

Abstract
Background
A 49‐year‐old male presented with late‐onset demyelinating peripheral neuropathy, cerebellar atrophy, and cognitive deficit. Nerve biopsy revealed intra‐axonal inclusions suggestive of polyglucosan bodies, raising the suspicion of adult polyglucosan bodies disease (OMIM 263570).
Methods and Results
While known genes associated with polyglucosan bodies storage were negative, whole‐exome sequencing identified an unreported monoallelic variant, c.397G>T (p.Val133Phe), in the histidyl‐tRNA synthetase (HARS) gene. While we did not identify mutations in genes known to be associated with polygucosan body disease, whole‐exome sequencing revealed an unreported monoallelic variant, c.397G>T in the histidyl‐tRNA synthetase (HARS) gene, encoding a substitution (Val133Phe) in the catalytic domain. Expression of this variant in patient cells resulted in reduced aminoacylation activity in extracts obtained from dermal fibroblasts, without compromising overall protein synthesis.
Interpretation
Genetic variants in the genes coding for the different aminoacyl‐tRNA synthases are associated with various clinical conditions. To date, a number of HARS variant have been associated with peripheral neuropathy, but not cognitive deficits. Further studies are needed to explore why HARS mutations confer a neuronal‐specific phenotype.
Introduction
Aminoacyl‐tRNA synthetases (ARSs) are a group of ubiquitously expressed enzymes essential for protein synthesis. Thirty‐seven different ARSs genes exist in humans. Among them, only three of them are functional in both cytoplasm and mitochondria, the rest acting exclusively either in the cytoplasm or in mitochondria.1, 2, 3 Mutations in 33 out of the 37 ARSs genes have been implicated in a large number of genetic disorders affecting multiple organs and tissues, including the nervous system.3, 4, 5, 6, 7, 8 Bi‐allelic mutations in ARSs typically cause severe, early‐onset, recessive diseases. Most of these mutations show loss‐of‐function effects but the full consequences for protein synthesis have in most cases not been assessed. In contrast, it is still unclear if peripheral neuropathies associated with dominant ARS mutations specifically affect the peripheral nervous system through loss‐of‐function, gain‐of‐function, or a combinatorial mechanism. Notably, the vast majority of neuropathy‐associated ARS alleles exhibit loss‐of‐function.2, 8, 9, 10, 11, 12 The associated phenotypic spectrum is broad and encompasses both axonal and demyelinating motor and sensory neuropathies, as well as pure motor axonal neuropathy.9, 11, 13
To date, five genes encoding a cytoplasmic ARS enzyme have been implicated in autosomal dominant neuropathy, a subgroup of the so‐called Charcot‐Marie‐Tooth (CMT) neuropathies.14 The reason why mono‐allelic ARS mutations seem to affect preferentially the nervous system remains unclear, but may relate to the specialized process of local translation occurring in the distal axon. These ARSs‐neuropathy‐related genes are typically characterized by a decrease in aminoacylation activity when assessed in vivo and in vitro.2, 10 Although some CMT mutations apparently have minimal effect on the activity of the mutant enzyme when assayed in vitro, protein synthesis measurements in the context of a model organism (Drosophila melanogaster) show a decrease nonetheless.12, 15 This, in turn, raises the possibility of altered tRNA‐charging having a preferential impact on the peripheral nerve and its function during development and in adulthood.4
Among ARSs CMT genes, the histidyl‐tRNA synthetase (HARS) gene (OMIM*142810) is unique in that deleterious variants in the cytoplasmic isoform of the enzyme can lead to autosomal dominant CMT disease (OMIM #616625),11 but also to Usher syndrome type B3 (OMIM *614504). In this inherited neurological syndrome, a single HARS variant in homozygous state is reported to be the cause of early onset of blindness and deafness.16, 17 In this report, we describe an unusual association between a novel variant in the HARS gene and a clinical phenotype initially suggestive of adult polyglucosan body disease (APBD, OMIM #263570), a condition affecting both the central and the peripheral nervous system, in an adult individual. Our case highlights the spectrum of neurological diseases linked to dominant and recessive ARS alleles.
