastatic Breast Cancer After Endocrine Failure) randomized phase III trial [68, 167]. The authors developed a method to assess tumor DNA purity in plasma via a targeted amplicon panel against ~ 1000 SNPs in regions commonly lost in breast cancer, and also performed dPCR for PIK3CA and ESR1 mutations [68]. Using this method, they identified plasma samples with tumor purity in plasma > 10% at day 1 and end of treatment, and performed WES on these samples (from 14 patients) and PyClone [168] analysis to detail clonal evolution non-invasively, including treatment-resistant subclonal RB1 and FGFR2 mutations that were confirmed by ddPCR [68]. The authors used their exome sequencing data to design a targeted panel for error-corrected amplicon-based NGS to assess the remainder of their cohort, thus more broadly defining the genomic landscape and clonal evolution in response to breast cancer treatment with palbociclib and fulvestrant, and validated several of their findings with ddPCR [68]. Overall, the combinatorial approach employed by O’Leary et al. [68] capably utilized the complementary strengths of WES, targeted sequencing, and ddPCR, to enable a thorough tumor evolution analysis on an clinically meaningful cohort.
Another combinatorial approach is to combine ctDNA detection with circulating protein biomarkers. Cohen et al. [169] utilized a multiplex PCR-based NGS assay to measure ctDNA using a 61-amplicon panel, and combined it with protein biomarker assessment in a combined assay called CancerSEEK. The immunoassay platform for protein biomarkers was optimized to increase sensitivity while maintaining high specificity in the detection of eight different cancer types (ovarian, liver, gastric, pancreatic, esophageal, colorectal, lung, and breast). When applied to 1005 patients with non-metastatic cancer, CancerSEEK had a median sensitivity and specificity of 70% and > 99%, respectively [169]. Then, using machine learning to predict the cancer’s tissue of origin, the assay localized the primary tumor to two anatomic sites in a median of 83% of patients and to a single site in a median of 63% of patients [169]. Using a similar approach, Cohen et al. [74] combined Safe-SeqS for KRAS mutations with carbohydrate antigen 19–9 (CA-19–9) as well as other protein biomarkers including carcinoembryonic antigen (CEA), hepatocyte growth factor (HGF), and osteopontin (OPN) to develop a non-invasive blood test for the detection of resectable pancreatic ductal adenocarcinoma. The five-component combinatorial panel led to a sensitivity of 64%, compared with 30% when KRAS was utilized alone [74]. Together, these studies illustrate the potential for combinatorial noninvasive assays to offer more sensitive cancer detection.
5. Application of ctDNA Molecular Residual Disease Detection to Solid Tumors
Analysis of ctDNA has shown promising clinical potential as a way to detect MRD for solid tumors following curative-intent treatment and in advance of clinical or radiographic disease relapse [63]. Furthermore, in small studies MRD detection by ctDNA profiling correlated with worse prognosis in patients with different types of solid tumor malignancy [6, 63, 64, 81, 82, 83, 87, 91].
In Sects. 5.1–5.5, recent developments in ctDNA MRD detection for lung, breast, colon, pancreatic, and bladder cancer are discussed with a focus on how early post-treatment ctDNA detection may inform and potentially improve adjuvant treatment strategies (Figs. 2 and 4).
Fig. 4:
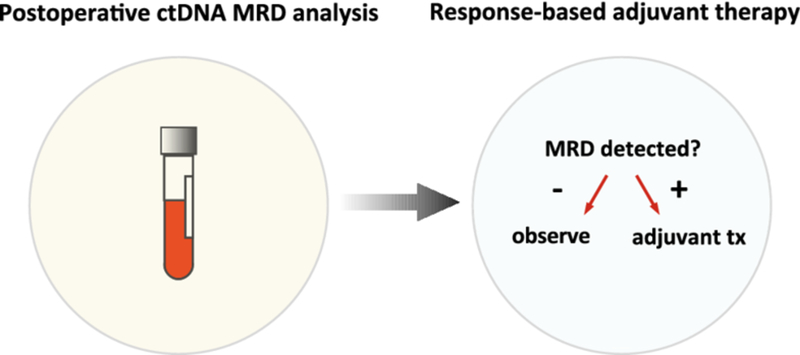
ctDNA MRD as a potential tool for adjuvant therapy clinical decision-making.
Postoperative ctDNA MRD analysis may be useful for identifying patients who are candidates for adjuvant systemic treatment versus those who can be safely observed, a paradigm that is being tested in the c-TRAK-TN trial in patients with resectable triple negative breast cancer (NCT03145961 [230]). ctDNA circulating tumor DNA, MRD molecular residual disease, tx treatment
5.1. Lung Cancer
In the treatment of lung cancer, routine clinical surveillance involves radiographic imaging [170]. However, standard imaging methods can only detect macroscopic disease recurrence [171], can be equivocal because inflammation or fibrosis are difficult to distinguish from tumor tissue especially after radiotherapy [172, 173], and outcomes are typically poor after clinical disease progression [174].
Chaudhuri et al. [63] hypothesized that ctDNA measured shortly after curative-intent treatment could identify patients with localized lung cancer who are at high risk for relapse, and post-treatment ctDNA could outperform standard-of-care imaging surveillance. The authors used CAPP-Seq to show that ctDNA detected within 4 months of treatment completion (defined as the MRD landmark) robustly identified patients who eventually relapsed with 94% sensitivity and 100% specificity, due in part to the detection of multiple somatic mutations (both driver and passenger) per patient [63]. ctDNA levels at the MRD landmark were confidently detected to mutant allele fractions as low as 0.003% [63]. Compared with ctDNA MRD-negative patients, ctDNA MRD-positive patients had significantly worse overall survival (p < 0.001, hazard ratio [HR] 14.3, 95% confidence interval [CI] 3.2–64.1), while computed tomography (CT) imaging at the same MRD landmark was not prognostic [63]. ctDNA MRD detection had a positive predictive value of 100% and a negative predictive value of 93%. Furthermore, post-treatment ctDNA detection preceded radiographic progression by a median of 5.2 months in 72% of patients who developed recurrence [63].
In addition to using ctDNA levels to prognosticate, molecular genomic information from ctDNA analysis could potentially guide patient-directed therapy. As reported by Chaudhuri et al. [63], some patients in the study cohort had activating EGFR mutations detectable in ctDNA at the MRD landmark. In these patients, ctDNA MRD detection might have enabled earlier targeted intervention with a first-generation EGFR tyrosine kinase inhibitor, such as erlotinib [175, 176]. The authors also showed that quantification of non-synonymous mutations in the CAPP-Seq Selector space allowed for estimation of exome-wide TMB from ctDNA [63], a finding also demonstrated by Gandara et al. [177] with the FoundationOne® CDx NGS assay [178] using cfDNA derived from the POPLAR and OAK advanced NSCLC immunotherapy trials [179, 180]. This is clinically relevant because elevated TMB has been shown to be a biomarker for response to immune checkpoint blockade in NSCLC [181, 182, 183].
Abbosh et al. [64, 91] also demonstrated the ability to detect MRD shortly after definitive-intent NSCLC treatment completion using a personalized UMI-based multiplex PCR NGS approach in the TRACERx study. The authors showed that 13 of the 14 early-stage NSCLC patients with post-surgical relapse diagnosed radiographically were also positive for the presence of ctDNA in plasma either before or at the time of diagnosed recurrence [64]. Five of the patients with ctDNA detection that preceded radiographic confirmation of tumor recurrence by over 100 days had a median clonal mutant allele fraction of < 0.1%, suggesting highly sensitive methods are necessary to robustly detect MRD [63, 64, 91]. The authors also used a modified version of PyClone to perform phylogenetic ctDNA analyses to track the subclonal nature of lung cancer relapse and progression, including a case where the subclonal mutation implicated in driving cancer relapse was a HER2 amplification event, which may have been amenable to targeted therapy [64]. Overall, the highly sensitive methods employed by Abbosh et al. [64, 91] and Chaudhuri et al. [63] suggest that ctDNA MRD can be robustly detected in patients with localized lung cancer after treatment, potentially enabling earlier clinical intervention and more informed treatment selection for relapse.
5.2. Breast Cancer
By using mutation tracking in postsurgical blood samples, Nicholas Turner’s group used a ddPCR approach to detect ctDNA and predict relapse in non-metastatic breast cancer patients treated with chemotherapy, surgery, and in some cases radiotherapy [83]. Among patients who eventually relapsed, 50% had detectable ctDNA MRD in a single postoperative sample drawn at 2–4 weeks after surgery [83]. In 37 analyzed patients, ctDNA MRD-positive patients exhibited significantly worse disease-free survival than ctDNA MRD-negative patients (p < 0.0001, HR 25.1; 95% CI 4.08–130.5) [83]. Serial mutation tracking in 43 patients beyond the postoperative timepoint increased the sensitivity of relapse prediction to 80% (p < 0.0001, HR 12.0, 95% CI 3.36–43.07), with ctDNA detected at a median of 7.9 months earlier than clinical relapse [83], thus opening a window to potentially intervene sooner with second-line treatment.
