

Abstract
Objective
Charcot‐Marie‐Tooth disease (CMT) is a clinically and genetically heterogeneous group of inherited neuropathies. Mutations in more than 90 genes have been implicated in CMT; however, the mutational spectrum of CMT in Chinese population remains obscure. This study aims to provide a comprehensive overview of the frequency of mutations in Taiwanese patients with CMT and look for genotype‐phenotype correlations.
Methods
Mutational analyses were performed on 427 unrelated Taiwanese patients with CMT by polymorphic microsatellite markers analysis or real‐time fluorescent PCR for PMP22 duplication, Sanger sequencing for GJB1 mutations, and targeted sequencing covering 124 genes causing or relevant to inherited neuropathies. We also correlated the genotypes with the phenotypic features, such as age at disease onset and ulnar motor nerve conduction velocity.
Results
Pathogenic mutations were identified in 312 patients (73.1%; 312/427), including 208 patients with a PMP22 duplication, 40 patients with a GJB1 mutation, and 64 patients with a mutation in one of other 18 CMT genes. A confirmed molecular diagnosis was achieved in 84.4% (266/315) of the patients with demyelinating CMT and 41.1% (46/112) of the patients with axonal CMT. Mutations in MPZ, MFN2, or NEFL are the most frequent disease causes in patients with infantile‐onset CMT (≤2 years), while PMP22 duplications and mutations in GJB1, MFN2, or MPZ are the frequent causes among patients with childhood‐ or adolescence‐onset CMT (3–9 years).
Interpretation
This study provides a genotype‐phenotype landscape of CMT in Taiwan and highlights the unique spectrum of CMT genes frequencies among patients of Chinese origin.
Introduction
Charcot‐Marie‐Tooth disease (CMT), also known as hereditary motor and sensory neuropathy, is a clinically and genetically heterogeneous group of inherited neuropathies characterized by progressive distal muscle atrophy and weakness, distal sensory loss, foot deformities, and depressed tendon reflexes.1, 2 It is one of the most common inherited neurological disorders with a prevalence of approximately one in 2500 individuals.3, 4 There were several kinds of CMT classification from historical perspectives.5 In clinical practice, one widely used classification is dichotomizing CMT into demyelinating and axonal forms using the median or ulnar motor nerve conduction velocity (MNCV) with a cut‐off value of 38 m/sec.6 Actually, most patients categorized into demyelinating CMT group have dysmyelination due to the genetic defect.7, 8 Sometimes, patients with CMT having an upper limb MNCV between 25 and 45 m/sec are classified as intermediate CMT.9 CMT can also be categorized by the inheritance pattern and electrophysiological features.1 CMT patients mostly are inherited in autosomal dominant manner (CMT1 for the demyelinating forms and CMT2 for the axonal forms), in autosomal recessive manner (CMT4) or in X‐linked manner (CMTX).
To date, mutations in more than 90 genes have been implicated in CMT,10, 11 and only a few of the genes, such as PMP22, GJB1, MFN2, and MPZ, account for a significant percentage of CMT cases when mutated.12, 13, 14, 15 It is still not fully clear about the contributions of other CMT‐related genes, especially those in which mutations have just identified in one or two families or sporadic cases in past few years. Although a number of studies have investigated the frequencies and spectrum of mutations in one or some particular genes in CMT patients of different ethnicities, only a few provided a comprehensive landscape of the mutation spectrum and frequencies of multiple genes. Most of the studies were performed in Caucasian cohorts with CMT12, 13, 14, 15, 16 and two studies were conducted in the Japanese population.17, 18 Except for a small‐scale study investigating 82 Chinese patients with CMT,19 no extensive mutational analysis has been performed in CMT patients of Han Chinese populations yet.
To fill this knowledge gap and increase the understanding of rare CMT subtypes, we assessed 427 unrelated Taiwanese CMT patients of Han Chinese origin for known CMT‐related genes using traditional methods and next‐generation sequencing (NGS) techniques. Clinical and neurophysiological features of these CMT patients were also analyzed. The aim of this study is to provide a comprehensive overview of the spectrum and frequencies of mutations in Taiwanese patients with CMT and look for genotype‐phenotype correlations.
Subjects and Methods
Patient cohort
Between 1996 and 2018, 427 index patients with CMT were consecutively recruited from the Neurology Clinics of Taipei Veterans General Hospital. The diagnosis and classification of CMT were based on the clinical manifestations, family history and the electrophysiological features.1 During this period, there were 81 unrelated patients with clinical presentations and genetic mutations compatible with the diagnostic guideline of hereditary neuropathy with liability to pressure palsies (HNPP).20 Eighty of them carried a PMP22 deletion and one patient harbored the PMP22 p.C42R mutation that had been reported previously.21 Although HNPP is traditionally categorized as a subtype of CMT, we did not include the 81 patients in the present study because of the discrepant phenotypes between HNPP and other CMT subtypes.
Nerve conduction studies were performed by standard techniques utilizing a Medelec MS25 electromyograph (Mistro, Surrey, U.K.) with surface electrode stimulations and recordings. Distal and proximal motor latencies and compound muscle action potential amplitudes were recorded from the median, ulnar, peroneal, and tibial nerves. Sensory nerve action potential amplitudes and distal latencies were recorded from median, ulnar, and sural nerves. MNCV was calculated by standard techniques. Forearm ulnar MNCV with a cutoff value of 38 m/sec was used to distinguish between demyelinating and axonal CMT.6
We further categorized the patients into four groups according to the age at disease onset: infancy (≤2 years), childhood/adolescence (3–19 years), early adulthood (20–39 years) and late adulthood (≥40 years). All the patients were of Han Chinese descent. To investigate the relationship between genotype and electrophysiological features, the patients were also divided into four groups according to their forearm ulnar MNCV: (1) MNCV ≤15 m/sec, (2) MNCV between 15 and 25 m/sec, (3) MNCV between 25 and 38 m/sec, and (4) MNCV >38 m/sec.
Written informed consent was obtained from all of the participants or their parents on behalf of those who were younger than 18 years. This study was approved by the Institutional Review Board of Taipei Veterans General Hospital.
