

Abstract
LTX-401 is a novel oncolytic compound designed for the local treatment of solid tumors. In the present study, we have examined the applicability and efficacy of LTX-401 in a rat model JM1 hepatocellular carcinoma, with particular interest in its ability to induce antitumor immunity. LTX-401 induces necrotic cell death followed by the release of immunogenic cell death mediators such as high-mobility group box 1 protein, ATP, and cytochrome c. When injected into subcutaneous and orthotopic JM1 tumors, LTX-401 treatment resulted in a strong antitumoral effect followed by complete tumor regression in the majority of animals. Additionally, LTX-401 could affect the growth of distal tumor deposits simulating metastases, hence indicating immune-mediated abscopal responses. Furthermore, LTX-401 treatment induced tumor-specific immune responses as seen by protection against tumor rechallenge and increased production of interferon-gamma (IFN-γ) by splenic cells in response to stimulation with tumor cells. Taken together, our data demonstrate that the oncolytic compound LTX-401 provides local tumor control followed by protective immune responses and may be exploited as a novel immunotherapeutic agent in hepatocellular carcinoma.
Keywords: oncolytic compound, LTX-401, immunotherapy, antitumor agent, hepatocellular carcinoma, ICD
Introduction
Liver cancer is estimated to be the third most common cause of cancer-related mortality globally, with incidence rates more than tripled since 1980.1, 2 Hepatocellular carcinoma (HCC) is the most frequent type of primary liver cancer, accounting for ∼90% of hepatic malignancies.3 If diagnosed at an early stage, HCC is potentially curable by surgical procedures such as resection, transplantation, and minimally invasive ablation techniques. However, less than 20% of patients are eligible for such treatments.4, 5 The vast majority of patients with HCC are diagnosed in late stages of the disease, hence leading to poor prognosis because of limited treatment options. Sorafenib, a multi-kinase inhibitor, has long been the only approved systemic treatment for advanced-stage HCC. More recently, however, the multi-kinase inhibitor lenvatinib also gained approval as a first-line treatment of patients with unresectable HCC. Despite being shown to confer some mortality benefit, lenvatinib extended median survival time by only 1.3 months compared with sorafenib,6 hence illustrating the impending need to pursue new and improved therapeutic approaches. Currently, different classes of immunotherapeutic agents are being investigated in patients with advanced-stage HCC.7, 8 Among these, monoclonal antibodies specifically targeting the immunoinhibitory PD-1 and PD-L1 axis have shown great potential in clinical trials, with reports of clinically meaningful and durable responses.9, 10
Anticancer peptides (ACPs), otherwise known as oncolytic peptides, represent a novel class of immunotherapeutics displaying broad-spectrum cytotoxic activity that is unaffected by multidrug resistance mechanisms.11, 12, 13 Cell death often occurs through primary necrosis, owing predominantly to the membranolytic mode of action. As a result, ACP-induced cancer cell death may potentially liberate tumor antigens and immunomodulatory components to prime tumor-specific T cells and promote antitumor immunity. This scenario is seemingly linked to the induction of immunogenic cell death (ICD), which is characterized by the cell-surface exposure and/or release of damage-associated molecular pattern molecules (DAMPs) such as calreticulin, ATP, and high-mobility group box 1 protein (HMGB1), which attract dendritic cell precursors to the proximity of dying cancer cells and stimulate the uptake of dead cell-associated antigens followed by optimal tumor antigen cross-presentation to ignite cytotoxic T lymphocyte responses.14, 15, 16, 17
Because oncolytic peptides are now being optimized for the treatment of cancer, we attempted to develop smaller derivates with bulky and lipophilic moieties for improved stability and anticancer activity. The resulting amphipathic β(2,2)-amino acid derivative LTX-401 (Figure 1), formerly known as BAA-1,18 resembles larger oncolytic peptides with respect to hydrophobicity and cationicity, thereby enabling it to interact with and permeabilize cancer cell membranes. LTX-401 also exerts cytotoxic activity against normal cell lines, but displays selectivity to human red blood cells.19 Considering its intended use as a locally administered agent, the selectivity toward normal cells is of less importance compared with systemic interventions. Local administration of LTX-401 into subcutaneously (s.c.) established B16 melanomas and MCA205 sarcomas has been found to induce focal tumor necrosis, followed by lymphoid infiltration and subsequent complete regression.19, 20 In the present study, we expand upon our previous findings and investigate the applicability and efficacy of LTX-401 against both s.c. and orthotopic tumors in an immunocompetent rat model of HCC.
Figure 1.
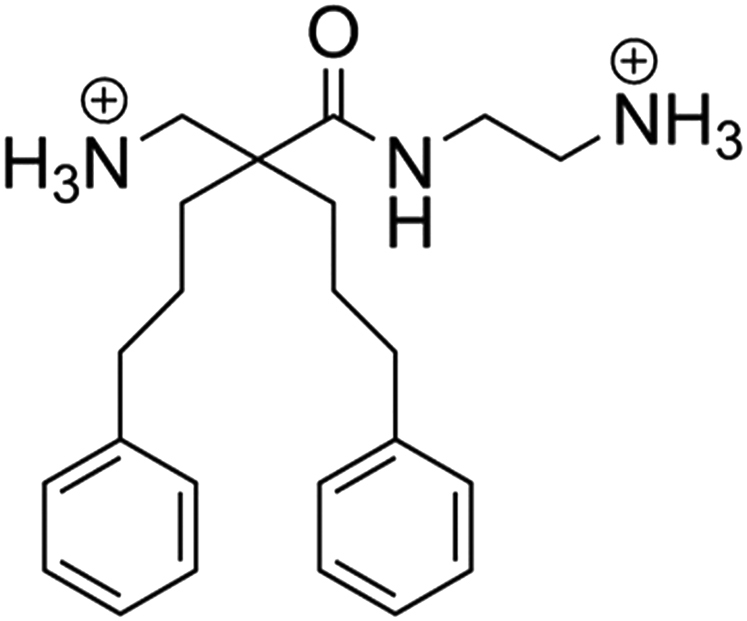
Chemical Structure of LTX-401 [N-(2-Aminoethyl)-2-(Aminomethyl)-5-Phenyl-2-(3-Phenylpropyl)Pentanamide]
Results
LTX-401 Rapidly Kills JM1 Cells
The MTT cell viability assay21 was employed in order to evaluate the cytotoxic activity and time course of killing (kinetics) by LTX-401 against JM1 cells. Both concentrations tested led to similar killing kinetics after 30 min of incubation, with cellular survival rate just beneath 50% (Figure 2). Within 90 min, LTX-401 had effectively killed nearly 100% of the cells, leaving a small subset of JM1 cells still metabolically active. In contrast, the chemotherapeutic agents doxorubicin and gemcitabine required a significantly longer incubation period to exert a similar cytotoxic effect against JM1 cells (Figure S1). Together, these data demonstrate that LTX-401 is a fast-acting and potent anticancer compound.
