

Abstract
Pathogenic loss‐of‐function variants in SCN1B are linked to Dravet syndrome (DS). Previous work suggested that neuronal pathfinding defects underlie epileptogenesis and SUDEP in the Scn1b null mouse model of DS. We tested this hypothesis by inducing Scn1b deletion in adult mice that had developed normally. Epilepsy and SUDEP, which occur by postnatal day 21 in Scn1b null animals, were observed within 20 days of induced Scn1b deletion in adult mice, suggesting that epileptogenesis in SCN1B‐DS does not result from defective brain development. Thus, the developmental brain defects observed previously in Scn1b null mice may model other co‐morbidities of DS.
Introduction
Voltage gated sodium channels (VGSCs) are heterotrimers, composed of one pore‐forming α subunit and two nonpore‐forming β subunits.1 β1 subunits are ubiquitously expressed throughout the nervous system with wide subcellular localization in neurons where they associate with many, if not all, VGSC α subunits.2 β1 subunits regulate VGSC α subunit trafficking, expression, and gating, associate with and modulate some voltage‐gated potassium channels, and play nonconducting roles in neuronal pathfinding and fasciculation as immunoglobulin superfamily cell adhesion molecules (Ig‐CAMs).2
Variants in VGSC genes are linked to the developmental and epileptic encephalopathies (DEEs).3 Dravet syndrome (DS) is a severe DEE predominantly linked to haploinsufficiency of SCN1A, encoding the VGSC α subunit Nav1.1. Results from mouse models suggest that epilepsy in DS initiates from disinhibition through reduced sodium current density in cortical and hippocampal parvalbumin‐positive GABAergic interneurons.4 While the majority of DS patients have de novo, heterozygous variants in SCN1A, inherited, homozygous loss‐of‐function variants in SCN1B, encoding the VGSC β1/β1B subunits, also result in DS, or a DS‐like DEE.5, 6 Scn1b null mice are a DS model, with severe seizures, ataxia, and 100% lethality prior to weaning.5 Scn1b null mice have neuronal pathfinding defects in brain that precede the onset of seizures.7 Based on these results, we proposed that epilepsy in this model is of neurodevelopmental origin due to the loss of β1‐mediated Ig‐CAM function.7 In the related Scn1b‐p.C121W homozygous mouse model, which has a similar DS‐like phenotype, neuronal hyperexcitability was postulated to arise from the developmental loss of β1‐mediated Ig‐CAM function.8 In support of this hypothesis, variants in other neurodevelopmental CAMs, for example, protocadherin 7 and 19,3, 8 are also associated with epilepsy.
Because β1 subunits are multifunctional Ig‐CAMs and channel modulators, it is important to understand what roles altered development play in epileptogenesis in response to Scn1b loss‐of‐function. Here, we test the hypothesis that altered neuronal development is necessary for epileptogenesis in the Scn1b model of DS. We show that severe seizures and SUDEP occur following Scn1b deletion in adult mice that had developed normally, suggesting that epileptogenesis in SCN1B‐DS does not result from defective brain development.
Methods
Animals
Experiments were performed in accordance with NIH guidelines and approved by the University of Michigan Institutional Animal Care and Use Committee. Scn1b Fl/Fl mice, on the C57BL/6J background,9 were crossed with Slick‐H mice (JAX Tg(Thy1‐cre/ERT2,‐EYFP)HGfng/PyngJ, stock #012708) received on the (C57BL/6J x CBA)F1 background and backcrossed to C57BL/6J, to generate Slick‐H/Scn1b Fl/Fl and Slick‐H/Scn1b E/E mice. Tamoxifen (TMX, Sigma‐Aldrich) was dissolved in sunflower oil + 2% ethanol at a final concentration of 10 μg/μL. 10 μL of TMX solution or vehicle per g mouse weight were administered intraperitoneal (IP) once per day for 4 consecutive days.
Immunohistochemistry
P60 Slick‐H mice, treated with TMX 10 days prior, were anesthetized with isoflurane and transcardially perfused with ~10 mL of PBS followed by ~10 mL of 4% paraformaldehyde (PFA). Tissues were postfixed overnight in 4% PFA, then cryoprotected in 10% sucrose followed by 30% sucrose overnight, flash frozen in 2‐methylbutane, and stored at −80°C. 20 μm coronal brain sections were cut on a Leica cryostat and stored at −20°C until processing for immunohistochemistry.