Patient and Methods
Illustrative case: patient description
A 49‐year old man of Spanish origin was referred to our Nerve‐Muscle Unit because of difficulty in performing daily life activities due to slowly progressive incoordination and muscle weakness. His neonatal and pediatric history was unremarkable. A mild stuttering was noted in adolescence and remained stable until the late 40s. Around the age of 44, his wife noticed a “strange way of walking”. Over the next 5 years, the patient developed progressive leg weakness with difficulty walking and going down the stairs. Running and playing soccer became impossible because of leg weakness and muscle pain. At the same time, he presented with progressive motor incoordination and difficulty with manual dexterity: his handwriting gradually worsened, doing up buttons and manipulating small objects became more and more cumbersome. At the social level, the patient became increasingly withdrawn, avoiding social interaction and engaging less in conversation as word finding and sustaining attention become laborious with time. Family history was negative for any neurologic conditions.
Neurological examination revealed moderate dysarthria and stuttering without any other cranial nerve deficit, moderate amyotrophy in distal lower limbs with associated mild distal muscle weakness (rated 4/5 according to the Medical Research Council Scale), genu valgus, pes cavus, hammer toes (Fig. 1A–C), a staggering gait, and marked axial ataxia. Bilateral Achilles tendon areflexia was also found. Muscle strength and deep tendon reflexes were preserved in the trunk and upper limbs. No sensory deficit was found.
Figure 1.
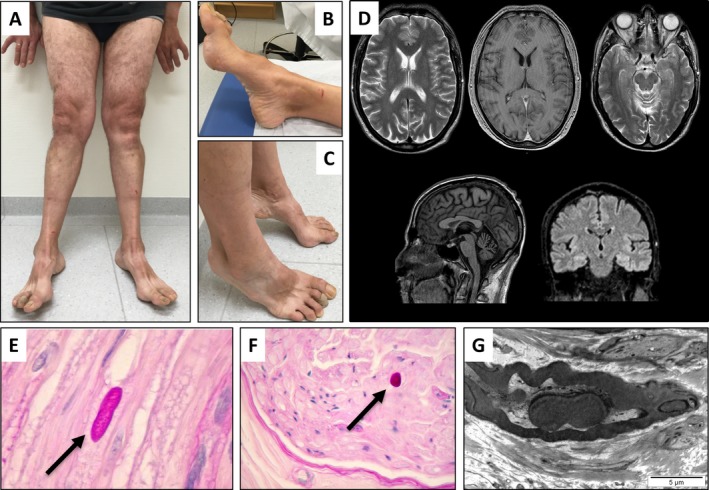
(A–G) Clinical phenotype of the patient. Genu valgus with a wide‐based gait (A). Pes cavus with amyotrophy of the intrinsic foot muscles (B–C). Coronal and sagittal T‐1 and T‐2 weighted brain MRI showing mild cerebellar atrophy (D). Histopathology (E–F) and electron microscopy (G) of left sural nerve biopsy with intra‐axonal inclusions suggestive of polyglucosan bodies. Shown by the arrows.
Neuropsychological testing revealed mild to moderate impairment of multiple cognitive functions, namely an important attention deficit, a deficit in executive function (frontal assessment battery score of 14/18), a slight apraxia, short‐term memory loss (rated using the Wechsler Memory Scale III), and emotional blunting.
Materials, methods and results
Laboratory workup showed only increased plasma creatine kinase levels (621 U/L, 25 < normal < 190). The rest was unremarkable, including levels of vitamin B12, B1, B6, and E. Methylmalonic acid, acylcarnitine profile, glycosylated hemoglobin, and thyroid‐stimulating hormone were all within the reference range. Brain MRI was normal with the exception of mild cerebellar atrophy (Fig. 1D). Nerve conduction studies revealed reduced motor nerve conduction velocities (median, ulnar, and tibial nerves were tested bilaterally). Sensory nerve action potential parameters and velocity were normal in all four limbs except for the median nerve (Table 1). Amplitude of motor responses was reduced for the peroneal and tibial nerves. Median nerve distal motor latency was prolonged at the wrist bilaterally. These results were consistent with a late onset demyelinating motor neuropathy. Left deltoid muscle biopsy showed minor changes with randomly distributed scattered rounded or atrophic muscle fibers (data not shown). Sural nerve biopsy showed periodic acid‐Schiff (PAS)‐positive intra‐axonal deposits suggestive of polyglucosan bodies or focally folded myelin sheaths characteristic of CMT type 1B (Fig. 1E–G).