Figure 2.

Killing Kinetics of JM1 Cells Treated with LTX-401
Cells were treated with 54 and 108 μM LTX-401 for designated time points (5, 15, 30, 60, 90, 120, and 240 min) and assessed for viability using the MTT assay. Data represent the mean ± SD of three independent experiments with triplicates for each.
LTX-401 Induces Morphological Signs of Necrotic Cell Death and Promotes Release of DAMPs from JM1 Cells
Due to the rapid mechanism of action, we hypothesized that LTX-401 induced primary necrosis. Following 60 min of incubation, electron microscopic examination of LTX-401-treated JM1 cells revealed increased cellular volume caused by swelling (oncosis), formation of intracellular vacuoles, and diminished plasma membrane integrity without drastic morphological changes in nuclei (Figures 3E and 3F) compared with control cells (Figures 3A and 3B). Moreover, distorted cell morphology was even observed after 5 min of incubation with LTX-401 that manifested itself in loss or reduction of cell surface (microvillus-like pseudopods) and moderate oncosis (Figures 3C and 3D). Higher magnification (×30,000) also revealed clear alterations in normal morphology of intracellular organelles, particularly in mitochondria, whose inner micro-compartments (cristae) were severely swollen and disintegrated (Figure 3H) compared with control cells (Figure 3G). Altogether, these data point toward the ability of LTX-401 to induce necrotic cancer cell death.
Figure 3.

Treatment with LTX-401 Leads to Ultrastructural Changes in JM1 Cells
Cells were treated with 108 μM LTX-401 for various time points (5 and 60 min) before being fixed in a PHEM-buffered malachite green fixative and prepared for electron microscopy studies. Untreated control cells (A, B, and G) were preserved in serum-free RPMI 1640 (vehicle control) until experimental endpoint (60 min) and compared with cells treated for 5 (C and D) and 60 min (E, F, and H).
Next, we wanted to investigate the capacity of LTX-401 to stimulate features of ICD by analyzing the release of selected DAMPs into the culture supernatants of LTX-401-treated JM1 cells. HMGB1, a highly conserved non-histone chromatin binding protein, occupies a critical role as a proinflammatory mediator because of its ability to engage multiple surface receptors on immune cells once released into the extracellular milieu.22 Here, HMGB1 was detected in cell culture supernatants (after 60 min) by western blot analysis when treated with 108 μM LTX-401 (Figure 4A). Control cells preserved in serum-free RPMI 1640 showed complete detainment of the protein only within the lysates.
Figure 4.

JM1 Cells Release DAMPs and ROS When Treated with LTX-401
(A) JM1 cells release HMGB1 from the lysate (L) to supernatant (S) after being stimulated with 108 μM LTX-401. (B) ATP is released from JM1 cells into supernatant following treatment with 54 μM LTX-401. (C) Release of cytochrome c into supernatant of LTX-401-treated JM1 cells (108 μM). (D) JM1 cells release ROS after being treated with 271 μM LTX-401 for 45 min. Data are expressed as fold-change in ROS release relative to control. For (B)–(D), data depict the mean ± SEM. *p < 0.05, **p < 0.01, ****p < 0.0001 by Kruskal-Wallis test (B and C) and Student’s t test (D).
As with HMGB1, the release of ATP from dying cells constitutes one of the major hallmarks of ICD.23 When incubated with 54 μM LTX-401, the extracellular concentration of ATP quickly increased after 60 min of treatment (Figure 4B), as measured by means of a bioluminescence reaction (luciferase assay). Moreover, a peak was observed at 90 min into LTX-401 treatment before declining toward 2 h of incubation.
A cytochrome c ELISA assay was carried out to further assess the capability of LTX-401 to release mitochondrial DAMPs. Translocation of cytochrome c into the extracellular space has been reported to trigger inflammation by modulating the production of pro-inflammatory cytokines and chemokines via activation of the NF-κB pathway.24 A significant amount of cytochrome c was found in cell culture supernatants after 1 h of incubation with 108 μM LTX-401 (Figure 4C). Cytochrome c was not detectable in the supernatant fraction of untreated control cells. Altogether, these studies indicate that LTX-401 is able to stimulate features of ICD.
Treatment with LTX-401 Leads to the Release of ROS
Based on the observed effect on intracellular organelles, we hypothesized that LTX-401 would promote the production of reactive oxygen species (ROS). When incubating JM1 cells with 271 μM LTX-401, we observed a 2-fold increase in ROS production as measured by fluorescence intensity relative to control values (Figure 4D). Although lower doses of LTX-401 could induce the release of ROS from JM1 cells, a higher dose was chosen to indicate a significant effect. It is likely that the release of ROS is secondary to plasma membrane rupture, as ROS is primarily located in mitochondria. The reason for the differences in LTX-401 concentration across various in vitro studies was based on initial pilot studies demonstrating that different concentrations were needed to induce the release of DAMPs.