While the ddPCR approach to ctDNA MRD detection is highly sensitive, it requires patient-specific assay customization via a priori knowledge of tumor-specific somatic mutations [19, 60, 83]. The authors thus also applied a hybrid capture-based NGS approach to assess ctDNA pre-and posttreatment in five patients, and showed examples of emergent mutations detected post-treatment that were not identified pre-treatment [83]. The Turner group further optimized this technology and applied it to advanced-stage breast cancer samples from the PALOMA-3 randomized phase III trial [68, 167], and used it to analyze clonal evolution non-invasively, including the identification of treatment-resistant subclonal RB1 and FGFR2 mutations that were confirmed by ddPCR [68].
5.3. Colon Cancer
In colon cancer, the Roche AVENIO® targeted hybrid capture-based NGS platform has been applied to patients with advanced disease to identify resistance mechanisms to anti-EGFR monoclonal antibodies and to predict time to treatment failure [76]. The use of ctDNA to predict recurrence risk could not only benefit high-risk patients at risk for relapse, but also may spare low-risk patients from potentially serious morbidities associated with adjuvant systemic therapy [184]. While up to 40% of patients with stage II colorectal cancer undergo adjuvant therapy after resection [185], the absolute risk reduction is estimated to be only 3–5% [186]. Using the Safe-SeqS platform to detect ctDNA MRD after surgery in stage II colon cancer patients, Tie et al. [82] showed that detectable ctDNA MRD measured 4–10 weeks postoperatively in a population of 230 patients portended eventual radiographic recurrence with a hazard ratio of 18 (95% CI 7.9–40) and significantly outperformed standard-of-care clinicopathologic characteristics. This approach demonstrated a very high specificity, with 97% of patients with negative ctDNA postoperatively exhibiting no eventual relapse, while sensitivity was more modest at ~ 40% [82]. The lower sensitivity may have been because only a single mutant allele (identified by SafeSeqS [111] applied to formalin-fixed paraffin-embedded tumor) was tracked in the plasma of each patient [82]. This notwithstanding, results remained highly significant when ctDNA was measured shortly after adjuvant chemotherapy in patients who received it [82]. On serial follow-up, ctDNA detection preceded imaging recurrence by a median of 5.5 months, significantly earlier than the median 2-month lead time observed with the CEA protein biomarker [82]. In a subsequent prospective study of locally advanced rectal cancer evaluating 159 patients, Tie et al. [81] showed that ctDNA measured after neoadjuvant chemoradiation was also highly prognostic (p < 0.001). These results support the use of ctDNA MRD analysis in colorectal cancer after treatment as a potential biomarker to guide risk-adapted adjuvant therapy.
5.4. Pancreatic Cancer
Pancreatic cancer carries a poor prognosis with the majority of patients succumbing to disease even when they are candidates for curative-intent surgery [187]. Genomically, KRAS mutations are very common in pancreatic cancer and plasma KRAS mutations have been shown to improve pre-treatment detection compared to CA-19–9 alone [74]. To address whether ctDNA can be utilized to detect relapse after surgical resection, Sausen et al. [87] used dPCR to measure ctDNA after surgical resection of pancreatic adenocarcinoma and demonstrated a significant improvement in progression-free survival in patients with no post-treatment ctDNA detected compared to those with post-treatment ctDNA detected. Post-treatment ctDNA was detected on average 3.1 months after surgery, approximately 6.5 months earlier than progression scored by CT imaging [87]. It is unclear whether ctDNA MRD status also translated to an overall survival difference, and which patients received adjuvant chemotherapy. Regardless, these data are promising and indicate potential prognostic utility for ctDNA MRD in patients with resectable pancreatic cancer.
5.5. Bladder Cancer
As liquid biopsy is often focused on sampling ctDNA in plasma for solid tumor MRD detection, the analysis of ctDNA in other bodily fluids may be more robust for certain cancer types [8, 60]. For example, urine contains cfDNA arising from circulation that passes into the urinary system by glomerular filtration, as well as DNA arising from cells that make up the urinary tract [3, 8]. Springer et al. [7] developed UroSEEK to non-invasively detect bladder cancer by analyzing the genetic material in pelleted urine sediment. Applied to a cohort of 570 patients at risk for developing bladder cancer, UroSEEK demonstrated a significant increase in the sensitivity of cancer detection (83%) compared to conventional cytology (43%) [7]. UroSEEK positivity preceded the diagnosis of bladder cancer by an average of 2.3 months, and by more than 1 year in eight cases [7]. Applied to a separate surveillance cohort of 322 patients with bladder cancer where urine was collected following surgery, UroSEEK identified recurrent cancer on average 7 months earlier than standard-of-care, with a clinical sensitivity of 68% and specificity of 80% [7]. In contrast, surveillance cytology had a clinical sensitivity of only 25% [7].
More recently, Dudley et al. [6] focused on the cell-free component of urine (liquid component, excluding pellet) for bladder cancer detection. The authors demonstrated that a targeted hybrid capture-based NGS technique that they termed urine tumor DNA CAPP-Seq (uCAPP-Seq) enables sensitive detection of early-stage bladder cancer, with up to 93% of patients having detectable urinary tumor DNA (utDNA) pre-treatment, and a specificity of 96–100% when assessed in 67 healthy adults [6]. utDNA was also assessed in the post-treatment surveillance setting, and was detected in 91% of patients who ultimately recurred, with utDNA detection preceding clinical progression in 92% of patients by a median of 2.7 months [6]. Post-treatment uCAPP-Seq significantly outperformed standard-of-care urine cytology surveillance, which was positive in only 37.8% of patients who developed recurrence [6]. Moreover, uCAPP-Seq detected 100% of recurrence cases identified by cytology as well as 82% of recurrence cases missed by cytology [6]. Overall, the Dudley et al. [6] data and the Springer et al. [7] data suggest that DNA derived from urine can be used to identify bladder cancer earlier and more sensitively than standard-of-care urine cytology, and facilitate non-invasive detection, genotyping, and monitoring.
6. Cell-Free Viral DNA as a Tumor Biomarker
It is well-documented that oncogenic viruses can lead to various human cancers, such as human papillomavirus virus (HPV) causing cervical [188, 189], anal [190], and oropharyngeal cancer (OPC) [191], Epstein-Barr virus (EBV) causing nasopharyngeal cancer [192], and hepatitis B virus (HBV) and hepatitis C virus (HCV) causing hepatocellular carcinoma [193, 194, 195, 196]. In most cases, viral genome integration represents a key step in carcinogenesis [197, 198, 199]. Due to the crucial role of viral integration in carcinogenesis, some have proposed measuring circulating viral DNA in plasma and serum as a prognostic biomarker in cancer diagnosis [200, 201, 202, 203, 204, 205, 206, 207, 208, 209], treatment monitoring [208, 210], and residual disease detection [207, 208, 211, 212].
In nasopharyngeal carcinoma, the clinical utility of using circulating EBV DNA in diagnosis and prognosis has been robustly demonstrated with multiple large cohorts of patients [204, 205, 206, 207, 211, 212, 213]. For instance, in Leung et al. [213], pre-treatment circulating EBV DNA load was found to be an independent prognostic factor to Union for International Cancer Control (UICC) staging, and combining EBV DNA data with UICC staging data led to improved risk discrimination, especially in patients with early-stage disease [213]. In Lin et al. [207], patients with pre-treatment plasma EBV DNA concentrations of at least 1500 copies/mL had worse overall survival and relapse-free survival, and patients with persistently detectable plasma EBV DNA one week after completion of radiotherapy had significantly worse overall survival and relapse-free survival [207]. Based on these studies, circulating EBV DNA has become one of the leading cfDNA blood tests for evaluating nasopharyngeal carcinoma in China, Hong Kong, and Taiwan, where this cancer is endemic [214].
In OPC, HPV-positive disease is associated with improved overall survival and progression-free survival compared to HPV-negative disease [215, 216]. Dahlstrom et al. [217] showed that in HPV-positive OPC, there was a non-significant trend in pre-treatment circulating HPV DNA detection portending worse progression-free survival. Hanna et al. [218] used ddPCR to assess five HPV strains at multiple timepoints throughout immunotherapy or chemotherapy treatment of 22 patients with recurrent, metastatic HPV-positive OPC [218]. The authors demonstrated that tumor burden measured by imaging strongly correlated with HPV levels in the cfDNA, that median plasma HPV cfDNA levels negatively correlated with survival, and that all participants demonstrated a corresponding change in their HPV cfDNA levels at a median of 16 days before restaging scans confirmed treatment response or progression [218]. To better elucidate the heterogeneity of HPV-positive OPC, Gupta et al. [219] are leading an ongoing study that has revealed two broad patterns of HPV DNA in the blood: (1) a favorable-risk cohort of patients characterized by very high levels of pre-treatment HPV DNA in the blood, which drop to undetectable after treatment; and (2) an unfavorable-risk cohort of patients with lower pre-treatment levels of HPV DNA that rapidly increase after initiation of therapy. These studies illustrate the molecular heterogeneity of HPV-positive OPC, suggesting the amount of circulating HPV DNA in the blood as well as clearance kinetics could aid further risk-stratification in a cohort of patients who already have a favorable prognosis based on molecular testing [215, 216, 217, 218, 219].
