

Abstract
Objective
GDP‐mannose pyrophosphorylase B (GMPPB) related phenotype spectrum ranges widely from congenital myasthenic syndrome (CMS), limb‐girdle muscular dystrophy type 2T (LGMD 2T) to severe congenital muscle‐eye‐brain syndrome. Our study investigates the clinicopathologic features of a patient with novel GMPPB mutations and explores the pathogenetic mechanism.
Methods
The patient was a 22‐year‐old woman with chronic proximal limb weakness for 9 years without cognitive deterioration. Weakness became worse after fatigue. Elevated serum creatine kinase and decrements on repetitive nerve stimulation test were recorded. MRI showed fatty infiltration in muscles of lower limbs and shoulder girdle on T1 sequence. Open muscle biopsy and genetic analysis were performed.
Results
Muscle biopsy showed myogenic changes. Two missense mutations in GMPPB gene (c.803T>C and c.1060G>A) were identified in the patient. Western blotting and immunostaining showed GMPPB and α‐dystroglycan deficiency in the patient's muscle. In vitro, mutant GMPPB forming cytoplasmic aggregates completely colocalized with microtubule‐associated protein 1 light chain 3‐II (LC3‐II), a classical marker of autophagosome. Degradation of GMPPB was accompanied by an upregulation of LC3‐II, which could be restored by lysosomal inhibitor leupeptin.
Interpretation
We identified two novel GMPPB mutations causing overlap phenotype between LGMD 2T and CMS. We provided the initial evidence that mutant GMPPB colocalizes with autophagosome at subcellular level. GMPPB mutants degraded by autophagy‐lysosome pathway is associated with LGMD 2T. This study shed the light into the enzyme replacement which could become one of the therapeutic targets in the future study.
Introduction
GDP‐mannose pyrophosphorylase B (GMPPB) related phenotype spectrum ranges widely from congenital myasthenic syndrome (CMS), limb‐girdle muscular dystrophy‐type 2T (LGMD 2T) to severe congenital muscle‐eye‐brain syndrome associated with α‐dystroglycanopathy.1, 2, 3, 4 GMPPB catalyzes the early step of glycosylation pathway in most of the human tissues, including muscle and brain.1 Under GMPPB catalyzation, the sugar donor GDP‐mannose is synthesized from mannose‐1‐phosphate and GTP.5 The GDP‐mannose then participates directly or indirectly into N‐glycosylation, O‐mannosylation, C‐mannosylation, and glycosylphosphatidylinositol‐anchor formation.1 In the neuromuscular junction, the precursors of dystroglycan undergo N‐ and O‐glycosylation and proteolytic process to generate two subunits (α‐dystroglycan and β‐dystroglycan), prior to working as a central component of dystrophin‐glycoprotein complex.6, 7, 8 The latter complex is responsible for linking the extracellular matrix to the cytoskeleton.7, 8 To date, 49 mutations in GMPPB gene have been documented to cause diseases, most of which are associated with muscular disease, including LGMD 2T (also known as LGMD R19 GMPPB‐related9) or overlapping with CMS. However, a small fraction of GMPPB mutations may lead to severe congenital phenotypes, such as mental retardation, epilepsy, cerebellar dystrophy, microcephaly, and retinal dysfunction. Hitherto, the exact pathogenesis of GMPPB‐related α‐dystroglycanopathy is elusive and the management remains symptomatic, however, a part of patients may respond to medications used to treat CMS.2
Here we identify two novel mutations in GMPPB gene in one patient presenting the overlap between LGMD 2T and CMS. On the basis of thorough clinical, pathological, and genetic analysis, we aimed to functionally investigate the pathogenesis of GMPPB‐related spectrum.
Materials and Methods
Participants
We identified a patient fulfilling the diagnosis of LGMD and CMS according to proximal muscle weakness and decrements in repetitive nerve stimulation (RNS) test at low rate stimulation. The patient and her parents were clinically examined.
Standard protocol approvals, registrations, and patient consents
The ethics committee of Rui Jin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China approved the study. All participants provided written informed consent.
Mutation analysis
Genomic DNA was extracted using a standard phenol/chloroform extraction protocol. Healthy individuals (n = 200) of matched geographic ancestry were included as normal controls. Exome sequencing was performed for the patient, using Agilent SureSelect v6 reagents for capturing exons and Illumina HiSeq X Ten platform. Alignment to human genome assembly hg19 (GRCh37) was carried out followed by recalibration and variant calling. Population allele frequencies from public databases of normal human variation (dbSNP, ESP6500, and 1000 g) were used to initially filter the data to exclude variants greater than 1‰ frequency. The variants were further interpreted and classified according to the American College of Medical Genetics and Genomics (ACMG) Standards and Guidelines.10 In this segment, two neurogeneticists analyzed the inheritance pattern, allele frequency (from: 1000 g, ESP6500, dbSNP, ExAC and 200 in‐house ethnically matched healthy controls), amino acid conservation, pathogenicity prediction [PolyPhen‐2 (http://genetics.bwh.harvard.edu/pph2), SIFT (http://sift.jcvi.org), and Mutationtaster (http://www.mutationtaster.org). Putative pathogenic variants were further confirmed by Sanger sequencing.