Molecular genetic analysis
In patients with demyelinating CMT, we first investigated PMP22 duplication by polymorphic microsatellite markers analysis or real‐time fluorescent PCR,22, 23 and then screened for mutations in GJB1 (RefSeq NM_000109764) by direct nucleotide sequencing. In patients with axonal CMT, we first sequenced GJB1 because of its small size and a proportion of patients with GJB1 mutations having a forearm ulnar NCV above 38 m/sec. For most of the patients without PMP22 duplication and GJB1 mutations, further mutational analysis was performed by utilizing a high‐throughput targeted NGS panel for detecting mutations in other CMT‐related genes. For a minor group of the patients recruited before 2014, the mutation analyses were conducted by direct nucleotide sequencing of PMP22 (RefSeq NM_000304.3), MPZ (RefSeq NM_000530.5), MFN2 (RefSeq NM_014874.3), NEFL (RefSeq NM_006158.4), AARS (RefSeq NM_001605.2), HSPB1 (RefSeq NM_001540.5), and GDAP1 (RefSrq NM_018972.3) as employed in our previous studies.21, 24, 25, 26
The targeted NGS panel covering the coding exons of 124 genes associated with inherited neuropathies (Table S1) was designed by using NimbleGen Design website (http://www.nimblegen.com/products/nimbledesign/). The NimbleGen SeqCap EZ Choice Library system (Roche NimbleGen, Madison, WI) was used to enrich the targeted regions. The enriched samples were sequenced using the HiSeq2000 platform (Illumina, San Diego, CA) with a paired‐end 100bp protocol. All sequenced reads were mapped to the Human Genome version 19 (hg19/GRCh37). The BaseSpace pipeline (https://basespace.illumina.com/) and the Illumina VariantStudio software (http://variantstudio.software.illumina.com/) were used for variant calling and annotation. After annotation, only rare nonsynonymous variants with minor allele frequencies lesser than 0.1% for dominant CMT and 1% for recessive CMT disease genes in the Taiwan Biobank (https://taiwanview.twbiobank.org.tw/index), which contains whole genome sequences of 1517 Taiwanese healthy controls, were taken for further analysis. Sanger sequencing was performed to confirm the potentially pathogenic variants.
Bioinformatics analyses
Previously reported pathogenic mutations were confirmed by literature reviews and querying the Inherited Neuropathy Variant Browser (http://hihg.med.miami.edu/code/http/cmt/public_html/index.html#/) and the Human Gene Mutation Database Professional (https://portal.biobase-international.com/hgmd/pro). To evaluate the pathogenicity of the novel variants, we surveyed the variants in the genome Aggregation Database (gnomAD; http://gnomad.broadinstitute.org).27 The novel and potential pathogenic variants were further checked among another 500 neurologically healthy individuals of Han Chinese origin. Segregation analysis was also done in those patients whose family members were available.
The pathogenicity of these variants were predicted in silico by Mutation Taster (http://www.mutationtaster.org),28 PolyPhen‐2 (http://genetics.bwh.harvard.edu/pph2/),29 combined annotation dependent depletion (CADD) (http://cadd.gs.washington.edu),30 and SIFT (http://sift.jcvi.org/).31 The UniProt website (http://www.uniprot.org) was used to evaluate evolutionary conservation of the mutated amino acid by aligning amino‐acid sequences of the orthologues from several species.32
Statistical analyses
Descriptive statistics were performed, and the data were represented as mean ± standard deviation (SD) for age of disease onset and ulnar MNCV. The undetectable nerve conduction parameters were excluded from mean and SD analyses. Statistical analysis was performed with the statistical software package SPSS for Windows (version 19.0; SPSS, Inc., Chicago, IL). A P < 0.05 was defined as statistically significant.
Results
Characteristics of study participants
Among the 427 unrelated CMT patients, 248 are male and 179 are female. The average age of disease onset was 23.8 ± 17.4 (range 1–72) years. Three hundred fifteen patients (73.8%) had demyelinating CMT and 112 patients (26.2%) had an axonal polyneuropathy. Family history of CMT was present in 58.6% of patients, in whom 48.6%, 7.3%, and 2.7% of cases were inherited in an autosomal dominant, X‐linked, and autosomal recessive manner, respectively.
Distribution of CMT subtypes
We identified the pathogenic mutations in 312 patients (73.1%; 312/427), including 208 patients with a PMP22 duplication, 40 patients with a GJB1 mutation, and 64 patients with a mutation in one of other 18 CMT‐related genes (Table 1). Up to 84.4% (266/315) of the patients with demyelinating CMT and 41.1% (46/112) of the patients with axonal CMT were confirmed to harbor a mutation responsible for their diseases. Among the 427 unrelated CMT patients, the most common genetic causes for the CMT patients were PMP22 duplication (48.7%), GJB1 mutations (9.4%), MPZ mutations (3.3%), MFN2 mutations (3.3%), and NEFL mutations (1.9%) (Fig. 1A and Table 1). These five groups of mutations accounted for 91.0% of all the CMT patients who had achieved a molecular diagnosis and 66.5% of all the CMT patients in the present cohort. Each of the remaining CMT‐related genes (i.e. PMP22, SH3TC2, EGR2, GNB4, LITAF, GARS, HSPB1, GDAP1, IGHMBP2, BSCL2, KIF5A, LRSAM1, AARS, TFG, and MORC2) accounted for <1% of total CMT patients each (Fig. 1B and Table 1).
Table 1.
Distribution of CMT subtypes and associated clinical/neurophysiological features.