For immunohistochemistry, sections at the same locations relative to Bregma for each animal were dried and postfixed for 10 min with 4% PFA, washed three times for 5 min each with 0.05 mol/L phosphate buffer (PB), and incubated in PBTGS blocking buffer (10% goat serum, 0.3% Triton X‐100, 0.1 mol/L PB) for ≥2 h in a humidified chamber. Sections were then incubated with primary antibodies (diluted in PBTGS) overnight in a humidified chamber and washed 3 times for 10 min with 0.1 mol/L PB. Primary antibodies were anti‐GABA (rabbit, Sigma‐Aldrich, 1:400), anti‐GFP (chicken, Invitrogen, 1:400), and anti‐CTIP2 to identify cortical layer V/VI neurons (rat, Abcam, 1:250). From this point forward, all steps were performed in the dark to minimize photobleaching of secondary antibodies. Sections were incubated with goat anti‐chicken AlexaFluor‐488, goat anti‐rat AlexaFluor‐647, and/or goat anti‐rabbit AlexaFluor‐594‐conjugated secondary antibodies (Invitrogen, diluted in PBTGS) for 2 h, washed three times for 10 min in 0.1% PB, incubated with DAPI for 20 min, dried, and then coverslips were mounted using ProLong Gold anti‐fade reagent (Invitrogen).
Sections were imaged using a Nikon A1R confocal microscope with Nikon NIS‐Elements AR software located in the UM Department of Pharmacology using a 20 × 0.75 NA dry objective. Confocal images spanning 10 μm were acquired at 0.9 μm intervals and flattened using maximum signal for analysis. Images were processed and analyzed using NIH‐ImageJ, and figures were assembled using Adobe Photoshop.
EEG Implantation
Slick‐H/Scn1b Fl/Fl, Slick‐H negative/Scn1b Fl/Fl , and Slick‐H/Scn1b E/E mice were implanted at P42 to 70 with 6 EEG electrodes. For surgery, each mouse was anesthetized with isoflurane or ketamine/xylazine and placed in a stereotaxic adapter. The electrodes were implanted over the left and right front lobes, the left and right parietal lobes, the cerebellum, and the sinus cavity using mounting screws. The electrodes were connected to a 6‐pin electrode pedestal and the headcap was secured using dental cement. After 7 days of recovery, simultaneous EEG recording and video monitoring were performed on Ceegraph Vision; Bio‐logic System Corporation and Natus recording systems. Following baseline recording for 2 days, mice were removed from recording for injection with TMX and recording was resumed at 7 days postinitial TMX injection. Signals were acquired at 256 Hz with simultaneous video monitoring. Data were filtered with a 1 Hz low pass filter, a 70 Hz high pass filter, and a notch filter at 60 Hz. Seizures were assessed manually.
Results
Scn1b null DS mice exhibit severe seizures beginning at ~postnatal day (P)10 and 100% lethality prior to ~P21.10 Our previous work suggested that aberrant brain development due to loss of β1‐mediated cell adhesion may underlie epileptogenesis in this model.7 To test this hypothesis, we crossed Scn1b Fl/Fl mice9 with a tamoxifen (TMX)‐inducible Thy1‐Cre (Slick‐H) strain.11 Slick‐H mice express Cre recombinase in response to TMX administration in Thy1‐ expressing neurons, and are thus useful for separation of locally projecting GABA‐positive (+) neurons from locally and nonlocally projecting excitatory neurons. Constitutive Thy1‐driven YFP expression in Slick‐H mouse cortex showed laminar restriction within pyramidal neurons of layers 3, 5, and 6 (Fig. 1A), as shown previously.11 We co‐stained with anti‐GABA to examine whether GABA+ neurons are also targeted by Thy1 promoter‐driven Cre recombinase in this strain. Neurons that were YFP+ were seldom GABA+ and vice versa (Fig. 1A, B, and F). We found similar results within the hippocampus, with dense labeling of the dentate gyrus and pyramidal neurons throughout the CA1, CA2, and CA3 regions, as well as in the subiculum, with limited overlap of YFP+ and GABA+ neurons (Fig. 1C and D).
Figure 1.
