

Abstract
The Chinese concave‐eared frog (Odorrana tormota) is a rare and threatened species with remarkable sexual dimorphism. Intestinal microbes are understood to play important roles in animal physiology, growth, ecology, and evolution. However, little is known about the intestinal microbes in female and male frogs, as well as the contributing effect by gut infesting nematodes to the co‐habiting bacteria and their function in degradation food rich in chitin. Here, this study analyzed the microbiota of the intestinal tract of both female and male, healthy as well as nematode‐infested concave‐eared frogs using high throughput 16S rRNA sequencing and metagenomic techniques. The results showed that the bacterial composition of the microbiota at the phylum level was dominated by Firmicutes, Verrucomicrobia, Bacteroidetes, and Proteobacteria. The study also revealed that the community composition below the class level could be represent sex differences, particularly with regard to Enterobacteriales, Enterobacteriaceae, Peptostreptococcaceae, and Rikenellaceae, among others. Carbohydrate‐active enzyme‐encoding genes and modules were identified in related gut bacteria by metagenomic analysis, with Bacteroidia, Clostridia, and gammaproteobacteria predicted to be the main classes of chitin‐decomposing bacteria in the frog intestine. In addition, the abundance of some bacteria significantly increased or decreased in nematode‐infected hosts compared with healthy individuals, including Verrucomicrobia, Verrucomicrobiae, Negativicutes, Actinobacteria, and Bacilli, among others. This indicates that nematode infection may affect the richness and composition of some gut bacteria.
Keywords: intestinal microorganism, metagenome, nematode infection, Odorrana tormota, sexual dimorphism
1. INTRODUCTION
The gastrointestinal tract is the primary site where microorganisms interact with the host species. The intestinal microbiota can develop a natural defense barrier exerting different protective, structural, and metabolic effects on the host epithelium (Gaskins, Croix, Nakamura, & Nava, 2008; Ivanov & Littman, 2011). The diversity of the frog gut microbiota was influenced by hibernation, metamorphosis, environmental pollution, and other factors (Van der, Cohen, & Nace, 1974; Jennifer, Loesche, & Nace, 1982; Kohl, Cary, Karasov, & Dearing, 2015). At present, research on intestinal microbes has generally been limited to a few frog species (Huang, Chang, Huang, Gao, & Liao, 2018; Kohl, Cary, Karasov, & Dearing, 2013; Vences et al., 2016; Wiebler, Kohl, Lee, & Costanzo, 2018).
Diet, as an important environmental factor, serves as both a source of bacteria and a change in the nutritional environment of the intestines (Costello, Stagaman, Dethlefsen, & Bohannan, 2012; David et al., 2014; Janssen & Kersten, 2015; Vences et al., 2016). Different diets can vary in their macronutrient content and therefore they might favor certain bacterial communities of the host (Knutie et al., 2017). For amphibian groups, most species show sexual dimorphism, with females larger than males (Shine, 1979).The males are likely to be limited by the size of the body as well as to the feeding organs, making it impossible to hunt larger volumes of food (Houston, 1973; Toft, 1980). Indeed, according to the theory of optimal foraging, larger frogs tend to prey on larger rather than smaller foods (Hirai, 2002; Lima & Moreira, 1993). Sex difference also affects the intestinal microbial composition (Costello et al., 2012; Freire, Basit, Choudhary, & Chee, 2011; Koren et al., 2012; Kovacs et al., 2011; Markle et al., 2014). To date, the effect of sex on the gut microbiota of amphibians has not been adequately explored.
The Chinese concave‐eared frog (Odorrana tormota) is the first non‐mammalian vertebrate shown to be able to communicate using ultrasound (Feng et al., 2006). It is only found in eastern China, mainly in the southern mountains of Anhui Province and the western mountains of Zhejiang Province (Fei, 1999; Feng, Zhang, Shu, & Yao, 2015). Because of its limited and fragmented distribution, the wild population is classified as a vulnerable species by the International Union for the Conservation of Nature and Natural Resources. O. tormota demonstrates sexual dimorphism, with females being significantly larger than males. Males have an average snout‐to‐vent length (SVL) of 32.5 mm, whereas females average 56 mm (Feng, Narins, & Xu, 2002). Adult frogs mainly feed on insects, including Lepidoptera, Arachnida, Hymenoptera, and Orthoptera species, as well as damselflies (Fei, 1999). The ratio of intestine length to SVL varies between 0.44 and 0.91, which is the lowest known ratio among the Anura (Wu, Xiong, Lei, & Jiang, 2012). Therefore, how the concave‐eared frog obtains enough energy from hydrolyzing chitin, the major component of the insect shell, in such a short gut needs to be further examined, as does the role of gut microbes in this process. Additionally, very little is known about the effects of pathogens, such as intestinal parasites, on the gut microbiota of most frogs.
Therefore, this study was aimed at comparing the gut bacterial communities between male and female Chinese concave‐eared frogs using a 16S rRNA‐based sequencing method. Additionally, metagenomic analysis was used to explore the potential function of the gut bacteria, especially the role of the gut bacteria in the biodegradation of chitin by frogs. Furthermore, the microbial communities of healthy and nematode‐infected individuals were compared with the aim of evaluating the effects of intestinal parasites on the gut microbial communities of frogs.
2. MATERIALS AND METHODS
2.1. Experimental animals and sample collection
Fifteen concave‐eared frogs, including seven females and eight males, were collected from Banqiao Provincial Natural Reserve, Anhui Province, China, during the 2017 breeding season. All individuals were separately placed into plastic boxes containing plant leaves and water from their natural environment and transported to the laboratory for further analyses. After being starved for 3 days, intestinal contents were collected from the midgut and small intestines as described in Mashoof, Goodroe, Du, and Eubanks (2013). All samples were then stored at −80°C until further processing. Among the 15 frogs, four (one female and three males) were found to be nematode‐infected after dissection.
To assess the effects of sex on the gut microbiota, five male individuals (RTM1–RTM5) and six female individuals (RTF1–RTF6) were compared. To assess the effects of nematode infection on the gut microbiota while controlling for the influence of sex, all three infected male individuals (Infect2–Infect4) and the five normal male individuals (RTM1–RTM5) were separated into infected and uninfected groups for further study. All frogs were determined to be 2 years of age on the basis of skeletochronology (Tsiora & Kyriakopoulou‐Sklavounou, 2002; Supporting Information Figure S1).
2.2. DNA extraction
A FastDNA SPIN Kit for soil (MoBio Laboratories, Carlsbad, CA) was used to extract DNA from the samples according to the manufacturer’s instructions. DNA quality was examined by 1% agarose gel electrophoresis and measured by spectrophotometry. All DNA samples were stored at −20°C until further processing.
