

Abstract
Obstruction of a coronary artery causes ischemia of heart tissue leading to myocardial infarction. Prolonged oxygen deficiency provokes tissue necrosis, which can result in heart failure and death of the patient. Therefore, restoration of coronary blood flow (reperfusion of the ischemic area) by re-canalizing the affected vessel is essential for a better patient outcome. Paradoxically, sudden reperfusion also causes tissue injury, thereby increasing the initial ischemic damage despite restoration of blood flow (=ischemia/reperfusion injury, IRI). Myocardial IRI is a complex event that involves various harmful mechanisms (e.g., production of reactive oxygen species and local increase in calcium ions) as well as inflammatory cells and signals like chemokines and cytokines. An involvement of platelets in the inflammatory reaction associated with IRI was discovered several years ago, but the underlying mechanisms are not yet fully understood. This mini review focusses on platelet contributions to the intricate picture of myocardial IRI. We summarize how upregulation of platelet surface receptors and release of immunomodulatory mediators lead to aggravation of myocardial IRI and subsequent cardiac damage by different mechanisms such as recruitment and activation of immune cells or modification of the cardiac vascular endothelium. In addition, evidence for cardioprotective roles of distinct platelet factors during IRI will be discussed.
Keywords: myocardial infarction, ischemia reperfusion injury, platelets, reperfusion, ischemia
Introduction
Myocardial infarction (MI) is the leading cause of death in the western world (1). The role of platelets in the progression of coronary plaques and the thrombotic occlusion of coronary vessels leading to ischemia and MI is well understood (2). Besides this, microembolization, and platelet accumulation within the affected microcirculation of the myocardium during late ischemia and reperfusion (IR) lead to a secondary tissue damage (2). Rapid restoration of blood flow after MI is the primary goal of state-of-the-art therapy in order to limit the damage of cardiac tissue caused by the lack of oxygen supply. Paradoxically, reperfusion itself also causes adverse effects and increases infarct size (1, 3). This phenomenon is called ischemia/reperfusion injury (IRI) and might account for nearly 50% of the final myocardial damage in acute MI (1). IRI can cause myocardial stunning, the no-reflow phenomenon, reperfusion arrhythmia and also lethal reperfusion injury (reviewed in (4). As underlying mechanisms oxidative stress, calcium overload and pH shifts have been described (1). In this context, mitochondrial damage including disruption of ATP production and opening of the mitochondrial permeability transition pore with subsequent necrotic and apoptotic cell death play an important role (5). Additionally, ischemia–reperfusion induced alterations in the regulation of cytosolic osmolality and cell volume cause cellular and interstitial edema that are associated to microvascular obstruction, cell dysfunction and death (6, 7). Reperfusion has repeatedly been shown to be an inflammatory state that is accompanied by leukocyte infiltration (8, 9). While a certain degree of inflammation is necessary for cardiac repair after IRI it may also develop in an unwanted direction and extend injury (9, 10). Already since the 1980s an involvement of platelets in the inflammatory reaction associated to IRI has been recognized. The damage caused by IR in the vascular endothelium causes activation of circulating platelets, resulting in the release of their immunomodulatory contents (11). The degree of platelet activation is related to the duration of the preceding ischemia and the extent of reperfusion injury (5). This mini review aims to summarize mechanisms of platelet contributions to myocardial IRI.
The Immunomodulatory Equipment of Platelets
Platelets are small anucleate cells that circulate through the bloodstream in large numbers. Beyond their primary role in hemostasis platelets have been recognized as immunomodulatory cells that fulfill their tasks through a variety of mechanisms ranging from secretable factors to stably or variably expressed surface receptors (12). Specifically, platelets secrete or expose adhesion proteins like fibrinogen, fibronectin, von Willebrand factor, thrombospondin or P-selectin that are involved in cell-cell interactions (e.g., with leukocytes or endothelial cells) during inflammation (13–20). Mitogenic factors released by platelets such as platelet-derived growth factor (PDGF) and transforming growth factor beta (TGFβ) target monocytes, macrophages as well as T-cells, for example to achieve wound healing or immunosuppression (21–24). And chemokines like RANTES/C-C motif ligand (CCL)5, platelet factor 4 (PF4)/ C-X-C motif ligand (CXL)4 or serotonin (5-hydroxytryptamine) are involved in leukocyte recruitment to sites of inflammation (25–29). Moreover, platelets can trigger the complement activation and play a role in localizing inflammatory areas (30). Already with these few examples of immunomodulatory platelet functions in mind, it is obvious to implicate these cells as key contributors to the myocardial IR associated inflammatory response.
Experimental Models of Ischemia/Reperfusion Injury
In the past years a number of experimental models of IRI were applied in various species by different research groups. The following table briefly summarizes the models mentioned in this review with their respective modes of platelet activation (Table 1).
Table 1.
Experimental models of ischemia/reperfusion injury.