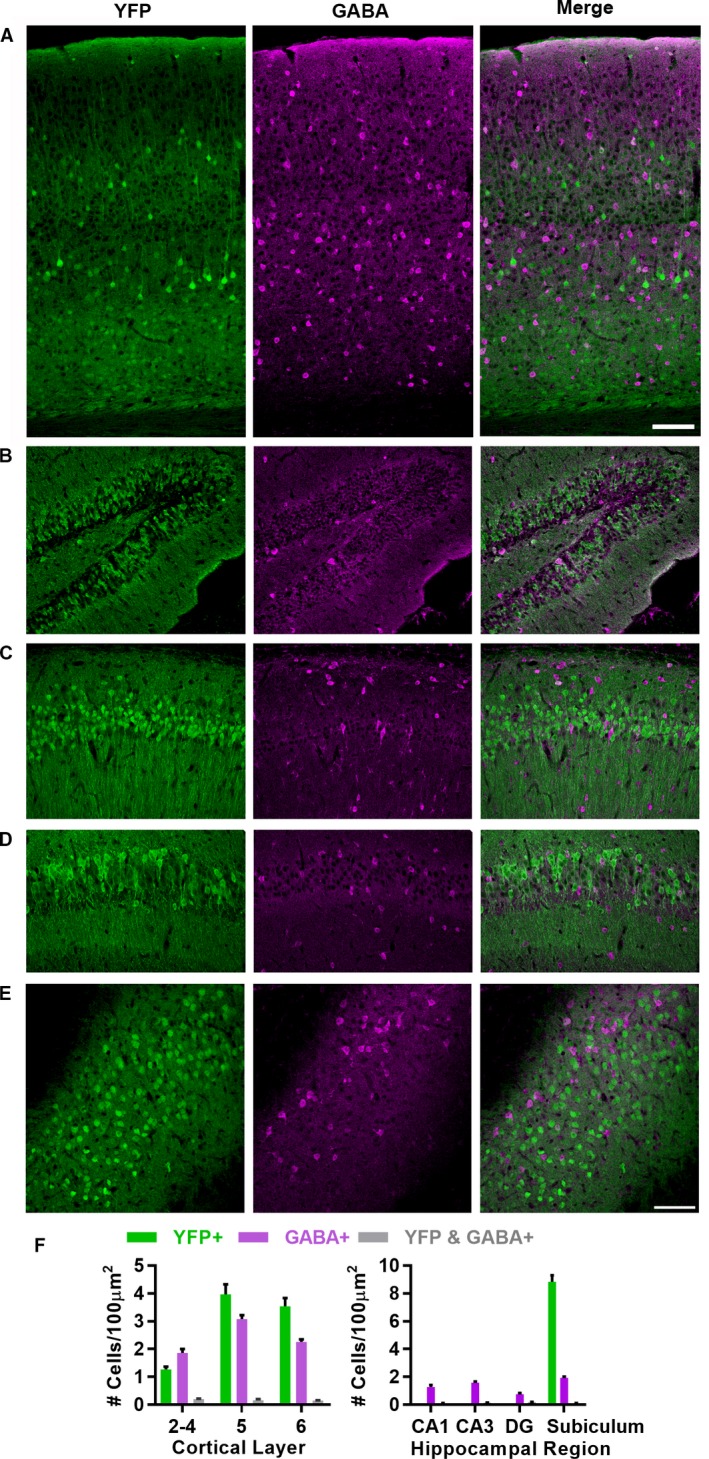
Slick‐H YFP expression in cortex, hippocampus, and subiculum. (A) Slick‐H mice express Cre and a YFP reporter (green) in projection neurons of the visual cortex with limited overlap with GABA+ cells (magenta). (B–E) Slick‐H mice express Cre and a YFP reporter in projection neurons of the hippocampus with limited overlap with GABA+ cells in the dentate gyrus (DG) (B), CA1 (C), CA3 (D), and subiculum (E). (F) Quantification of overlap between YFP and GABA+ cells across cortical layers (left) and hippocampal regions (right). YFP+ cell density in DG, CA1, and CA3 not quantified due to unclear cell boundaries with high density. Scale bars (in A and E) = 100 μm.