Table 1.
Motor and sensory nerve conduction studies in the patient
| Median motor | Ulnar motor | Tibial motor | Median sensory | Radial sensory | Sural sensory | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | CV | A | CV | A | CV | A | CV | A | CV | A | CV | |
| R | 61 | 26.7 | 8 | 40 | 0.2 | 28.7 | 22 | 31.6 | 10 | 47.8 | 11.2 | 48 |
| L | 51 | 36.6 | 7.7 | 40 | 0.2 | 33.1 | – | – | – | – | – | – |
Prolonged distal motor latency at 6.9 and 5.3 msec, respectively (normal < 4 msec); –: not performed; A, amplitude; CV, conduction velocity, (normal > 49 m/sec in upper limbs; >40 m/sec in lower limbs); R, right; L, left.
Based on the association of a progressive motor neuropathy with cognitive difficulties and the presence of this unusual histology of the peripheral nerve, a late‐onset form of glycogen debranching enzyme deficiency known as APBD was supposed. However, fibroblast glycogen branching enzyme activity was active (0.93 μmol/min/mg Prot, normal 0.97–1.63) making the diagnosis of APBD unlikely. Total patient DNA was extracted using standard procedures, for further analysis of GBE1, the APBD‐associated gene. Bidirectional Sanger sequencing of GBE1 gene did not reveal any pathogenic variant (Laboratory for Molecular Diseases, Lausanne University Hospital, Switzerland). Whole‐Exome Sequencing (WES) was then carried out on Illumina HiSeq2500 sequencer, with Agilent Sure Select XT Human All Exon V5 capture kit, after appropriate informed consent. Data processing and variants annotation and filtering were made as already described.18 We first analyzed candidate genes known to be associated with polyglucosan bodies storage (Division of Genetic Medicine, Lausanne University Hospital, Switzerland). Screening of GYS1, GYG1, RBCK1, PFKM, EPM2A, EPM2B, PRDM8, and PRKAG2 genes, first by Sanger sequencing, and then by WES did not reveal deleterious variants. We then focused on all rare, nonsynonymous, high quality variants in the patient that were not present or with a low frequency among healthy control cohorts from the Exome Aggregation Consortium (ExAC) database (http://exac.broadinstitute.org) and in the overlapping and updated genome Aggregate Database (gnomAD; http://gnomad.broadinstitute.org).19 The analysis was thus reduced to only a few variants (Table S1).
Among the variants remaining after the filtering procedure, we identified a heterozygote variant in the histidyl‐tRNA synthetase (HARS) gene, c.397G>T (p.Val133Phe), not previously detected in our in‐house cohort of WES patients nor in any public database so far. This variant is likely to be deleterious to protein function as it affects a highly conserved amino acid present in the catalytic domain of the protein, as do the other reported pathogenic neuropathy variants (Fig. 2A–C). It is predicted to be deleterious by various in silico software programs (CADD,20 VEST3,21 MutationTaster (http://www.mutationtaster.org/) and SIFT (http://sift.jcvi.org/). It clusters with other known pathogenic variants in a small region of the protein (Fig. 2A–B). The variant was then confirmed by Sanger sequencing in the patient and shown to be absent in maternal DNA (paternal DNA was not available). Although the variant is located in the consensus acceptor splice site of exon 5, in‐silico predictive software does not predict a major impact on splicing, similarly to the other surrounding known pathogenic HARS variants (Fig. 2B).
Figure 2.