| Model | Species | Characteristics | Modes of platelet activation | References |
|---|---|---|---|---|
| LAD ligation | Mouse, rat, rabbit, minipig, and sheep | Intact heart with physiological blood flow Localized ischemia induction via ligation of LAD No occlusive thrombus formation Reperfusion with whole blood via reopening of LAD ligature | Accumulation of procoagulants due to stasis Activated endothelium due to stasis, hypoxia and subsequent reperfusion Cytokines and chemokines released in the ischemic myocardium To some extent by the invasive surgical procedure | (31–44) |
| Isolated working heart preparations | Guinea pig and rat | Perfusion with physiological cell-free buffer Induction of global ischemia and reperfusion with cell-free buffer Optional perfusion with washed platelets or supernatants of aggregated platelets | Activated endothelium Cytokines, chemokines released in ischemic myocardium Optional pre-activation by chemical agents Optional pre-activation by IRI in AMI patients | (45–48) |
| Isolated working heart preparations, low flow ischemia | Guinea pig | Perfusion with physiological cell-free buffer Induction of global low flow ischemia (1 ml/min) and subsequent reperfusion Optional perfusion with platelets | Exogenous thrombin activation during low flow ischemia and beginning of reperfusion | (49) |
| Coronary thrombosis induction | Dog | Coronary thrombus induction by electrolytic injury Localized ischemia Reperfusion by thrombolysis with tissue plasminogen activator | Injured endothelium (e.g., GPVI interaction with exposed subendothelial collagen, PSGL-1/P-selectin interaction etc.) Shear stress Activated endothelium due to ischemia and reperfusion Cytokines, chemokines released in ischemic myocardium | (50) |
Platelet Membrane Proteins in IRI
Collagen Receptor
The combination of glycoprotein (GP) VI with Fc receptor (FcR)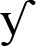 forms the platelet collagen receptor. Takaya et al. demonstrated in 2005 that collagen-dependent activation of platelets can drive the extension of myocardial IRI. In their study, knock-out (k.o.) of FcR improved outcome of mice after myocardial IR with the infarct size being significantly smaller than in wildtype (WT) mice. Mechanistically the FcR−/− mice displayed less platelet aggregation and occlusive microthrombi, less platelet spleen tyrosine kinase (Syk) activation as well as reduced myeloperoxidase (Mpo) activity in the injured area (51). A more recent study confirmed a role of the collagen receptor in IRI by targeting GPVI with a monoclonal antibody derivative in a murine in vivo model of left anterior descending artery (LAD) ligation followed by reperfusion. Anti-GPVI treatment significantly reduced infarct size vs. control, which was again primarily based on an improved microperfusion (52).
forms the platelet collagen receptor. Takaya et al. demonstrated in 2005 that collagen-dependent activation of platelets can drive the extension of myocardial IRI. In their study, knock-out (k.o.) of FcR improved outcome of mice after myocardial IR with the infarct size being significantly smaller than in wildtype (WT) mice. Mechanistically the FcR−/− mice displayed less platelet aggregation and occlusive microthrombi, less platelet spleen tyrosine kinase (Syk) activation as well as reduced myeloperoxidase (Mpo) activity in the injured area (51). A more recent study confirmed a role of the collagen receptor in IRI by targeting GPVI with a monoclonal antibody derivative in a murine in vivo model of left anterior descending artery (LAD) ligation followed by reperfusion. Anti-GPVI treatment significantly reduced infarct size vs. control, which was again primarily based on an improved microperfusion (52).
Adenosine Diphosphate (ADP) Receptor
The platelet P2Y12 receptor is responsible for mediating the sustained ADP-dependent platelet aggregation response. In a dog coronary thrombosis model, ticagrelor, a reversibly binding P2Y12 receptor antagonist, was shown to significantly decrease infarct size and rapidly restore myocardial tissue perfusion by inhibition of ADP-mediated platelet aggregation and recruitment (50). Supporting results were demonstrated by infusing platelets from acute MI (AMI) patients (untreated or treated with 10 μM of the P2Y12 inhibitor cangrelor) into isolated rat hearts that were subjected to 40 min of ischemia with subsequent 60 min of reperfusion. Platelets from AMI patients significantly augmented myocardial injury while P2Y12 blockage with cangrelor reduced infarct size and attenuated the adverse effects of platelet infusion on cardiac function (48). Besides direct inhibition of aggregation, protective effects of cangrelor during IRI could in part be mediated by inhibition of platelet P-selectin expression and platelet-leukocyte interactions (53). Furthermore, in a rabbit model of myocardial IR, coapplication of individual inhibitors revealed a role for adenosine A2B receptors, ERK, Akt, redox signaling, and mitochondrial KATP channels in mediating the protective cangrelor effect independent of its antiaggregatory properties (37).
Glycoprotein-IIb/IIIa-Receptor
Given its important role in platelet activation GPIIb/IIIa is predestined as a contributor to IRI. Indeed, inhibition of GPIIb/IIIa reduces platelet induced aggravation of IRI in isolated rat hearts. Infusion of platelets from AMI patients worsened myocardial injury in hearts subjected to IR, as measured by left ventricular (LV) developed pressure, higher maximal LV end-diastolic pressure and coronary resistance as well as increased lactate dehydrogenase (LDH) release and infarct size. Pretreatment of platelets with the GPIIb/IIIa inhibitor abciximab greatly attenuated these effects (48). Mechanistically, in addition to interrupting platelet adherence to the reperfused endothelium through GPIIb/IIIa-fibrinogen binding, abciximab also interferes with other mechanisms of platelet adhesion including the vitronectin receptor or leukocyte Macrophage-1 antigen (Mac-1) (48, 54, 55). In contrast, a publication from 2016 did not confirm a significant effect of GPIIb/IIIa antagonization on infarct size using a specific monoclonal antibody derivative in a murine in vivo model of left coronary artery ligation (52). Yet, another study confirmed the adverse effect of GPIIb/IIIa dependent intracoronary platelet retention during low flow ischemia on cardiac function using the GPIIb/IIIa inhibitor tirofiban. This was partially attributed to tirofiban-induced blockage of platelet adherence to the vessel wall mediated by an interaction of the GPIIb/IIIa-receptor and von Willebrand-factor (56). However, in this study no GPIIb/IIIa effect on cardiac function was observed during the reperfusion phase (56), suggesting that conflicting results between studies might at least partially be based on different time points of inhibitor application during the course of IRI, especially since platelet adhesion to reperfused endothelium and platelet-mediated myocardial damage have been described to occur very early after reperfusion (48). Additionally, the use of different experimental models which induce different modes of platelet activation are a decisive factor for experimental outcome (see Table 1).