Myopathology
We performed open biopsy on the right deltoid in the patient. The muscle tissue was frozen and then cut at 7 μm sections. These sections were stained according to standard histological and enzyme histochemical procedures with hematoxylin and eosin (HE), modified Gomori Trichrome (MGT), periodic acidic Schiff (PAS), oil red O (ORO), nicotinamide adenine dinucleotide tetrazolium reductase, succinate dehydrogenease, cytochrome C oxidase, and esterase.
Sections were also immunostained with the following primary antibodies with standard procedures: dystrophin (DYS1: Rod domain; DYS2: C‐terminus; DYS3: N‐terminus, Novocastra, Newcastle‐upon‐Tyne, UK), dysferlin (NCLHamlet, Novocastra), MHC‐I (ab52922, abcam), anti‐α‐dystroglycan (IIH6C4, Millipore), anti‐GMPPB (HPA014657, Sigma). Additionally, anti‐β‐Spectrin (NCL‐SPEC1, Novocastra) was exploited as sarcolemma marker. A nondisease control was set and labeled at the same magnification and exposure.
Protein of muscle tissue lysate was extracted and the expression level of GMPPB was detected by western blotting with anti‐GMPPB.
Cell culture transfection and Western blotting
HEK 293T cell line was obtained from the Cell Bank of Chinese Academy of Sciences (www.cellbank.org.cn) and maintained in Dulbecco's Modified Eagle Medium (DMEM) with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (PS) at 37°C in a humidified incubator with 5% CO2. One day before transfection, cells were plated at 150,000 cells per well in 6‐well culture dish. The next day, cells were transfected with 2.5 μg of EGFP control plasmid DNA or GMPPB‐EGFP wild‐type (GMPPB‐WT) or mutant (c.803T>C and c.1060G>A) plasmid DNA using Lipofectamine 3000 transfection reagent (Invitrogen). Moreover, another four mutations reported previously as causative were set as positive controls (c.781C>T, c.877C>T, c.1034T>C, and c.1108G>C2, 4, 11, 12). Forty‐eight hours later, the cells were split in radioactive immunoprecipitation assay (RIPA) buffer (Beyotime) to extract protein for western blot analysis. Cell lysates were diluted in an equivalent volume of 6X SDS‐PAGE Sample Loading Buffer (Beyotime) for protein denaturation. For cell lysates, equal volumes were run on 12% SDS polyacrylamide gels. Total GMPPB levels were detected using the Anti‐GFP antibody (1:2500, GFP‐1010, AVES). GAPDH primary antibody (1:1000, 2118‐14C10, Cell Signaling Technology) was used to ensure equal protein loading. Blots were then incubated with antichicken and antirabbit secondary HRP‐conjugated antibodies (1:5000, Beyotime) and bands were detected by enhanced chemiluminescence using Western Blot Enhancer reagents (Thermo Scientific).
For the analysis of the localization and the impact of mutant GMPPB on muscle cells, C2C12 myoblast cells (Cell Bank of Chinese Academy of Sciences) were also transfected with the constructs mentioned above. For the pharmacological treatment assay, C2C12 cells were incubated for 12 or 18 h in the presence or absence of lysosomal inhibitor (100 μg/mL leupeptin Sigma‐Aldrich). After treatment, cells were lysed and subjected to western blot analysis of GMPPB protein by Anti‐GFP and Anti‐GAPDH antibodies mentioned above. In addition, microtubule‐associated protein 1 light chain 3 (LC3), the marker of autophagy, was detected by anti‐LC3B (D11) (rabbit [3868], 1:1000 for western blotting, Cell Signaling Technology).
Immunofluorescence
HEK 293T and C2C12 cells transfected with the respective expression constructs were washed in PBS and fixed using 4% paraformaldehyde for immunofluorescence test. Cells were blocked with 10% normal donkey serum and 0.3% Triton X‐100 in PBS for 60 min, incubated with primary antibody mentioned above (anti‐EGFP or anti‐LC3B) and anti‐LAMP1 (D2D11, 1:200, Cell Signaling Technology) in blocking solution at 4°C overnight and incubated with Alexa Fluor 488 or 594 secondary antibodies (1:1000, Life). DAPI (4′,6‐diami‐dino‐2‐phenylindole) (1:10,000, Life) was used for nucleic acid staining. Images were taken with a Zeiss 710 confocal microscope.
Quantitative real‐time PCR
Total RNA was extracted from HEK 293T cells transfected with relative plasmids using a standard method with TRIZOL Reagent (Invitrogen), and reverse‐transcribed using the PrimeScript1 RT reagent Kit (Takara) according to the manufacture's instruction. To determine whether the GMPPB variant affects the mRNA level, we designed three pairs of primers (Table S1), namely 2F/3R (at exon 2 and 3, respectively), 5F/5R (at exon 5), and 9F/9R (at exon 9) to verify the amount of GMPPB mRNA.