In cervical cancer, HPV DNA testing from a cervical swab was shown to have greater sensitivity for the detection of cervical intraepithelial neoplasia than the Papanicolaou screening test [220], the standard-of-care for cervical cancer screening [221]. Campitelli et al. demonstrated the ability to detect tumor-specific HPV viral integration sites in the serum of 11 stage II to IV cervical cancer patients, and showed their correlation with tumor response to therapy in two patients [208]. Additionally, Han et al. [222] reported that in a cohort of 19 women with HPV-positive cervical cancer who received curative-intent chemoradiation, detectable plasma HPV DNA at the end of therapy preceded the clinical diagnosis of metastasis and was associated with inferior progression-free survival. Furthermore, 3-month plasma HPV DNA detection was more accurate than 3-month FDG-PET (fluorodeoxyglucose–positron emission tomography) imaging for identifying MRD in these patients [222].
While multiple studies have detailed the role of circulating viral DNA in the evaluation of various virus-induced cancers, limited progress has been made in using circulating HBV and HCV DNA in the setting of hepatocellular carcinoma. It may be difficult to use circulating viral DNA in the diagnosis and monitoring of hepatocellular carcinoma because acute and/or chronic hepatitis may be difficult to distinguish from hepatocellular carcinoma [223]. In fact, this challenge limits the use of most viral ctDNA assays, especially for diagnostic screening [32]. Therefore, it is important to establish clinically meaningful cut-off levels and potentially incorporate viral ctDNA data with other tumor biomarkers to improve the sensitivity and specificity of these assays [32].
7. Discussion and Future Perspectives
Growing evidence supports the use of ctDNA as a solid tumor MRD biomarker, with the potential to guide therapeutic decision-making. These studies have predominantly utilized blood plasma, although research suggests that analyses of other bodily fluids such as urine or saliva may hold great potential [7, 8, 16]. Due to the low ctDNA concentrations associated with MRD, detection techniques in the clinical workflow must consistently detect mutations in plasma at mutant allele fractions of < 0.1% and should incorporate strategies to control for sequencing artifacts [91]. Such strategies include multi-mutation detection and tracking, UMIs to reduce the effect of PCR errors, and approaches to reduce stereotypic noise such as background polishing [64, 73, 84, 111].
While preliminary data on the clinical utility of ctDNA in MRD detection is promising, the studies demonstrating this are mostly small, restricted to limited applications, and need to be validated using large independent cohorts [47, 60, 61]. Only with these further studies can we move forward to the next important question, which is whether acting on a ctDNA MRD-positive result can improve clinical outcomes, or if ctDNA MRD can be used to more precisely guide adjuvant therapy. Prospective trials testing the clinical utility of ctDNA MRD-based therapy will be the ultimate determinant of the significance of this research.
Accordingly, multiple trials are being set up in the ctDNA space, with a ClinicalTrials.gov search revealing that 358 studies involve ctDNA-based observation or intervention. When querying only actively recruiting studies, there are 204 such studies. These studies include, to name but a few, NCT02887612 [224], a study from Sun Yat-sen University in China that is querying ctDNA for the prediction of postoperative relapse in early and intermediate stage gastric cancer; NCT03691012 [225], a study from Johns Hopkins University and the Walter and Eliza Hall Institute of Medical Research measuring ctDNA as a marker of residual disease and response to adjuvant chemotherapy in ovarian cancer; and NCT03774758 [226], a study from the University of California in San Francisco that will follow patients undergoing lung cancer screening with ctDNA testing.
Further research also is required to explore the pre-analytical considerations and analytical validity of ctDNA analyses. For instance, more studies are necessary to address the pre-analytical variables that may affect ctDNA testing, including specimen type and quantity, sampling timepoints, handling variables, storage conditions and duration, and patient-related biologic factors [227]. Additionally, the accuracy, sensitivity, specificity, and robustness of different ctDNA detection methods remain to be optimized and refined, with closer attention needed for variants near an assay’s reported lower limit of detection. Cross-validation studies across different platforms using standardized samples are necessary to assess head-to-head performance [228, 229]. Since mutant allele fraction calculations may become inflated or deflated in some approaches versus others, such studies will also help normalize these variations across platforms.
Discordance between different ctDNA assays can also be related to the different sets of mutations queried. Given the low sensitivity of WES or WGS for ctDNA detection [19, 60], assays typically target specific loci and differences in the targeted sequencing space can account for some of the differences observed between commercial assays. Finally, differences in library preparation techniques, UMIs, duplex sequencing, variant calling, and targeted error correction may account for some of the differences observed between platforms.
Aside from factoring in the aforementioned pre-analytical variables and analytical validity, future ctDNA analyses including MRD detection must focus on demonstrating clinical validity and utility in oncologic care. Potential clinical applications include risk-stratification of patients based on molecular treatment response, tracking mutations associated with acquired therapeutic resistance and clonal evolution, and selecting appropriate targeted therapy. By optimizing risk versus benefit analyses using ctDNA-based prognostication, treatment de-escalation could spare low-risk patients from the toxicity of unnecessary adjuvant therapy. On the other hand, treatment intensification with aggressive adjuvant therapy in high-risk patients could maximize efficacy while disease burden and clonal heterogeneity are minimal [19, 91]. By using this risk-stratification strategy, prospective studies aiming to demonstrate clinical benefit of treatment could thus maximize their therapeutic index.
Future studies should focus on whether intervention at the early ctDNA MRD timepoint improves clinical outcomes. Currently, c-TRAK-TN is a multicenter phase II study of patients with resectable triple negative breast cancer (NCT03145961 [230]), which aims to assess whether ctDNA screening for one year after completing primary treatment enables the detection of MRD. Patients with detectable ctDNA MRD will be randomly assigned in a 2:1 ratio to receive either the immune checkpoint inhibitor pembrolizumab or undergo continued observation (Fig. 4). The results of this trial will provide insight into the utility of ctDNA MRD for the selection of patients for adjuvant systemic therapy. The clinical trial NRG-HN001 is applying a similar approach to locoregionally advanced nasopharyngeal cancer treated with curative-intent chemoradiation, with adjuvant chemotherapy given based on post-treatment MRD detection; however, it restricts analysis to only plasma EBV viral DNA. Similar clinical trials are needed to demonstrate the clinical utility of ctDNA MRD in other disease types and settings.
It will also be important to demonstrate that ctDNA can be cleared following effective adjuvant treatment, and that ctDNA clearance corresponds with improved patient survival. Abbosh et al. [64] demonstrated an example of this with patient CRUK0013, who had early-stage lung adenocarcinoma with detectable ctDNA after surgery that was cleared following adjuvant chemoradiation. The patient remained ctDNA undetectable on surveillance, and this correlated with long-term disease-free survival [64]. Similarly, Tie et al. [82] demonstrated three examples of colon cancer patients who were ctDNA MRD-positive after surgery that was cleared by adjuvant chemotherapy, and these patients remained recurrence-free long-term [82]. It will be important to systematically design future studies measuring ctDNA post-operatively (or post-definitive radiotherapy) and again during adjuvant systemic treatment to confirm the capability to track clearance of postoperative ctDNA MRD, and correlate this with clinical outcomes.
Large studies that apply ctDNA based genomic profiling to clinical cohorts are already underway. Zill et al. [108] compared genomic alteration patterns in ctDNA in a cohort of 21,807 advanced cancer patients with those in published sequencing studies compiled in TCGA [108]. Their comparison revealed significant correlation, except for treatment-induced secondary mutations that conferred resistance, such as EGFRT790M and EGFRC797S [231, 232, 233], which were only observed in ctDNA. This likely reflected cohort differences, with TCGA patients being pre-treatment and the ctDNA analysis patients typically being post-treatment with the assay performed to query suspected therapeutic resistance. This observation supports data discussed earlier [68, 74, 75, 76, 77], illustrating that ctDNA can identify resistant subclones with the potential to guide clinical decision-making.
8. Conclusion
The emergence of highly sensitive ctDNA assays has the potential to revolutionize cancer management. Future studies will be necessary to determine how to best integrate complementary information from standard solid tumor biopsies, clinical examination, and medical imaging with oncogenomic and MRD information from ctDNA-based liquid biopsies. Ultimately, the implementation of ctDNA MRD analysis into the clinical workflow has the potential to guide clinical decision-making and improve patient outcomes in this era of personalized precision medicine.
Key Points.
Circulating tumor DNA (ctDNA) is shed into the bloodstream and other bodily fluids and serves as a potential biomarker for post-treatment molecular residual disease (MRD) for solid tumor malignancies such as lung, breast, colon, pancreatic, and bladder cancer.
Different techniques can detect ctDNA with high sensitivity and specificity, including digital PCR, multiplex PCR-based next-generation sequencing (NGS), targeted hybrid capture-based NGS, and combinatorial approaches.
Interpretation of ctDNA needs to account for sources of somatic mosaicism such as clonal hematopoiesis of indeterminate potential (CHIP) that result in genetic variants in cell-free DNA unrelated to a solid tumor malignancy.
Acknowledgments
Funding
This work was funded by the NCI under award number K08CA238711 (A. A. Chaudhuri), a Cancer Research Foundation Young Investigator Award (A. A. Chaudhuri), the Washington University SPORE in Pancreatic Cancer Career Enhancement Program (A. A. Chaudhuri), and the Conquer Cancer Foundation ASCO Young Investigator Award (A. A. Chaudhuri), supported by Takeda Pharmaceuticals. Any opinions, findings and conclusions expressed in this material are those of the authors and do not necessarily reflect those of the American Society of Clinical Oncology®, Conquer Cancer®, or Takeda®.