P-Selectin
P-selectin is an adhesion molecule expressed on the surface of activated platelets and mediates cell-cell interactions involving platelets, e.g., platelet-neutrophil-complexes which have been associated with many inflammatory conditions (14, 57). Platelet P-selectin expression is increased after IR in several animal models as well as in humans (48, 58). Infusion of platelets activated in this manner into isolated rat hearts subjected to ischemia and reperfusion strongly increases myocardial LDH release representing cardiomyocyte damage (58). In loss of function approaches it was demonstrated that the chemical or small molecule dependent blockade of platelet P-selectin has beneficial effects on platelet mediated reperfusion injury after myocardial IR in pigs and rats, respectively (38, 43). Additionally, a study using genetically modified mice confirmed these results as significantly smaller infarct sizes after myocardial IR were observed in P-selectin k.o. mice or mice transfused with P-selectin k.o. platelets as compared to wild-type (32). Based on these studies the role of activated platelets in the process of myocardial IRI seems to depend at least in part on their activation status as represented by platelet-derived P-selectin. Mechanistically, enhanced P-selectin expression on platelets increases adherence to the reperfused endothelium as well as postischemic leukocyte adhesion, thereby aggravating the inflammatory reaction associated to IRI. However, in terms of leukocyte recruitment it needs to be taken into account that endothelial P-selectin expression can contribute to the observed effects of P-selectin antagonization in some of the reported experimental models, too (59).
G-Proteins
G proteins are involved in transmitting signals from a variety of stimuli outside of a platelet to its interior. Gαq k.o. in mice eliminates platelet function in terms of aggregation and secretion of cytokines. In these mice infarct size to area at risk ratio was significantly smaller as compared to WT after 30 min of regional myocardial ischemia (LAD ligation) followed by 24 h of reperfusion. Additionally, Gαq k.o. improved fractional shortening in this model. The beneficial effects were resembled by transplantation of Gαq k.o. bone marrow into WT mice (34). The effect of Gαq k.o. outperformed the protection of sole inhibition of platelet aggregation and was accompanied by reduced expression of the fibrinogen receptor CD41 and P-selectin as well as secretion of platelet-derived growth factor after platelet activation (34). Likewise, the platelet Gi protein Gαi2 is an essential mediator of thrombo-inflammatory organ damage in mice. This was shown in mice lacking Gαi2 in megakaryocytes and platelets (Gnai2fl/fl/PF4-Cre) that developed a dramatically reduced reperfusion injury that correlated with diminished formation of ADP-dependent platelet neutrophil complexes after myocardial IR (35).
Na+/H+ Exchanger Isoform 1 (NHE1)
NHE1 is an integral membrane protein that removes one intracellular H+ for one extracellular Na+ protecting cells from intracellular acidification. NHE1 activation in cardiomyocytes is known to contribute to injury and arrhythmias during IR by promoting calcium overload via the sodium-calcium exchanger (60–62). But not only cardiomyocyte NHE1 activation is a driver of IRI. NHE1 is also expressed on platelets and is involved in the regulation of the platelet's intracellular pH, platelet volume as well as cell signaling and platelet activation (63, 64). In a rat model of myocardial IR the NHE1 blocker KR-32568 dose-dependently inhibited NHE-1-mediated rabbit platelet swelling and dose dependently reduced infarct size when applied 10 min before ischemia (39).
Mediators Released by Platelets
As part of their pleiotropic actions platelets can rapidly secrete a wide array of inflammatory mediators, either from their granules or in a granule independent manner (12) which are reasonable candidates for mediating inflammation during IRI.
Reactive Oxygen Species (ROS)
ROS occur as byproducts of certain enzymatic reactions (e.g., catalyzed by xanthine oxidases, cytochrome P450 or NADPH oxidase). ROS play important roles in cell signaling and homeostasis but are also involved in the pathogenesis of several diseases including cardiovascular disease (65, 66). In guinea pig hearts exposed to low-flow ischemia with following reperfusion activated human platelets administered in the beginning of reperfusion significantly reduced the recovery of external heart work (REHW). Coapplication of the radical scavenger enzyme superoxide dismutase improved REHW during reperfusion indicating a role of ROS in the provoked IRI. Interestingly, by coapplication of the GPIIb/IIIa-blocker tirofiban the authors could show that the platelet-induced ROS-dependent myocardial dysfunction in their experimental model was independent of intracoronary platelet adhesion (49, 56, 66). In a follow up study, by applying a platelet pretreatment with diphenyliodonium chloride Seligmann et al. elegantly proved that the shown cardiodepressive effects were mediated by ROS released from platelets and not the heart itself (49). ROS-induced effects on reperfused myocardium are based on several mechanisms including calcium overload by interference with myocardial calcium transport, damage to membranes and proteins, as well as opening of the mitochondrial permeability transition pore and subsequent apoptosis (4, 49).