Results
Clinical findings
The patient was a 22‐year‐old female with recurrent exertional muscle fatigue and walking slowly for 9 years. She was born of full‐term spontaneous vaginal delivery with normal birth weight (3.3 kg). She achieved sitting, standing, and walking alone at 8, 10, and 12 months old, respectively. Initially, muscle weakness was demonstrated by difficulties in climbing stairs, standing up from squatting and sitting up from lying. Then, muscle weakness became continuous and progressed slowly to proximal upper limbs, especially in combing hair and dressing, which could be aggravated by labor and mildly relieved after resting. During school days (after the age of 13), poor performances of physical education examinations were recorded, such as running, ball games, and rope skipping. At the age of 22, she had normal strength in neck flexion (5/5 on a medical research council scale graded 0–5), reduced strength in iliopsoas (3+/5), proximal upper limbs and proximal lower limbs (4+/5), and normal strength in distal limbs. Muscle tone and tendon reflex in four limbs were reduced. Ptosis, nystagmus, dysarthria, ataxia, myokymia, muscle pain, and dyspnea were not noticed. In the exercise fatigue test, the patient was asked to elevate her right lower limb repeatedly, followed by muscle strength reexamination, showing further deterioration of strength in iliopsoas (3/5), quadriceps femoris (3/5), and posterior femoral muscles (4/5). Then we recorded the time spent on standing up from squatting before and after neostigmine treatment (intramuscular, 1 mg): before injection = 8 sec, 10 min after injection = 5 sec, 20 min after injection = 3 sec, 30 min after injection = 4 sec. We also reexamined the muscle strength with partial alleviation of strength 10 min after injection: iliopsoas (4−/5), quadriceps femoris (5/5), and posterior femoral muscles (5/5); 20 min after injection: iliopsoas (4/5), quadriceps femoris (5/5), and posterior femoral muscles (5/5); 30 min after injection: iliopsoas (4−/5), quadriceps femoris (4+/5), and posterior femoral muscles (4+/5). She walked slowly with mild waddling gait. Mental psychological and cognitive tests were normal. During repeated hospitalizations, elevating serum creatine kinase (CK) 1288–2007 IU/L (normal 22–269 IU/L) and lactate dehydrogenase (LDH) 281.9–843 IU/L (normal 98–192 IU/L) were recorded. The patient's AST was 63 U/L (8–40 U/L) and ALT was 58 U/L (5–40 U/L). The electromyogram showed motor unit potential (MUP) with small, short multiphase in deltoid, biceps brachii, and iliopsoas. As shown in Figure 1A, before medical treatment, RNS tests performed on bilateral deltoids showed 45.2% and 36.0% amplitude decrement on the left and right side, respectively. After administration with pyridostigmine bromide (30 mg, 40 min), the decremental responses still existed, with 39.4% on the left side and 36.0% on the right side. Nerve conduction was in normal range. Muscle MRI showed mild to moderate increased signal intensity mainly involving the posterior muscles in lower limbs, muscles of pelvic girdle and shoulder girdle on the T1‐weighted sequences (Fig. 1B–C). The degree of muscle fatty infiltration was quantified as semimembranosus = 1 point, biceps femoral muscle = 2 points, rectus femoris gracilis = 1 point, left anterior tibial muscle = 2 points, right anterior tibial muscle = 3 points, left peroneal muscle = 2 points, right peroneal muscle = 3 points, trapezius = 1 point, and teres major muscle = 2 points.13, 14 The patient reported moderate improvement on muscle weakness after prescribing with 30 mg of pyridostigmine bromide for three times per day. One year after regular treatment with pyridostigmine bromide, reexamination showed the muscle strength with iliopsoas (4−/5), quadriceps femoris (4+/5), and posterior femoral muscles (4+/5).
Figure 1.
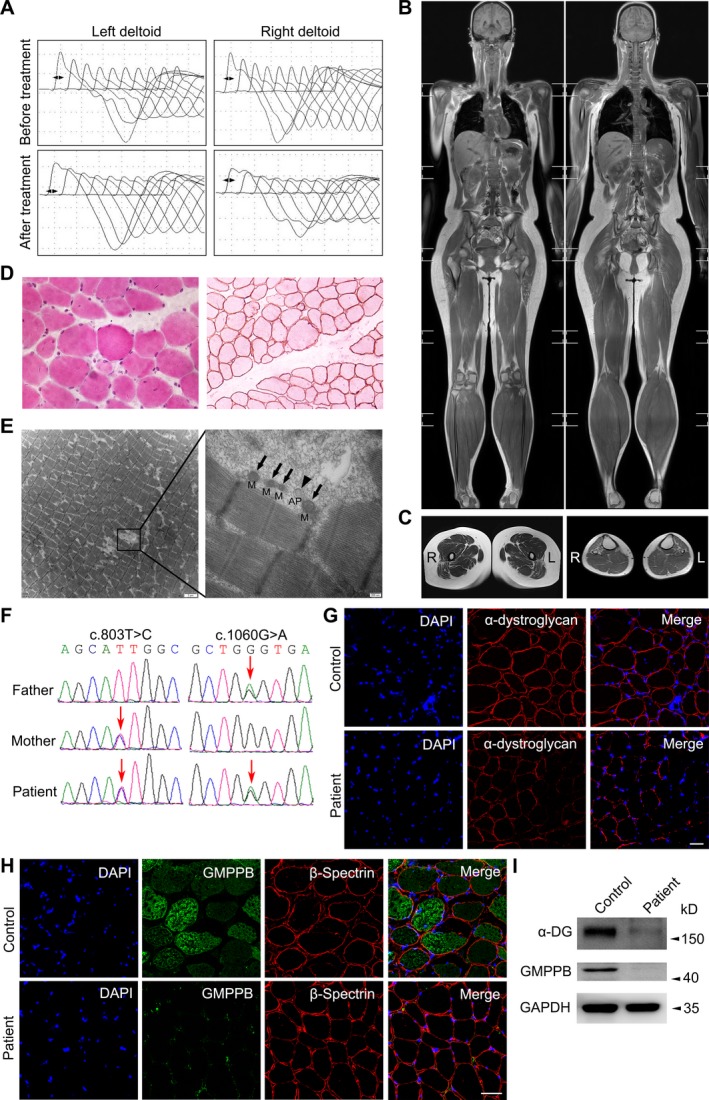