Footnotes
Conflict of interest
Aadel A. Chaudhuri has received speaker honoraria and travel support from Varian Medical Systems, Roche Sequencing Solutions, and Foundation Medicine, Inc., has research support from Roche Sequencing Solutions, has served as a consultant for Tempus Labs and for Oscar Health, and is a scientific advisor for Roche Sequencing Solutions and for Geneoscopy, LLC. Jonathan C. Dudley has served as a consultant for Merck. Re-I Chin, Kevin Chen, Abul Usmani, Chanelle Chua, Peter K. Harris, Michael S. Binkley, and Tej D. Azad declare that they have no conflicts of interest that might be relevant to the contents of this review.
References
- 1.Mandel P, Métais P. Les acides nucléiques du plasma sanguin chez l’ homme. C R Seances Soc Biol Fil. 1948;142:241–3. [PubMed] [Google Scholar]
- 2.Vietsch EE, Wellstein A. Circulating DNA in cancer diagnosis and prognosis In: Dammacco F, Silvestris F, editors. Oncogenomics. London: Elsevier Inc.; 2019Google Scholar [Google Scholar]
- 3.Botezatu I, Serdyuk O, Potapova G, et al. Genetic analysis of DNA excreted in urine: a new approach for detecting specific genomic DNA sequences from cells dying in an organism. Clin Chem. 2000;46:1078–84. [PubMed] [Google Scholar]
- 4.Chan KCA, Leung SF, Yeung SW, et al. Quantitative analysis of the transrenal excretion of circulating EBV DNA in nasopharyngeal carcinoma patients. Clin Cancer Res. 2008;14:4809–13. [DOI] [PubMed] [Google Scholar]
- 5.Burnham P, Dadhania D, Heyang M, et al. Urinary cell-free DNA is a versatile analyte for monitoring infections of the urinary tract. Nat Commun. 2018;9:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dudley JC, Schroers-Martin J, Lazzareschi DV, et al. Detection and surveillance of bladder cancer using urine tumor DNA. Cancer Discov. 2018. 10.1158/2159-8290.CD-18-0825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Springer SU, Chen C-H, Rodriguez Pena MDC, et al. Non-invasive detection of urothelial cancer through the analysis of driver gene mutations and aneuploidy. Elife. 2018;7:1–27.Google Scholar [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu T, Li J. Clinical applications of urinary cell-free DNA in cancer: current insights and promising future. Am J Cancer Res. 2017;7:2318–32. [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Y, Springer S, Zhang M, et al. Detection of tumor-derived DNA in cerebrospinal fluid of patients with primary tumors of the brain and spinal cord. Proc Natl Acad Sci USA. 2015;112:9704–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pan W, Gu W, Nagpal S, et al. Brain tumor mutations detected in cerebral spinal fluid. Clin Chem. 2015;61:514–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Mattos-Arruda L, Mayor R, Ng CKY, et al. Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nat Commun. 2015;6:1–6.Google Scholar [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sriram KB, Relan V, Clarke BE, et al. Pleural fluid cell-free DNA integrity index to identify cytologically negative malignant pleural effusions including mesotheliomas. BMC Cancer. 2012;12:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soh J, Toyooka S, Aoe K, et al. Usefulness of EGFR mutation screening in pleural fluid to predict the clinical outcome of gefitinib treated patients with lung cancer. Int J Cancer. 2006;119:2353–8. [DOI] [PubMed] [Google Scholar]
- 14.Husain H, Nykin D, Bui N, et al. Cell-free DNA from ascites and pleural effusions: molecular insights into genomic aberrations and disease biology. Mol Cancer Ther. 2017;16:948–55. [DOI] [PubMed] [Google Scholar]
- 15.Mithani SK, Smith IM, Zhou S, et al. Mitochondrial resequencing arrays detect tumor-specific mutations in salivary rinses of patients with head and neck cancer. Clin Cancer Res. 2007;13:7335–40. [DOI] [PubMed] [Google Scholar]
- 16.Wei F, Lin C-C, Joon A, et al. Noninvasive saliva-based EGFR gene mutation detection in patients with lung cancer. Am J Respir Crit Care Med. 2014;190:1117–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jahr S, Hentze H, Englisch S, et al. DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001;61:1659–65. [PubMed] [Google Scholar]
- 18.Choi JJ, Reich CF, Pisetsky DS. The role of macrophages in the in vitro generation of extracellular DNA from apoptotic and necrotic cells. Immunology. 2005;115:55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chaudhuri AA, Binkley MS, Osmundson EC, et al. Predicting radiotherapy responses and treatment outcomes through analysis of circulating tumor DNA. Semin Radiat Oncol. 2015;25:305–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thierry AR, El Messaoudi S, Gahan PB, et al. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 2016;35:347–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stroun M, Lyautey J, Lederrey C, et al. About the possible origin and mechanism of circulating DNA apoptosis and active DNA release. Ann N Y Acad Sci. 2006;906:161–8. [DOI] [PubMed] [Google Scholar]
- 22.Anker P, Stroun M, Maurice PA. Spontaneous extracellular synthesis of DNA released by human blood lymphocytes. Cancer Res. 1976;36:2832–9. [PubMed] [Google Scholar]
- 23.Bronkhorst AJ, Wentzel JF, Aucamp J, et al. Characterization of the cell-free DNA released by cultured cancer cells. Biochim Biophys Acta. 2016;1863:157–65. [DOI] [PubMed] [Google Scholar]
- 24.Tsumita T, Iwanaga M. Fate of injected deoxyribonucleic acid in mice. Nature. 1963;198:1088–8 [DOI] [PubMed] [Google Scholar]
- 25.Korabecna M, Opatrna S, Wirth J, et al. Cell-free plasma DNA during peritoneal dialysis and hemodialysis and in patients with chronic kidney disease. Ann N Y Acad Sci. 2008;1137:296–301. [DOI] [PubMed] [Google Scholar]
- 26.Lo YM, Zhang J, Leung TN, et al. Rapid clearance of fetal DNA from maternal plasma. Am J Hum Genet. 1999;64:218–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tamkovich SN, Cherepanova AV, Kolesnikova EV, et al. Circulating DNA and DNase activity in human blood. Ann N Y Acad Sci. 2006;1075:191–6. [DOI] [PubMed] [Google Scholar]
- 28.Yao W, Mei C, Nan X, et al. Evaluation and comparison of in vitro degradation kinetics of DNA in serum, urine and saliva: a qualitative study. Gene. 2016;590:142–8. [DOI] [PubMed] [Google Scholar]
- 29.Elshimali YI, Khaddour H, Sarkissyan M, et al. The clinical utilization of circulating cell free DNA (CCFDNA) in blood of cancer patients. Int J Mol Sci. 2013;14:18925–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lui YYN, Chik KW, Chiu RWK, et al. Predominant hematopoietic origin of cell-free DNA in plasma and serum after sex-mismatched bone marrow transplantation. Clin Chem. 2002;48:421–7. [PubMed] [Google Scholar]
- 31.Snyder MW, Kircher M, Hill AJ, et al. Cell-free DNA comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Cell. 2016;164:57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schwarzenbach H, Hoon DSB, Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer. 2011;11:426–37. [DOI] [PubMed] [Google Scholar]
- 33.Lehmann-Werman R, Neiman D, Zemmour H, et al. Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc Natl Acad Sci USA. 2016;113:E1826–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chang CPY, Chia RH, Wu TL, et al. Elevated cell-free serum DNA detected in patients with myocardial infarction. Clin Chim Acta. 2003;327:95–101. [DOI] [PubMed] [Google Scholar]
- 35.Bustamante A, Mancha F, Macher HC, et al. Circulating cell-free DNA is a predictor of short-term neurological outcome in stroke patients treated with intravenous thrombolysis. J Circ Biomarkers. 2016;5:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O’Connell GC, Petrone AB, Tennant CS, et al. Circulating extracellular DNA levels are acutely elevated in ischaemic stroke and associated with innate immune system activation. Brain Inj. 2017;31:1369–75. [DOI] [PubMed] [Google Scholar]
- 37.De Vlaminck I, Martin L, Kertesz M, et al. Noninvasive monitoring of infection and rejection after lung transplantation. Proc Natl Acad Sci USA. 2015;112:13336–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lo YMD, Rainer TH, Chan LYS, et al. Plasma DNA as a prognostic marker in trauma patients. Clin Chem. 2000;46:319–23. [PubMed] [Google Scholar]
- 39.Weerakoon KG, McManus DP. Cell-Free DNA as a diagnostic tool for human parasitic infections. Trends Parasitol. 2016;32:378–91. [DOI] [PubMed] [Google Scholar]
- 40.Margraf S, Lögters T, Reipen J, et al. Neutrophil-derived circulating free DNA (CF-DNA/NETs): a potential prognostic marker for posttraumatic development of inflammatory second hit and sepsis. Shock. 2008;30:352–8. [DOI] [PubMed] [Google Scholar]
- 41.Zhong XY, Von Mühlenen I, Li Y, et al. Increased concentrations of antibody-bound circulatory cell-free DNA in rheumatoid arthritis. Clin Chem. 2007;53:1609–14. [DOI] [PubMed] [Google Scholar]
- 42.Tug S, Helmig S, Menke J, et al. Correlation between cell free DNA levels and medical evaluation of disease progression in systemic lupus erythematosus patients. Cell Immunol. 2014;292:32–9. [DOI] [PubMed] [Google Scholar]
- 43.Lo D, Sargent IL, Redman CW, et al. Presence of fetal DNA in maternal plasma and serum. Lancet. 1997;350:485–7. [DOI] [PubMed] [Google Scholar]
- 44.Tug S, Helmig S, Deichmann ER, et al. Exercise-induced increases in cell free DNA in human plasma originate predominantly from cells of the haematopoietic lineage. Exerc Immunol Rev. 2015;21:164–73. [PubMed] [Google Scholar]