LTX-401 Induces Complete Regression of Subcutaneous JM1 Tumors
In order to investigate the direct antitumor effects of LTX-401, syngeneic Fisher 344 rats with day 7 or 8 s.c. JM1 tumors (60–90 mm3) were treated with either single doses of LTX-401 (n = 10) or vehicle control (n = 5) for 3 consecutive days (Figure 5A). Seven out of 10 tumor-bearing rats were cured by LTX-401 treatment (Figures 5B and 5C), whereas all animals in the control group had to be euthanized by day 23 post-inoculation because of tumor overburden and/or severe ulceration. Animals cured by LTX-401 treatment (n = 7) were rechallenged with 1 × 105 viable JM1 cells in the contralateral flank of the abdomen 4 weeks after complete regression. All animals resisted tumor development despite initially having slight tumor growth. In contrast, naive recipients (n = 4) all rapidly developed tumor (Figures 5D and 5E). In order to examine systemic protective immune responses, animals surviving second tumor challenge (s.c.) were additionally implanted orthotopically with live JM1 cells in the inferior right liver lobe. All animals previously cured by LTX-401 (n = 7) were resistant against intrahepatic tumor growth, whereas control animals (n = 4) all developed tumors and had to be sacrificed within 26 days post-inoculation because of excess tumor burden and general health condition (Figures 5F and 5G).
Figure 5.

Therapeutic Efficacy of LTX-401 against Subcutaneous JM1 Tumors
(A) Schematic depiction of the treatment schedule in a syngeneic tumor model in immunocompetent Fisher 344 rats inoculated subcutaneously with 1 × 105 JM1 hepatoma cells. Subcutaneous JM1 tumors were injected intratumorally with either (B) sterile 0.9% NaCl (control) or 0.4 mg LTX-401 once a day for 3 consecutive days (7, 8, and 9) after tumor cell inoculation. (C) Survival cures of animals treated with LTX-401. (D) Animals surviving primary tumor challenge were rechallenged subcutaneously with 1 × 105 viable JM1 cells contralateral to the first tumor site and measured for tumor growth. (E) Survival curves of subcutaneous rechallenge. (F) Four weeks after subcutaneous rechallenge, surviving animals were given intrahepatic rechallenge by the direct injection of 1 × 105 viable JM1 into the inferior right lobe of the liver and measured for tumor growth. (G) Survival curves of intrahepatic rechallenge. The percentages of surviving animals were analyzed using a log rank Mantel-Cox test. ****p < 0.0001. s.c., subcutaneous.
LTX-401 Treatment Mediates Abscopal Effects
To examine the ability of LTX-401 to affect a distal tumor deposit, we applied a two-tumor model in which the size of an intrahepatic tumor was measured after treating a s.c. established tumor (Figure 6A). Three consecutive injections of LTX-401 led to a complete response at the initial tumor site (Figure 6B) and was sufficient in controlling the growth of an untreated intrahepatic tumor compared with control animals treated with sterile saline (Figures 6C and 6D). However, the abscopal effect was statistically more significant at day 18 (9 days after the last LTX-401 injection) compared to day 21 (s.c. tumor endpoint). Taken together, these data indicate that LTX-401 is capable of inducing a partial immune-mediated abscopal effect.
Figure 6.

Partial Abscopal Response after Local Therapy with LTX-401
(A) Schematic depiction of the abscopal tumor model. Animals were inoculated subcutaneously with 1 × 105 JM1 cells on day 0 followed by intrahepatic inoculation with 1 × 105 JM1 cells on day 6. The subcutaneous tumor was treated with 0.4 mg LTX-401 per injection or sterile 0.9% NaCl (control). (B) Subcutaneous (treated) tumor size at day 21 (tumor endpoint). Intrahepatic (untreated) tumor size at (C) day 18 and (D) day 21. One dot represents one animal. *p < 0.05, **p < 0.01, ***p < 0.001 by Mann-Whitney test.
LTX-401 Treatment Induces Complete Regression of Orthotopic JM1 Liver Tumors and Generates Long-Term Protective Immune Responses
Next, we wanted to investigate the direct antitumor effects of LTX-401 against orthotopic JM1 liver tumors, hence mimicking clinical HCC. Rats bearing JM1 tumors (30–60 mm3) in the inferior left liver lobe were given two injections of 1.5 mg LTX-401 (n = 9) on days 6 and 8, whereas control animals (n = 5) received vehicle solution (Figure 7A). Five out of nine intrahepatic tumor-bearing rats were cured by LTX-401 treatment (Figures 7B and 7C). Control animals were sacrificed by day 24 post-inoculation because of tumor overburden and general health condition. We did not experience any drug-related or treatment-related adverse events by intratumoral injection of LTX-401 in these studies. Ultrasound monitoring post-treatment revealed extensive tumor necrosis that slowly regressed the following months (Figure 7G). Tumor-free animals (n = 5) were rechallenged with 1 × 105 viable JM1 cells s.c. 4 weeks after complete regression of intrahepatic tumors. All animals resisted rechallenge, indicating systemic protection, except one animal that started to develop s.c. tumor 70 days after tumor inoculation (Figures 7D and 7E). We next measured interferon-gamma (IFN-γ) production in response to (tumor) restimulation as a marker for T cell activity. Splenocytes harvested from animals that resisted s.c. rechallenge (n = 4) demonstrated increased IFN-γ production in response to irradiated JM1 cells compared with splenocytes taken from naive control animals (Figure 7F).
Figure 7.