Clinical, genetic, and pathological characterizations. (A) Low rates (3–5 Hz) of repetitive nerve stimulation of bilateral deltoids at baseline in patient before and after oral administration with pyridostigmine bromide (30 mg, 40 min). (B–C) Muscle MRI in whole‐body and lower limbs on the T1‐weighted sequences. Coronal view showed fatty tissue replacement of the muscles in pelvic girdle and lower limbs. Muscles in upper limbs were well preserved except that bilateral trapezius and teres major muscles were mildly affected. The semimembranosus, biceps femoral muscle, rectus femoris gracilis of the thigh and anterior tibial muscles were preferentially affected. (D) Muscle pathology of the patients showed a marked variation in fiber size with hypertrophy, atrophy, hypercontracted fibers as well as regeneration by HE 400 × (left panel). Immunohistochemical staining of R‐dystrophin 200 × was normal (right panel). (E) The patient's longitudinal cutting muscle specimens under electronic microscopy (M = mitochondria, AP = autophagosome). (F) Sequence chromatograms of GMPPB gene of the patient. Mutations and the corresponding normal sequences are shown. It displays two missense mutations of GMPPB gene (arrows) were inherited from mother (c.803T>C) and father (c.1060G>A), respectively. (G) Immunofluorescence of α‐dystroglycan exhibited a patchy deficiency in the patient (lower panel) in comparison with normal control (upper panel). The scale bar represents 50 μm. (H) Immunofluorescence staining of GMPPB in frozen muscle sections from patient (lower panel) and control (upper panel). The scale bar represents 50 μm. (I) The levels of α‐dystroglycan (α‐DG) and GMPPB in the patient's and healthy control's muscle tissue by western blotting.
Myopathological findings
Muscle pathology of the patients showed a marked variation in fiber size with atrophy, hypertrophy, hypercontracted fibers, as well as regeneration by HE (Fig. 1D, left panel), without an increase in lipid by ORO or abnormal glycogen storage by PAS. Immunohistochemical staining of R‐dystrophin (Fig. 1D, right panel), N‐dystrophin, and C‐dystrophin was normal. Under electronic microscopy, the myofibrillar arrangement is slightly disordered with dissolution and degeneration of individual myofibrils. Occasionally, autophagosome can be observed on longitudinally cut muscle specimens, without autophagic vacuoles or other disease‐specific pathological changes (Fig. 1E). Immunofluorescence disclosed the normal distribution of α‐dystroglycan on sarcolemma and cytoplasmic distribution mode of GMPPB in muscle fiber from nondisease control (Fig. 1G and H). While the patient's muscle exhibited a mosaic deficiency of α‐dystroglycan and GMPPB could be hardly detected (Fig. 1G and H). Likewise, the results of western blotting suggested the significant lower levels of α‐dystroglycan and GMPPB protein in the patient's muscle comparing with the control's (Fig. 1I).
Genetic findings
We identified two compound heterozygous variants of GMPPB gene (NM_013334) in the patient, c.803T>C (p.Ile268Thr) and c.1060G>A (p.Gly354Ser). Both of her parents, respectively, carried one of the two heterozygous variations (Fig. 1F), which were not identified in 200 healthy controls, dbSNP (http://www.ncbi.nlm.nih.gov/snp), 1000 Genome Project (http://browser.1000genomes.org), NHLBI Exome Sequencing Project (ESP) Exome Variant Server (http://evs.gs.washington.edu/EVS), Exome Aggregation Consortium (ExAC) or gnomAD (http://gnomad-old.broadinstitute.org). p.Ile268Thr was predicted to be probably damaging by PolyPhen2 (probability score 0.999, sensitivity: 0.14, specificity: 0.99), damaging by SIFT (SIFT score: 0.000), and disease causing by Mutationtaster (probability score 0.999). p.Gly354Ser was predicted to be probably damaging by PolyPhen2 (probability score 1.000, sensitivity: 0.00, specificity: 1.00), damaging by SIFT (SIFT score: 0.001), and disease causing by Mutationtaster (probability score 0.999).
Mutant protein detection
The protein level of GMPPB‐WT/Mut was examined by western blotting after transfecting the GMPPB constructs into HEK 293T cells, showing the c.1060G>A was about 78.8% of the WT, and the c.803T>C was even lower (57.1%) through three independent repeated experiments (Fig. 2A). In immunofluorescence staining, the c.803T>C and c.1060G>A mutant GMPPB tended to form punctate aggregates compared with the diffuse distribution of wild‐type (Fig. 2B).
Figure 2.