Serotonin
Serotonin (5-hydroxytryptamine) is a biogenic amine present in circulation and non-neuronal cells as peripheral hormone and in the central nervous system as neurotransmitter. In the periphery it is stored in high concentrations in dense granules of platelets. Myocardial IRI is accompanied by elevated serotonin plasma levels in mice (33). In 1994 Hohlfeld et al. demonstrated that nexopamil, a combined Ca2+ and serotonin antagonist, reduced infarct size and improved functional cardiac parameters in minipigs subjected to 1 h of LAD occlusion with a subsequent 3 h reperfusion. Besides calcium channel blocking activity, inhibition of ischemia-induced neutrophil activation and enhanced endogenous PGI2 formation were claimed to be factors contributing to the beneficial effects of nexopamil (42). Later, serotonin's harmful mode of action in IRI was partially attributed to oxidative stress caused by mitochondrial MAO-A activity. MAO-A is responsible for serotonin degradation with H2O2 production. Evidence was presented that the oxidative stress induced by this enzymatic reaction was responsible for receptor-independent apoptotic effects of serotonin in cardiomyocytes and postischemic myocardial injury (40). We found recently that absence of platelet serotonin improves outcome of mice after myocardial ischemia and reperfusion, i.e., a 30% smaller infarct size and less compromised LV function and ejection fraction, which were accompanied by reduced neutrophil infiltration within the infarcted tissue. Mechanistically, platelet-derived serotonin induced neutrophil degranulation with release of myeloperoxidase and H2O2 as well as increased surface expression of the adhesion molecule CD11b, leading to enhanced inflammation in the infarct area and reduced myocardial salvage (33).
Platelet Activating Factor (PAF)
PAF is a phosphoglyceride produced by platelets, leukocytes and endothelial cells which acts as an autocrine and paracrine mediator on different cell types, e.g., cardiomyocytes, endothelial cells and platelets (67). During IRI high quantities of PAF (1–10 nmol/L) are released and can exert negative effects on coronary and cardiac functions, including arrhythmogenic effects (68–70). Negative effects of PAF can be mediated either by the generation of secondary mediators, or through the activation of inflammatory cells like platelets and neutrophils (67). For example, it was demonstrated that administration of a specific PAF receptor antagonist immediately before reflow in an intact sheep model reduces myocardial reperfusion injury—an impact which was partially attributed to its anti-platelet effect (44). Furthermore, PAF has been shown to stimulate NHE1 in neutrophils and platelets (63, 71). Negative consequences of NHE1 activation in the context of IRI involve platelet swelling and calcium overload in cardiomyocytes (see above). However, PAF seems to play a dual role in IR depending on its local concentration. Potential cardioprotective effects of PAF are described later in this review.
Leukocyte-Platelet-Interactions in IRI
It has long been known that the interaction between platelets and neutrophils is associated to MI (57). Already in 1997 Neumann et al. showed that in peripheral venous blood samples of patients with AMI leukocyte platelet adhesion was increased and claimed that this was part of the regulation of the inflammatory response in acute MI (72). A causative connection between platelet neutrophil interactions and IRI was supported by several studies showing worsened cardiac functions in isolated heart models of global ischemia and reperfusion after simultaneous perfusion with both neutrophils and platelets compared to perfusion with either platelets or neutrophils alone (45, 73) as well as attenuation of these adverse effects by inhibition of platelet neutrophil complex formation (74). These relationships seem logical, especially as neutrophils interact strongly with platelets to regulate the performance of their immune cell functions (75). However, there are opposing studies as well. Seligmann et al. did not observe an additional effect of simultaneous applications of platelets and neutrophils over sole application of either of both in a study using isolated guinea pig hearts (76). In contrast, indications for a role of platelet neutrophil interactions in IRI were also found by different inhibition approaches of the P-selectin-P-selectin glycoprotein ligand 1(PSGL1) axis that led to alleviation of myocardial IRI. Several animal studies in different species demonstrated beneficial effects of P-Selectin neutralization, e.g., via small molecule inhibition (38), chemical blockade (43), or antibody blocking (77) on IRI which were all associated with an antiplatelet effect accompanied by less neutrophil infiltration or platelet-neutrophil adhesion in the infarcted region. In addition to P-selectin, also platelet GPIIb/IIIa is claimed to contribute to postischemic leukocyte adhesion (36) and also the disintegrin-dependent attenuation of platelet induced myocardial IRI was shown to be accompanied by reduced neutrophil infiltration (41).
Platelet-Endothelial-Interactions in IRI
IR induces cellular responses on microvascular endothelial cells and circulating platelets become activated (5). At the same time the adhesion and activation of platelets goes along with the release of various proinflammatory and promitogenic substances which change chemotactic, adhesive and proteolytic properties of the endothelial cells in the local surrounding (2). The inhibition of GPVI-mediated platelet-endothelial interaction via recombinant soluble GPVI-Fc was shown to reduce platelet degranulation and the release of proinflammatory cytokines. This lead to decreased infarct size and improved cardiac function due to a reduced inflammatory response of the infarcted myocardium in a mouse model of IRI (31). Furthermore, CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells (78) and dual neutrophil and platelet infiltration leads to enhanced P-selectin expression on the coronary microvascular endothelium in rat hearts subjected to IR (73).
Cardioprotective Effects of Platelets in IRI
Besides the negative effects induced by platelet adhesion and aggregation as well as the platelet dependent leukocyte infiltration into the infarcted myocardium, constituents released by platelets may have beneficial effects on the integrity of the coronary endothelium and on cardiac function after IRI (79, 80). In isolated rat hearts perfusion with platelets or supernatant of aggregated platelets was shown to exhibit cardioprotective effects (46) that were partially attributed to serotonin, thromboxane A2 or adenin nucleotides and their ability to induce the release of cardiac microvascular endothelial NO and its associated tissue protecting effects (81, 82). The platelet α granule contents transforming growth factor-beta 1 (TGF-β 1) and stromal cell-derived factor (SDF)1-α were also shown to be cardioprotective (83–85)—an effect most probably mediated by enhanced cardiomyocyte proteinkinase C (PKC) activity as a prosurvival signaling mechanism (85). Also platelet-derived sphingosine-1-phosphate (S1P) seems to facilitate protection from IRI. Although platelet derived S1P can have both pro- and anti-aggregatory effects via G-protein coupled receptors on platelets it was shown to directly induce myocardial protection. S1P signals through S1P receptors of cardiomyocytes with concomitant activation of pro-survival signaling, namely the reperfusion injury salvage kinase (RISK) and the survivor activating factor enhancement (SAFE) pathway (68). SAFE and RISK were both shown to be protective on cardiomyocytes when acutely activated at the time of reperfusion, most probably through inhibition of the opening of the mitochondrial permeability transition pore (86, 87). PAF which was already mentioned as an IRI causing factor earlier in this review is cardioprotective in very low concentrations. This effect involves activation of the RISK pathway, including protein kinase C, AKT, and nitric oxide synthase (47, 67). Another described protective mechanism of platelets during IRI is the process of mitophagy which removes damaged mitochondria. Hypoxic mitophagy in platelets leads to extensive degradation of mitochondria and reduces IRI by diminishing platelet activity (5).