Therapeutic Efficacy of LTX-401 against Orthotopic JM1 Liver Tumors
(A) Schematic depiction of the treatment schedule in a syngeneic tumor model in immunocompetent Fisher 344 rats inoculated intrahepatically with 1 × 105 JM1 hepatoma cells. Intrahepatic JM1 tumors were injected intratumorally with (B) either sterile 0.9% NaCl (control) or 1.5 mg LTX-401 on two occasions (days 6 and 8) after tumor cell inoculation. (C) Survival cures of animals treated with LTX-401. (D) Animals cured by LTX-401 treatment were 4 weeks later rechallenged subcutaneously with 1 × 105 viable JM1 cells and monitored for tumor growth, whereas (E) indicates survival outcome after rechallenge. (F) A total of 1.0 × 106 splenocytes harvested from long-term survivors (LTX-401) and naive control animals were co-cultured with 2.5 × 105 irradiated JM1 cells and incubated for 40 h at 37°C. The number of IFN-γ-producing cells in response to tumor cell stimulation was evaluated using an ELISpot assay. The number of spots was counted in triplicate and counted automatically on an ELISpot counter. Top images depict representative wells. (G) Ultrasound images depicting tumor and liver pre-treatment, 4 weeks post-treatment showing necrosis and fibrotic tissue, and 3 months post-treatment. Data depict the mean ± SEM and Student’s t test. The percentages of surviving animals were analyzed using a log rank Mantel-Cox test. ***p < 0.001. n, necrosis; s.c., subcutaneous; T, tumor.
Histopathological Examination
In the untreated rats, well-circumscribed tumors were seen in the s.c. fat tissue or in the liver tissue (Figures 8A and 8C). The immune infiltrate consisted of NK cells and CD3+ T cells whereof the majority were CD8+ T cells as shown by immunohistochemistry (IHC) (Figures 8E–8P). Tumors injected with LTX-401 were largely necrotic (Figures 8B and 8D), and some necrosis was also observed in adjacent liver tissue, likely caused by leakage from the tumor. Surrounding the areas with induced necrosis, a rim of granulation tissue was seen. The inflammatory infiltrate at the border of the tumors and necrotic tissue was similar to the untreated rats. Masson-trichrome staining showed no fibrosis in control tumors and only low levels of collagen at the border of the necrotic tissue in LTX-401-treated animals (Figures 8Q and 8R). Animals bearing intrahepatic tumor that were cured by LTX-401 treatment displayed no remaining tumor tissue, and Masson-trichrome staining revealed livers with small areas of collagen-rich fibrosis with thin and thick-walled vessels and collections of pigment macrophages, but without prominent inflammation.
Figure 8.

Histopathological Examinations of JM1 Tumors
Representative images of tumor area from subcutaneous and intrahepatic localized tumors, taken from both control animals (Ctrl) and animals treated with LTX-401 (7 days post-treatment). (A–D) Sections stained with hematoxylin, eosin, and saffron (HES). (B and D) LTX-401-treated tumors displayed large areas of necrotic tumor tissue with small remaining nodules of vital tumor tissue. (Q and R) Masson-trichrome staining (M-T) of the liver tumors showed no fibrosis in control tumors and only low levels of collagen at the border of the necrotic tissue in LTX-401-treated animals. (E–H) Sections immunostained for CD3+ T cells, (I–L) CD8+ T cells, and (M–P) natural killer cells. Arrow points at border of tumor. (A–D, Q, and R) Original magnification ×20; scale bars, 500 μm. Insets in upper right corner show higher original magnification of the tumors: ×400. (E–P) Original magnification ×200; scale bars, 50 μm. gt, granulation tissue; n, necrosis; T, tumor.
Discussion
The complete failure of tumor-debulking therapies, such as systemic chemotherapy, has gradually shifted the treatment of advanced HCC toward molecular-targeted therapies. However, survival benefits vary significantly among patients likely because of the complexity of hepatocarcinogenesis and heterogeneity of HCC.25 There is increasing evidence to support that solid tumors are composed of heterogenous tumor cell populations expressing different tumor antigens. As such, tumor heterogeneity contributes to drug resistance and disease recurrence following therapy and imposes a substantial obstruction to the development of effective cancer treatments.26, 27, 28 Targeted therapies may also aid in the selection of more invasive and resistant clones by eliminating the therapy-sensitive sub-clones.29 A novel approach for tackling tumor heterogeneity can involve immunotherapy. Augmentation of immune responses against tumor antigens is likely one of the most effective ways to therapeutically influence the host-cancer interaction. Hence therapies designed to maximize the liberation of tumor antigens in an immunogenic fashion could prove beneficial for mounting immune responses directed against several antigens, thus addressing heterogeneity. Here, we demonstrate that the novel anticancer compound LTX-401 exerts a membrane perturbating (oncolytic) effect on JM1 hepatoma cells with subsequent release of immunogenic mediators such as HMGB1, ATP, and cytochrome c. These observations suggest that LTX-401 is capable of releasing the full spectrum of tumor antigens, which explains why intratumoral treatment of both s.c. and orthotopic tumors led to complete regression and long-term protective immune responses. Other anti-cancer agents known to induce ICD and tumor antigen release include doxorubicin, which is often applied in chemoembolization procedures against intermediate-stage HCC. However, recurrence rates are high and once progressing into advanced HCC, chemotherapeutic regimens are typically not well tolerated because of underlying hepatic dysfunction.