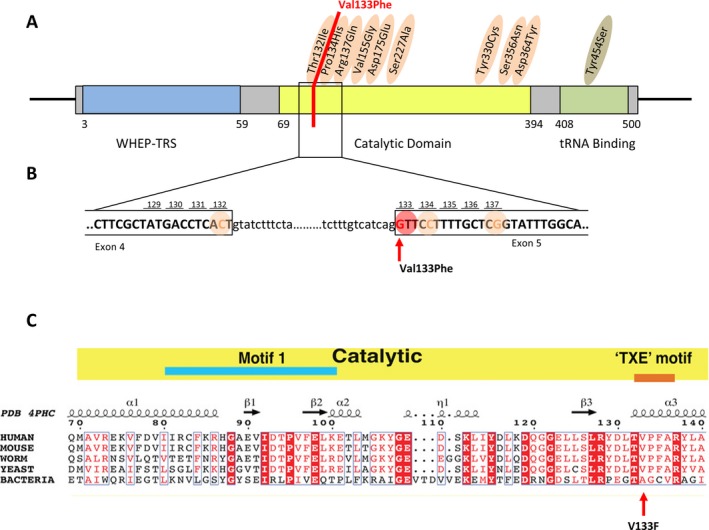
(A–C) Localization of reported pathogenic HARS variants on its protein domains. In orange, variants associated with Charcot‐Marie‐Tooth disease11 and in green with Usher syndrome.16, 17 In red, localization of our newly discovered variant (A). HARS cDNA sequence around our newly discovered variant (B). Alignment of HARS sequences from distantly related genera. p.Val133Phe (V133F) is strictly conserved among all eukaryotes, and replaced by a smaller residue (Alanine) in E. coli (C).
The p.V133F substitution associated with this mutation is located within the HARS catalytic domain, on helix α3 in the active site. This location is proximal to seven different substitutions that have previously been linked to CMT(Fig. 3A–B). With respect to primary sequence, Val133 lies between Thr132 and Pro134. Previously, the p.T132I and p.P134H substitutions were linked to CMT in well‐characterized families, each with multiple subjects.11 Of note, several of the CMT patients with these mutations had late onset of the disease, like our case.10 In addition, three‐dimensional modeling suggests that the V133F substitution introduces a steric clash with the nearby residue Tyr330, potentially altering its interactions with histidine. Valine 133 is strictly conserved in eukaryotes, and is replaced by alanine in E. coli, highlighting the likely steric constraints at this position. The p.Y330C substitution is also linked to CMT, and the substitution decreases the k cat/K M(ATP) for aminoacylation by ~500‐fold.2 Based on the proximity of V133F to these other CMT‐associated mutants, the substitution is likely to be pathogenic.
Figure 3.
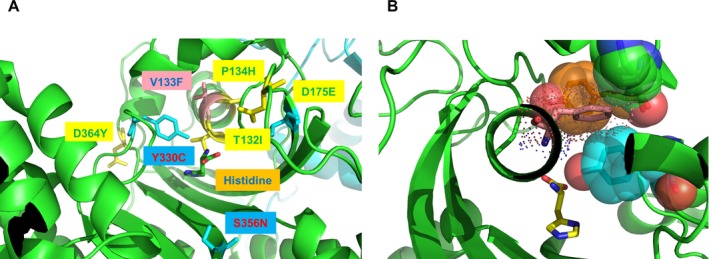
(A–B) Location of the peripheral neuropathy‐associated substitution V133F in the active site of human histidyl‐tRNA synthetase (HARS). Histidyl‐tRNA synthetase (PDB 4PHC) is shown as a dimer in ribbon cartoon representation with the first monomer in green and the second monomer in cyan. Neuropathy‐associated HARS residues are shown in the 3D structure in stick representation where substitutions reported in Brozkova et al. are shown in yellow, those reported in Abbott et al. are shown in cyan, and the substitution reported here is shown in salmon2, 11 (A). Modeling of the p.Val133Phe (V133F) substitution (van der Waals radius in dot representation) indicates steric clash with Tyr107, Lys106, and Tyr330, all depicted in CPK space filling representation. Note that Y330C is one of the substitutions associated with Charcot–Marie tooth syndrome, as described in Abbott et al..2 In this modeling depiction, the resulting structure was not subjected to energy minimization, which would likely produce local side chain rearrangements (B).