| Genetic subtype | N | % in patients with demyelinating or axonal CMT | % in all patients with CMT | Age of disease onset, year (mean ± SD) | Ulnar MNCV, m/sec (mean ± SD) |
|---|---|---|---|---|---|
| Demyelinating CMT | 315 | 100 | 73.8 | ||
| PMP22 duplication | 208 | 66.0 | 48.7 | 26.5 ± 17.3 (3–64) | 19.1 ± 5.4 (5.7–36.8) |
| GJB1 | 32 | 10.2 | 7.5 | 19.8 ± 8.5 (7–40) | 32.8 ± 2.9 (25.0–38.0) |
| Male | 29 | 19.6 ± 8.8 (7–40) | 32.5 ± 2.9 (25.0–38.0) | ||
| Female | 3 | 21.7 ± 5.7 (17–28) | 35.5 ± 0.4 (35.0–35.8) | ||
| MPZ | 12 | 3.8 | 2.8 | 15.5 ± 16.5 (1–42) | 15.0 ± 6.9 (5.1–33.3) |
| PMP22 1 | 4 | 1.3 | 0.9 | 13.5 ± 8.7 (1–20) | 11.3 ± 8.0 (5.9–20.5) |
| SH3TC2 | 3 | 1.0 | 0.7 | 26.3 ± 20.6 (5–46) | 26.9 ± 1.8 (25.5–28.9) |
| NEFL | 2 | 0.6 | 0.5 | 1.5 ± 0.7 (1–2) | 21.1 ± 1.5 (20.0–22.1) |
| EGR2 | 2 | 0.6 | 0.5 | 1.0 | 4.7 |
| GNB4 | 2 | 0.6 | 0.5 | 25.0 ± 28.3 (5–45) | 25.6 ± 9.5 (18.8–32.3) |
| LITAF | 1 | 0.3 | 0.2 | 40.0 | 31.0 |
| Unknown | 49 | 15.6 | 11.5 | 23.2 ± 16.7 (1–64) | 25.9 ± 8.8 (8.1–43.0) |
| Axonal CMT | 112 | 100 | 26.2 | ||
| MFN2 | 14 | 12.5 | 3.3 | 13.7 ± 17.6 (1–72) | 47.6 ± 9.2 (30.0–61.4) |
| GJB1 | 8 | 7.1 | 1.9 | 25.9 ± 15.1 (7–45) | 45.3 ± 6.4 (38.3–57.0) |
| Male | 4 | 26.8 ± 18.6 (7–45) | 44.4 ± 5.6 (38.3–51.4) | ||
| Female | 4 | 24.7 ± 12.9 (10–34) | 46.2 ± 7.8 (38.9–57.0) | ||
| NEFL | 6 | 5.4 | 1.4 | 17.2 ± 14.7 (1–40) | 41.9 ± 4.3 (36.8–48.7) |
| MPZ | 2 | 1.8 | 0.5 | 53.0 ± 11.3 (45–61) | 51.0 ± 1.4 (50.0–52.0) |
| GARS | 2 | 1.8 | 0.5 | 1.5 ± 0.7 (1–2) | 43.5 |
| HSPB1 | 2 | 1.8 | 0.5 | 33.5 ± 19.1 (20–47) | 39.5 ± 7.1 (34.5–44.5) |
| GDAP1 | 2 | 1.8 | 0.5 | 1.5 ± 0.7 (1–2) | 48.1 |
| IGHMBP2 | 2 | 1.8 | 0.5 | 11.5 ± 14.8 (1–22) | 38.8 ± 16.8 (26.9–50.6) |
| BSCL2 | 2 | 1.8 | 0.5 | 14.5 ± 13.4 (5–24) | 48.5 ± 4.9 (45.0–52.0) |
| KIF5A | 2 | 1.8 | 0.5 | 15.0 ± 7.1 (10–20) | 48.1 ± 1.6 (47.0–49.2) |
| LRSAM1 | 1 | 0.9 | 0.2 | 65.0 | 63.2 |
| AARS | 1 | 0.9 | 0.2 | 30.0 | 42.1 |
| TFG | 1 | 0.9 | 0.2 | 32.0 | 63.5 |
| MORC2 | 1 | 0.9 | 0.2 | 1.0 | 33.0 |
| Unknown | 66 | 58.9 | 15.5 | 26.5 ± 19.4 (1–70) | 47.7 ± 7.6 (32.0–62.8) |
CMT, Charcot‐Marie‐Tooth disease; SD, standard deviation; MNCV, motor nerve conduction velocities.
PMP22 point mutation.
Figure 1.

Genetic spectrum of CMT among the Han Chinese population in Taiwan. (A) The common genetic causes, and (B) rare genetic causes in our CMT cohort. CMT, Charcot‐Marie‐Tooth disease; dup., duplication.
Among the demyelinating CMT patients, the most common genetic causes are PMP22 duplication (66%), GJB1 mutations (10.2%), and MPZ mutations (3.8%) (Table 1). These three groups of mutations accounted for 94.7% (252/266) of the demyelinating CMT patients whose mutations had been identified in the present study. Among the axonal CMT patients, the most common genetic causes are MFN2 mutations (12.5%), GJB1 mutations (7.1%), and NEFL mutations (5.4%). These three groups of mutations accounted for 60.9% (28/46) of the axonal CMT patients with an identified mutation.
Among the 81 pathogenic mutations in CMT‐related genes identified in the present study, 12 mutations were novel (Table 2). These novel mutations are p.F51C, p.Y135D, p.R183F and p.T185Pfs*11 in GJB1, p.[L139del];[L1048P] in SH3TC2, p.N98Y in NEFL, p.E356K in EGR2, p.R280P in MFN2, p.[L40R];[R595W] and p.[A786Pfs*45];[711+1G>C] in IGHMBP2, p.Q764* in KIF5A, and p.E680* in LRSAM1. The following points support the pathogenicity of these mutations (Table 3). First, all the 12 novel mutations are absent in the 500 healthy Taiwanese controls. Among these mutations, the nine dominant CMT mutations were also absent from both gnomAD and Taiwan Biobank database containing genome sequence of 1517 healthy Taiwanese. The three recessively inherited CMT with compound heterozygous mutations were absent or present in a very low allele frequency in the both databases. Second, these mutations alter the evolutionarily conserved amino acid residues of the mutated proteins and their pathogenicity was supported by in silico prediction, using Mutation Taster, PolyPhen‐2, CADD score, and SIFT programs. Third, six of the novel missense mutations change an amino acid residue where a different missense change determined to be pathogenic has been seen before, including GJB1 p.F51C,33 GJB1 p.Y135D,34 GJB1 p.R183F,35, 36 NEFL p.N98Y,37, 38 MFN2 p.R280P,39 and IGHMBP2 p.R595W40. Five of these novel mutations lead to protein length changes, such as GJB1 p.T185Pfs*11, SH3TC2 L139del, LRSAM1 p.E680*, IGHMBP2 p.A786Pfs*45, and KIF5A p.Q764*.
Table 2.