2.3. 16S rRNA gene amplification, sequencing, and processing of sequencing data
16S ribosomal RNA (rRNA) genes were amplified from all samples for barcode‐based sequencing. We amplified the V3–V4 region of the bacterial 16S rRNA gene using the universal forward primer 338F (5′‐ACTCCTACGGGAGGCAGCAG‐3′) and the reverse primer 806R (5′‐GGACTACHVGGGTWTCTAAT‐3′; Xu, Wang, Gai, & Xia, 2016). Purified amplicons were pooled in equimolar concentrations and paired‐end sequenced (2 × 300) on an Illumina MiSeq platform (Illumina, San Diego) by Majorbio Bio‐Pharm Technology Co. Ltd. (Shanghai, China) according to standard protocols.
Operational taxonomic units (OTUs) were clustered with a 97% similarity cutoff using Usearch (version 7.0 https://drive5.com/uparse/) and chimeric sequences were identified and removed using UCHIME. The taxonomy of each 16S rRNA gene sequence was analyzed using the RDP Classifier algorithm (https://rdp.cme.msu.edu/) against the Silva (SSU123) 16S rRNA database using a confidence threshold of 70%.
2.4. Metagenomic sequencing, quality control, and genome assembly
To characterize and compare the microbial communities in the intestines of the male and female concave‐eared frogs, two metagenomic DNA samples were sequenced. For the male sample, equal quantities of total DNA were isolated from five individual frogs and pooled, while for the female sample, equal quantities of total DNA were isolated from six individual frogs and pooled. DNA was fragmented to an average size of about 300 bp for paired‐end library construction using a Covaris M220 ultrasonicator. Paired‐end libraries were prepared using a TruSeq DNA Sample Prep Kit (Illumina). Adapters containing the full complement of sequencing primer hybridization sites were ligated to the blunt‐end fragments. Paired‐end sequencing was performed on a HiSeq4000 platform (Illumina) at Majorbio Bio‐Pharm Technology using a HiSeq 3000/4000 PE Cluster Kit and HiSeq 3000/4000 SBS Kits according to the manufacturer’s instructions. Each read was then trimmed using Sickle (https://github.com/najoshi/scickle). Reads that aligned with the Xenopus tropicalis and Nanorana parkeri genomes, as determined by BWA (https://bio-bwa.sourceforge.net), and any hits associated with the reads and their mated reads were removed. The resultant high‐quality reads were then used for further analysis. The Illumina reads were assembled into contigs using IDBA‐UD (Peng, Leung, Yiu, & Chin, 2012) with default parameters.
2.5. Gene prediction, taxonomy, and functional annotation
Genes were predicted within the contigs using MetaGeneMark (Zhu, Lomsadze, & Borodovsky, 2010). A non‐redundant gene catalog was constructed with CD‐HIT (Li & Godzik, 2006) using a sequence identity cutoff of 0.95, with a minimum coverage cutoff of 0.9 for the shorter sequences. This catalog contained 982,379 microbial genes (Supporting Information Table S1). Gene reads were characterized using BLASTX (Altschul, Madden, Schäffer, & Zhang, 1997) comparisons against the integrated NCBI non‐redundant (nr) protein database (E‐values <10−5). The LCA‐based algorithm implemented in MEGAN (Huson, Auch, Qi, & Schuster, 2007) was used to determine the taxonomic level of each gene. MetaGene Annotator (Noguchi, Park, & Takagi, 2006) was applied to the assembled contigs to identify open reading frames (ORFs) longer than 100 bp. ORFs were translated using the Bacterial Genetic Code. BLASTP (Version 2.2.28+, https://blast.ncbi.nlm.nih.gov/Blast.cgi) was used for taxonomic annotations by aligning non‐redundant gene catalogs against the NCBI nr database with an e‐value cutoff of 1e−5. Clusters of orthologous groups (COG) analysis of the annotated ORFs was performed using BLASTP against the eggNOG database (v4.5) with an e‐value cutoff of 1e−5 (Jensen et al., 2008; Tatusov, Fedorova, Jackson, & Jacobs, 2003). Carbohydrate‐active enzymes were annotated using hmmscan (https://hmmer.janelia.org/search/hmmscan) against the CAZy database (v5.0; https://www.cazy.org/) with an e‐value cutoff of 1e−5 (Rice, Longden, & Bleasby, 2000).
2.6. Statistical analysis
Several α‐diversity measurements were calculated for each sample. The Shannon index, Simpson’s index, and the Good’s coverage index were calculated to estimate diversity. Chao1 was also calculated to estimate OTU richness. All diversity metrics were then compared using the Mann–Whitney U test.
To identify taxa with different abundance between healthy and nematode‐infected frogs, the LDA Effect Size (LEfSe) algorithm was used through an online Galaxy interface (https://huttenhower.sph.harvard.edu/galaxy/root). This performed non‐parametric factorial Kruskal–Wallis sum‐rank tests and linear discriminant analysis (LDA) to determine whether these features are consistent with the expected behavior of the different biological classes (Segata et al., 2011).
To compare community compositions between groups, analysis of similarities (ANOSIM) and non‐metric multidimensional scaling (NMDS) was conducted to investigate dissimilarities between healthy and nematode‐infected individuals. ANOSIM was conducted using a Bray–Curtis index of similarity with 9999 permutations. R values indicate the biological importance of differences, ranging between −1 and 1. The closer R was to 1, the greater the difference between groups than within groups. NMDS analysis was performed in the R “vegan” package (Oksanen, Kindt, Legendre, & Hara, 2007) using a Bray–Curtis index.
3. RESULTS
3.1. Concave‐eared frog dataset
Overall, the dataset consisted of 546,643 high‐quality 16S rRNA gene sequences, with an average of 439 sequences for each of the 15 samples (Supporting Information Table S2). OTUs were delineated at a 97% similarity level, leaving 406,905 sequences for further analysis (Supporting Information Table S3). The p‐value of <0. 05 indicated that the difference between groups was significantly larger than that within groups.
3.2. Gut microbiota of female vs. male concave‐eared frogs
An average of 36,414 ± 3,763 (mean ± SD) high quality, classifiable 16S rRNA gene sequences from the gut microbial communities of the Chinese concave‐eared frogs were obtained, with average counts per sample ranging from 35,719 ± 2,315 to 36,994 ± 999 (Mean ± SD). The sequences were classified into 2,289 OTUs based on 97% sequence identity. The gut microbial communities of both female and male frogs were dominated at the phylum level by Firmicutes (23.16% and 32.67%, respectively), Verrucomicrobia (24.24% and 26.23%), Bacteroidetes (27.25% and 11.61%), Proteobacteria (13.82% and 18.77%), and Fusobacteria (10.50% and 2.82%), and there was no significant difference in the relative abundance of the phyla and classes between sexes (p > 0.05 for all; Figure 1). The abundance of gut microbial communities in each individual at the phyla level was shown in Supporting Information Figure S2. All phylogenetic indices (Shannon index, Simpson’s index, Chao1 index, and the Good’s coverage index) confirmed that there was no significant difference in gut microbial diversity between males and females at the phylum and class levels (Mann–Whitney U test, p > 0.05 for all indices; Supporting Information Figure s3); however, inter‐sex differences were identified at lower taxonomic levels. For example, at the order level, significantly more reads were assigned to Enterobacteriales in female samples (10.12%) than in male samples (2.69%). At the family level, the relative abundance of Enterobacteriaceae was significantly higher in females than in males, whereas the opposite was observed for Rikenellaceae. Additionally, several microbial families also exhibited marked differences between sexes (Table 1).