Conclusions
The pathophysiology of IRI and the contribution of the involved immune cells to its progression is complex and only partially understood. However, during the past years critical roles of platelets in the origin and course of IRI as well as several underlying molecular mechanisms have been unraveled and Figure 1 summarizes most of them. Although single-agent approaches targeting these mechanisms have not yet entered clinical practice, a better understanding of platelet mechanisms in IRI could provide the basis for new and effective treatment strategies aimed at further improving protection of the myocardium during reperfusion in the future.
Figure 1.

Overview of relevant platelet derived mediators and their influence on myocardial ischemia/reperfusion injury (IRI). Endothelial damage caused by IRI leads to activation of platelets. This is accompanied by upregulation of surface proteins and the release of immunomodulatory contents that influence the progression of IRI via different mechanisms. Platelet receptors that are involved in IRI aggravation are glycoprotein (GP) IIb/IIIa, GPVI and P2Y12. Additionally, platelet membrane proteins, such as sodium-hyodrogen-exchanger 1 (NHE1), Gαq, Gαi2, and P-selectin, or secretable factors, e.g., reactive oxygen species (ROS) and serotonin, worsen the cardiac outcome after myocardial infarction. Cardioprotective effects are for example exerted by platelet-derived sphingosine-1-phosphate (S1P), low concentrations of platelet activating factor (PAF) and transforming growth factor beta 1(TGFβ1).
Author Contributions
NS did literature research, designed figures, and wrote the first draft of the manuscript. DD contributed the idea of the manuscript, wrote sections of the manuscript, and provided critical feedback. CB supported writing and provided critical feedback. All authors contributed to manuscript revision, read, and approved the submitted version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- 1.Hausenloy DJ, Yellon DM. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest. (2013) 123:92–100. 10.1172/JCI62874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gawaz M. Role of platelets in coronary thrombosis and reperfusion of ischemic myocardium. Cardiovasc Res. (2004) 61:498–511. 10.1016/j.cardiores.2003.11.036 [DOI] [PubMed] [Google Scholar]
- 3.Prasad A, Stone GW, Holmes DR, Gersh B. Reperfusion injury, microvascular dysfunction, and cardioprotection: the “dark side” of reperfusion. Circulation. (2009) 120:2105–12. 10.1161/CIRCULATIONAHA.108.814640 [DOI] [PubMed] [Google Scholar]
- 4.Yang CF. Clinical manifestations and basic mechanisms of myocardial ischemia/reperfusion injury. Ci Ji Yi Xue Za Zhi. (2018) 30:209–15. 10.4103/tcmj.tcmj_33_18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang W, Chen C, Wang J, Liu L, He Y, Chen Q. Mitophagy in cardiomyocytes and in platelets: a major mechanism of cardioprotection against ischemia/reperfusion injury. Physiology (Bethesda). (2018) 33:86–98. 10.1152/physiol.00030.2017 [DOI] [PubMed] [Google Scholar]
- 6.Garcia-Dorado D, Andres-Villarreal M, Ruiz-Meana M, Inserte J, Barba I. Myocardial edema: a translational view. J Mol Cell Cardiol. (2012) 52:931–9. 10.1016/j.yjmcc.2012.01.010 [DOI] [PubMed] [Google Scholar]
- 7.Hausenloy DJ, Chilian W, Crea F, Davidson SM, Ferdinandy P, Garcia-Dorado D, et al. The coronary circulation in acute myocardial ischaemia/reperfusion injury–a target for cardioprotection. Cardiovasc Res. (2019) 115:1143–55. 10.1093/cvr/cvy286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Menger MD, Vollmar B. Pathomechanisms of ischemia-reperfusion injury as the basis for novel preventive strategies: is it time for the introduction of pleiotropic compounds? Transplant Proc. (2007) 39:485–8. 10.1016/j.transproceed.2007.01.022 [DOI] [PubMed] [Google Scholar]
- 9.Bonaventura A, Montecucco F, Dallegri F. Cellular recruitment in myocardial ischaemia/reperfusion injury. Eur J Clin Invest. (2016) 46:590–601. 10.1111/eci.12633 [DOI] [PubMed] [Google Scholar]
- 10.Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodeling. Nat Rev Cardiol. (2014) 11:255–65. 10.1038/nrcardio.2014.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Golebiewska EM, Poole AW. Platelet secretion: from haemostasis to wound healing and beyond. Blood Rev. (2015) 29:153–62. 10.1016/j.blre.2014.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duerschmied D, Bode C, Ahrens I. Immune functions of platelets. Thromb Haemost. (2014) 112:678–91. 10.1160/TH14-02-0146 [DOI] [PubMed] [Google Scholar]