To investigate the developmental dependence of epilepsy and SUDEP in response to Scn1b deletion, we administered TMX to Slick‐H/Scn1b Fl/Fl mice over various time ranges (P14–19, P42–117, and P > 100) to ask whether induced Scn1b deletion in adult mice that had developed normally resulted in seizures and death. Regardless of age of administration, TMX treatment resulted in severe epilepsy and death in 100% of Slick‐H/Scn1b Fl/Fl mice compared to controls (Slick‐H negative Scn1b Fl/Fl mice injected with vehicle or Slick‐H/Scn1b E/E mice injected with TMX) within 20 days (Fig. 2A). All TMX‐treated Slick‐H/Scn1b Fl/Fl mice exhibited frequent spontaneous, behavioral seizures. To verify epileptic activity electrographically, we implanted three Slick‐H/Scn1b Fl/Fl mice, two Slick‐H/Scn1b Fl/Fl vehicle control mice and two Slick‐H negative Scn1b Fl/Fl control mice with EEG electrodes. Two days of baseline recording were followed by one IP administration of TMX or vehicle per day for four consecutive days to delete Scn1b. EEG recording was resumed 7 days after the initial TMX treatment (post‐TMX). We observed no seizures in Slick‐H/Scn1b Fl/Fl or control mice for the duration of baseline recording. In the Slick‐H/Scn1b Fl/Fl group, seizures were detected as early as 7 days post‐TMX (Fig. 2B and C). Two mice exhibited severe epilepsy and were euthanized for ethical reasons at day 8 and 9, respectively. The third mouse had a more gradual increase in seizure severity and was euthanized at 20 days post‐TMX due to morbidity. These results demonstrate that, although developmental brain defects are present in Scn1b‐DS mice, epileptogenesis and SUDEP are not the result of impaired brain development.
Figure 2.
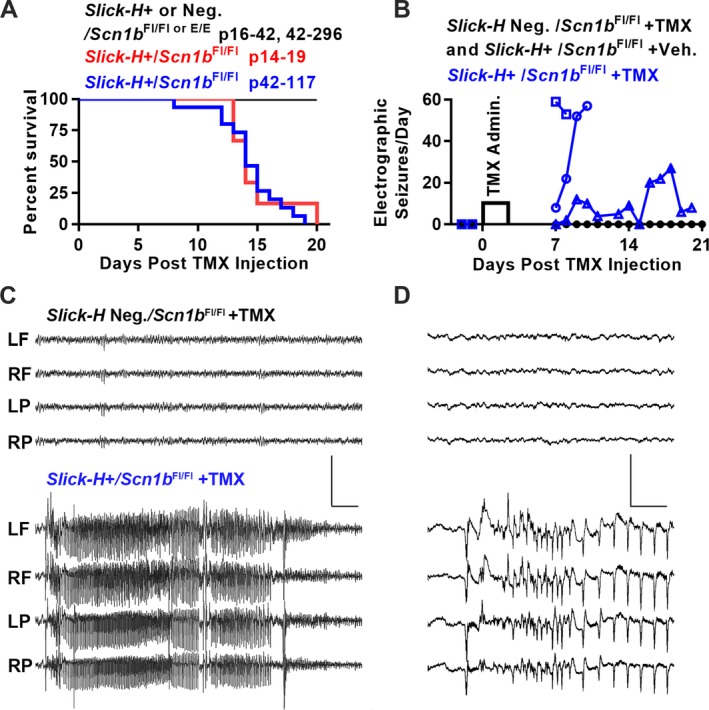
Inducible deletion of Scn1b in adult projection neurons results in epilepsy and SUDEP. (A) Kaplan‐Meier survival curve of Slick‐H/Scn1b Fl/Fl mice injected with TMX at P14–19 (n = 5) or P42–117 (n = 15) compared to control mice: P14–19 Slick‐H/Scn1b E/E adolescent (n = 4) and Slick‐H negative Scn1b Fl/Fl P42–117 mice (n = 7). Survival of adult and adolescent Slick‐H/Scn1b Fl/Fl mice following TMX administration was not different (P = 0.71, Mantel‐Cox test). All Slick‐H/Scn1b Fl/Fl mice treated with TMX exhibited numerous behavioral seizures. (B) TMX‐induced Scn1b deletion in adult mice results in behavioral and electrographic seizures within 8 days of initial TMX administration. Control mice (black, n = 4) exhibited no seizures throughout the experiment while Slick‐H/Scn1b Fl/Fl mice that were administered TMX (blue squares, circles, and triangles represent individual mice; n = 3) experienced as many as 60 seizures/day. Slick‐H mice were euthanized at days 8, 9, and 20 due to morbidity. (C) Representative EEG traces of Slick‐H/Scn1b Fl/Fl and Slick‐H/Scn1b E/E mice. Left(L)/Right(R) and Frontal(F)/Parietal(P) screw electrode placement. Scale bar = 2000 μV and 3 sec. (D) Expanded time scale of traces in panel C showing seizure onset. Scale bar = 2000 μV and 0.25 sec.