Figure 1.
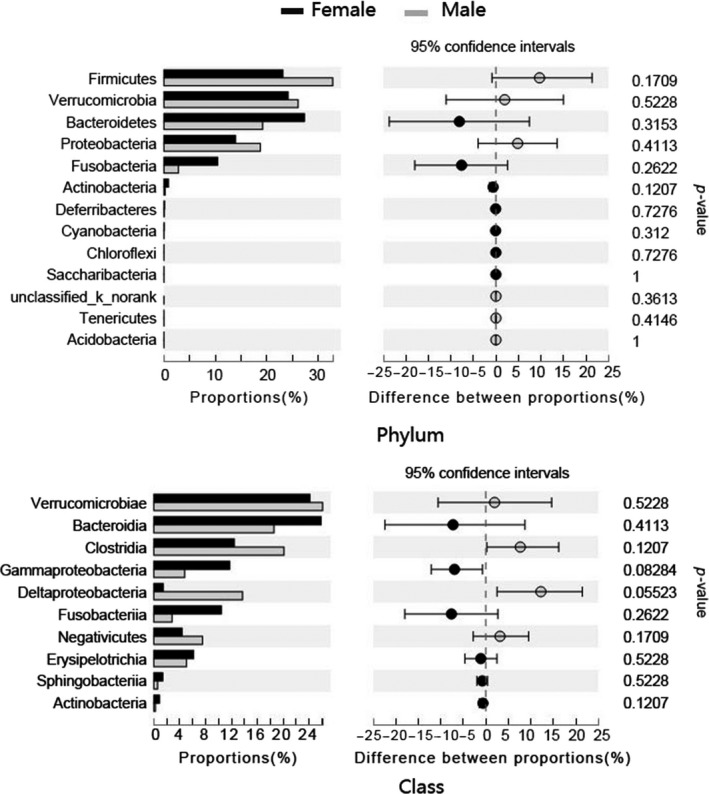
Comparison of the taxonomic compositions of the gut microbiota of male and female Chinese concave‐eared frogs. Relative abundances (percentage) of the microbiota at the phylum and class levels for female and male samples are presented (Mann–Whitney U test)
Table 1.
Differences in taxonomic composition of the intestinal microbiota of male vs. female Chinese concave‐eared frogs
| Rank | Classification | Female (%) | Male (%) | p‐Value |
|---|---|---|---|---|
| Order | Enterobacteriales | 10.120 | 2.691 | 0.022 |
| Family | Enterobacteriaceae | 10.120 | 2.691 | 0.022 |
| Family | Peptostreptococcaceae | 1.005 | 0.037 | 0.008 |
| Family | Rikenellaceae | 0.017 | 0.305 | 0.008 |
| Genus | Unclassified of Erysipelotrichaceae | 0.253 | 1.507 | 0.036 |
| Genus | Robinsoniella | 0.997 | 0.117 | 0.008 |
| Genus | Erysipelatoclostridium | 0.223 | 0.075 | 0.075 |
| Genus | Alistipes | 0.011 | 0.235 | 0.034 |
Significant differences in microbial composition (relative abundance, %) at the genus, family, and order levels between male and female Chinese concave‐eared frogs are indicated. p‐values < 0.05 indicate significance, as calculated using a Mann–Whitney U test.
3.3. Functional analysis of metagenomic datasets
Metagenomic data analysis confirmed most of the dominant microbial phyla as determined by 16S rRNA sequencing, except for Fusobacteria, with Bacteroidetes, Firmicutes, Verrucomicrobia, and Proteobacteria identified as the four most dominant phyla (Supporting Information Figure S4). Among the 5,349,836 annotated genes and modules in the metagenomic dataset, 47.47% were identified as glycoside hydrolases (GH), 18.96% were assigned to carbohydrate‐binding module (CBM) families, 18.84% were glycosyltransferases (GT), and 10.41% belonged to carbohydrate esterase (CE) families. The GH catalytic modules contained 2,539,430 sequences belonging to 93 GH families, while the CBM modules included 1,014,146 sequences from 60 families. Enzymes related to chitin degradation are shown in Table 2. Phylogenetic analysis of these contigs indicated that the dominant phyla of chitin‐degrading bacteria were Bacteroidetes (39.53%), Firmicutes (37.50%), and Proteobacteria (17.18%). Specifically, Bacteroidia (38.63%), Clostridia (29.46%), and gammaproteobacteria (14.08%) appeared to be the main classes of chitin‐decomposing bacteria in the frog intestine (Supporting Information Table S4).
Table 2.