Our study also demonstrates the ability of LTX-401 to mediate (partial) abscopal effects, which, undoubtedly, is one of the major goals of local immunotherapy. However, we did not observe a complete regression in the distant intrahepatic tumor, which is likely due to established local immunosuppression that counteracts antitumor immunity.30 Nevertheless, an abscopal effect implies that LTX-401 induced tumor antigen release and ICD within the treated lesion to prime (systemic) antitumor T cell responses. The latter is also supported by experiments where animals cured by LTX-401 treatment, either at s.c. or intrahepatic site, were resistant to tumor rechallenge, thus indicating that a systemic tumor-specific immunological memory was generated. As shown by ELISpot assay, the presence of markedly higher IFN-γ production by splenocytes harvested from cured animals accentuates the capacity of LTX-401 to induce antitumor immune responses. An increased expression of Th1 cytokines, such as IFN-γ, is known to positively correlate with prognosis and survival in several malignancies,31, 32 and is applied as a marker to reveal tumor-specific lymphocytes in response to tumor antigen stimulation. Despite observing a slight increase of T cell and NK cell infiltration in s.c. tumors after treatment, we did not detect a significant change of immune infiltrate in intrahepatic tumors. This might be attributed to the fact that the carcinogen-induced JM1 tumor model33 is relatively immunogenic, thus attracting immune cells to the tumor parenchyma. Moreover, as seen in intrahepatic control tumors, large areas of the center core were necrotic. Rapidly growing tumors are frequently deprived of oxygen (hypoxia), which may result in necrotic cell death and tumor-elicited inflammation.34 However, evidence suggests that hypoxia-induced leukocyte recruitment promotes cancer progression and increases metastatic potential, as well as immunosuppression.35, 36 In view of these considerations, we speculate that LTX-401 is capable of reshaping the tumor microenvironment, hence tipping the balance toward effective antitumor immune responses. The observation that LTX-401 induces T cell infiltration and complete regression of poorly immunogenic B16 melanomas19 provides additional support for this hypothesis. However, further immune cell phenotyping studies are required to characterize the changes in the tumor microenvironment following LTX-401 treatment.
Direct percutaneous-based treatment options like radiofrequency or ethanol ablation are already standard of care in selected primary tumors or metastases in the liver with a diameter less than 30 mm. These modalities use direct intratumoral puncture or injection under imaging guidance. Although LTX-401 was administered into orthotopic liver tumors without the guidance of radiological techniques, we did not experience any surgery- or treatment-related difficulties or adverse events. One might speculate a variable precision level caused by lack of ultrasonic guidance could lead to suboptimal local dosage and explain the therapeutic failure in some animals. In a clinical setting, however, LTX-401 would have been amenable to injection under radiographic guidance to specifically elicit maximal effect and necrosis of the tumor while minimizing necrosis of surrounding healthy liver tissue, which is the only potential clinical toxicity expected of this therapy considering the intratumoral route of administration. A histopathological assessment of intrahepatic tumors revealed both unresorbed necrosis and granulation (healing) tissue post-treatment, which was in accordance with ultrasound scans showing the presence of a well-defined round shape without typical tumor morphology 2 weeks after treatment. Ultrasound scans and tissue samples taken 3 months after tumor treatment displayed healthy and tumor-free livers, indicating regeneration of liver parenchymal tissue in the area of tumor necrosis. Finally, toxicology studies conducted in rats have demonstrated that LTX-401 is generally well tolerated at both intravenous and direct intrahepatic administration, and that high doses (>10 mg/kg) are required to identify dose-limiting toxicity.
Conclusions
Taken together, the present study demonstrates that LTX-401 has the potential to stimulate anticancer immune responses and may be applied therapeutically as a novel antitumor and immunotherapeutic approach in HCC. Further studies are required to assess the potential additive or synergistic activity in combination with therapies blocking immune checkpoints.
Materials and Methods
Reagents
LTX-401 (molecular weight [MW]net = 367.53) was synthetized and provided by Synthetica AS (Oslo, Norway). LTX-401 was dissolved in serum-free RPMI 1640 unless stated otherwise.
Cell Cultures
JM1 HCC cells33, 37 were cultured in DMEM (Sigma) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (GIBCO) and kept under standard cell culture conditions (37°C, <95% humidity, and 5% CO2).
Kinetic Analysis
Cells were incubated with LTX-401 solutions (both 2 × IC504h = 54 μM and 4 × IC504h value = 108 μM; where IC504h is the half maximum inhibitory concentration after 4 h) for 5, 15, 30, 60, 90, 120, and 240 min before being washed once with serum-free RPMI 1640 and further incubated in a 10% MTT solution (diluted in serum-free RPMI 1640) for an additional 2 h. Lastly, acidified isopropanol was added to facilitate formazan crystal solubilization. Absorbance was measured at 570 nm on a spectrophotometric microtiter plate reader (Thermomax Molecular Devices, NJ, USA). Cell survival was calculated as the A570 nm of treated cells relative to the negative control (100% viable cells) using the mean of two or three independent experiments and expressed as a 50% inhibitory concentration (IC50). For experiments comparing the cytotoxic activity of LTX-401 against doxorubicin and gemcitabine, the 2 × IC5048h was chosen to display differences in killing kinetics because the chemotherapeutic agents required longer incubation periods to reach IC50 values. IC50 values were determined by dilution series; 2.7–272 μM for LTX-401 and 0.01–100 μM for doxorubicin and gemcitabine. Recordings were made at 4, 24, and 48 h. The reason for the differences in LTX-401 concentration across various in vitro studies was based on initial pilot studies demonstrating that different concentrations were needed to induce the release of DAMPs.
Electron Microscopy
Samples were treated according to previously applied protocol19 involving the use of malachite green fixation and microwave-assisted processing and embedding for transmission electron microscopy. In short, cells were treated with LTX-401, harvested at various time points (5, 30, and 60 min), and prepared for electron microscopy. Control cells were treated with vehicle control (serum-free RPMI 1640) until experimental endpoint (60 min).
Detection of Extracellular HMGB1
JM1 HCC cells were seeded at a density of 2.0 × 105 cells/well in DMEM supplemented with 10% FBS and left to adhere overnight under standard cell culture conditions. Extracellular HMGB1 following incubation with LTX-401 was detected by western blot analysis as previously described.19 In short, LTX-401 was diluted in vehicle (serum-free RPMI 1640), whereas control cells were preserved in vehicle solution only.