Pathogenic mutations identified in Taiwanese CMT patients.
| Gene | Ever reported in literatures or public database1 | Novel |
|---|---|---|
| Demyelinating CMT | ||
| GJB1 | p.M1I, p.L6S, p.I20F, p.S26L, p.S49Y, p.V91M, p.I101Rfs*8, p.L144del, p.F153L, p.Y160C, p.R164Q, p.C173Y, p.R183C, p.R183H, p.E186K p.A197V, p.R215P, | p.F51C, p.Y135D p.R183F, p.T185Pfs*11 |
| MPZ | p.V58D, p.S63F, p.T65I, p.R98C, p.R98H, p. G123S, p.D128G, p.I135M, p.Q187Pfs*63, p.S233fs | – |
| PMP22 | p.Q86*, p.I104Ffs*7, c.319+G>A | – |
| SH3TC2 | p.W245*, p.E657K | p.[L139del];[L1048P] |
| NEFL | p.N98S | p.N98Y |
| EGR2 | p.E412K | p.E356K |
| GNB4 | p.G53D, p.K89E | – |
| LITAF | p.G112S | – |
| Axonal CMT | ||
| MFN2 | p.R94Q, p.T159_Q162del, p.L218P, p.L233V, p.R280H, p.R364W, p.R400P, p.L724P, p.W740C, p.E744M | p.R280P |
| GJB1 | p.S138G, p.M162L, p.R220*, p.D278V, c.‐459C>T, c.‐529T>C | |
| NEFL | p.P8R, p.P22S, p.E396K | – |
| MPZ | p.T124M | – |
| GARS | p.D146Y, p.M238R | – |
| HSPB1 | p.R127L, p.T164A | – |
| GDAP1 | p.[H256R];[R282H], p.[H256R];[118‐1G>C] | – |
| IGHMBP2 | – | p.[L40R];[R595W], p.[A786Pfs*45];[711+1G>C], |
| BSCL2 | p.N88S, p.S90L | – |
| KIF5A | p.R280C | p.Q764* |
| LRSAM1 | – | p.E680* |
| AARS | p.N71Y | – |
| TFG | p.G269V | – |
| MORC2 | p.S25L | – |
CMT, Charcot‐Marie‐Tooth disease.
Previously reported pathogenic mutations were confirmed by literature reviews and querying the Inherited Neuropathy Variant Browser (http://hihg.med.miami.edu/code/http/cmt/public_html/index.html#/) and the Human Gene Mutation Database Professional (https://portal.biobase-international.com/hgmd/pro).
Table 3.
Bioinformatics prediction and population analyses of the novel pathogenic mutations.
| Gene | Nucleotide changes | Amino acid changes | Bioinformatics prediction | Population genetics1 | Conservation | ||||
|---|---|---|---|---|---|---|---|---|---|
| Mutation taster | PolyPhen2 | CADD score | SIFT | gnomAD | Taiwan Biobank | ||||
| GJB1 | c.152T>G | p.F51C | Disease causing | Probably damaging | 23.3 | Damaging | Not found | Not found | Zebrafish |
| GJB1 | c.403T>G | p.Y135D | Disease causing | Probably damaging | 26.0 | Damaging | Not found | Not found | Zebrafish |
| GJB1 | c.547_548delinsTT | p.R183F | N/A | Probably damaging | 27.6 | Damaging | Not found | Not found | Zebrafish |
| GJB1 | c.552del | p.T185Pfs*11 | Disease causing | Probably damaging | 23.6 | N/A | Not found | Not found | Zebrafish |
| NEFL | c.292A>T | p.N98Y | N/A | Probably damaging | 27.1 | Damaging | Not found | Not found | Zebrafish |
| EGR2 | c.1066G>A | p.E356K | Disease causing | Probably damaging | 27.5 | Damaging | Not found | Not found | C. elegans |
| MFN2 | c.839G>C | p.R280P | Disease causing | Probably damaging | 33 | Damaging | Not found | Not found | C. elegans |
| KIF5A | c.2290C>T | p.Q764* | Disease causing | N/A | 43 | N/A | Not found | Not found | Drosophila |
| LRSAM1 | c.2038G>T | p.E680* | Disease causing | N/A | 48 | N/A | Not found | Not found | Zebrafish |
| SH3TC2 | c.[415_417del]; [3143T>C] | p.[L139del]; [L1048P] | Disease causing | N/A | 22.2 | N/A | 1/246236 | Not found | Zebrafish |
| Disease causing | Probably damaging | 28.3 | Damaging | 14/246082 | 1/1517 | Zebrafish | |||
| IGHMBP2 | c.[119T>G]; [1783C>T] | p.[L40R]; [R595W] | Disease causing | Probably damaging | 28.1 | Damaging | Not found | Not found | Zebrafish |
| Disease causing | Probably damaging | 35 | Damaging | 4/245652 | Not found | Zebrafish | |||
| IGHMBP2 | c.[2354del]; [711+1G>C] | p.[A786Pfs*45];[711+1G>C] | Disease causing | Benign | 23.9 | N/A | 5/276040 | Not found | N/A |
| N/A | N/A | N/A | N/A | N/A | N/A | N/A | |||
CADD, combined annotation dependent depletion; gnomAD, genome aggregation database; N/A, not applicable.
Minor allele count/total allele count.
Genotype‐phenotype correlations
To investigate the genotype‐phenotype relationship within this CMT cohort, we analyzed the mutation spectrum in patients with different age of disease onset and in patients with different scopes of ulnar MNCV.
The age of disease onset was ascertained in 352 patients by self‐reporting the time when the parents noticed any motor abnormalities in their children, or when the patients themselves began to be aware of their motor or sensory dysfunctions. Most of the CMT patients (43.8%) have a childhood‐ or adolescence‐onset disease (3–19 years), around one‐fourth (26.4%) with an early adulthood‐onset disease (20–39 years), another one‐fourth (23.3%) with a late adulthood‐onset disease (≥40 years), and only 6.5% of them have disease onset at infancy (≤2 years). In the patients with infantile‐onset CMT, mutations in MPZ, MFN2, or NEFL are the most frequent disease causes and mutations in each gene account for 13.0% of the infantile‐onset patients (Fig. 2). Among the patients with childhood‐ or adolescence‐onset CMT, PMP22 duplications (42.2%) and mutations in GJB1 (12.3%), MFN2 (5.8%), or MPZ (3.2%) are the frequent causes of diseases. In the early adulthood‐onset CMT group, PMP22 duplications (40.9%) and GJB1 mutations (18.3%) are the major etiologies. For CMT patients whose first symptom occurs during late adulthood, PMP22 duplication was the most common cause (51.2%), followed by MPZ mutations (6.1%), and then GJB1 mutations (3.7%).