Glycoside hydrolase (GH) and carbohydrate‐binding module (CBM) profiles of intestinal microbiota in Chinese concave‐eared frogs in relation to chitin degradation. Data are presented as the sum number of genes encoding the corresponding enzyme
| RTF | RTM | Total | Known chitin degradation activities | |
|---|---|---|---|---|
| Chitinases | ||||
| GH18 | 109 | 57 | 163 | Chitinase; lysozyme; endo‐β‐N‐acetylglucosaminidase; peptidoglycan hydrolase with endo‐β‐N‐acetylglucosaminidase; others |
| GH19 | 8 | 16 | 24 | Chitinase; lysozyme |
| GH23 | 137 | 116 | 253 | Lysozyme type G; peptidoglycan lyase; chitinase |
| Chitosanase | ||||
| GH7 | 0 | 1 | 1 | Endo‐β‐1,4‐glucanase; reducing end‐acting cellobiohydrolase; chitosanase; endo‐β‐1,3‐1,4‐glucanase |
| GH8 | 12 | 4 | 16 | Chitosanase; cellulase; licheninase; endo‐1,4‐β‐xylanase; reducing‐end xylose‐releasing exo‐oligoxylanase |
| GH46 | 8 | 1 | 9 | Chitosanase |
| GH75 | 3 | 1 | 4 | Chitosanase |
| GH80 | 1 | 0 | 1 | Chitosanase |
| Lysozyme | ||||
| GH18 | 106 | 57 | 163 | Chitinase; lysozyme; endo‐β‐N‐acetylglucosaminidase; peptidoglycan hydrolase with endo‐β‐N‐acetylglucosaminidase; others |
| GH19 | 8 | 16 | 24 | Chitinase; lysozyme |
| GH22 | 0 | 2 | 2 | Lysozyme type C; lysozyme type i; α‐lactalbumin |
| GH24 | 31 | 39 | 70 | Lysozyme |
| Cellulases | ||||
| GH5 | 57 | 27 | 84 | Endo‐β‐1,4‐glucanase/cellulase; endo‐β‐1,4‐xylanase; β‐glucosidase; β‐mannosidase; others |
| GH8 | 12 | 4 | 16 | Chitosanase; cellulase; licheninase; endo‐1,4‐β‐xylanase; reducing‐end‐xylose releasing exo‐oligoxylanase |
| N‐acetylglucosaminidase | ||||
| GH18 | 106 | 57 | 163 | Chitinase; lysozyme; endo‐β‐N‐acetylglucosaminidase; peptidoglycan hydrolase with endo‐β‐N‐acetylglucosaminidase; others |
| GH20 | 209 | 91 | 300 | β‐hexosaminidase; lacto‐N‐biosidase; β‐1,6‐N‐acetylglucosaminidase; β‐6‐SO3‐N‐acetylglucosaminidase |
| GH73 | 98 | 58 | 156 | Lysozyme; mannosyl‐glycoprotein endo‐β‐N‐acetylglucosaminidase; peptidoglycan hydrolase with endo‐β‐N‐acetylglucosaminidase specificity |
| GH84 | 43 | 23 | 66 | N‐acetyl β‐glucosaminidase; hyaluronidase; [protein]‐3‐O‐(GlcNAc)‐l‐Ser/Thr β‐N‐acetylglucosaminidase |
| GH85 | 10 | 4 | 14 | Endo‐β‐N‐acetylglucosaminidase |
| GH89 | 38 | 22 | 60 | α‐N‐acetylglucosaminidase |
| GH111 | 1 | 0 | 1 | Keratan sulfate hydrolase (endo‐β‐N‐acetylglucosaminidase) |
| GH116 | 16 | 13 | 29 | β‐glucosidase; β‐xylosidase; acid β‐glucosidase/β‐glucosylceramidase; β‐N‐acetylglucosaminidase |
| Chitin‐binding function | ||||
| CBM2 | 2 | 10 | 12 | Several of these modules have been shown to also bind chitin or xylan; others |
| CBM3 | 3 | 0 | 3 | In one instance binding to chitin has been reported; others |
| CBM5 | 983 | 19 | 992 | Chitin‐binding described in several cases; others |
| CBM12 | 23 | 17 | 40 | The majority of these modules is found among chitinases where the function is chitin‐binding; others |
| CBM14 | 3 | 2 | 5 | The chitin‐binding function has been demonstrated in several cases; others |
| CBM18 | 0 | 1 | 1 | The chitin‐binding function has been demonstrated in many cases. These modules are found attached to a number of chitinase catalytic domains, but also in non‐catalytic proteins either in isolation or as multiple repeats; others |
| CBM19 | 0 | 2 | 2 | Modules of 60–70 residues with chitin‐binding function |
| CBM73 | 5 | 1 | 6 | Modules of approx 65 residues found on various enzymes active of chitin. Chitin‐binding function demonstrated for the Cellvibrio japonicus CjLPMO10A protein |
The distribution of the genome among the general functional categories was assessed based on BLAST matches against the COG database. When the metagenomic data were included, the following categories were identified: carbohydrate transport and metabolism [G], amino acid transport and metabolism [E], inorganic ion transport and metabolism [P], energy production and conversion [C], coenzyme transport and metabolism [H], nucleotide transport and metabolism [F], and lipid transport and metabolism [I] (Supporting Information Figure s5). Some genes were categorized as “unknown function” (30.09% for female and 27.73% for male).
3.4. Comparison of gut microbiota between healthy and nematode‐infected individuals
The diversity of the gut bacterial communities of the nematode‐infected frogs was not significantly different from that of the healthy frogs, as confirmed by Shannon index, Simpson’s index, Chao1 index, and the Good’s coverage index analyses (Mann–Whitney U test, p > 0.05 for all indices).
Investigation of the nematode‐infected frog samples showed that frog intestinal microbial communities exhibited a significant reduction in the relative abundance of Verrucomicrobia, Verrucomicrobiae, and Negativicutes, and a significant increase in the relative abundance of Bacilli and Actinobacteria at the phylum and class levels (Figure 2).
Figure 2.
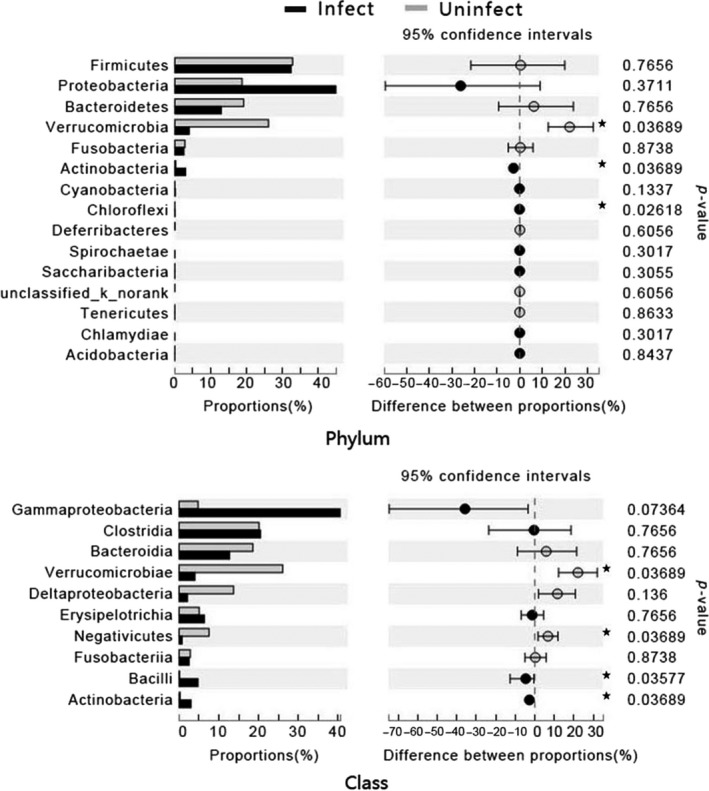
Comparison of the taxonomic compositions of the gut microbiota of the nematode‐infected and uninfected Chinese concave‐eared frogs. The relative abundances (percentage) of the microbiota at the phylum and class levels are presented. Asterisks indicate significant differences (Mann–Whitney U test: *p ≤ 0.05)
A supervised comparison using LEfSe was then performed to statistically define (at a log LDA threshold of 2.0) the differences in microbial composition between healthy and nematode‐infected frogs. This also confirmed that bacteria from the family Verrucomicrobiaceae, the genus Akkermansia, the order Verrucomicrobiales, and the class Verrucomicrobiae were more abundant in healthy individuals, while those from the genus Enterobacter, the class Bacilli, and the phylum Actinobacteria were more abundant in the infected individuals (Figure 3a). A clear distinction in the gut bacterial community structures of infected and uninfected frogs was also revealed by ANOSIM (R = 0.4154, p = 0.043) and NMDS analyses (Figure 3b).