Luciferin-Luciferase Detection of Extracellular ATP
JM1 HCC cells were seeded into 96-well plates at a density of 1.0 × 104 cells/well, left to adhere overnight, and treated with 54 μM LTX-401 for designated time points (10, 30, 60, 90, and 120 min) the following day. LTX-401-induced ATP secretion was determined as previously described.19 In short, LTX-401 was diluted in vehicle (serum-free RPMI 1640), whereas control cells were preserved in vehicle solution only.
Detection of Extracellular Cytochrome c
For the detection of extracellular cytochrome c, JM1 HCC cells were cultured and treated with LTX-401 as described previously for the luciferin-luciferase assay. In short, LTX-401 was diluted in vehicle (serum-free RPMI 1640), whereas control cells were preserved in vehicle solution only. Supernatants were diluted 1/5 in serum-free RPMI 1640 before the amount of cytosolic cytochrome c after treatment with LTX-401 was quantified using an ELISA-based cytochrome c detection kit (R&D Systems, USA) according to the manufacturer’s protocol.
ROS Measurement
JM1 cells were seeded out in clear-bottomed black-sided 96-well plates at a concentration of 2.5 × 104 cells/well. Cells were treated with 271 μM LTX-401 (100 μg/mL) for 45 min and analyzed for ROS release using the 2′,7′-dichlorofluorescin diacetate (DCFDA) Cellular ROS Detection Assay Kit from Abcam (ab113851). LTX-401 was dissolved in serum-free RPMI 1640 without phenol red (as recommended in the Assay Kit instructions), and DCFDA was used at a concentration of 25 μM. Negative control was cells incubated with serum-free RPMI 1640 without phenol red, and the positive control was 150 μM tert-butyl hydroperoxide (TBHP).
Animals
Six- to ten-week-old male Fisher 344 rats (170–250 g) were obtained from Taconic Biosciences (Denmark) and housed in cages (containing one to two rats) specifically designed for rats, with a 12 h/12 h day-night cycle, and allowed ad libitum access to high-quality rodent chow and water. All animals were monitored daily by both the animal care staff and researchers for adequacy of food, water, and general overall health conditions. Rats were placed individually in an induction chamber, and anesthesia was induced with 5% isoflurane (Isobar Vet; 100%; Nomeco, Copenhagen, Denmark) at 2 L/min mixed with purified oxygen until loss of righting reflex. Thereafter, rats were placed on their back in a nose cone, and anesthesia was maintained with a 0.5 L/min 2%–3% isoflurane-oxygen mix during tumor cell inoculation, tumor treatment, and surgery. Experiments were in compliance with local and European Ethical Committee guidelines and were approved by the Norwegian National Animal Research Authority (NARA approval ID: 6995).
Tumor Treatment
Syngeneic JM1 HCC cells were harvested, washed in serum-free RPMI 1640, and inoculated s.c. into the right side of the abdomen in Fisher 344 rats (1 × 105 JM1 cells per rat/50 μL RPMI 1640). Palpable tumors (60–90 mm3) were injected with single doses with LTX-401 (0.4 mg/50 μL saline) for 3 consecutive days. Control animals received saline only (0.9% NaCl in sterile H2O). Subcutaneous tumors were measured using an electronic caliper every 3 or 4 days. Animals were euthanized with pentobarbital (0.1 mL/100 g intraperitoneally) if weight loss exceeded 15% of total body weight or if tumor size exceeded 5,000 mm3. In addition, the following were implemented as humane endpoints: signs of unacceptable pain, infection, severe ulceration, or surgical complications.
Secondary Tumor Challenge and Abdominal Surgery
Animals displaying complete regression of s.c. JM1 tumors were given a second tumor challenge (s.c.) with JM1 cells (1 × 105 JM1 cells per rat/50 μL RPMI 1640) into the left flank of the abdomen 4 weeks after curative LTX-401 treatment. Animals surviving the s.c. tumor rechallenge were additionally given an intrahepatic rechallenge by opening the abdominal cavity through standard midline incision, while releasing the liver from its ligamentous attachments. Next, an intrahepatic HCC tumor was established by the direct implantation of JM1 cells into the inferior right lobe of the liver remnant using a 27G needle. In another set of experiments, LTX-401 (1.5 mg/50 μL saline) was injected directly into orthotopic HCC tumors 6 and 8 days after tumor cell inoculation, following the exact procedures as previously described. In experiments examining the abscopal effects of LTX-401 treatment, a s.c. tumor was established at day 0. Next, an intrahepatic HCC tumor was established on day 6. The s.c. tumor was injected in single doses with LTX-401 (0.4 mg/50 μL saline) on days 7, 8, and 9. Care was taken to avoid post-surgical pain and discomfort by the administration of 0.03–0.05 mg/kg buprenorphine s.c. (Temgesic; Reckitt), one dose before surgery and the second dose 6 h later. Intrahepatic tumor was monitored and measured in vivo by ultrasound imaging (Vevo 3100 system; FUJIFILM VisualSonics). Tumor volumes were calculated using the following formula: a × b2 (a being the width at the widest point of the tumor, and b the width perpendicular to a).
ELISpot Assay
In brief, harvested splenocytes were plated at a density of 1.0 × 106 cells and co-cultured with 2.5 × 105 irradiated JM1 cells and incubated for 40 h at 37°C. Spots were developed according to the manufacturer’s instructions (R&D Systems) and counted automatically on a Immunospot analyzer (Cellular Technology) as a means of IFN-γ production.