Discussion
The results of this study demonstrate that epileptogenesis resulting from Scn1b loss‐of‐function is not due to impaired neuronal development. Furthermore, our data show that deletion of Scn1b in forebrain excitatory, but not inhibitory, neurons recapitulates the DS phenotype in mice. In previous work, targeted deletion of Scn1a in mouse inhibitory neurons phenocopied DS.12 In contrast, targeted deletion of Scn1a in excitatory neurons failed to recapitulate the DS phenotype and instead ameliorated disease severity when coupled with deletion in interneurons.13 Importantly, our work demonstrates that diverse mechanisms can lead to common neurological disease phenotypes.
Author Contributions
JMH and CC performed TMX injection and monitoring of mouse survival. HAO performed all immunohistochemical staining and image analyses. BCC performed EEG surgeries. GO‐F analyzed EEG data. MBJ monitored mice for EEG recordings. SJA provided facilities and instruction for EEG surgeries. JMP provided facilities and personnel for EEG monitoring. JMH, HAO, and LLI wrote the manuscript. LLI was responsible for overseeing all experiments.
Conflict of Interest
None.
Acknowledgments
The authors acknowledge the expert technical assistance of C. Liu. This work was funded by R37 NS076752 to LLI, a University of Michigan Rackham Merit Fellowship to JMH, and a pre‐doctoral fellowship from the Michigan Brain Institute to JMH.
Funding Information
This work was funded by R37 NS076752 to LLI, a University of Michigan Rackham Merit Fellowship to JMH, and a pre‐doctoral fellowship from the Michigan Brain Institute to JMH.
Funding Statement
This work was funded by National Institute of Neurological Disorders and Stroke grant NS076752.
References
- 1. O'Malley HA, Isom LL. Sodium channel β subunits: emerging targets in channelopathies. Annu Rev Physiol 2015;77:481-504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hull JM, Isom LL. Voltage‐gated sodium channel β subunits: the power outside the pore in brain development and disease. Neuropharmacology. 2018;132:43-57. 10.1016/j.neuropharm.2017.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Steel D, Symonds JD, Zuberi SM, Brunklaus A. Dravet syndrome and its mimics: beyond SCN1A . Epilepsia 2017;58:1807-1816. [DOI] [PubMed] [Google Scholar]
- 4. Yu FH, Mantegazza M, Westenbroek RE, et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci 2006;9:1142–1149. [DOI] [PubMed] [Google Scholar]
- 5. Patino GA, Claes LR, Lopez‐Santiago LF, et al. A functional null mutation of SCN1B in a patient with Dravet syndrome. J Neurosci 2009;29:10764-10778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ramadan W, Patel N, Anazi S, et al. Confirming the recessive inheritance of SCN1B mutations in developmental epileptic encephalopathy. Clin Genet 2017;92(3):327-331. 10.1111/cge.12999. [DOI] [PubMed] [Google Scholar]
- 7. Brackenbury WJ, Yuan Y, O'Malley HA, et al. Abnormal neuronal patterning occurs during early postnatal brain development of Scn1b‐null mice and precedes hyperexcitability. Proc Natl Acad Sci USA 2013;110:1089-1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lal D, Ruppert AK, Trucks H, et al. Burden analysis of rare microdeletions suggests a strong impact of neurodevelopmental genes in genetic generalised epilepsies. PLoS Genet 2015;11:e1005226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen C, Dickendesher TL, Oyama F, et al. Floxed allele for conditional inactivation of the voltage‐gated sodium channel β1 subunit Scn1b . Genesis 2007;45:547-553. [DOI] [PubMed] [Google Scholar]
- 10. Chen C, Westenbroek RE, Xu X, et al. Mice lacking sodium channel β1 subunits display defects in neuronal excitability, sodium channel expression, and nodal architecture. J Neurosci 2004;24:4030-4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Heimer‐McGinn V, Young P. Efficient inducible Pan‐neuronal cre‐mediated recombination in SLICK‐H transgenic mice. Genesis 2011;49:942–949. [DOI] [PubMed] [Google Scholar]
- 12. Cheah CS, Yu FH, Westenbroek RE, et al. Specific deletion of NaV1.1 sodium channels in inhibitory interneurons causes seizures and premature death in a mouse model of Dravet syndrome. Proc Natl Acad Sci USA 2012;109:14646–14651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ogiwara I, Iwasato T, Miyamoto H, et al. Nav1.1 haploinsufficiency in excitatory neurons ameliorates seizure‐associated sudden death in a mouse model of Dravet syndrome. Hum Mol Genet. 2013;22:4784–4804. [DOI] [PMC free article] [PubMed] [Google Scholar]