Figure 3.
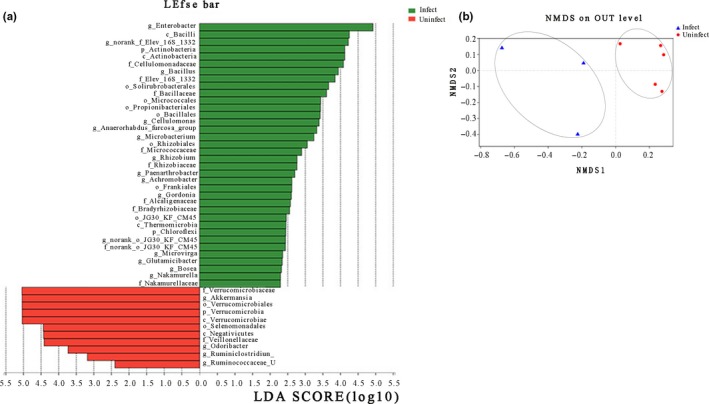
(a) Bacterial taxa that were differentially abundant in the gut microbiota profiles of nematode‐infected and uninfected Chinese concave‐eared frogs visualized using a log LDA score above 2.00. (b) NMDS analysis showing differences in gut microbiota between nematode‐infected and uninfected Chinese concave‐eared frogs
4. DISCUSSION
There is a significant degree of variation in the dominant members of the gut microbial communities of vertebrates. For example, the communities of teleost fish are rich in Proteobacteria (Sullam et al., 2012), while tetrapod communities are dominated by Firmicutes and Bacteroidetes (Costello et al., 2012; Kohl & Yahn, 2016). In the present study, in addition to Firmicutes and Bacteroidetes, the phyla Verrucomicrobia and Proteobacteria were also abundant in the Chinese concave‐eared frog, as confirmed by both 16S rRNA sequencing and metagenomic analysis. Similar results have previously been obtained in other frog species (Huang et al., 2018; Vences et al., 2016), suggesting that Firmicutes, Bacteroidetes, Verrucomicrobia, and Proteobacteria are the dominant phyla in amphibian gastrointestinal tracts. These results are also consistent with the previous suggestion that animals housed in similar environments and with similar predation conditions tend to harbor similar microbial groups at the higher taxonomic levels (Kovacs et al., 2011).
Diet category or host trophic level (carnivorous, omnivorous, and herbivorous) is the major factor driving the composition and metabolism of gut microbiota (Han et al., 2016; Ley et al., 2008). Sexual dimorphism is a common phenomenon in amphibians (Shine, 1979). Former researches have showed that frogs with large body size tend to hunt for larger preys, while the small ones might tend to prey on smaller foods (Hirai, 2002; Houston, 1973; Lima & Moreira, 1993; Toft, 1980). The Chinese concave‐eared frog has a larger and smaller body sizes for females and males respectively, hence the types of food available to both sexes may slightly vary within the same region. The current study showed lack of similarity in the microbial diversity and relative abundance of communities between male and female concave‐eared frogs at the phylum and class levels based on 16S rRNA sequence data. However, significant differences in the gut microbial composition between sexes were observed at some of the lower taxonomic levels. The difference in composition of intestinal microbes at low levels may have resulted from weak differences in predation between sexes. Based on our current findings, we could also not determine whether these differences were caused by hormone–microbe interactions, sex‐specific immune responses, or other factors (Bolnick, Snowberg, Hirsch, & Lauber, 2014; Markle et al., 2014).
Insects, which constitute the staple diet of frogs, are rich in protein and chitin. Studies indicate that chitin degradation depends on several specific enzymes (Beier & Bertilsson, 2013). Goodrich and Morita (1977) found a direct correlation between the chitin content of the natural diet and bacterial chitinase activity in the stomach of marine piscivorous fish species. Microbial chitinases can degrade chitin into its monomeric or oligomeric components, thereby degrading the major component of the insect outer skeleton (Suganthi, Senthilkumar, Arvinth, & Chandrashekara, 2017). Chitinolytic enzymes in the digestive systems of marine fishes are derived from both the prey and enteric bacteria (Gutowska, Drazen, & Robison, 2004), while chitinases in the frog gut can be produced in the stomach (Fujimoto et al., 2004). However, to date, little is known about the chitinolytic activity of bacteria in the frog gut (Delsuc et al., 2014; Vences et al., 2016). The present study indicates that bacterial members of the frog gut microbiota can digest chitin using chitin‐degrading enzymes, shown by the presence of genes assigned to GH families and CAZy modules. Furthermore, the COG functional category profiles from the frog intestinal metagenomes showed an abundance of sequences associated with carbohydrate transport and metabolism, as well as many chitinolytic enzymes associated with Bacteroides. A large proportion of the proteins produced by Bacteroides species are used to break down polysaccharides and metabolize sugars (Xu, 2003). These enzymes play a fundamental role in the processing of complex molecules into simpler forms in the host intestine. The ability to harvest alternative energy sources from food might allow Bacteroides species to be more competitive than other bacteria in the frog intestine. Therefore, intestinal microbes may be a complementary pathway for frog digestion of chitin.
Parasitic nematodes, known as helminths, cause a wide range of diseases in humans and animals, and it is estimated that >10% of the world’s population is at risk of helminth infection every year (Crompton, 1999). The intestinal microbiota composition may reflect the state of the immune system and health of the host species (Round & Mazmanian, 2009). However, Lukeš, Stensvold, Jirků‐Pomajbíková, and Wegener Parfrey (2015) promoted the idea of some parasites being beneficial to the host rather than culprits of disease. For example, a mutualistic relationship exists between bullfrog tadpoles (Rana catesbeiana) and a tadpole‐specific gastrointestinal nematode (Gyrinicola batrachiensis; Pryor & Bjorndal, 2010). As yet, the complex interactions between helminths, gut microbiota, and the host have not been adequately studied in wild species (Kreisinger, Bastien, Hauffe, Marchesi, & Perkins, 2015). Therefore, in the current study, we examined the association between nematode infection and gut microbiota diversity and composition in wild concave‐eared frogs. We found that while nematode infection was not associated with changes in the overall gut microbiota diversity, there did appear to be an effect on the microbial community composition. This result is consistent with findings in wild mice, where helminth infection did not affect the diversity of the gut microbiota (Kreisinger et al., 2015). In addition, the gut microbial communities of the nematode‐infected and healthy frogs in the current study were clearly separated by ANOSIM (R = 0.5827, p = 0.002) and NMDS analyses. Interestingly, the infected frogs seemed to exhibit higher inter‐individual variation, especially in terms of community structure. These results may demonstrate that nematode infection can increase heterogeneity of microbial communities among individuals.