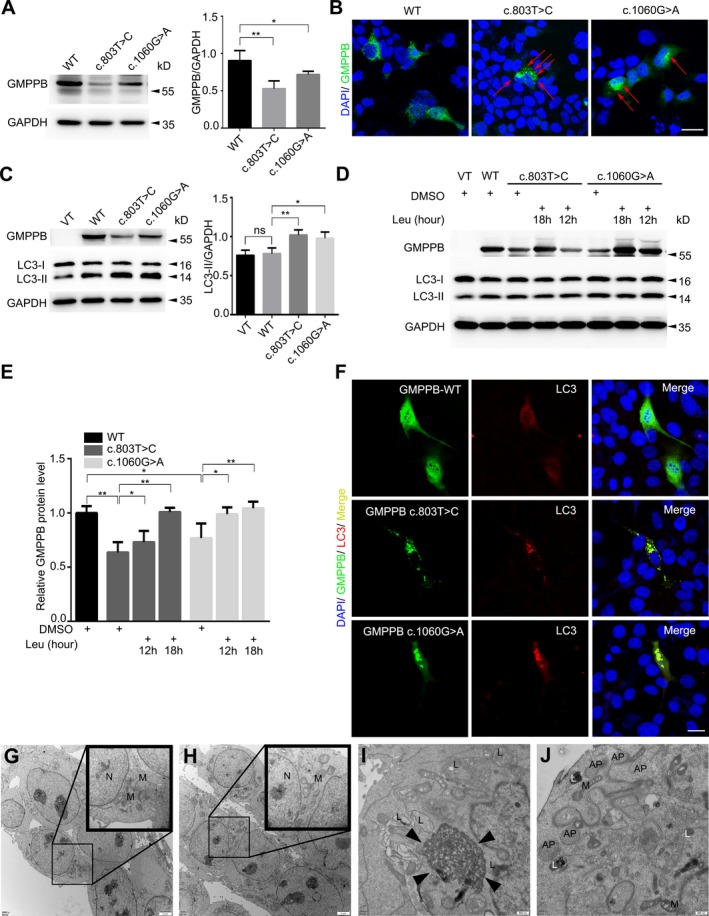
Effect of GMPPB variants on protein level and localization in vitro. (A) In HEK 293T cells, western blotting showed that the expression level of c.803T>C and c.1060G>A was 57.1% and 78.8% of WT, respectively (n = 3 separate transfections, * 0.01 ≤ P < 0.05, **P < 0.01). (B) GMPPB‐EGFP transfected HEK 293T cells showing the presence of WT in diffuse distribution but the mutant tended to form punctate aggregates (red arrows). The scale bar represents 20 μm. (C) The LC3‐II/GAPDH level in C2C12 cells expressing the two mutations were higher than the vector (VT) and WT groups (n = 3 separate transfections, ns = nonsignificance, * 0.01 ≤ P < 0.05, **P < 0.01). (D) In mutant groups, the lower level of GMPPB and higher level of LC3‐II were restored after lysosome inhibitor leupeptin (Leu) addition for 12 or 18 h. (E) The statistics of GMPPB restoration by leupeptin (n = 4 separate transfections, *0.01 ≤ P < 0.05, **P < 0.01). (F) The diffuse distribution of both GMPPB and LC3 in WT expressed cells. The colocalization of mutant GMPPB aggregates (green) with LC3 aggregates (red) in Mut expressed cells. The scale bar represents 20 μm. (G–J) HEK 293T cells transfected with VT (G), WT (H), c.803T>C (I) and c.1060G>A (J) under electronic microscopy (N = nucleus, M = mitochondria, L = lysosome, AP = autophagosome, Black triangle = cytoplasmic protein accumulation).
Furthermore, we also tested the level of LC3 in C2C12 cells transfected with GMPPB‐WT/Mut (Fig. 2C, left panel). Since LC3‐II is more stable and more closely related to autophagosomes than LC3‐I, the consensus on autophagy detection is that levels of LC3‐II should be compared to internal parameter (such as actin or GAPDH), but not to LC3‐I.15 In blotting, the LC3‐II/GAPDH in two mutant groups was higher than the WT or vector (VT) group (Fig. 2C).To further determine whether the mutant GMPPB was degraded via autophagy‐lysosome pathway, we treated the cells with lysosomal inhibitor leupeptin for 12 h and observed that the decreased c.803T>C and c.1060G>A were restored and with the leupeptin maintained for longer time (18 h), the restoration was further promoted (Fig. 2D). In addition, both mutant GMPPB and LC3, appearing as aggregates, completely colocalized with each other in the mutant groups in immunolabeling. While in the WT group, both GMPPB and LC3 spread uniformly in cytoplasm (Fig. 2F). In comparison with VT (Fig. 2G) and WT groups (Fig. 2H), the cells transfected with c.803T>C (Fig. 2I) and c.1060G>A (Fig. 2J) have an increased number of lysosomes (L) and autophagosomes (AP) under electronic microscopy. However, the results of qPCR did not show statistical difference between WT c.803T>C or c.1060G>A (Fig. S1A). In vitro, GMPPB aggregates were surrounded or colocalized with lysosomes labeled by LAMP1 (Fig. S1B). The four mutations selected from previous literatures also had similar behavioral characteristics (Fig. S1B–D).