While the seven previously characterized HARS CMT variants display a loss‐of‐function effect in yeast and/or cell‐free enzymatic assays,2, 11 the aminoacylation function of these ARS‐CMT variants has not been directly assessed in patient cells. Using a previously described method,22 we examined aminoacylation activity directly in cell lysates collected from V133F patient fibroblasts. We observed that the rate of tRNAHis aminoacylation was significantly reduced (~25%) in patient cell lysates, relative to extracts obtained from control fibroblasts (Fig. 4). The reduction in HARS activity was not associated with altered protein expression, as determined by western blotting and immunofluorescence imaging (Fig. 5). Furthermore, HARS protein localized to the cytoplasm with a slight perinuclear enrichment in both control and patient cells (Fig. 5). Using the OPP‐puromycin labeling assay,23 we found that the rate of protein synthesis was similar in control and patient fibroblasts (Fig. 6), which is unsurprising given the tissue‐specific phenotype observed.
Figure 4.
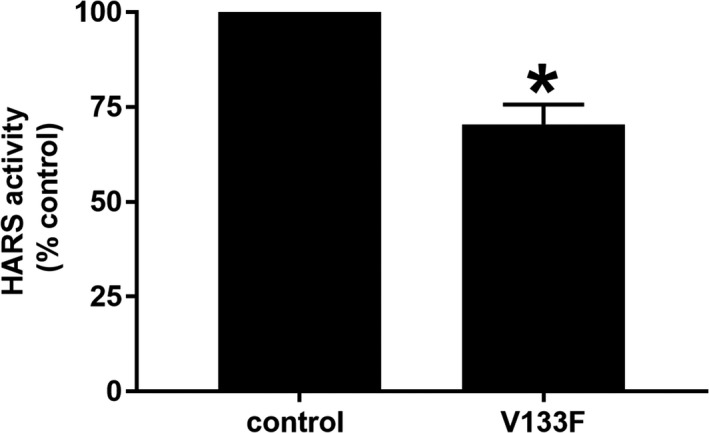
Analysis of HARS activity in control and V133F patient dermal fibroblasts. HARS activity in cell extracts was determined by calculating the slope of product formed over time. Activity was significantly reduced (~25%) in V133F patient cells. Data are displayed as mean ± SEM (n = 3). * indicates P < 0.05 by two‐tailed unpaired t‐test
Figure 5.
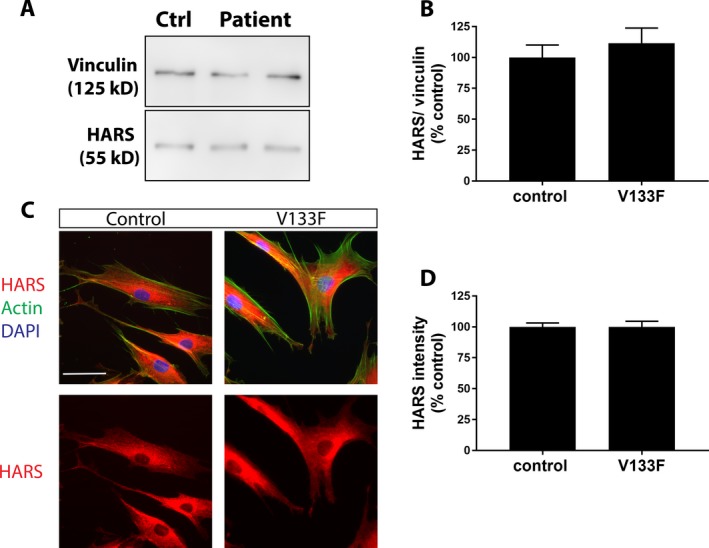
(A–D) HARS western blotting and immunofluorescence microscopy in control and V133F patient dermal fibroblasts. Expression of HARS at the protein level is similar in control and V133F patient cells as determined by western blotting (A, B) and immunofluorescence imaging (C, D). HARS protein localizes primarily perinuclearly, and throughout the cytoplasm in both control and patient cells (C). Data are displayed as mean ± SEM (western blot n = 3; imaging n = 48 control cells, 52 patient cells). Scale bar = 50 μm.