- 45.Atamaniuk J, Vidotto C, Tschan H, et al. Increased concentrations of cell-free plasma DNA after exhaustive exercise. Clin Chem. 2004;50:1664–8. [DOI] [PubMed] [Google Scholar]
- 46.Breitbach S, Tug S, Simon P. Circulating cell-free DNA: an up-coming molecular marker in exercise physiology. Sport Med. 2012;42:565–86. [DOI] [PubMed] [Google Scholar]
- 47.Corcoran RB, Chabner BA. Application of cell-free DNA analysis to cancer treatment. N Engl J Med. 2018;379:1754–65. [DOI] [PubMed] [Google Scholar]
- 48.Lo YMD, Chan KCA, Sun H, et al. Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci Transl Med. 2010;2:1–13. [DOI] [PubMed] [Google Scholar]
- 49.Chandrananda D, Thorne NP, Bahlo M. High-resolution characterization of sequence signatures due to non-random cleavage of cell-free DNA. BMC Med Genom. 2015;8:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jiang P, Lo YMD. The long and short of circulating cell-free DNA and the ins and outs of molecular diagnostics. Trends Genet. 2016;32:360–71. [DOI] [PubMed] [Google Scholar]
- 51.Underhill HR, Kitzman JO, Hellwig S, et al. Fragment length of circulating tumor DNA. PLoS Genet. 2016;12:1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mouliere F, El Messaoudi S, Gongora C, et al. Circulating cell-free DNA from colorectal cancer patients may reveal high KRAS or BRAF mutation load. Transl Oncol. 2013;6:319–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mouliere F, Robert B, Peyrotte E, et al. High fragmentation characterizes tumour-derived circulating DNA. PLoS One. 2011;6:e23418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mouliere F, Chandrananda D, Piskorz AM, et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci Transl Med. 2018;10:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu SCY, Chan KCA, Zheng YWL, et al. Size-based molecular diagnostics using plasma DNA for noninvasive prenatal testing. Proc Natl Acad Sci USA. 2014;111:8583–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lun FM, Tsui NB, Chan KA, et al. Noninvasive prenatal diagnosis of monogenic diseases by digital size selection and relative mutation dosage on DNA in maternal plasma. Proc. Natl. Acad. Sci. 2008;105:19920–19925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Minarik G, Repiska G, Hyblova M, et al. Utilization of benchtop next generation sequencing platforms ion torrent PGM and miseq in noninvasive prenatal testing for chromosome 21 trisomy and testing of impact of in silico and physical size selection on its analytical performance. PLoS One. 2015;10:1–12.Google Scholar [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ulz P, Thallinger GG, Auer M, et al. Inferring expressed genes by whole-genome sequencing of plasma DNA. Nat Genet. 2016;48:1273–8. [DOI] [PubMed] [Google Scholar]
- 59.Stroun M, Anker P, Lyautey J, et al. Isolation and characterization of DNA from the plasma of cancer patients. Eur J Cancer Clin Oncol. 1987;23:707–12. [DOI] [PubMed] [Google Scholar]
- 60.Wan JCM, Massie C, Garcia-Corbacho J, et al. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat Rev Cancer. 2017;17:223–38. [DOI] [PubMed] [Google Scholar]
- 61.Heitzer E, Haque IS, Roberts CES, et al. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat Rev Genet. 2019;20(2):71–88. [DOI] [PubMed] [Google Scholar]
- 62.Sirera R, Bremnes RM, Cabrera A, et al. Circulating DNA is a useful prognostic factor in patients with advanced non-small cell lung cancer. J Thorac Oncol. 2011;6:286–90. [DOI] [PubMed] [Google Scholar]
- 63.Chaudhuri AA, Chabon JJ, Lovejoy AF, et al. Early detection of molecular residual disease in localized lung cancer by circulating tumor DNA profiling. Cancer Discov. 2017;7:1394–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Abbosh C, Birkbak NJ, Wilson GA, et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature. 2017;545:446–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nong J, Gong Y, Guan Y, et al. Circulating tumor DNA analysis depicts subclonal architecture and genomic evolution of small cell lung cancer. Nat Commun. 2018;9:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Diehl F, Schmidt K, Choti MA, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14:985–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.To EWH, Chan KCA, Leung S, et al. Rapid clearance of plasma Epstein-Barr virus DNA after surgical treatment of nasopharyngeal carcinoma. Clin Cancer Res. 2003;9:3254–9. [PubMed] [Google Scholar]
- 68.O’Leary B, Cutts RJ, Liu Y, et al. The genetic landscape and clonal evolution of breast cancer resistance to palbociclib plus fulvestrant in the PALOMA-3 trial. Cancer Discov. 2018;8:1390–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bettegowda C, Sausen M, Leary RJ, et al. Detection of circulating tumor DNA in early-and late-stage human malignancies. Sci Transl Med. 2014;6:224ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Newman AM, Bratman SV, To J, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014;20:548–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chung JH, Pavlick D, Hartmaier R, et al. Hybrid capture-based genomic profiling of circulating tumor DNA from patients with estrogen receptor-positive metastatic breast cancer. Ann Oncol. 2017;28:2866–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Scherer F, Kurtz DM, Newman AM, et al. Distinct biological subtypes and patterns of genome evolution in lymphoma revealed by circulating tumor DNA. Sci Transl Med. 2016;8:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Phallen J, Sausen M, Adleff V, et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci Transl Med. 2017;9:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cohen JD, Javed AA, Thoburn C, et al. Combined circulating tumor DNA and protein biomarker-based liquid biopsy for the earlier detection of pancreatic cancers. Proc Natl Acad Sci USA. 2017;114:10203–7.Google Scholar [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chabon JJ, Simmons AD, Lovejoy AF, et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun. 2016;7:1–14.Google Scholar [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Khan KH, Cunningham D, Werner B, et al. Longitudinal liquid biopsy and mathematical modeling of clonal evolution forecast time to treatment failure in the PROSPECT-C phase II colorectal cancer clinical trial. Cancer Discov. 2018;8(10):1270–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Oxnard GR, Thress KS, Alden RS, et al. Association between plasma genotyping and outcomes of treatment with osimertinib (AZD9291) in advanced non-small-cell lung cancer. J Clin Oncol. 2016;34:3375–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Thress KS, Paweletz CP, Felip E, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med. 2015; 21:560–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Murtaza M, Dawson SJ, Tsui DWY, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013;497:108–12. [DOI] [PubMed] [Google Scholar]
- 80.Taniguchi K, Uchida J, Nishino K, et al. Quantitative detection of EGFR mutations in circulating tumor DNA derived from lung adenocarcinomas. Clin Cancer Res. 2011;17:7808–15. [DOI] [PubMed] [Google Scholar]
- 81.Tie J, Cohen JD, Wang Y, et al. Serial circulating tumour DNA analysis during multimodality treatment of locally advanced rectal cancer: a prospective biomarker study. Gut. 2019;68(4):663–71. 10.1136/gutjnl-2017-315852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tie J, Wang Y, Tomasetti C, et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci Transl Med. 2016;8:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Garcia-Murillas I, Schiavon G, Weigelt B, et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci Transl Med. 2015;7:1–7. [DOI] [PubMed] [Google Scholar]
- 84.Newman AM, Lovejoy AF, Klass DM, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol. 2016;34:547–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dawson S-J, Tsui DWY, Murtaza M, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013;368:1199–209. [DOI] [PubMed] [Google Scholar]
- 86.Kurtz DM, Scherer F, Jin MC, et al. Circulating tumor DNA measurements as early outcome predictors in diffuse large B-cell lymphoma. J Clin Oncol. 2018;36(28):2845–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sausen M, Phallen J, Adleff V, et al. Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nat Commun. 2015;6:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Endesfelder D, Math D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.De Mattos-Arruda L, Weigelt B, Cortes J, et al. Capturing intra-tumor genetic heterogeneity by de novo mutation profiling of circulating cell-free tumor DNA: a proof-of-principle. Ann Oncol. 2014;25:1729–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jamal-Hanjani M, Wilson GA, McGranahan N, et al. Tracking the evolution of non-small-cell lung cancer. N Engl J Med. 2017;376:2109–21. [DOI] [PubMed] [Google Scholar]
- 91.Abbosh C, Birkbak NJ, Swanton C. Early stage NSCLC: challenges to implementing ctDNA-based screening and MRD detection. Nat Rev Clin Oncol. 2018;15(9):577–86. [DOI] [PubMed] [Google Scholar]
- 92.Swanton C, Venn O, Aravanis A, et al. Prevalence of clonal hematopoiesis of indeterminate potential (CHIP) measured by an ultra-sensitive sequencing assay: exploratory analysis of the Circulating Cancer Genome Atlas (CCGA) study. J Clin Oncol. 2018;36:12003. [Google Scholar]
- 93.Hu Y, Ulrich BC, Supplee J, et al. False-positive plasma genotyping due to clonal hematopoiesis. Clin Cancer Res. 2018;24:4437–43. [DOI] [PubMed] [Google Scholar]