Discussion
We described a Chinese female patient presenting limb‐girdle muscle weakness with mild fluctuation, without intellectual disability or epilepsy, due to compound heterozygous GMPPB mutations (c.803T>C and c.1060G>A). Her parents each carrying one of the two variants were not clinically affected. Similarly, in 2017, Luo et al. documented four cases harboring distinct features of LGMD as well as neuromuscular junction defect.11 However, in 2015, Belaya et al. described eight cases with predominant muscle weakness fluctuation.2 Among these, the myasthenic component could benefit from pyridostigmine treatment.
The GMPPB gene, encoding GDP‐mannose pyrophosphorylase B, has two transcript variants NM_013334 and NM_021971, with eight and nine exons, respectively. The first seven exons of both transcripts are identical. The eighth exon in NM_013334 is 396 bases long encoding 131 amino acids. The middle 27 amino acids are spliced out in NM_021971 to separate exon 8 and 9. Thus, NM_013334 encodes protein with 387 amino acids (Isoform 1 in Fig. 3), whereas NM_021971 encodes protein with 360 amino acids (Isoform 2 in Fig. 3). It has been confirmed that Isoform 2 is expressed at much higher levels than Isoform 1 in the skeletal muscle and the central nervous system.1 To date, no pathogenic mutations have been identified in the 27 amino acids that are specific to Isoform 1. Thus, Isoform 2 is commonly used for the annotation of variants described in most of the related studies.1, 2, 11, 16, 17 Thus far, a total of 51 mutations in GMPPB have been associated with the disease, including 42 missense variants, 5 nonsense, 2 frameshift and 2 splicing (Fig. 3). Although no clear genotype–phenotype correlations have been clarified and mutations may locate through the whole length of the protein, a total of 15 mutations can be classified with relatively high risk to cause severe phenotype with not only myopathy but also mental retardation, cerebellar involvement, and epilepsy (red in Fig. 3).1, 2, 4, 12, 16, 18 As shown in Figure 3, GMPPB consists of two important domains, N‐terminal catalytic domain and Left‐handed parallel beta‐Helix (LbH) domain.1 Comparing with other parts of the protein, causative mutations mostly concentrated in LbH domain (32% per bp) indicating an evolutionary highly conservative sequence with essential physiological function.1, 2, 3, 12, 16, 18, 19
Figure 3.
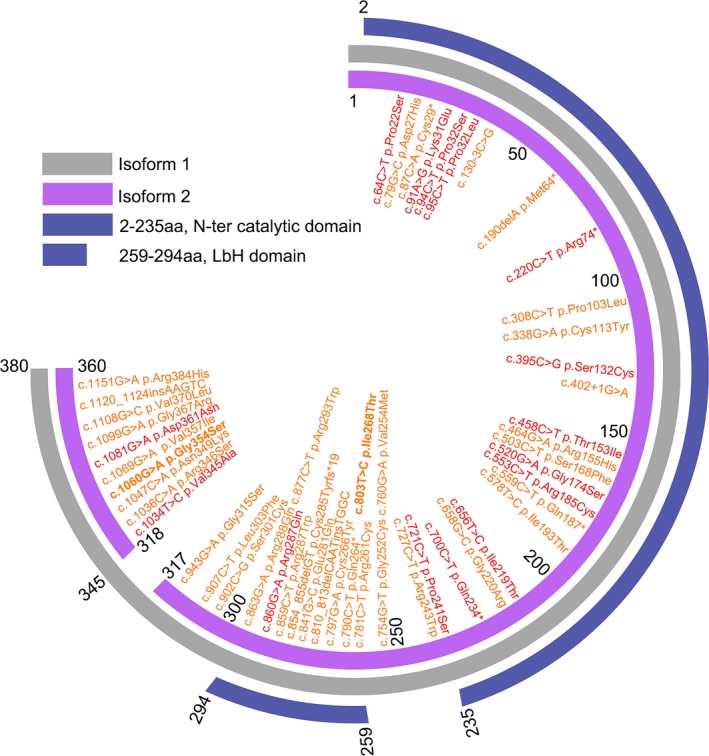
The schematic diagram of GMPPB structure with mutations. GMPPB contains two transcript isoforms. Isoform 1 (NM_013334) in gray consists of 387 amino acids. Isoform 2 (NM_021971) in purple consists of 360 amino acids. All the mutations are illustrated on basis of Isoform 1. Mutations identified with severe phenotype are in red. Other mutations in orange are related with pure myopathy (LGMD or CMS). The mutations first identified in this paper are in bold font.
Thus far, several compelling reports have confirmed GMPPB mutation could cause the protein aggregates and expression impairment.1, 2, 11 However, it remains elusive about the accurate location of GMPPB aggregates and the mechanism of mutant degradation. In this work, we likewise observed low levels of GMPPB and α‐dystroglycan immunostain in the patient's muscle. In vitro, two mutations exhibited varied degrees of aggregation and degradation accompanied by the upregulation of LC3‐II, which could be blocked and reversed by lysosomal inhibitor. Furthermore, the insoluble aggregates of GMPPB mutants completely colocalized with LC3‐II (the membrane type of LC3). In eukaryotes, autophagy–lysosome and ubiquitin–proteasome system (UPS) are two major quality control and recycling mechanisms responsible for cellular homeostasis.20, 21, 22 The UPS is responsible for the degradation of short‐lived proteins and soluble un/mis‐folded proteins, whereas autophagy‐lysosome eliminates long‐lived proteins, insoluble protein aggregates and degenerated organelles and intracellular parasites.22, 23, 24, 25 In addition, another four mutations documented before (c.781C>T, c.877C>T, c.1034T>C, and c.1108G>C, NM_013334), also have similar behaviors with the mutations reported in this paper. In this work, we support the mutant GMPPB can be distinguished by autophagy–lysosome and lead to protein degradation through lysosomal‐degradation pathway.