Figure 6.
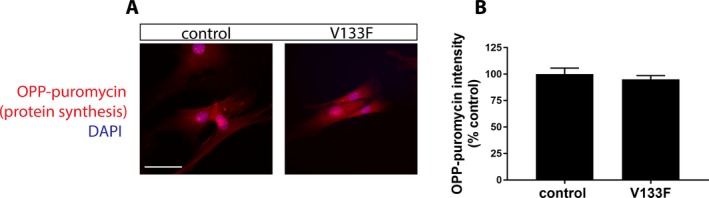
OPP‐puromycin/click‐chemistry labeling assay of protein synthesis rate. Rate of protein synthesis as determined by OPP‐puromycin fluorescence intensity is unaltered in V133F patient fibroblasts (A, B). Data are displayed as mean ± SEM (n = 47 control cells, 40 patient cells). Scale bar = 50 μm.
Discussion
The association of late onset motor neuropathy, cognitive dysfunction, and the presence of rare intra‐axonal inclusions in nerve biopsy initially suggested a diagnosis of APBD. Instead of a genetic defect in one of the candidate genes related to polyglucosan storage diseases, WES revealed an unreported variant in the HARS gene, namely c.397G>T (p.Val133Phe). The following observations support its role in the pathogenesis of the clinical picture in our patient. Firstly, the variant is extremely rare, consistent with other pathogenic ARS variants associated with CMT. Secondly, the affected residue is situated proximal to other active site residues abutting the histidine‐binding site which have previously been shown to cause CMT.2, 11 Thirdly, the mutation was shown to reduce histidine‐specific aminoacylation in cells, despite the retention of wild‐type HARS levels in patient cells (Fig. 5). Interestingly, the ~25% decrease in aminoacylation associated with the dominant negative mutation did not reduce overall protein synthesis, as measured by the puromycin click‐chemistry assay (Fig. 6). These results are consistent with a scenario in which the aminoacylation decreases linked to ARS CMT mutations specifically impact specialized tissues with high protein synthesis demands (i.e. neurons), but not cells that may only require a “housekeeping” level of protein synthesis. Interestingly, mere haploinsufficiency of a given ARS allele does not lead to disease.24, 25 Accordingly, future studies including segregation and additional functional studies in neurons will help in defining the pathogenic role of this HARS gene variant in inherited neuropathy.
Only a small number of pathogenic missense variants in the HARS gene have been reported, and the one detected in our patient maps in close proximity to the seven that have been characterized in detail (Figs. 2 and 3A).10, 11 All are presumed to have an impact on aminoacylation function, and all have been associated with peripheral neuropathy. However, no cognitive deficit like the one present in our patient, nor PAS positive (“polyglucosan”) inclusions in nerve biopsy have been reported in HARS‐related neuropathy patients so far. As inclusion bodies may be observed in the nerves of healthy individuals in rare instances, in particular in the elderly, without clinical consequences,26 it cannot be excluded that this feature may in fact be unrelated to the neuropathy.
Next generation sequencing has showed us that individuals bearing two or more genetic conditions are not rare; and that the distinct conditions may interact to form a more complex phenotype.27, 28 While the principle of looking for a unifying diagnosis remains valid, physicians should consider the possibility of multiple coexisting diagnoses. This is particularly valid in cases where clinical features are difficult to be explained by a single diagnosis. In our case and despite a thorough molecular analysis, no co‐existing diagnosis explaining the entire clinical spectrum was identified.