The relative abundance of symbionts and pathogenic microbes also reflects the health status of the host species (Sekirov, Russell, Antunes, & Finlay, 2010). We found that the relative abundance of Verrucomicrobia was markedly reduced in nematode‐infected frogs compared with healthy frogs. Additionally, the relative abundance of some bacteria, such as Actinobacteria and Bacilli, increased in the infected frogs compared with the uninfected group. Actinobacteria have been associated with disease in humans (Abusleme et al., 2013), while Bacilli are highly abundant in the guts of several animal species and may enhance digestion by complementing the digestive enzymes in the gut, thereby improving nutrition through the provision of vitamins and amino acids to the host (Gandotra, Kumar, Naga, & Bhuyan, 2018; Voirol, Frago, Kaltenpoth, Hilker, & Fatouros, 2018). Therefore, our findings indicate that nematode infection of vulnerable Chinese concave‐eared frogs has complex implications for the gut microbiota, including loss of some beneficial microbes and increases in the abundance of some disease‐associated microbial taxa. As the functions of these bacteria in the concave‐eared frog have not been adequately described owing to the relatively low number of samples in this study, more work is needed to fill the gaps in our understanding of the interaction between helminths and the gut microbiota of this vulnerable species.
CONFLICTS OF INTEREST
The authors declare that there are no competing interests.
AUTHORS CONTRIBUTION
Hailong Wu, Bangding Xiao, Yilin Shu, and Pei Hong designed experiments. Dong Tang, Yilin Shu, Hui Qing, Huan Wang, and Oscar Omondi Donde collected samples and carried out experiments. Pei Hong and Yilin Shu analyzed experimental results and wrote the manuscript.
ETHICS STATEMENT
All samples used in this study were collected with the permission of the Management Bureau of the Banqiao Provincial Natural Reserve. The animal experiments were performed under an animal ethics approval granted by Anhui Normal University.
DATA ACCESSIBILITY
The 16S rRNA gene sequences and metagenome sequences from the frog gut microbiota samples reported in this study have been submitted to the NCBI Sequence Read Archive under accession numbers SRP131550 (https://www.ncbi.nlm.nih. gov/sra/SRP131550) and SRP130863 (https://www.ncbi.nlm.nih.gov/sra/SRP130863), respectively.
Supporting information
ACKNOWLEDGMENTS
We thank staff at the Banqiao Provincial Natural Reserve for help with sampling. This research was financially supported by the National Natural Science Foundation of China (no. 31370537), and by the Doctoral Fund of the Ministry of Education of China (no. 20133424110006).
Shu Y, Hong P, Tang D, et al. Comparison of intestinal microbes in female and male Chinese concave‐eared frogs (Odorrana tormota) and effect of nematode infection on gut bacterial communities. MicrobiologyOpen. 2019;8:e749 10.1002/mbo3.749
Contributor Information
Bangding Xiao, Email: bdxiao@ihb.ac.cn.
Hailong Wu, Email: whlong@mail.ahnu.edu.cn.
REFERENCES
- Abusleme, L. , Dupuy, A. K. , Dutzan, N. , Silva, N. , Burleson, J. A. , Strausbaugh, L. D. , … Diaz, P. I. (2013). The subgingival microbiome in health and periodontitis and its relationship with community biomass and inflammation. ISME Journal, 7, 1016–1025. 10.1038/ismej.2012.174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul, S. F. , Madden, T. L. , Schäffer, A. A. , & Zhang, J. (1997). Gapped BLAST and PSI‐BLAST_ a new generation of protein database search programs. Nucleic Acids Research, 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier, S. , & Bertilsson, S. (2013). Bacterial chitin degradation‐mechanisms and ecophysiological strategies. Frontiers in Microbiology, 4, 149 10.3389/fmicb.2013.00149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolnick, D. I. , Snowberg, L. K. , Hirsch, P. E. , & Lauber, C. L. (2014). Individual diet has sex‐dependent effects on vertebrate gut microbiota. Nature Communications, 5, 4500 10.1038/ncomms5500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello, E. K. , Stagaman, K. , Dethlefsen, L. , & Bohannan, B. J. (2012). The application of ecological theory toward an understanding of the human microbiome. Science, 336, 1255–1262. 10.1126/science.1224203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton, D. (1999). How much human helminthiasis is there in the world? Journal of Parasitology, 85, 397–403. 10.2307/3285768 [DOI] [PubMed] [Google Scholar]
- David, L. A. , Maurice, C. F. , Carmody, R. N. , Gootenberg, D. B. , Button, J. E. , … Turnbaugh, P. J. (2014). Diet rapidly and reproducibly alters the human gut microbiome. Nature, 505, 559–563. 10.1038/nature12820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delsuc, F. , Metcalf, J. L. , Wegener, P. L. , Song, S. J. , González, A. , & Knight, R. (2014). Convergence of gut microbiomes in myrmecophagous mammals. Molecular Ecology, 23, 1301–1317. 10.1111/mec.12501 [DOI] [PubMed] [Google Scholar]
- Fei, L. (1999). Identification of Chinese amphibious animal (pp. 230–244). Zhengzhou: Henan Science and Technology Press. [Google Scholar]
- Feng, A. S. , Narins, P. M. , & Xu, C. H. (2002). Vocal acrobatics in a Chinese frog, Amolops tormotus . Naturwissenschaften, 89, 352–356. 10.1007/s00114-002-0335-x [DOI] [PubMed] [Google Scholar]
- Feng, A. S. , Narins, P. M. , Xu, C. H. , Lin, W. Y. , Yu, Z.‐L. , Qiu, Q. , … Shen, J.‐X. (2006). Ultrasonic communication in frogs. Nature, 440, 333–336. 10.1038/nature04416 [DOI] [PubMed] [Google Scholar]
- Feng, J. , Zhang, J. H. , Shu, Y. L. , & Yao, L. (2015). The geographic distribution pattern and habitat selection of the Chinese concave‐eared in southern Anhui Province. Acta Ecologica Sinica, 35, 5638–5647. [Google Scholar]
- Freire, A. C. , Basit, A. W. , Choudhary, R. P. , & Chee, W. (2011). Does sex matter? The influence of gender on gastrointestinal physiology and drug delivery. International Journal of Pharmaceutics, 415, 15–28. 10.1016/j.ijpharm.2011.04.069 [DOI] [PubMed] [Google Scholar]
- Fujimoto, W. , Kimura, K. , Suzuki, M. , Syuto, B. , Onuma, M. , & Iwanaga, T. (2004). Quantitative changes in serum concentration of bovine gut chitinase in Theileria infection. Journal of Veterinary Medical Science, 66, 291–294. 10.1292/jvms.66.291 [DOI] [PubMed] [Google Scholar]
- Gandotra, S. , Kumar, A. , Naga, K. , & Bhuyan, P. M. (2018). Bacterial community structure and diversity in the gut of Muga silkworm, Antheraea assamensis (Lepidoptera: Saturniidae) from India. Insect Molecular Biology, 27, 603–619. [DOI] [PubMed] [Google Scholar]