- 13.Wagner DD, Frenette PS. The vessel wall and its interactions. Blood. (2008) 111:5271–81. 10.1182/blood-2008-01-078204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zarbock A, Polanowska-Grabowska RK, Ley K. Platelet-neutrophil-interactions: linking hemostasis and inflammation. Blood Rev. (2007) 21:99–111. 10.1016/j.blre.2006.06.001 [DOI] [PubMed] [Google Scholar]
- 15.Moore KL, Stults NL, Diaz S, Smith DF, Cummings RD, Varki A, et al. Identification of a specific glycoprotein ligand for P-selectin (CD62) on myeloid cells. J Cell Biol. (1992) 118:445–56. 10.1083/jcb.118.2.445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ruggeri ZM, Mendolicchio GL. Adhesion mechanisms in platelet function. Circ Res. (2007) 100:1673–85. 10.1161/01.RES.0000267878.97021.ab [DOI] [PubMed] [Google Scholar]
- 17.Coller BS, Peerschke EI, Scudder LE, Sullivan CA. A murine monoclonal antibody that completely blocks the binding of fibrinogen to platelets produces a thrombasthenic-like state in normal platelets and binds to glycoproteins IIb and/or IIIa. J Clin Invest. (1983) 72:325–38. 10.1172/JCI110973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Savage B, Almus-Jacobs F, Ruggeri ZM. Specific synergy of multiple substrate-receptor interactions in platelet thrombus formation under flow. Cell. (1998) 94:657–66. 10.1016/S0092-8674(00)81607-4 [DOI] [PubMed] [Google Scholar]
- 19.Ginsberg MH, Forsyth J, Lightsey A, Chediak J, Plow EF. Reduced surface expression and binding of fibronectin by thrombin-stimulated thrombasthenic platelets. J Clin Invest. (1983) 71:619–24. 10.1172/JCI110808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wencel-Drake JD, Painter RG, Zimmerman TS, Ginsberg MH. Ultrastructural localization of human platelet thrombospondin, fibrinogen, fibronectin, and von Willebrand factor in frozen thin section. Blood. (1985) 65:929–38. [PubMed] [Google Scholar]
- 21.Barrientos S, Stojadinovic O, Golinko MS, Brem H, Tomic-Canic M. Growth factors and cytokines in wound healing. Wound Repair Regen. (2008) 16:585–601. 10.1111/j.1524-475X.2008.00410.x [DOI] [PubMed] [Google Scholar]
- 22.Linder BL, Chernoff A, Kaplan KL, Goodman DS. Release of platelet-derived growth factor from human platelets by arachidonic acid. Proc Natl Acad Sci USA. (1979) 76:4107–11. 10.1073/pnas.76.8.4107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lo RES, Lecocq M, Uwambayinema F, Yakoub Y, Delos M, Demoulin JB, et al. Platelet-derived growth factor-producing CD4+ Foxp3+ regulatory T lymphocytes promote lung fibrosis. Am J Respir Crit Care Med. (2011) 184:1270–81. 10.1164/rccm.201103-0516OC [DOI] [PubMed] [Google Scholar]
- 24.Fava RA, Casey TT, Wilcox J, Pelton RW, Moses HL, Nanney LB. Synthesis of transforming growth factor-beta 1 by megakaryocytes and its localization to megakaryocyte and platelet alpha-granules. Blood. (1990) 76:1946–55. [PubMed] [Google Scholar]
- 25.Antczak AJ, Singh N, Gay SR, Worth RG. IgG-complex stimulated platelets: a source of sCD40L and RANTES in initiation of inflammatory cascade. Cell Immunol. (2010) 263:129–33. 10.1016/j.cellimm.2010.03.009 [DOI] [PubMed] [Google Scholar]
- 26.Von Hundelshausen P, Koenen RR, Sack M, Mause SF, Adriaens W, Proudfoot AE, et al. Heterophilic interactions of platelet factor 4 and RANTES promote monocyte arrest on endothelium. Blood. (2005) 105:924–30. 10.1182/blood-2004-06-2475 [DOI] [PubMed] [Google Scholar]
- 27.Von Hundelshausen P, Weber KS, Huo Y, Proudfoot AE, Nelson PJ, Ley K, et al. RANTES deposition by platelets triggers monocyte arrest on inflamed and atherosclerotic endothelium. Circulation. (2001) 103:1772–7. 10.1161/01.CIR.103.13.1772 [DOI] [PubMed] [Google Scholar]
- 28.Scheuerer B, Ernst M, Durrbaum-Landmann I, Fleischer J, Grage-Griebenow E, Brandt E, et al. The CXC-chemokine platelet factor 4 promotes monocyte survival and induces monocyte differentiation into macrophages. Blood. (2000) 95:1158–66. [PubMed] [Google Scholar]
- 29.Duerschmied D, Suidan GL, Demers M, Herr N, Carbo C, Brill A, et al. Platelet serotonin promotes the recruitment of neutrophils to sites of acute inflammation in mice. Blood. (2013) 121:1008–15. 10.1182/blood-2012-06-437392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Del Conde I, Cruz MA, Zhang H, Lopez JA, Afshar-Kharghan V. Platelet activation leads to activation and propagation of the complement system. J Exp Med. (2005) 201:871–9. 10.1084/jem.20041497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schonberger T, Ziegler M, Borst O, Konrad I, Nieswandt B, Massberg S, et al. The dimeric platelet collagen receptor GPVI-Fc reduces platelet adhesion to activated endothelium and preserves myocardial function after transient ischemia in mice. Am J Physiol Cell Physiol. (2012) 303:C757–66. 10.1152/ajpcell.00060.2012 [DOI] [PubMed] [Google Scholar]