The first report of disease‐causing variant in an ARS gene was published in 2003.29 Since then, an increasing number of phenotype–genotype associations have been reported.3, 4 Surprisingly, many of these genetic defects are associated with diseases of the nervous system, including encephalopathy, cerebellar ataxia, and peripheral neuropathy. This data underlines the importance of ARSs in neuronal development and function and suggests that a broader set of ARSs genes may play an important role in inherited neurological diseases.
However, it remains to be determined whether HARS deleterious variants are more pleiotropic than hitherto assumed and that their clinical expression may include PAS‐positive intra‐axonal inclusions and/or involvement of the central nervous system. Recruitment of more patients with HARS variants and functional studies to predict their pathogenicity are necessary to better define the HARS gene and its impact on the phenotypic expression.
Conflict of Interest
The authors declare that they have no conflict of interest.
Author Contributions
PM, BRB, CF, BCX, LMC, AJL, CT, ASF, MRB, and CR were involved in analysis and interpretation of the data. CT, ASF, PM, BRB, TK, CK, JG, CF and TP were involved in drafting or revising the manuscript. All authors approved the final version of the manuscript.
Supporting information
Table S1. Number of variants at the different filtering steps.
Acknowledgment
We thank J.‐M. Vallat from the Department of Neurology, CHU Limoges, Dupuytren University Hospital, France for his helpful review of the histolopathology.
Funding information
The work carried out by C. F. and P. M. on this project was supported by NIGMS 5R01GM054899‐20.
BRB, PT and PM contributed equally.
Funding Statement
This work was funded by NIGMS grant 5R01GM054899-20.
References
- 1. Antonellis A, Green ED. The role of aminoacyl‐tRNA synthetases in genetic diseases. Annu Rev Genomics Hum Genet 2008;9:87–107. [DOI] [PubMed] [Google Scholar]
- 2. Abbott JA, Meyer‐Schuman R, Lupo V, et al. Substrate interaction defects in histidyl‐tRNA synthetase linked to dominant axonal peripheral neuropathy. Hum Mutat 2018;39:415–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rajendran V, Kalita P, Shukla H, et al. Aminoacyl‐tRNA synthetases: structure, function, and drug discovery. Int J Biol Macromol 2018;111:400–414. [DOI] [PubMed] [Google Scholar]
- 4. Oprescu SN, Griffin LB, Beg AA, Antonellis A. Predicting the pathogenicity of aminoacyl‐tRNA synthetase mutations. Methods 2017;113:139–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sissler M, Gonzalez‐Serrano LE, Westhof E. Recent advances in mitochondrial aminoacyl‐tRNA synthetases and disease. Trends Mol Med 2017;23:693–708. [DOI] [PubMed] [Google Scholar]
- 6. Antonellis A, Oprescu SN, Griffin LB, et al. Compound heterozygosity for loss‐of‐function FARSB variants in a patient with classic features of recessive aminoacyl‐tRNA synthetase‐related disease. Hum Mutat 2018;39:834–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Musante L, Puttmann L, Kahrizi K, et al. Mutations of the aminoacyl‐tRNA‐synthetases SARS and WARS2 are implicated in the etiology of autosomal recessive intellectual disability. Hum Mutat 2017;38:621–636. [DOI] [PubMed] [Google Scholar]
- 8. Tsai PC, Soong BW, Mademan I, et al. A recurrent WARS mutation is a novel cause of autosomal dominant distal hereditary motor neuropathy. Brain 2017;140:1252–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Meyer‐Schuman R, Antonellis A. Emerging mechanisms of aminoacyl‐tRNA synthetase mutations in recessive and dominant human disease. Hum Mol Genet 2017;26(R2):R114–R127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vester A, Velez‐Ruiz G, McLaughlin HM, et al. A loss‐of‐function variant in the human histidyl‐tRNA synthetase (HARS) gene is neurotoxic in vivo. Hum Mutat 2013;34:191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Safka Brozkova D, Deconinck T, Griffin LB, et al. Loss of function mutations in HARS cause a spectrum of inherited peripheral neuropathies. Brain 2015;138(Pt 8):2161–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jordanova A, Irobi J, Thomas FP, et al. Disrupted function and axonal distribution of mutant tyrosyl‐tRNA synthetase in dominant intermediate Charcot‐Marie‐Tooth neuropathy. Nat Genet 2006;38:197–202. [DOI] [PubMed] [Google Scholar]