Conclusion
This work identified two novel GMPPB mutations causing an overlap between LGMD 2T and CMS, without mental retardation and epilepsy. Moderate improvement was gained after pyridostigmine bromide treatment (30 mg, three times/day). The mutations lead to abnormal GMPPB distribution and reduced expression, as well as α‐dystroglycan deficiency. This study provides the initial evidence that mutant GMPPB colocalizes with autophagosome and can be degraded through autophagy–lysosome pathway. Considering GMPPB‐related LGMD 2T is generally due to enzyme deficiency and has intimate associations with lysosomal degradation, we suppose the enzyme replacement could become one of the therapeutic targets in the future, especially for the patients with severe phenotype.
Author Contributions
Tian: data acquisition, analysis and interpretation of data, study design, statistical analysis, drafting the manuscript.
Zhou: funding, data acquisition, analysis and interpretation of data, statistical analysis.
Zhan: data acquisition.
Zhu: data acquisition.
Yang: electronic microscopy detection and observation.
S. D. Chen: funding, study design and conceptualization, data acquisition, analysis and interpretation of data, manuscript revision.
Luan: funding, study design and conceptualization, data acquisition, analysis and interpretation of data, manuscript revision.
L. Cao: funding, study design and conceptualization, data acquisition, analysis and interpretation of data, statistical analysis, manuscript revision.
Conflicts of Interest
Nothing to report.
Disclosure
Zhou is in charge of Natural Science Foundation of Science and Technology of Shanghai (No. 15ZR1426700).
S. D. Chen is in charge of National Natural Science Foundation of China (No. 81430022).
Luan is in charge of National Natural Science Foundation of China (No. 81200965), the Research Fund for the Doctoral Program of Higher Education (No. 20110073120088), and Guang Ci Qing Nian Grant (No. GCQN‐2017‐A03).
L. Cao is in charge of National Natural Science Foundation of China (No. 81870889 and No. 81571086), National Key R&D Program of China (No. 2017YFC1310200), Shanghai Municipal Education Commission‐Gaofeng Clinical Medicine Grant (No. 20161401), and Interdisciplinary Project of Shanghai Jiao Tong University (No. YG2016MS64).
The other coauthors report no disclosures relevant to the manuscript.
Supporting information
Table S1. Quantitative real‐time PCR primers used in the study.
Figure S1. The detection of GMPPB expression and lysosome‐related markers in HEK 293T cells transfected with GMPPB‐WT/Mut.
Acknowledgments
The authors acknowledge the physicians and the patient for their cooperation. The authors thank Liu for his technology suggestions. This study was supported by the grants from the National Natural Science Foundation of China (No. 81571086, No. 81870889, No. 81200965, and No. 81430022), National Key R&D Program of China (No. 2017YFC1310200), Shanghai Municipal Education Commission‐Gaofeng Clinical Medicine Grant Support (No. 20161401), Interdisciplinary Project of Shanghai Jiao Tong University (No. YG2016MS64), the Research Fund for the Doctoral Program of Higher Education (No. 20110073120088), Natural Science Foundation of Science and Technology of Shanghai (No. 15ZR1426700), and Guang Ci Qing Nian Grant (No. GCQN‐2017‐A03).
Funding Information
This study was supported by the grants from the National Natural Science Foundation of China (No. 81571086, No. 81870889, No. 81200965, and No. 81430022), National Key R&D Program of China (No. 2017YFC1310200), Shanghai Municipal Education Commission‐Gaofeng Clinical Medicine Grant Support (No. 20161401), Interdisciplinary Project of Shanghai Jiao Tong University (No. YG2016MS64), the Research Fund for the Doctoral Program of Higher Education (No. 20110073120088), Natural Science Foundation of Science and Technology of Shanghai (No. 15ZR1426700), and Guang Ci Qing Nian Grant (No. GCQN‐2017‐A03).
Data Analysis: Wo‐Tu Tian, Li Cao, Department of Neurology, Rui Jin Hospital & Rui Jin Hospital North, Shanghai Jiao Tong University School of Medicine, Shanghai, China.
Funding Statement
This work was funded by National Natural Science Foundation of China grants 81571086, 81870889, 81200965, and 81430022; National Key R&D Program of China grant 2017YFC1310200; Shanghai Municipal Education Commission‐Gaofeng Clinical Medicine Grant Support grant 20161401; Interdisciplinary Project of Shanghai Jiao Tong University grant YG2016MS64; Research Fund for the Doctoral Program of Higher Education grant 20110073120088; Natural Science Foundation of Science and Technology of Shanghai grant 15ZR1426700; Guang Ci Qing Nian grant GCQN‐2017‐A03.
Contributor Information
Sheng‐Di Chen, Email: ruijincsd@126.com.
Xing‐Hua Luan, Email: green_lxh@hotmail.com.
Li Cao, Email: caoli2000@yeah.net.