- 32.Xu Y, Huo Y, Toufektsian MC, Ramos SI, Ma Y, Tejani AD, et al. Activated platelets contribute importantly to myocardial reperfusion injury. Am J Physiol Heart Circ Physiol. (2006) 290:H692–9. 10.1152/ajpheart.00634.2005 [DOI] [PubMed] [Google Scholar]
- 33.Mauler M, Herr N, Schoenichen C, Witsch T, Marchini T, Härdtner C, et al. Platelet serotonin aggravates myocardial ischemia/reperfusion injury via neutrophil degranulation. Circulation. (2019) 139:918–31. 10.1161/CIRCULATIONAHA.118.033942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weig HJ, Bott-Flugel L, Stadele C, Winter K, Schmidt R, Gawaz M, et al. Impaired platelet function reduces myocardial infarct size in Galphaq knock-out mice in vivo. J Mol Cell Cardiol. (2008) 44:143–50. 10.1016/j.yjmcc.2007.09.018 [DOI] [PubMed] [Google Scholar]
- 35.Devanathan V, Hagedorn I, Köhler D, Pexa K, Cherpokova D, Kraft P, et al. Platelet Gi protein Galphai2 is an essential mediator of thrombo-inflammatory organ damage in mice. Proc Natl Acad Sci USA. (2015) 112:6491–6. 10.1073/pnas.1505887112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kupatt C, Wichels R, Horstkotte J, Krombach F, Habazettl H, Boekstegers P. Molecular mechanisms of platelet-mediated leukocyte recruitment during myocardial reperfusion. J Leukoc Biol. (2002) 72:455–61. 10.1189/jlb.72.3.455 [DOI] [PubMed] [Google Scholar]
- 37.Yang XM, Liu Y, Cui L, Yang X, Liu Y, Tandon N, et al. Platelet P2Y(1)(2) blockers confer direct postconditioning-like protection in reperfused rabbit hearts. J Cardiovasc Pharmacol Ther. (2013) 18:251–62. 10.1177/1074248412467692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oostingh GJ, Pozgajova M, Ludwig RJ, Krahn T, Boehncke WH, Nieswandt B, et al. Diminished thrombus formation and alleviation of myocardial infarction and reperfusion injury through antibody- or small-molecule-mediated inhibition of selectin-dependent platelet functions. Haematologica. (2007) 92:502–12. 10.3324/haematol.10741 [DOI] [PubMed] [Google Scholar]
- 39.Roh HY, Jung IS, Park JW, Yun YP, Yi KY, Yoo SE, et al. Cardioprotective effects of [5-(2-methyl-5-fluorophenyl)furan-2-ylcarbonyl]guanidine (KR-32568) in an anesthetized rat model of ischemia and reperfusion heart injury. Pharmacology. (2005) 75:37–44. 10.1159/000086192 [DOI] [PubMed] [Google Scholar]
- 40.Bianchi P, Kunduzova O, Masini E, Cambon C, Bani D, Raimondi L, et al. Oxidative stress by monoamine oxidase mediates receptor-independent cardiomyocyte apoptosis by serotonin and postischemic myocardial injury. Circulation. (2005) 112:3297–305. 10.1161/CIRCULATIONAHA.104.528133 [DOI] [PubMed] [Google Scholar]
- 41.Hung YC, Kuo YJ, Huang SS, Huang TF. Trimucrin, an Arg-Gly-Asp containing disintegrin, attenuates myocardial ischemia-reperfusion injury in murine by inhibiting platelet function. Eur J Pharmacol. (2017) 813:24–32. 10.1016/j.ejphar.2017.07.039 [DOI] [PubMed] [Google Scholar]
- 42.Hohlfeld T, Braun M, Strobach H, Schror K. Protection of reperfused ischemic pig myocardium by nexopamil, a new combined Ca2+ and serotonin antagonist. J Cardiovasc Pharmacol. (1994) 23:922–31. 10.1097/00005344-199406000-00010 [DOI] [PubMed] [Google Scholar]
- 43.Barrabes JA, Garcia-Dorado D, Mirabet M, Inserte J, Agullo L, Soriano B, et al. Antagonism of selectin function attenuates microvascular platelet deposition and platelet-mediated myocardial injury after transient ischemia. J Am Coll Cardiol. (2005) 45:293–9. 10.1016/j.jacc.2004.09.068 [DOI] [PubMed] [Google Scholar]
- 44.Ko W, Lang D, Hawes AS, Zelano JA, Isom OW, Krieger KH. Platelet-activating factor antagonism attenuates platelet and neutrophil activation and reduces myocardial injury during coronary reperfusion. J Surg Res. (1993) 55:504–15. 10.1006/jsre.1993.1176 [DOI] [PubMed] [Google Scholar]
- 45.Gotz AK, Zahler S, Stumpf P, Welsch U, Becker BF. Intracoronary formation and retention of micro aggregates of leukocytes and platelets contribute to postischemic myocardial dysfunction. Basic Res Cardiol. (2005) 100:413–21. 10.1007/s00395-005-0540-9 [DOI] [PubMed] [Google Scholar]
- 46.Yang B, Mehta P, Mehta JL. Platelet-mediated cardioprotective effect against ischemia-reperfusion injury in isolated rat hearts: role of platelet number and contribution of supernatant of aggregated platelets. J Cardiovasc Pharmacol Ther. (1998) 3:23–8. [DOI] [PubMed] [Google Scholar]
- 47.Penna C, Alloatti G, Cappello S, Gattullo D, Berta G, Mognetti B, et al. Platelet-activating factor induces cardioprotection in isolated rat heart akin to ischemic preconditioning: role of phosphoinositide 3-kinase and protein kinase C activation. Am J Physiol Heart Circ Physiol. (2005) 288:H2512–20. 10.1152/ajpheart.00599.2004 [DOI] [PubMed] [Google Scholar]