Figure 2.
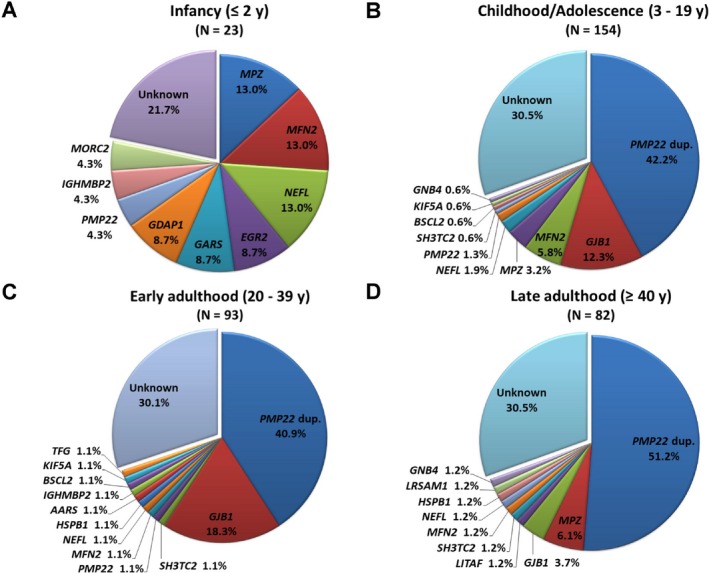
The mutational distribution of CMT‐related genes based on age at disease onset. The frequencies of genetic diagnoses in patients with (A) infantile‐onset CMT (≤2 years), (B) childhood‐ or adolescence‐onset CMT (3–19 years), (C) early adulthood‐onset CMT (20–39 years), and (D) late adulthood‐onset CMT (≥40 years). CMT, Charcot‐Marie‐Tooth disease; dup., duplication.
When the CMT patients were grouped based on their ulnar MNCV, 14.6% of the CMT patients have MNCV ≤15 m/sec, 36.2% have MNCV between 15 and 25 m/sec, 24.6% have MNCV between 25 and 38 m/sec, and 24.6% have MNCV faster than 38 m/sec. In the CMT patients with ulnar MNCV ≤15 m/sec, PMP22 duplications and MPZ mutations are the two major causes and account for 67.9% and 15.1% of the patients, respectively (Fig. 3). Among the patients with ulnar MNCV between 15 and 25 m/sec, PMP22 duplication is the sole major etiology, accounting for 84% of the cases. In the patients with ulnar MNCV between 25 and 38 m/sec, the major etiologies are GJB1 mutations (32.6%) and PMP22 duplications (23.6%). In patients with ulnar MNCV faster than 38 m/sec, the major disease causes are mutations in MFN2 (12.4%), GJB1 (9.0%), and NEFL (4.5%).
Figure 3.
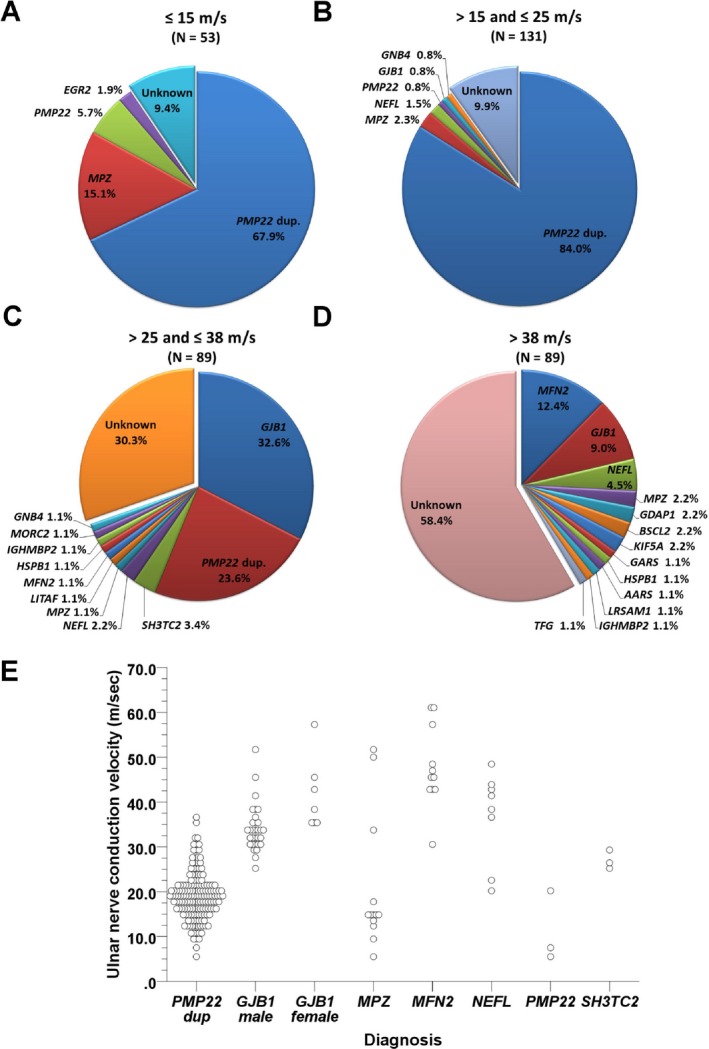
The mutational distribution of CMT‐related genes based on ulnar MNCV. The frequencies of genetic diagnoses in the CMT patients with (A) ulnar MNCV ≤15 m/sec, (B) ulnar MNCV between 15 and 25 m/sec, (C) ulnar MNCV between 25 and 38 m/sec, and (D) ulnar MNCV faster than 38 m/sec. (E) The distribution of ulnar MNCV in relation to each CMT‐related genes. CMT, Charcot‐Marie‐Tooth disease; dup., duplication.
Discussion
This study demonstrated the genotypic and phenotypic profiles of CMT in a Han Chinese population by investigating 427 unrelated CMT patients in Taiwan. Eighty‐one different pathogenic mutations were identified in 312 patients (73.1%; 312/427), including 208 patients with a PMP22 duplication, 40 patients with a GJB1 mutation, and 64 patients with a mutation in one of other 18 CMT genes. Among the pathogenic mutations identified in the present study, 12 were novel. This is the first large‐scale genetic research on CMT in the Chinese population. In comparison with similar studies in Caucasian or Japanese populations (Table 4), the present study revealed that CMT in Taiwanese population had a higher proportion of PMP22 duplication and relatively more common NEFL mutations. The present study also showed the prioritization of genetic testing for CMT patients of Han Chinese descent by using the age of disease onset and ulnar MNCV to separate the patients into specific subgroups.