- 13. Harripaul R, Vasli N, Mikhailov A, et al. Mapping autosomal recessive intellectual disability: combined microarray and exome sequencing identifies 26 novel candidate genes in 192 consanguineous families. Mol Psychiatry 2018;23:973–984. [DOI] [PubMed] [Google Scholar]
- 14. Wei N, Zhang Q, Yang XL. Neurodegenerative Charcot‐Marie‐Tooth disease as a case study to decipher novel functions of aminoacyl‐tRNA synthetases. J Biol Chem 2019;294(14):5321–5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Froelich CA, First EA. Dominant Intermediate Charcot‐Marie‐Tooth disorder is not due to a catalytic defect in tyrosyl‐tRNA synthetase. Biochemistry 2011;50:7132–7145. [DOI] [PubMed] [Google Scholar]
- 16. Puffenberger EG, Jinks RN, Sougnez C, et al. Genetic mapping and exome sequencing identify variants associated with five novel diseases. PLoS ONE 2012;7:e28936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Abbott JA, Guth E, Kim C, et al. The usher syndrome type IIIB histidyl‐tRNA synthetase mutation confers temperature sensitivity. Biochemistry 2017;56:3619–3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Royer‐Bertrand B, Castillo‐Taucher S, Moreno‐Salinas R, et al. Mutations in the heat‐shock protein A9 (HSPA9) gene cause the EVEN‐PLUS syndrome of congenital malformations and skeletal dysplasia. Sci Rep 2015;5:17154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein‐coding genetic variation in 60,706 humans. Nature 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kircher M, Witten DM, Jain P, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014;46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Carter H, Douville C, Stenson PD, et al. Identifying Mendelian disease genes with the variant effect scoring tool. BMC Genom 2013;14(Suppl 3):S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Siekierska A, Stamberger H, Deconinck T, et al. Biallelic VARS variants cause developmental encephalopathy with microcephaly that is recapitulated in vars knockout zebrafish. Nat Commun 2019;10:708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu J, Xu Y, Stoleru D, Salic A. Imaging protein synthesis in cells and tissues with an alkyne analog of puromycin. Proc Natl Acad Sci USA 2012;109:413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Seburn KL, Nangle LA, Cox GA, et al. An active dominant mutation of glycyl‐tRNA synthetase causes neuropathy in a Charcot‐Marie‐Tooth 2D mouse model. Neuron 2006;51:715–726. [DOI] [PubMed] [Google Scholar]
- 25. Storkebaum E, Leitao‐Goncalves R, Godenschwege T, et al. Dominant mutations in the tyrosyl‐tRNA synthetase gene recapitulate in Drosophila features of human Charcot‐Marie‐Tooth neuropathy. Proc Natl Acad Sci USA 2009;106:11782–11787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cavanagh JB. Corpora‐amylacea and the family of polyglucosan diseases. Brain Res Brain Res Rev 1999;29:265–295. [DOI] [PubMed] [Google Scholar]
- 27. Posey JE, Harel T, Liu P, et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N Engl J Med 2017;376:21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Voinea C, Gonzalez Rodriguez E, Beigelman‐Aubry C, et al. Hepatosplenomegaly, pneumopathy, bone changes and fronto‐temporal dementia: Niemann‐Pick type B and SQSTM1‐associated Paget's disease in the same individual. J Bone Miner Metab 2019;37:378–383. [DOI] [PubMed] [Google Scholar]
- 29. Antonellis A, Ellsworth RE, Sambuughin N, et al. Glycyl tRNA synthetase mutations in Charcot‐Marie‐Tooth disease type 2D and distal spinal muscular atrophy type V. Am J Hum Genet 2003;72:1293–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Number of variants at the different filtering steps.