Immunohistochemistry
Tumor samples were harvested and prepared for histological examination as previously described.38 Slides were incubated overnight at 4°C with primary antibody, rabbit polyclonal anti-CD3 (clone A0452; Dako), mouse monoclonal anti-CD8 (clone OX-8; Abcam), or mouse monoclonal anti-NK (clone ANK61; Santa Cruz). As a secondary antibody, the anti-rabbit horseradish peroxidase SignalStain Boost IHC (immunohistochemistry) Detection Reagent (Cell Signaling Technology) or anti-mouse-HRP Envision+ System from Dako was used. A matched isotype control was used as a control for non-specific background staining.
Statistical Analysis
Results are presented as a mean ± SEM or SD of at least two independent experiments. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays were conducted twice with three parallels, and cytochrome c assays were conducted twice with two parallels, whereas ATP assays were conducted three times with two parallels. ELISpot IFN-γ assay was conducted with splenocytes plated in triplicates. Multiple comparisons (ATP and cytochrome c release assays) were analyzed using Kruskal-Wallis test, whereas unpaired events (abscopal tumor model experiments) were analyzed using Mann-Whitney test. Two sets of data (ROS and ELISpot IFN-γ assays) were analyzed using Student’s t test. Survival curves were generated with the Kaplan-Meier method and compared using the Mantel-Cox test. All graphs and statistical analyses were performed using Prism 6 software (GraphPad). p values ≤ 0.05 were considered to be statistically significant.
Author Contributions
Concept and Design: B.M., K.A.C., J.S., Ø.R., B.S., and P.-D.L.; Experiments and Procedures: B.M., K.A.C., J.S., B.S., and P.-D.L.; Preparation of the Manuscript, Tables, and Figures: B.M., K.A.C., B.S., Ø.R., and P.-D.L.
Conflicts of Interest
B.M. is an employee of Lytix Biopharma AS. Ø.R., B.S., and K.A.C. are employees and shareholders in Lytix Biopharma AS.
Acknowledgments
This project was supported by the Norwegian Research Council (project 257967) and Lytix Biopharma. The authors would like to acknowledge the Section of Comparative medicine for technical assistance regarding animal experiments.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omto.2019.05.002.
Supplemental Information
References
- 1.Liu H., Chen W., Zhi X., Chen E.J., Wei T., Zhang J., Shen J., Hu L.Q., Zhao B., Feng X.H. Tumor-derived exosomes promote tumor self-seeding in hepatocellular carcinoma by transferring miRNA-25-5p to enhance cell motility. Oncogene. 2018;37:4964–4978. doi: 10.1038/s41388-018-0309-x. [DOI] [PubMed] [Google Scholar]
- 2.Yao T., Chen Q., Shao Z., Song Z., Fu L., Xiao B. Circular RNA 0068669 as a new biomarker for hepatocellular carcinoma metastasis. J. Clin. Lab. Anal. 2018;32:e22572. doi: 10.1002/jcla.22572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jelic S., Sotiropoulos G.C., ESMO Guidelines Working Group Hepatocellular carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2010;21(Suppl 5):v59–v64. doi: 10.1093/annonc/mdq166. [DOI] [PubMed] [Google Scholar]
- 4.Pievsky D., Pyrsopoulos N. Profile of tivantinib and its potential in the treatment of hepatocellular carcinoma: the evidence to date. J. Hepatocell. Carcinoma. 2016;3:69–76. doi: 10.2147/JHC.S106072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin S., Hoffmann K., Schemmer P. Treatment of hepatocellular carcinoma: a systematic review. Liver Cancer. 2012;1:144–158. doi: 10.1159/000343828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kudo M., Finn R.S., Qin S., Han K.H., Ikeda K., Piscaglia F., Baron A., Park J.W., Han G., Jassem J. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018;391:1163–1173. doi: 10.1016/S0140-6736(18)30207-1. [DOI] [PubMed] [Google Scholar]
- 7.Waidmann O., Trojan J. Novel drugs in clinical development for hepatocellular carcinoma. Expert Opin. Investig. Drugs. 2015;24:1075–1082. doi: 10.1517/13543784.2015.1058776. [DOI] [PubMed] [Google Scholar]
- 8.Trojan J., Zangos S., Schnitzbauer A.A. Diagnostics and Treatment of Hepatocellular Carcinoma in 2016: Standards and Developments. Visc. Med. 2016;32:116–120. doi: 10.1159/000445730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.El-Khoueiry A.B., Melero I., Crocenzi T.S., Welling T.H., Yau T.C., Yeo W., Chopra A., Grosso J., Lang L., Anderson J. Phase I/II safety and antitumor activity of nivolumab in patients with advanced hepatocellular carcinoma (HCC): CA209-040. J. Clin. Oncol. 2015;33(Suppl 18):LBA101. [Google Scholar]
- 10.El-Khoueiry A.B., Sangro B., Yau T., Crocenzi T.S., Kudo M., Hsu C., Kim T.Y., Choo S.P., Trojan J., Welling T.H. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389:2492–2502. doi: 10.1016/S0140-6736(17)31046-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaspar D., Veiga A.S., Castanho M.A. From antimicrobial to anticancer peptides. A review. Front. Microbiol. 2013;4:294. doi: 10.3389/fmicb.2013.00294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoskin D.W., Ramamoorthy A. Studies on anticancer activities of antimicrobial peptides. Biochim. Biophys. Acta. 2008;1778:357–375. doi: 10.1016/j.bbamem.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fadnes B., Uhlin-Hansen L., Lindin I., Rekdal Ø. Small lytic peptides escape the inhibitory effect of heparan sulfate on the surface of cancer cells. BMC Cancer. 2011;11:116. doi: 10.1186/1471-2407-11-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krysko D.V., Garg A.D., Kaczmarek A., Krysko O., Agostinis P., Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer. 2012;12:860–875. doi: 10.1038/nrc3380. [DOI] [PubMed] [Google Scholar]