- 94.Genovese G, Kahler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371:2477–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jaiswal S, Ebert BL. MDS is a stem cell disorder after all. Cancer Cell. 2014;25:713–4. [DOI] [PubMed] [Google Scholar]
- 97.Potter NE, Greaves M. Cancer: persistence of leukaemic ancestors. Nature. 2014;506:300–1. [DOI] [PubMed] [Google Scholar]
- 98.Damm F, Mylonas E, Cosson A, et al. Acquired initiating mutations in early hematopoietic cells of CLL patients. Cancer Discov. 2014;4:1088–101. [DOI] [PubMed] [Google Scholar]
- 99.Chung SS, Kim E, Park JH, et al. BRAFV600E mutations in hematopoietic stem cells of hairy cell leukemia patients suggests a stem cell origin. Sci Transl Med. 2014;99:404–5.Google Scholar [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Steensma DP, Bejar R, Jaiswal S, et al. Perspective: clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2018;126:9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Acuna-Hidalgo R, Sengul H, Steehouwer M, et al. Ultra-sensitive sequencing identifies high prevalence of clonal hematopoiesis-associated mutations throughout adult life. Am J Hum Genet. 2017;101:50–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zink F, Stacey SN, Norddahl GL, et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood. 2017;130:742–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Young AL, Challen GA, Birmann BM, et al. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat Commun. 2016;22(7):12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Xie M, Lu C, Wang J, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20:1472–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.McKerrell T, Park N, Moreno T, et al. Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis. Cell Rep. 2015;10:1239–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Coombs CC, Zehir A, Devlin SM, et al. Therapy-related clonal hematopoiesis in patients with non-hematologic cancers is common and associated with adverse clinical outcomes. Cell Stem Cell. 2017;21(374–382):e4.Google Scholar [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bailey MH, Tokheim C, Porta-Pardo E, et al. Comprehensive characterization of cancer driver genes and mutations. Cell. 2018;173(371–385):e18.Google Scholar [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zill OA, Banks KC, Fairclough SR, et al. The landscape of actionable genomic alterations in cell-free circulating tumor DNA from 21,807 advanced cancer patients. Clin Cancer Res. 2018;24:3528–38. [DOI] [PubMed] [Google Scholar]
- 109.McCulloch SD, Kunkel TA. The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res. 2008;18:148–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lynch M. Rate, molecular spectrum, and consequences of human mutation. Proc Natl Acad Sci USA. 2010;107:961–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kinde I, Wu J, Papadopoulos N, et al. Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci USA. 2011;108:9530–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Newton CR, Graham A, Heptinstall LE, et al. Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS). Nucleic Acids Res. 1989;17:2503–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wu DY, Ugozzoli L, Pal BK, et al. Allele-specific enzymatic amplification of beta-globin genomic DNA for diagnosis of sickle cell anemia. Proc Natl Acad Sci USA. 1989;86:2757–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kimura H, Kasahara K, Kawaishi M, et al. Detection of epidermal growth factor receptor mutations in serum as a predictor of the response to gefitinib in patients with non-small-cell lung cancer. Clin Cancer Res. 2006;12:3915–21. [DOI] [PubMed] [Google Scholar]
- 115.Khakoo S, Georgiou A, Gerlinger M, et al. Circulating tumour DNA, a promising biomarker for the management of colorectal cancer. Crit Rev Oncol Hematol. 2018;122:72–82. [DOI] [PubMed] [Google Scholar]
- 116.Milbury C, Li J, Makrigiorgos G. PCR-based methods for the enrichment of minority alleles and mutations. Clin Chem. 2009;55:632–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Vogelstein B, Kinzler KW. Digital PCR. Genetics. 1999;96:9236–41.Google Scholar [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gerdes L, Iwobi A, Busch U, et al. Optimization of digital droplet polymerase chain reaction for quantification of genetically modified organisms. Biomol Detect Quantif. 2016;7:9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hindson BJ, Ness KD, Masquelier DA, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011;83:8604–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Karlovich C, Goldman JW, Sun JM, et al. Assessment of EGFR mutation status in matched plasma and tumor tissue of NSCLC patients from a phase I study of rociletinib (CO-1686). Clin Cancer Res. 2016;22:2386–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Liu J, Huang S, Sun M, et al. An improved allele-specific PCR primer design method for SNP marker analysis and its application. Plant Methods. 2012;8:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kwok S, Kellogg DE, Mckinney N, et al. Effects of primer-template mismatches on the polymerase chain reaction: human immunodeficiency virus type 1 model studies. Nucleic Acids Res. 1990;18:999–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cha RS, Zarbl H, Keohavong P, et al. Mismatch amplification mutation assay (MAMA): application to the c-H-ras gene. Genome Res. 1992;2:14–20. [DOI] [PubMed] [Google Scholar]
- 124.Huggett JF, Whale A. Digital PCR as a novel technology and its potential implications for molecular diagnostics. Clin Chem. 2013;59:1691–3. [DOI] [PubMed] [Google Scholar]
- 125.Taly V, Pekin D, El Abed A, et al. Detecting biomarkers with microdroplet technology. Trends Mol Med. 2012;18:405–16. [DOI] [PubMed] [Google Scholar]
- 126.Dressman D, Yan H, Traverso G, et al. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc Natl Acad Sci USA. 2003;100:8817–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Nakano M, Komatsu J, Matsuura SI, et al. Single-molecule PCR using water-in-oil emulsion. J Biotechnol. 2003;102:117–24. [DOI] [PubMed] [Google Scholar]
- 128.Diehl F, Li M, He Y, et al. BEAMing: single-molecule PCR on microparticles in water-in-oil emulsions. Nat Methods. 2006;3:551–9. [DOI] [PubMed] [Google Scholar]
- 129.Taly V, Pekin D, Benhaim L, et al. Multiplex picodroplet digital PCR to detect KRAS mutations in circulating DNA from the plasma of colorectal cancer patients. Clin Chem. 2013;59:1722–31. [DOI] [PubMed] [Google Scholar]
- 130.Zhu G, Ye X, Dong Z, et al. Highly sensitive droplet digital PCR method for detection of EGFR-activating mutations in plasma cell-free DNA from patients with advanced non-small cell lung cancer. J Mol Diagnostics. 2015;17:265–72. [DOI] [PubMed] [Google Scholar]
- 131.Yung TKF, Chan KCA, Mok TSK, et al. Single-molecule detection of epidermal growth factor receptor mutations in plasma by microfluidics digital PCR in non-small cell lung cancer patients. Clin Cancer Res. 2009;15:2076–84. [DOI] [PubMed] [Google Scholar]
- 132.Sacher AG, Paweletz C, Dahlberg SE, et al. Prospective validation of rapid plasma genotyping as a sensitive and specific tool for guiding lung cancer care. JAMA Oncol. 2016;2:1014–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Diehl F, Li M, Dressman D, et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci USA. 2005;102:16368–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Schiavon G, Hrebien S, Garcia-Murillas I, et al. Analysis of ESR1 mutation in circulating tumor DNA demonstrates evolution during therapy for metastatic breast cancer. Sci Transl Med. 2015;7:148–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sanmamed MF, Fernandez-Landazuri S, Rodriguez C, et al. Quantitative cell-free circulating BRAFV600E mutation analysis by use of droplet digital PCR in the follow-up of patients with melanoma being treated with BRAF inhibitors. Clin Chem. 2015;61:297–304. [DOI] [PubMed] [Google Scholar]
- 136.Birkenkamp-Demtroder K, Nordentoft I, Christensen E, et al. Genomic alterations in liquid biopsies from patients with bladder cancer. Eur Urol. 2016;70:75–82. [DOI] [PubMed] [Google Scholar]
- 137.Christensen E, Nordentoft I, Vang S, et al. Optimized targeted sequencing of cell-free plasma DNA from bladder cancer patients. Sci Rep. 2018;8:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Alizadeh AA, Aranda V, Bardelli A, et al. Toward understanding and exploiting tumor heterogeneity. Nat Med. 2015;21:846–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Heitzer E, Ulz P, Belic J, et al. Tumor-associated copy number changes in the circulation of patients with prostate cancer identified through whole-genome sequencing. Genome Med. 2013;5:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Farris C, Trimarchi JM. Plasma-seq: a novel strategy for metastatic prostate cancer analysis. Genome Med. 2013;5:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kinde I, Papadopoulos N, Kinzler KW, et al. FAST-SeqS: a simple and efficient method for the detection of aneuploidy by massively parallel sequencing. PLoS One. 2012;7:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Douville C, Springer S, Kinde I, et al. Detection of aneuploidy in patients with cancer through amplification of long interspersed nucleotide elements (LINEs). Proc Natl Acad Sci USA. 2018;115:1871–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chan KCA, Jiang P, Zheng YWL, et al. Cancer genome scanning in plasma: detection of tumor-associated copy number aberrations, single-nucleotide variants, and tumoral heterogeneity by massively parallel sequencing. Clin Chem. 2013;59:211–24. [DOI] [PubMed] [Google Scholar]