Table 4.
Comparison of genetic distribution of CMT subtypes in diverse populations.
| Gene | This study | U.S.A.12 | U.K.13 | Germany14 | Spain16 | Japan17 | International (INC)15 |
|---|---|---|---|---|---|---|---|
| N = 427 | N = 787 (48 HNPP) | N = 425 | N = 589 (83 HNPP) | N = 438 | N = 354 | N = 1652 (36 HNPP) | |
| PMP22 dup | 48.7% | 36.9% | 39.5% | 35.6% | 42.0% | 15.0% | 37.2% |
| GJB1 | 9.4% | 10.2% | 10.8% | 9.3% | 12.8% | 7.1% | 6.5% |
| MPZ | 3.3% | 5.7% | 3.1% | 4.2% | 4.3% | 7.1% | 4.1% |
| MFN2 | 3.3% | 2.7% | 2.8% | 2.4% | 1.4% | 4.0% | 4.2% |
| NEFL | 1.9% | 0.5% | 0.5% | 0.0% | 0.9% | 2.3% | 0.66% |
| PMP22 1 | 0.9% | 0.6% | 1.4% | 0.8% | 0.5% | 2.8% | 1.0% |
| SH3TC2 | 0.7% | 0.4% | 1.2% | 0.0% | 6.2% | n.t. | 0.85% |
| EGR2 | 0.5% | 0.1% | 0.0% | 0.0% | 0.0% | 0.3% | 0.06% |
| GARS | 0.5% | 0.4% | n.t. | 0.4% | 0.9% | 0.3% | 0.12% |
| HSPB1 | 0.5% | n.t. | 0.5% | 0.0% | 1.6% | n.t. | 0.42% |
| GDAP1 | 0.5% | 0.6% | 0.5% | 0.0% | 9.6% | 0.3% | 0.54% |
| IGHMBP2 | 0.5% | n.t. | n.t. | n.t. | n.t. | n.t. | n.t. |
| BSCL2 | 0.5% | n.t. | 0.2% | n.t. | 0.0% | n.t. | 0.30% |
| KIF5A | 0.5% | n.t. | n.t. | n.t. | n.t. | n.t. | n.t. |
| GNB4 | 0.5% | n.t. | n.t. | n.t. | n.t. | n.t. | n.t. |
| LITAF | 0.2% | 0.6% | 0.9% | 0.0% | 0.0% | 0.0% | 0.12% |
| LRSAM1 | 0.2% | n.t. | n.t. | n.t. | 0.0% | n.t. | n.t. |
| AARS | 0.2% | n.t. | n.t. | n.t. | 0.0% | n.t. | n.t. |
| TFG | 0.2% | n.t. | n.t. | n.t. | n.t. | n.t. | n.t. |
| MORC2 | 0.2% | n.t. | n.t. | n.t. | n.t. | n.t. | n.t. |
| Unknown | 26.9% | 33.0% | 37.4% | 42.4% | 16.7% | 59.6% | 39.6% |
CMT, Charcot‐Marie‐Tooth disease; HNPP, Hereditary neuropathy with liability to pressure palsy; dup, duplication; n.t., not tested.
PMP22 point mutation.
In our CMT cohort, the most frequent genetic causes are PMP22 duplication and mutations in GJB1, MPZ, and MFN2. These genetic alterations comprised 88.5% of cases with positive molecular diagnosis in our study, which is in accordance with the findings in the U.S., U.K., and Germany studies.12, 13, 14 The PMP22 duplication accounted for 66.7% of the genetically confirmed CMT cases and was responsible for 48.7% of all CMT patients in our cohort. The proportion of PMP22 duplication among CMT patients in Taiwan was higher than that in U.S., U.K., Germany, Spain and Japan (15–42%).12, 13, 14, 15, 16, 17, 18 This phenomenon may come from ethnic differences. All patients that carried PMP22 duplications had ulnar MNCV <38 m/sec (Fig. 3E). The age of clinical onset varied from 3 to 64 years, and sporadic cases accounted for 24.2% of them. Therefore, onset at late adulthood and lack of family history did not exclude the possibility of CMT1A.
Mutations in GJB1 were the second most frequent cause of CMT and accounted for 12.8% of CMT patients with a confirmed pathogenic mutation and 9.4% of overall CMT patients in the study. The majority of the patients carrying a GJB1 mutation were male. The ulnar MNCV of the patients with GJB1 mutations varied widely, ranging from 25 to 57 m/sec and crossing the usually used cutoff value 38 m/sec for distinguishing between demyelinating and axonal CMT. Several mutations in GJB1 affected more than two index patients, including four cases with p.S26L, four with p.R164Q, and three with p.V91M, suggesting the possibility of multiple founders of GJB1 mutations in our cohort. Because GJB1 mutations are not rare in CMT and the coding region of GJB1 is not large, in our lab, we do traditional GJB1 sequencing for CMT patients without PMP22 duplication before considering the NGS targeted sequencing panel.
Among the patients with axonal CMT, we identified 37 distinct mutations in 14 genes. In Caucasian populations, GJB1, MFN2, and MPZ were the most common mutated genes in axonal CMT.12, 13, 14 However, our data suggested that MFN2, GJB1, and NEFL genes are the three main disease genes for axonal CMT in our population. The relatively high percentage of NEFL mutations in our CMT cohort (1.9%) with recurrent mutations of p.P8R and p.E396K (two and three pedigrees, respectively) suggested population‐specific founder effects of the NEFL mutations in Chinese population. NEFL mutations were also relatively prevalent in Japan, accounting for 0.9% and 2.3% of total CMT patients in two Japanese cohorts.17, 18
After a series of mutational analyses, including PMP22 dosage assay, Sanger sequencing, and targeted sequencing with NGS technique, a causal mutation could be identified in 73.1% of the patients in our CMT cohort. The diagnostic yield rate of demyelinating CMT was higher than that of axonal CMT (84.4% vs. 41.1%). Moreover, we identified 81 different mutations in 19 CMT‐related genes in our CMT cohort, and point mutations in 15 genes accounted for <1% of the patients each. The number of the mutated genes identified in this study was higher than similar studies. Possible explanations include the ethnic factor, different inclusion criteria, and different mutation‐detecting strategies.