- 15.Kroemer G., Galluzzi L., Kepp O., Zitvogel L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013;31:51–72. doi: 10.1146/annurev-immunol-032712-100008. [DOI] [PubMed] [Google Scholar]
- 16.Inoue H., Tani K. Multimodal immunogenic cancer cell death as a consequence of anticancer cytotoxic treatments. Cell Death Differ. 2014;21:39–49. doi: 10.1038/cdd.2013.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galluzzi L., Buqué A., Kepp O., Zitvogel L., Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017;17:97–111. doi: 10.1038/nri.2016.107. [DOI] [PubMed] [Google Scholar]
- 18.Ausbacher D., Svineng G., Hansen T., Strøm M.B. Anticancer mechanisms of action of two small amphipathic β(2,2)-amino acid derivatives derived from antimicrobial peptides. Biochim. Biophys. Acta. 2012;1818:2917–2925. doi: 10.1016/j.bbamem.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 19.Eike L.M., Mauseth B., Camilio K.A., Rekdal Ø., Sveinbjørnsson B. The Cytolytic Amphipathic β(2,2)-Amino Acid LTX-401 Induces DAMP Release in Melanoma Cells and Causes Complete Regression of B16 Melanoma. PLoS ONE. 2016;11:e0148980. doi: 10.1371/journal.pone.0148980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou H., Sauvat A., Gomes-da-Silva L.C., Durand S., Forveille S., Iribarren K., Yamazaki T., Souquere S., Bezu L., Müller K. The oncolytic compound LTX-401 targets the Golgi apparatus. Cell Death Differ. 2016;23:2031–2041. doi: 10.1038/cdd.2016.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 22.Pitt J.M., Kroemer G., Zitvogel L. Immunogenic and Non-immunogenic Cell Death in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2017;1036:65–79. doi: 10.1007/978-3-319-67577-0_5. [DOI] [PubMed] [Google Scholar]
- 23.Kepp O., Senovilla L., Vitale I., Vacchelli E., Adjemian S., Agostinis P., Apetoh L., Aranda F., Barnaba V., Bloy N. Consensus guidelines for the detection of immunogenic cell death. OncoImmunology. 2014;3:e955691. doi: 10.4161/21624011.2014.955691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eleftheriadis T., Pissas G., Liakopoulos V., Stefanidis I. Cytochrome c as a Potentially Clinical Useful Marker of Mitochondrial and Cellular Damage. Front. Immunol. 2016;7:279. doi: 10.3389/fimmu.2016.00279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farazi P.A., DePinho R.A. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat. Rev. Cancer. 2006;6:674–687. doi: 10.1038/nrc1934. [DOI] [PubMed] [Google Scholar]
- 26.Dagogo-Jack I., Shaw A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018;15:81–94. doi: 10.1038/nrclinonc.2017.166. [DOI] [PubMed] [Google Scholar]
- 27.McQuerry J.A., Chang J.T., Bowtell D.D.L., Cohen A., Bild A.H. Mechanisms and clinical implications of tumor heterogeneity and convergence on recurrent phenotypes. J. Mol. Med. (Berl.) 2017;95:1167–1178. doi: 10.1007/s00109-017-1587-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McGranahan N., Swanton C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell. 2017;168:613–628. doi: 10.1016/j.cell.2017.01.018. [DOI] [PubMed] [Google Scholar]
- 29.Burrell R.A., Swanton C. Tumour heterogeneity and the evolution of polyclonal drug resistance. Mol. Oncol. 2014;8:1095–1111. doi: 10.1016/j.molonc.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang R., Zhou T., Liu W., Zuo L. Molecular mechanism of bystander effects and related abscopal/cohort effects in cancer therapy. Oncotarget. 2018;9:18637–18647. doi: 10.18632/oncotarget.24746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burkholder B., Huang R.Y., Burgess R., Luo S., Jones V.S., Zhang W., Lv Z.Q., Gao C.Y., Wang B.L., Zhang Y.M., Huang R.P. Tumor-induced perturbations of cytokines and immune cell networks. Biochim. Biophys. Acta. 2014;1845:182–201. doi: 10.1016/j.bbcan.2014.01.004. [DOI] [PubMed] [Google Scholar]
- 32.Castro F., Cardoso A.P., Gonçalves R.M., Serre K., Oliveira M.J. Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front. Immunol. 2018;9:847. doi: 10.3389/fimmu.2018.00847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Novicki D.L., Jirtle R.L., Michalopoulos G. Establishment of two rat hepatoma cell strains produced by a carcinogen initiation, phenobarbital promotion protocol. In Vitro. 1983;19:191–202. doi: 10.1007/BF02618059. [DOI] [PubMed] [Google Scholar]
- 34.Kuraishy A., Karin M., Grivennikov S.I. Tumor promotion via injury- and death-induced inflammation. Immunity. 2011;35:467–477. doi: 10.1016/j.immuni.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vakkila J., Lotze M.T. Inflammation and necrosis promote tumour growth. Nat. Rev. Immunol. 2004;4:641–648. doi: 10.1038/nri1415. [DOI] [PubMed] [Google Scholar]
- 36.Qiu Y., Li P., Ji C. Cell Death Conversion under Hypoxic Condition in Tumor Development and Therapy. Int. J. Mol. Sci. 2015;16:25536–25551. doi: 10.3390/ijms161025536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shi J.H., Scholz H., Huitfeldt H.S., Line P.D. The effect of hepatic progenitor cells on experimental hepatocellular carcinoma in the regenerating liver. Scand. J. Gastroenterol. 2014;49:99–108. doi: 10.3109/00365521.2013.854406. [DOI] [PubMed] [Google Scholar]
- 38.Camilio K.A., Berge G., Ravuri C.S., Rekdal O., Sveinbjørnsson B. Complete regression and systemic protective immune responses obtained in B16 melanomas after treatment with LTX-315. Cancer Immunol. Immunother. 2014;63:601–613. doi: 10.1007/s00262-014-1540-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.