- 144.Welch JS, Larson DE, Wallis J, et al. Use of whole-genome sequencing to diagnose a cryptic fusion oncogene. JAMA. 2011;305:1577–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Leary RJ, Kinde I, Diehl F, et al. Development of personalized tumor biomarkers using massively parallel sequencing. Sci Transl Med. 2010;2:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.McKernan KJ, Peckham HE, Costa G, et al. Sequence and structural variation in a human genome uncovered by short-read, massively parallel ligation sequencing using two-base encoding. Genome Res. 2009;19:1527–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.McBride DJ, Orpana AK, Sotiriou C, et al. Use of cancer-specific genomic rearrangements to quantify disease burden in plasma from patients with solid tumors. Genes. Chromosomes Cancer. 2010;49:1062–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kirkizlar E, Zimmermann B, Constantin T, et al. Detection of clonal and subclonal copy-number variants in cell-free DNA from patients with breast cancer using a massively multiplexed PCR methodology. Transl Oncol. 2015;8:407–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Przybyl J, Chabon JJ, Spans L, et al. Combination approach for detecting different types of alterations in circulating tumor DNA in leiomyosarcoma. Clin Cancer Res. 2018;24:2688–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Leary RJ, Sausen M, Kinde I, et al. Detection of chromosomal alterations in the circulation of cancer patients with whole-genome sequencing. Sci Transl Med. 2012;4:162ra154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Heitzer E, Auer M, Hoffmann EM, et al. Establishment of tumor-specific copy number alterations from plasma DNA of patients with cancer. Int J Cancer. 2013;133:346–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Belic J, Koch M, Ulz P, et al. Rapid identification of plasma DNA samples with increased ctDNA levels by a modified FAST-SeqS approach. Clin Chem. 2015;61:838–49. [DOI] [PubMed] [Google Scholar]
- 153.Han X, Wang J, Sun Y. Circulating tumor DNA as biomarkers for cancer detection. Genom Proteom Bioinform. 2017;15:59–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Choi M, Scholl UI, Ji W, et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci USA. 2009;106:19096–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Forshew T, Murtaza M, Parkinson C, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med. 2012;4:136ra68. [DOI] [PubMed] [Google Scholar]
- 156.Gale D, Plagnol V, Lawson A, et al. Analytical performance and validation of an enhanced TAm-Seq circulating tumor DNA sequencing assay [abstract]. In: AACR 107th annual meeting, 16–20 Apr 2016, New OrleansGoogle Scholar [Google Scholar]
- 157.Jamal-Hanjani M, Hackshaw A, Ngai Y, et al. Tracking genomic cancer evolution for precision medicine: the Lung TRACERx study. PLoS Biol. 2014;12:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Gagan J, Van Allen EM. Next-generation sequencing to guide cancer therapy. Genome Med. 2015;7:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Newman AM, Bratman SV, Stehr H, et al. FACTERA: a practical method for the discovery of genomic rearrangements at breakpoint resolution. Bioinformatics. 2014;30:3390–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Bratman SV, Newman AM, Alizadeh AA, et al. Potential clinical utility of ultrasensitive circulating tumor DNA detection with CAPP-Seq. Expert Rev Mol Diagn. 2015;15:715–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lanman RB, Mortimer SA, Zill OA, et al. Analytical and clinical validation of a digital sequencing panel for quantitative, highly accurate evaluation of cell-free circulating tumor DNA. PLoS One. 2015;10:1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Odegaard JI, Vincent JJ, Mortimer S, et al. Validation of a plasma-based comprehensive cancer genotyping assay utilizing orthogonal tissue-and plasma-based methodologies. Clin Cancer Res. 2018;24:3539–49. [DOI] [PubMed] [Google Scholar]
- 163.FoundationOne® Liquid. Foundation Medicing Inc; 2018. https://www.foundationmedicine.com/genomic-testing/foundation-one-liquid.
- 164.Torga G, Pienta KJ. Patient-paired sample congruence between 2 commercial liquid biopsy tests. JAMA Oncol. 2018;4:868–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Alexandrov LB, Nik-zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Amant F, Verheecke M, Wlodarska I, et al. Presymptomatic identification of cancers in pregnant women during noninvasive prenatal testing. JAMA Oncol. 2015;1:814–9. [DOI] [PubMed] [Google Scholar]
- 167.Cristofanilli M, Turner NC, Bondarenko I, Ro J, Im S-A, Masuda N, Colleoni M, DeMichele A, Loi S, Verma S, Iwata H, Harbeck N, Zhang K, Theall KP, Jiang Y, Bartlett CH, Koehler M. Slamon D (2016) Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. The Lancet Oncology. 2016;17:425–39. [DOI] [PubMed] [Google Scholar]
- 168.Roth A, Khattra J, Yap D, et al. PyClone: statistical inference of clonal population structure in cancer. Nat Methods. 2014;11:396–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Cohen JD, Li L, Wang Y, et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science. 2018;359:926–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Ettinger DS, Wood DE, Aisner DL, et al. Non-small cell lung cancer, version 5.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Cancer Netw. 2017;15:504–35. [DOI] [PubMed] [Google Scholar]
- 171.Billè A, Ahmad U, Woo KM, et al. Detection of recurrence patterns after wedge resection for early stage lung cancer: rationale for radiologic follow-up. Ann Thorac Surg. 2016;102:1067–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kocak Z, Evans ES, Zhou SM, et al. Challenges in defining radiation pneumonitis in patients with lung cancer. Int J Radiat Oncol Biol Phys. 2005;62:635–8. [DOI] [PubMed] [Google Scholar]
- 173.Huang K, Dahele M, Senan S, et al. Radiographic changes after lung stereotactic ablative radiotherapy (SABR)—can we distinguish recurrence from fibrosis? A systematic review of the literature. Radiother Oncol. 2012;102:335–42. [DOI] [PubMed] [Google Scholar]
- 174.Hung JJ, Hsu WH, Hsieh CC, et al. Post-recurrence survival in completely resected stage I non-small cell lung cancer with local recurrence. Thorax. 2009;64:192–6. [DOI] [PubMed] [Google Scholar]
- 175.Shepherd F, Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353:123–32. [DOI] [PubMed] [Google Scholar]
- 176.Pérez-Soler R, Chachoua A, Hammond LA, et al. Determinants of tumor response and survival with erlotinib in patients with non-small-cell lung cancer. J Clin Oncol. 2004;22:3238–47. [DOI] [PubMed] [Google Scholar]
- 177.Gandara DR, Paul SM, Kowanetz M, et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat Med. 2018;24(9):1441–8. [DOI] [PubMed] [Google Scholar]
- 178.Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31:1023–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Fehrenbacher L, Spira A, Ballinger M, Kowanetz M, Vansteenkiste J, Mazieres J, Park K, Smith D, Artal-Cortes A, Lewanski C, Braiteh F, Waterkamp D, He P, Zou W, Chen DS, Yi J, Sandler A, Rittmeyer A. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. The Lancet. 2016;387:1837–46. [DOI] [PubMed] [Google Scholar]
- 180.Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, von Pawel J, Gadgeel SM, Hida T, Kowalski DM, Dols MC, Cortinovis DL, Leach J, Polikoff J, Barrios C, Kabbinavar F, Frontera OA, De Marinis F, Turna H, Lee J-S, Ballinger M, Kowanetz M, He P, Chen DS, Sandler Gandara DR. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. The Lancet. 2017;389:255–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Goodman AM, Kato S, Bazhenova L, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16(11):2598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Rizvi NA, Hellmann MD, Snyder A, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Hellmann MD, Ciuleanu T-E, Pluzanski A, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med. 2018;378:2093–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Hoeben KWJ, van Steenbergen LN, van de Wouw AJ, et al. Treatment and complications in elderly stage III colon cancer patients in the Netherlands. Ann Oncol. 2013;24:974–9. [DOI] [PubMed] [Google Scholar]
- 185.Benson AB, Venook AP, Al-Hawary MM, et al. NCCN guidelines insights: colon cancer, version 2.2018. J Natl Compr Cancer Netw. 2018;16:359–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Wirtzfeld DA, Mikula L, Gryfe R, et al. Concordance with clinical practice guidelines for adjuvant chemotherapy in patients with stage I-III colon cancer: experience in 2 Canadian provinces. Can J Surg. 2009;52:92–7. [PMC free article] [PubMed] [Google Scholar]
- 187.Perini MV, Montagnini AL, Jukemura J, et al. Clinical and pathologic prognostic factors for curative resection for pancreatic cancer. HPB (Oxford). 2008;10:356–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Walboomers JMM, Jacobs MV, Manos MM, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol. 1999;189:12–9. [DOI] [PubMed] [Google Scholar]