Forearm MNCV is a popular parameter to categorize CMT into demyelinating or axonal subtype. Moreover, Saporta et al.12 analyzed data from 787 CMT patients and proposed a genetic testing strategy for CMT based on different ranges of ulnar MNCV. In this study, we utilized ulnar MNCV and age of disease onset to separate the patients with identified mutations into 16 subgroups (Table S2). This information can help predict the genotype according to the MNCV and age of onset and provide a guide for the prioritization of genetic testing for CMT patients of Han Chinese origin. We acknowledged that clinical assessment scales like Charcot‐Marie‐Tooth neuropathy score41 and Charcot‐Marie‐Tooth disease Pediatric Scale42 could provide precious information toward phenotype‐genotype correlations; unfortunately, we have just started to use them to evaluate our CMT patients and only a limited number of our patients had such data. We also acknowledged that additional functional experiments are needed to confirm the causal roles of the 12 novel mutations observed in the present CMT cohort. Although their pathogenicity can be partially supported by in silico analysis and population data, there are still some degrees of uncertainty.
In conclusion, this study presents the mutational spectrum and genotype‐phenotype correlations of 427 patients of Han Chinese descent in Taiwan. There is substantial difference in the frequencies of PMP22 duplication and NEFL mutations between Taiwanese population and Caucasian populations. These findings broaden the spectrum of mutations causing CMT and are useful for optimal strategies of mutational analysis and genetic counseling of CMT for patients of Han Chinese origin.
Author Contributions
Yun‐Hsin Hsu: collecting and analyses of the data, drafting the manuscript, Kon‐Ping Lin: patient enrollment, collecting the clinical data. Yuh‐Cherng Guo: patient enrollment, colleting the clinical data. Yu‐Shuen Tsai: bioinformatics support, analyses of the next generation sequencing data. Yi‐Chu Liao: analyses and interpretation of the data, revising the manuscript for intellectual content. Yi‐Chung Lee: conceptualizing and designing the study, interpretation of the data, revising the manuscript for intellectual content.
Conflict of Interest
All authors read and approved the final manuscript. They declared no conflicts of interest.
Supporting information
Table S1. Genes in the targeted next‐generation sequencing panel.
Table S2. Genetic distribution in accordance with age of onset and ulnar MNCV (n = 227).
Acknowledgments
The authors thank for the Clinical and Industrial Genomic Application Development Service Center (C1) of the National Core Facility for Biopharmaceuticals (NCFB) for the NGS service and the bioinformatics support. The core is funded by NCFB of Ministry Science and Technology, Taiwan, ROC (MOST107‐2319‐B‐010‐002).
Yi‐Chu Liao and Yi‐Chung Lee are equal contribution.
Funding Information
This work was supported by the grants from Ministry of Science and Technology, Taiwan, ROC (MOST102‐2628‐B‐075‐006‐MY3 and MOST105‐2628‐B‐075‐002‐MY3), and Taipei Veterans General Hospital (V104C‐041 and V105C‐027), and Brain Research Center, National Yang‐Ming University from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan.
Funding Statement
This work was funded by Taipei Veterans General Hospital grants V104C-041 and V105C-027; Ministry of Science and Technology, Taiwan grants MOST102-2628-B-075-006-MY3 and MOST105-2628-B-075-002-MY3; Brain Research Center grant ; Ministry of Education grant .
Contributor Information
Yi‐Chu Liao, Email: ycliao5@vghtpe.gov.tw.
Yi‐Chung Lee, Email: ycli@vghtpe.gov.tw.
References
- 1. Shy ME, Lupski JR, Chance PF, et al. Hereditary motor and sensory neuropathies: an overview of clinical, genetic, electrophysiologic and pathologic features In: Dyck P. J. and Thomas P. K., eds. Peripheral Neuropathy. 4th ed Pp. 1623–1658. Philadelphia: Elsevier Saunders, 2005. [Google Scholar]
- 2. Rossor AM, Polke JM, Houlden H, Reilly MM. Clinical implications of genetic advances in Charcot‐Marie‐Tooth disease. Nat Rev Neurol 2013;9:562–571. [DOI] [PubMed] [Google Scholar]
- 3. Skre H. Genetic and clinical aspects of Charcot‐Marie‐Tooth's disease. Clin Genet 1974;6:98–118. [DOI] [PubMed] [Google Scholar]
- 4. Reilly MM, Murphy SM, Laura M. Charcot‐Marie‐Tooth disease. J Peripher Nerv Syst 2011;16:1–14. [DOI] [PubMed] [Google Scholar]
- 5. Mathis S, Goizet C, Tazir M, et al. Charcot‐Marie‐Tooth diseases: an update and some new proposals for the classification. J Med Genet 2015;52:681–690. [DOI] [PubMed] [Google Scholar]
- 6. Harding AE, Thomas PK. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain 1980;103:259–280. [DOI] [PubMed] [Google Scholar]
- 7. Brennan KM, Bai Y, Shy ME. Demyelinating CMT–what's known, what's new and what's in store? Neurosci Lett 2015;596:14–26. [DOI] [PubMed] [Google Scholar]
- 8. Houlden H, Reilly MM. Molecular genetics of autosomal‐dominant demyelinating Charcot‐Marie‐Tooth disease. Neuromolecular Med 2006;8:43–62. [DOI] [PubMed] [Google Scholar]
- 9. Berciano J, Garcia A, Gallardo E, et al. Intermediate Charcot‐Marie‐Tooth disease: an electrophysiological reappraisal and systematic review. J Neurol 2017;264:1655–1677. [DOI] [PubMed] [Google Scholar]
- 10. Timmerman V, Strickland AV, Zuchner S. Genetics of Charcot‐Marie‐Tooth (CMT) disease within the frame of the human genome project success. Genes (Basel) 2014;5:13–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rossor AM, Tomaselli PJ, Reilly MM. Recent advances in the genetic neuropathies. Curr Opin Neurol 2016;29:537–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Saporta AS, Sottile SL, Miller LJ, et al. Charcot‐Marie‐Tooth disease subtypes and genetic testing strategies. Ann Neurol 2011;69:22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Murphy SM, Laura M, Fawcett K, et al. Charcot‐Marie‐Tooth disease: frequency of genetic subtypes and guidelines for genetic testing. J Neurol Neurosurg Psychiatry 2012;83:706–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gess B, Schirmacher A, Boentert M, Young P. Charcot‐Marie‐Tooth disease: frequency of genetic subtypes in a German neuromuscular center population. Neuromuscul Disord 2013;23:647–651. [DOI] [PubMed] [Google Scholar]