- Gaskins, H. R. , Croix, J. A. , Nakamura, N. , & Nava, G. M. (2008). Impact of the intestinal microbiota on the development of mucosal defense. Clinical Infectious Diseases, 46, S80–S86. 10.1086/523336 [DOI] [PubMed] [Google Scholar]
- Goodrich, T. D. , & Morita, R. Y. (1977). Bacterial chitinase in the stomachs of marine fishes from Yaquina Bay, Oregon, USA. Marine Biology, 41, 355–360. 10.1007/BF00389101 [DOI] [Google Scholar]
- Gutowska, M. A. , Drazen, J. C. , & Robison, B. H. (2004). Digestive chitinolytic activity in marine fishes of Monterey Bay, California. Comparative Biochemistry and Physiology Part A Molecular Integrative Physiology, 139, 351–358. 10.1016/j.cbpb.2004.09.020 [DOI] [PubMed] [Google Scholar]
- Han, L. , Guo, X. , Ravi, G. , Lai, R. , Zeng, C. , Zhan, F. , & Wang, W. (2016). The gut microbiome and degradation enzyme activity of wild freshwater fishes influenced by their trophic levels. Scientific Reports, 6, 24340 10.1038/srep24340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai, T. (2002). Ontogenetic change in the diet of the pond frog, Rana nigromaculata . Ecological Research, 17, 639–644. 10.1046/j.1440-1703.2002.00521.x [DOI] [Google Scholar]
- Houston, W. W. K. (1973). The food of the common frog, Rana temporaria, on high moorland in northern England. Journal of Zoology, 171, 153–165. 10.1111/j.1469-7998.1973.tb02212.x [DOI] [Google Scholar]
- Huang, B. H. , Chang, C. W. , Huang, C. W. , Gao, J. , & Liao, P. C. (2018). Composition and functional specialists of the gut microbiota of frogs reflect habitat differences and agricultural activity. Frontiers in Microbiology, 8, 2670 10.3389/fmicb.2017.02670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson, D. H. , Auch, A. F. , Qi, J. , & Schuster, S. C. (2007). MEGAN analysis of metagenomic data. Genome Research, 17, 377–386. 10.1101/gr.5969107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov, I. I. , & Littman, D. R. (2011). Modulation of immune homeostasis by commensal bacteria. Current Opinion in Microbiology, 14, 106–114. 10.1016/j.mib.2010.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen, A. W. , & Kersten, S. (2015). The role of the gut microbiota in metabolic health. FASEB Journal, 29, 3111–3123. 10.1096/fj.14-269514 [DOI] [PubMed] [Google Scholar]
- Jennifer, G. , Loesche, W. J. , & Nace, G. W. (1982). Large intestine bacterial flora of nonhibernating and hibernating leopard frogs (Rana pipiens). Applied and Environment Microbiology, 44, 59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen, L. J. , Julien, P. , Kuhn, M. , von Mering, C. , Muller, T. , … Bork, P. (2008). eggNOG: Automated construction and annotation of orthologous groups of genes. Nucleic Acids Research, 36, D250–D254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutie, S. A. , Shea, L. A. , Kupselaitis, M. , Wilkinson, C. L. , Kohl, K. D. , & Rohr, J. R. (2017). Early‐life diet affects host microbiota and later‐life defenses against parasites in frogs. Integrative and Comparative Biology, 57, 732–742. 10.1093/icb/icx028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl, K. D. , Cary, T. L. , Karasov, W. H. , & Dearing, M. D. (2013). Restructuring of the amphibian gut microbiota through metamorphosis. Environmental Microbiology Reports, 5, 899–903. 10.1111/1758-2229.12092 [DOI] [PubMed] [Google Scholar]
- Kohl, K. D. , Cary, T. L. , Karasov, W. H. , & Dearing, M. D. (2015). Larval exposure to polychlorinated biphenyl 126 (PCB‐126) causes persistent alteration of the amphibian gut microbiota. Environmental Toxicology and Chemistry, 34, 1113–1118. 10.1002/etc.2905 [DOI] [PubMed] [Google Scholar]
- Kohl, K. D. , & Yahn, J. (2016). Effects of environmental temperature on the gut microbial communities of tadpoles. Environmental Microbiology, 18, 1561–1565. 10.1111/1462-2920.13255 [DOI] [PubMed] [Google Scholar]
- Koren, O. , Goodrich, J. K. , Cullender, T. C. , Spor, A. , Laitinen, K. , Kling Bäckhed, H. , … Ley, R. E. (2012). Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell, 150, 470–480. 10.1016/j.cell.2012.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs, A. , Ben‐Jacob, N. , Tayem, H. Halperin, E. , Iraqi, F. A. , & Gophna, U. (2011). Genotype is a stronger determinant than sex of the mouse gut microbiota. Microbial Ecology, 61, 423–428. 10.1007/s00248-010-9787-2 [DOI] [PubMed] [Google Scholar]
- Kreisinger, J. , Bastien, G. , Hauffe, H. C. , Marchesi, J. , & Perkins, S. E. (2015). Interactions between multiple helminths and the gut microbiota in wild rodents. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences, 370(1675), 20140295 10.1098/rstb.2014.0295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley, R. E. , Hamady, M. , Lozupone, C. , Turnbaugh, P. , Ramey, R. R. , Bircher, J. S. , … Gordon, J. I. (2008). Evolution of mammals and their gut microbes. Science, 320, 1647–1651. 10.1126/science.1155725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, W. , & Godzik, A. (2006). Cd‐hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics, 22, 1658–1659. 10.1093/bioinformatics/btl158 [DOI] [PubMed] [Google Scholar]
- Lima, A. P. , & Moreira, G. (1993). Effects of prey size and foraging mode on the ontogenetic change in feeding niche of Colostethus stepheni (Anura: Dendrobatidae). Oecologia, 95, 93–102. 10.1007/BF00649512 [DOI] [PubMed] [Google Scholar]
- Lukeš, J. , Stensvold, C. R. , Jirků‐Pomajbíková, K. , & Wegener Parfrey, L. Are human intestinal eukaryotes beneficial or commensals? PLoS Path 2015;11:e1005039 10.1371/journal.ppat.1005039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markle, J. G. , Frank, D. N. , Mortin‐Toth, S. , Robertson, C. E. , Feazel, L. M. , Rolle‐Kampczyk, U. , … Danska, J. S. (2014). Sex differences in the gut microbiome drive hormone‐dependent regulation of autoimmunity. Science, 339, 1084–1088. 10.1126/science.1233521 [DOI] [PubMed] [Google Scholar]
- Mashoof, S. , Goodroe, A. , Du, C. C. , & Eubanks, J. O. (2013). Ancient T‐independence of mucosal IgX/A: Gut microbiota unaffected by larval thymectomy in Xenopus laevis . Mucosal Immunology, 6, 358–368. 10.1038/mi.2012.78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi, H. , Park, J. , & Takagi, T. (2006). MetaGene: Prokaryotic gene finding from environmental genome shotgun sequences. Nucleic Acids Research, 34, 5623–5630. 10.1093/nar/gkl723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksanen, J. , Kindt, R. , Legendre, P. , & Hara, B. O. (2007). The VEGAN package: Community ecology package.