- 189.Muñoz N, Bosch FX, de Sanjosé S, et al. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med. 2003;348:518–27. [DOI] [PubMed] [Google Scholar]
- 190.Frisch M, Glimelius B, van den Brule AJC, et al. Sexually transmitted infection as a cause of anal cancer. N Engl J Med. 1997;337:1350–8. [DOI] [PubMed] [Google Scholar]
- 191.Chaturvedi AK, Engels EA, Pfeiffer RM, et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J Clin Oncol. 2011;29:4294–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.zur Hausen H, Schulte-Holthausen H, Klein G, et al. EBV DNA in biopsies of Burkitt tumours and anaplastic carcinomas of the nasopharynx. Nature. 1970;228:1056–8. [DOI] [PubMed] [Google Scholar]
- 193.Kim C-M, Koike K, Saito I, et al. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature. 1988;351:317–20. [DOI] [PubMed] [Google Scholar]
- 194.Beasley RP. Hepatitis B virus. The major etiology of hepatocellular carcinoma. Cancer. 2016;61:1942–56. [DOI] [PubMed] [Google Scholar]
- 195.Saito I, Miyamura T, Ohbayashi A, et al. Hepatitis C virus infection is associated with the development of hepatocellular carcinoma. Proc Natl Acad Sci USA. 1990;87:6547–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Moriya K, Fujie H, Shintani Y, et al. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat Med. 1998;4:1065–7. [DOI] [PubMed] [Google Scholar]
- 197.Peitsaro P, Johansson B, Syrjänen S. Integrated human papillomavirus type 16 is frequently found in cervical cancer precursors as demonstrated by a novel quantitative real-time PCR technique. J Clin Microbiol. 2002;40:886–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Wiest T, Schwarz E, Enders C, Flechtenmacher C, Bosch FX. Involvement of intact HPV16 E6/E7 gene expression in head and neck cancers with unaltered p53 status and perturbed pRb cell cycle control. Oncogene. 2002;21:1510–7. [DOI] [PubMed] [Google Scholar]
- 199.Raab-Traub N Epstein-Barr virus in the pathogenesis of NPC. Semin Cancer Biol. 2002;14:423–30.Google Scholar [DOI] [PubMed] [Google Scholar]
- 200.Mayrand M-H, Duarte-Franco E, Rodrigues I, et al. Human papillomavirus DNA versus Papanicolaou screening tests for cervical cancer. New Engl. J Med. 2010;357:1579–1588. [DOI] [PubMed] [Google Scholar]
- 201.Manos MM, Kinney WK, Hurley LB, et al. Identifying women with cervical neoplasia using human papillomavirus DNA testing for equivocal Papanicolaou results. JAMA. 1999;281:1605–10. [DOI] [PubMed] [Google Scholar]
- 202.Ndiaye C, Mena M, Alemany L, et al. HPV DNA, E6/E7 mRNA, and p16INK4a detection in head and neck cancers: a systematic review and meta-analysis. Lancet Oncol. 2014;15:1319–31. [DOI] [PubMed] [Google Scholar]
- 203.Reimers N, Kasper HU, Weissenborn SJ, et al. Combined analysis of HPV-DNA, p16 and EGFR expression to predict prognosis in oropharyngeal cancer. Int J Cancer. 2007;120:1731–8. [DOI] [PubMed] [Google Scholar]
- 204.Feinmesser R, Miyazaki I, Cheung R, et al. Diagnosis of nasopharyngeal carcinoma by DNA amplification of tissue obtained by fine-needle aspiration. N Engl J Med. 2015;326:17–21. [DOI] [PubMed] [Google Scholar]
- 205.Chan KCA, Hung ECW, Woo JKS, et al. Early detection of nasopharyngeal carcinoma by plasma Epstein-Barr virus DNA analysis in a surveillance program. Cancer. 2013;119:1838–44. [DOI] [PubMed] [Google Scholar]
- 206.Chan KCA, Woo JKS, King A, et al. Analysis of plasma Epstein-Barr virus DNA to screen for nasopharyngeal cancer. N Engl J Med. 2017;377:513–22. [DOI] [PubMed] [Google Scholar]
- 207.Lin J-C, Wang W-Y, Chen KY, et al. Quantification of plasma Epstein-Barr virus DNA in patients with advanced nasopharyngeal carcinoma. N Engl J Med. 2004;350:2461–70. [DOI] [PubMed] [Google Scholar]
- 208.Campitelli M, Jeannot E, Peter M, et al. Human papillomavirus mutational insertion: specific marker of circulating tumor DNA in cervical cancer patients. PLoS One. 2012;7:e43393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Jeannot E, Becette V, Campitelli M, et al. Circulating human papillomavirus DNA detected using droplet digital PCR in the serum of patients diagnosed with early stage human papillomavirus-associated invasive carcinoma. J Pathol Clin Res. 2016;2:201–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Lo YMD, Leung S, Chan LYS, et al. Kinetics of plasma Epstein-Barr virus DNA during radiation therapy for nasopharyngeal carcinoma. J Cancer Res. 2000;60:2351–5.Google Scholar [PubMed] [Google Scholar]
- 211.Wang W-Y, Twu C-W, Chen H-H, et al. Long-term survival analysis of nasopharyngeal carcinoma by plasma Epstein-Barr virus DNA levels. Cancer. 2013;119:963–70. [DOI] [PubMed] [Google Scholar]
- 212.Le Q, Zhang Q, Cao H, et al. An international collaboration to harmonize the quantitative plasma Epstein-Barr Virus (EBV) DNA assay for future biomarker-guided trials in nasopharyngeal carcinoma. Clin Cancer Res. 2013;19:2208–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Leung SF, Zee B, Ma BB, et al. Plasma Epstein-Barr viral deoxyribonucleic acid quantitation complements tumor-node-metastasis staging prognostication in nasopharyngeal carcinoma. J Clin Oncol. 2006;24:5414–8. [DOI] [PubMed] [Google Scholar]
- 214.Chua MLK, Wee JTS, Hui EP, et al. Nasopharyngeal carcinoma. Lancet. 2016;387:1012–24. [DOI] [PubMed] [Google Scholar]
- 215.Fakhry C, Westra WH, Li S, et al. Improved survival of patients with human papillomavirus-positive head and neck squamous cell carcinoma in a prospective clinical trial. J Natl Cancer Inst. 2008;100:261–9. [DOI] [PubMed] [Google Scholar]
- 216.Ang KK, Harris J, Wheeler R, et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N Engl J Med. 2010;363:24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Dahlstrom KR, Li G, Hussey CS, et al. Circulating human papillomavirus DNA as a marker for disease extent and recurrence among patients with oropharyngeal cancer. Cancer. 2015;121:3455–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Hanna GJ, Supplee JG, Kuang Y, et al. Plasma HPV cell-free DNA monitoring in advanced HPV-associated oropharyngeal cancer. Ann Oncol. 2018;29:1980–6. [DOI] [PubMed] [Google Scholar]
- 219.Gupta GP, Kumar S, Marron D, et al. Circulating tumor HPV16 DNA as a biomarker of tumor genomics and disease control in HPV-associated oropharyngeal squamous cell carcinoma [abstract]. Radiat Oncol. 2018;100(5):1310.Google Scholar [Google Scholar]
- 220.Koliopoulos G, Nyaga VN, Santesso N, et al. Cytology versus HPV testing for cervical cancer screening in the general population. Cochrane Database Syst Rev. 2017;8: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Koss LG. The Papanicolaou test for cervical cancer detection: a triumph and a tragedy. JAMA. 1989;261:737–43. [PubMed] [Google Scholar]
- 222.Han K, Leung E, Barbera L, et al. Circulating human papillomavirus DNA as a biomarker of response in patients with locally advanced cervical cancer treated with definitive chemoradiation. JCO Precis Oncol. 2018;2:1–8. [DOI] [PubMed] [Google Scholar]
- 223.El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–76. [DOI] [PubMed] [Google Scholar]
- 224.Sun Yat-sen University. ctDNA for prediction of relapse in gastric cancer [ClinicalTrials.gov identifier NCT02887612]. National Institutes of Health, ClinicalTrials.gov; https://clinicaltrials.gov Accessed 11 Feb 2019. [Google Scholar]
- 225.Johns Hopkins University and the Walter and Eliza Hall Institute of Medical Research. Circulating tumour DNA as a marker of residual disease and response to adjuvant chemotherapy in stage I-IV ovarian cancer [ClinicalTrials.gov identifier NCT03691012]. National Institutes of Health, ClinicalTrials.gov; https://clinicaltrials.gov Accessed 11 Feb 2019. [Google Scholar]
- 226.University of California, San Francisco. Biomarkers for risk stratification in lung cancer [ClinicalTrials.gov identifier NCT03774758]. National Institutes of Health, ClinicalTrials.gov; https://clinicaltrials.gov Accessed 11 Feb 2019. [Google Scholar]
- 227.Merker JD, Oxnard GR, Compton C, et al. Circulating tumor DNA analysis in patients with cancer: American Society of Clinical Oncology and College of American Pathologists joint review. J Clin Oncol. 2018;36:1631–41. [DOI] [PubMed] [Google Scholar]
- 228.Xu T, Kang X, You X, et al. Cross-platform comparison of four leading technologies for detecting EGFR mutations in circulating tumor DNA from non-small cell lung carcinoma patient plasma. Theranostics. 2017;7:1437–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Thress KS, Brant R, Carr TH, et al. EGFR mutation detection in ctDNA from NSCLC patient plasma: a cross-platform comparison of leading technologies to support the clinical development of AZD9291. Lung Cancer. 2015;90:509–15. [DOI] [PubMed] [Google Scholar]
- 230.Institute of Cancer Research, United Kingdom. A trial using ctDNA blood tests to detect cancer cells after standard treatment to trigger additional treatment in early stage triple negative breast cancer patients (c-TRAK-TN) [ClinicalTrials.gov identifier NCT03145961]. National Institutes of Health, ClinicalTrials.gov; https://clinicaltrials.gov Accessed 11 Feb 2019. [Google Scholar]
- 231.Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:0225–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Kobayashi S, Boggon T, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–92. [DOI] [PubMed] [Google Scholar]
- 233.Soria J-S, Ohe Y, Vansteenkiste J, et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. NEJM. 2018;378:113–25. [DOI] [PubMed] [Google Scholar]