References
- 1. Carss KJ, Stevens E, Foley AR, et al. Mutations in GDP‐mannose pyrophosphorylase B cause congenital and limb‐girdle muscular dystrophies associated with hypoglycosylation of alpha‐dystroglycan. Am J Hum Genet 2013;93:29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Belaya K, Rodríguez Cruz PM, Liu WW, et al. Mutations in GMPPB cause congenital myasthenic syndrome and bridge myasthenic disorders with dystroglycanopathies. Brain 2015;138:2493–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cabrera‐Serrano M, Ghaoui R, Ravenscroft G, et al. Expanding the phenotype of GMPPB mutations. Brain 2015;138:836–844. [DOI] [PubMed] [Google Scholar]
- 4. Jensen BS, Willer T, Saade DN, et al. GMPPB‐associated dystroglycanopathy: emerging common variants with phenotype correlation. Hum Mutat 2015;36:1159–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ning B, Elbein AD. Cloning, expression and characterization of the pig liver GDP‐mannose pyrophosphorylase. Evidence that GDP‐mannose and GDP‐Glc pyrophosphorylases are different proteins. Eur J Biochem 2000;267:6866–6874. [DOI] [PubMed] [Google Scholar]
- 6. Barresi R, Campbell KP. Dystroglycan: from biosynthesis to pathogenesis of human disease. J Cell Sci 2006;119:199–207. [DOI] [PubMed] [Google Scholar]
- 7. Ibraghimov‐Beskrovnaya O, Ervasti JM, Leveille CJ, et al. Primary structure of dystrophin‐associated glycoproteins linking dystrophin to the extracellular matrix. Nature 1992;355:696–702. [DOI] [PubMed] [Google Scholar]
- 8. Holt KH, Crosbie RH, Venzke DP, Campbell KP. Biosynthesis of dystroglycan: processing of a precursor propeptide. FEBS Lett 2000;468:79–83. [DOI] [PubMed] [Google Scholar]
- 9. Straub V, Murphy A, Udd B; LGMD workshop study group . 229th ENMC international workshop: limb girdle muscular dystrophies ‐ nomenclature and reformed classification Naarden, the Netherlands, 17‐19 March 2017. Neuromuscul Disord 2018;2:702–710. [DOI] [PubMed] [Google Scholar]
- 10. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Luo S, Cai S, Maxwell S, et al. Novel mutations in the C‐terminal region of GMPPB causing limb‐girdle muscular dystrophy overlapping with congenital myasthenic syndrome. Neuromuscul Disord 2017;27:557–564. [DOI] [PubMed] [Google Scholar]
- 12. Sarkozy A, Torelli S, Mein R, et al. Mobility shift of beta‐dystroglycan as a marker of GMPPB gene‐related muscular dystrophy. J Neurol Neurosurg Psychiatry 2018;89:762–768. [DOI] [PubMed] [Google Scholar]
- 13. Fischer D, Kley RA, Strach K, et al. Distinct muscle imaging patterns in myofibrillar myopathies. Neurology 2008;71:758–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Díaz‐Manera J, Llauger J, Gallardo E, Illa I. Muscle MRI in muscular dystrophies. Acta Myol 2015;34:95–108. [PMC free article] [PubMed] [Google Scholar]
- 15. Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016;12:1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Astrea G, Romano A, Angelini C, et al. Broad phenotypic spectrum and genotype‐phenotype correlations in GMPPB‐related dystroglycanopathies: an Italian cross‐sectional study. Orphanet J Rare Dis 2018;13:170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nigro V, Savarese M. Genetic basis of limb‐girdle muscular dystrophies: the 2014 update. Acta Myol 2014;33:1–12. [PMC free article] [PubMed] [Google Scholar]
- 18. Raphael AR, Couthouis J, Sakamuri S, et al. Congenital muscular dystrophy and generalized epilepsy caused by GMPPB mutations. Brain Res 2014;1575:66–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Oestergaard ST, Stojkovic T, Dahlqvist JR, et al. Muscle involvement in limb‐girdle muscular dystrophy with GMPPB deficiency (LGMD2T). Neurol Genet 2016;2:e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hershko A. Ubiquitin: roles in protein modification and breakdown. Cell 1983;34:11–12. [DOI] [PubMed] [Google Scholar]
- 21. Hershko A. The ubiquitin system for protein degradation and some of its roles in the control of the cell‐division cycle (Nobel lecture). Angew Chem Int Ed Engl 2005;44:5932–5943. [DOI] [PubMed] [Google Scholar]
- 22. Kocaturk NM, Gozuacik D. Crosstalk between mammalian autophagy and the ubiquitin‐proteasome system. Front Cell Dev Biol 2018;6:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Groll M, Huber R. Substrate access and processing by the 20S proteasome core particle. Int J Biochem Cell Biol 2003;35:606–616. [DOI] [PubMed] [Google Scholar]
- 24. Groll M, Huber R. Inhibitors of the eukaryotic 20S proteasome core particle: a structural approach. Biochim Biophys Acta 2004;1695:33–44. [DOI] [PubMed] [Google Scholar]
- 25. Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 2007;8:931–937. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Quantitative real‐time PCR primers used in the study.
Figure S1. The detection of GMPPB expression and lysosome‐related markers in HEK 293T cells transfected with GMPPB‐WT/Mut.