- 15. Fridman V, Bundy B, Reilly MM, et al. CMT subtypes and disease burden in patients enrolled in the Inherited Neuropathies Consortium natural history study: a cross‐sectional analysis. J Neurol Neurosurg Psychiatry 2015;86:873–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sivera R, Sevilla T, Vílchez JJ, et al. Charcot‐Marie‐Tooth disease: genetic and clinical spectrum in a Spanish clinical series. Neurology 2013;81:1617–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Abe A, Numakura C, Kijima K, et al. Molecular diagnosis and clinical onset of Charcot‐Marie‐Tooth disease in Japan. J Hum Genet 2011;56:364–368. [DOI] [PubMed] [Google Scholar]
- 18. Yoshimura A, Yuan JH, Hashiguchi A, et al. Genetic profile and onset features of 1005 patients with Charcot‐Marie‐Tooth disease in Japan. J Neurol Neurosurg Psychiatry 2019;90:195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sun B, Chen Z, Ling L, et al. Clinical and genetic spectra of Charcot‐Marie‐Tooth disease in Chinese Han patients. J Peripher Nerv Syst 2017;22:13–18. [DOI] [PubMed] [Google Scholar]
- 20. Dubourg O, Mouton P, Brice A, et al. Guidelines for diagnosis of hereditary neuropathy with liability to pressure palsies. Neuromuscul Disord 2000;10:206–208. [DOI] [PubMed] [Google Scholar]
- 21. Liao YC, Tsai PC, Lin TS, et al. Clinical and molecular characterization of PMP22 point mutations in Taiwanese patients with inherited neuropathy. Sci Rep 2017;7:15363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Latour P, Boutrand L, Levy N, et al. Polymorphic short tandem repeats for diagnosis of the Charcot‐Marie‐Tooth 1A duplication. Clin Chem 2001;47:829–837. [PubMed] [Google Scholar]
- 23. Ruiz‐Ponte C, Loidi L, Vega A, et al. Rapid real‐time fluorescent PCR gene dosage test for the diagnosis of DNA duplications and deletions. Clin Chem 2000;46:1574–1582. [PubMed] [Google Scholar]
- 24. Tsai PC, Yang DM, Liao YC, et al. Clinical and biophysical characterization of 19 GJB1 mutations. Ann Clin Transl Neurol 2016;3:854–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tsai PC, Chen CH, Liu AB, et al. Mutational analysis of the 5' non‐coding region of GJB1 in a Taiwanese cohort with Charcot‐Marie‐Tooth neuropathy. J Neurol Sci 2013;332:51–55. [DOI] [PubMed] [Google Scholar]
- 26. Lin KP, Soong BW, Yang CC, et al. The mutational spectrum in a cohort of Charcot‐Marie‐Tooth disease type 2 among the Han Chinese in Taiwan. PLoS ONE 2011;6:e29393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein‐coding genetic variation in 60,706 humans. Nature 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep‐sequencing age. Nat Methods 2014;11:361–362. [DOI] [PubMed] [Google Scholar]
- 29. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kircher M, Witten DM, Jain P, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014;46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non‐synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009;4:1073–1081. [DOI] [PubMed] [Google Scholar]
- 32. UniProt C . Activities at the Universal Protein Resource (UniProt). Nucleic Acids Res 2014;42(Database issue):D191–D198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang R, He J, Li JJ, et al. Clinical and genetic spectra in a series of Chinese patients with Charcot‐Marie‐Tooth disease. Clin Chim Acta 2015;451(Pt B):263–270. [DOI] [PubMed] [Google Scholar]
- 34. Nelis E, Haites N, Van Broeckhoven C. Mutations in the peripheral myelin genes and associated genes in inherited peripheral neuropathies. Hum Mutat 1999;13:11–28. [DOI] [PubMed] [Google Scholar]
- 35. Mandich P, Grandis M, Geroldi A, et al. Gap junction beta 1 (GJB1) gene mutations in Italian patients with X‐linked Charcot‐Marie‐Tooth disease. J Hum Genet 2008;53:529–533. [DOI] [PubMed] [Google Scholar]
- 36. Bort S, Nelis E, Timmerman V, et al. Mutational analysis of the MPZ, PMP22 and Cx32 genes in patients of Spanish ancestry with Charcot‐Marie‐Tooth disease and hereditary neuropathy with liability to pressure palsies. Hum Genet 1997;99:746–754. [DOI] [PubMed] [Google Scholar]
- 37. Yoshihara T, Yamamoto M, Hattori N, et al. Identification of novel sequence variants in the neurofilament‐light gene in a Japanese population: analysis of Charcot‐Marie‐Tooth disease patients and normal individuals. J Peripher Nerv Syst 2002;7:221–224. [DOI] [PubMed] [Google Scholar]
- 38. Sivera R, Sevilla T, Vilchez JJ, et al. Charcot‐Marie‐Tooth disease: genetic and clinical spectrum in a Spanish clinical series. Neurology 2013;81:1617–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zuchner S, Mersiyanova IV, Muglia M, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot‐Marie‐Tooth neuropathy type 2A. Nat Genet 2004;36:449–451. [DOI] [PubMed] [Google Scholar]
- 40. Yuan JH, Hashiguchi A, Yoshimura A, et al. Clinical diversity caused by novel IGHMBP2 variants. J Hum Genet 2017;62:599–604. [DOI] [PubMed] [Google Scholar]
- 41. Murphy SM, Herrmann DN, McDermott MP, et al. Reliability of the CMT neuropathy score (second version) in Charcot‐Marie‐Tooth disease. J Peripher Nerv Syst 2011;16:191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Burns J, Ouvrier R, Estilow T, et al. Validation of the Charcot‐Marie‐Tooth disease pediatric scale as an outcome measure of disability. Ann Neurol 2012;71:642–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Genes in the targeted next‐generation sequencing panel.
Table S2. Genetic distribution in accordance with age of onset and ulnar MNCV (n = 227).