- Peng, Y. , Leung, H. C. , Yiu, S. M. , & Chin, F. Y. (2012). IDBA‐UD: A de novo assembler for single‐cell and metagenomic sequencing data with highly uneven depth. Bioinformatics, 28, 1420–1428. 10.1093/bioinformatics/bts174 [DOI] [PubMed] [Google Scholar]
- Pryor, G. S. , & Bjorndal, K. A. (2010). Effects of the nematode Gyrinicola batrachiensis on development, gut morphology, and fermentation in bullfrog tadpoles (Rana catesbeiana): A novel mutualism. Journal of Experimental Zoology Part A: Comparative Experimental Biology, 303, 704–712. [DOI] [PubMed] [Google Scholar]
- Rice, P. , Longden, I. , & Bleasby, A. (2000). EMBOSS: The European molecular miology open software suite. Trends in Genetics, 16, 276–277. [DOI] [PubMed] [Google Scholar]
- Round, J. L. , & Mazmanian, S. K. (2009). The gut microbiota shapes intestinal immune responses during health and disease. Nature Reviews Immunology, 9, 313–323. 10.1038/nri2515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata, N. , Lzard, J. , Waldron, L. , & Gevers, D. , Miropolsky, L. , Garrett, W. S. , & Huttenhower, C. (2011). Metagenomic biomarker discovery and explanation. Genome Biology, 12, R60 10.1186/gb-2011-12-6-r60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekirov, I. , Russell, S. , Antunes, L. C. , & Finlay, B. B. (2010). Gut microbiota in health and disease. Physiological Reviews, 90, 859–904. 10.1152/physrev.00045.2009 [DOI] [PubMed] [Google Scholar]
- Shine R. (1979). Sexual Selection and Sexual Dimorphism in the Amphibia. Copeia, 1979, 297‐306. [Google Scholar]
- Suganthi, M. , Senthilkumar, P. , Arvinth, S. , & Chandrashekara, K. N. (2017). Chitinase from Pseudomonas fluorescens and its insecticidal activity against Helopeltis theivora . Journal of General and Applied Microbiology, 63, 222–227. [DOI] [PubMed] [Google Scholar]
- Sullam, K. E. , Essinger, S. D. , Lozupone, C. A. , O'Connor, M. P. , Rosen, G. l. , Knight, R. , … Russell, J. A. (2012). Environmental and ecological factors that shape the gut bacterial communities of fish: A meta‐analysis. Molecular Ecology, 21, 3363–3378. 10.1111/j.1365-294X.2012.05552.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov, R. L. , Fedorova, N. D. , Jackson, J. D. , & Jacobs, A. R. (2003). The COG database: An updated version includes eukaryotes. BMC Bioinformatics, 4, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toft, C. A. (1980). Feeding ecology of thirteen syntopic species of anurans in a seasonal tropical environment. Oecologia, 45, 131–141. 10.1007/BF00346717 [DOI] [PubMed] [Google Scholar]
- Tsiora, A. , & Kyriakopoulou‐Sklavounou, P. (2002). A skeletochronological study of age and growth in relation to adult size in the water frog Rana epeirotica . Zoology, 10, 55 10.1078/0944-2006-00049 [DOI] [PubMed] [Google Scholar]
- Van der, W. D. , Cohen, B. J. , & Nace, G. W. (1974). Colonization patterns of aerobic gram‐negative bacteria in the cloaca of Rana pipiens . Laboratory Animal Science, 24, 307–317. [PubMed] [Google Scholar]
- Vences, M. , Lyra, M. L. , Kueneman, J. G. , Bletz, M. C. , Archer, H. M. , Canitz, J. , … Glos, J. (2016). Gut bacterial communities across tadpole ecomorphs in two diverse tropical anuran faunas. Naturwissenschaften, 103, 1–14. 10.1007/s00114-016-1348-1 [DOI] [PubMed] [Google Scholar]
- Voirol, L. R. P. , Frago, E. , Kaltenpoth, M. , Hilker, M. , & Fatouros, N. E. (2018). Bacterial symbionts in Lepidoptera: Their diversity, transmission and impact on the host. Frontiers in Microbiology, 9, 556 10.3389/fmicb.2018.00556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiebler, J. M. , Kohl, K. D. , Lee, R. J. , & Costanzo, J. P. (2018). Urea hydrolysis by gut bacteria in a hibernating frog: Evidence for urea‐nitrogen recycling in Amphibia. Proceedings Biological Sciences, 16, 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, Y. H. , Xiong, R. C. , Lei, F. Z. , & Jiang, J. P. (2012). Dissection and histological observation of digestive system of Odorrana tormota . Sichuan Journal of Zoology, 31, 583–588. [Google Scholar]
- Xu, J. (2003). A Genomic view of the human‐bacteroides thetaiotaomicron symbiosis. Science, 299, 2074–2076. 10.1126/science.1080029 [DOI] [PubMed] [Google Scholar]
- Xu, N. T. , Wang, G. C. , Gai, H. Y. , & Xia, P. (2016). Effect of biochar additions to soil on nitrogen leaching, microbial biomass and bacterial community structure. European Journal of Soil Biology, 74, 1–8. 10.1016/j.ejsobi.2016.02.004 [DOI] [Google Scholar]
- Zhu, W. , Lomsadze, A. , & Borodovsky, M. (2010). Ab initio gene identification in metagenomic sequences. Nucleic Acids Research, 38, e132 10.1093/nar/gkq275 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The 16S rRNA gene sequences and metagenome sequences from the frog gut microbiota samples reported in this study have been submitted to the NCBI Sequence Read Archive under accession numbers SRP131550 (https://www.ncbi.nlm.nih. gov/sra/SRP131550) and SRP130863 (https://www.ncbi.nlm.nih.gov/sra/SRP130863), respectively.