- 48.Barrabes JA, Inserte J, Mirabet M, Quiroga A, Hernando V, Figueras J, et al. Antagonism of P2Y12 or GPIIb/IIIa receptors reduces platelet-mediated myocardial injury after ischaemia and reperfusion in isolated rat hearts. Thromb Haemost. (2010) 104:128–35. 10.1160/TH09-07-0440 [DOI] [PubMed] [Google Scholar]
- 49.Seligmann C, Prechtl G, Kusus-Seligmann M, Daniel WG. A myocardial ischemia- and reperfusion-induced injury is mediated by reactive oxyg en species released from blood platelets. Platelets. (2013) 24:37–43. 10.3109/09537104.2012.658107 [DOI] [PubMed] [Google Scholar]
- 50.Wang K, Zhou X, Huang Y, Khalil M, Wiktor D, Van Giezen JJ, et al. Adjunctive treatment with ticagrelor, but not clopidogrel, added to tPA enables sustained coronary artery recanalisation with recovery of myocardium perfusion in a canine coronary thrombosis model. Thromb Haemost. (2010) 104:609–17. 10.1160/TH09-12-0823 [DOI] [PubMed] [Google Scholar]
- 51.Takaya N, Katoh Y, Iwabuchi K, Hayashi I, Konishi H, Itoh S, et al. Platelets activated by collagen through the immunoreceptor tyrosine-based activation motif in the Fc receptor gamma-chain play a pivotal role in the development of myocardial ischemia-reperfusion injury. J Mol Cell Cardiol. (2005) 39:856–64. 10.1016/j.yjmcc.2005.07.006 [DOI] [PubMed] [Google Scholar]
- 52.Pachel C, Mathes D, Arias-Loza AP, Heitzmann W, Nordbeck P, Deppermann C, et al. Inhibition of platelet GPVI protects against myocardial ischemia-reperfusion injury. Arterioscler Thromb Vasc Biol. (2016) 36:629–35. 10.1161/ATVBAHA.115.305873 [DOI] [PubMed] [Google Scholar]
- 53.Storey RF, Judge HM, Wilcox RG, Heptinstall S. Inhibition of ADP-induced P-selectin expression and platelet-leukocyte conjugate formation by clopidogrel and the P2Y12 receptor antagonist AR-C69931MX but not aspirin. Thromb Haemost. (2002) 88:488–94. 10.1055/s-0037-1613242 [DOI] [PubMed] [Google Scholar]
- 54.Neumann FJ, Zohlnhofer D, Fakhoury L, Ott I, Gawaz M, Schomig A. Effect of glycoprotein IIb/IIIa receptor blockade on platelet-leukocyte interaction and surface expression of the leukocyte integrin Mac-1 in acute myocardial infarction. J Am Coll Cardiol. (1999) 34:1420–6. 10.1016/S0735-1097(99)00350-2 [DOI] [PubMed] [Google Scholar]
- 55.Tam SH, Sassoli PM, Jordan RE, Nakada MT. Abciximab (ReoPro, chimeric 7E3 Fab) demonstrates equivalent affinity and functional blockade of glycoprotein IIb/IIIa and alpha(v)beta3 integrins. Circulation. (1998) 98:1085–91. 10.1161/01.CIR.98.11.1085 [DOI] [PubMed] [Google Scholar]
- 56.Seligmann C, Simsek Y, Schimmer M, Leitsch T, Bock A, Schultheiss HP. Human thrombocytes are able to induce a myocardial dysfunction in the ischemic and reperfused guinea pig heart mediated by free radicals-role of the GPIIb/IIIa-blocker tirofiban. Life Sci. (2002) 71:2319–29. 10.1016/S0024-3205(02)02021-0 [DOI] [PubMed] [Google Scholar]
- 57.Ren F, Mu N, Zhang X, Tan J, Li L, Zhang C, et al. Increased platelet-leukocyte aggregates are associated with myocardial no-reflow in patients with ST elevation myocardial infarction. Am J Med Sci. (2016) 352:261–6. 10.1016/j.amjms.2016.05.034 [DOI] [PubMed] [Google Scholar]
- 58.Mirabet M, Garcia-Dorado D, Inserte J, Barrabés JA, Lidón RM, Soriano B, et al. Platelets activated by transient coronary occlusion exacerbate ischemia-reperfusion injury in rat hearts. Am J Physiol Heart Circ Physiol. (2002) 283:H1134–41. 10.1152/ajpheart.00065.2002 [DOI] [PubMed] [Google Scholar]
- 59.Weyrich AS, Ma XY, Lefer DJ, Albertine KH, Lefer AM. In vivo neutralization of P-selectin protects feline heart and endothelium in myocardial ischemia and reperfusion injury. J Clin Invest. (1993) 91:2620–9. 10.1172/JCI116501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Karmazyn M, Sostaric JV, Gan XT. The myocardial Na+/H+ exchanger: a potential therapeutic target for the prevention of myocardial ischaemic and reperfusion injury and attenuation of postinfarction heart failure. Drugs. (2001) 61:375–89. 10.2165/00003495-200161030-00006 [DOI] [PubMed] [Google Scholar]
- 61.Karki P, Fliegel L. Overexpression of the NHE1 isoform of the Na(+)/H(+) exchanger causes elevated apoptosis in isolated cardiomyocytes after hypoxia/reoxygenation challenge. Mol Cell Biochem. (2010) 338:47–57. 10.1007/s11010-009-0337-5 [DOI] [PubMed] [Google Scholar]
- 62.Wang Y, Meyer JW, Ashraf M, Shull GE. Mice with a null mutation in the NHE1 Na+-H+ exchanger are resistant to cardiac ischemia-reperfusion injury. Circ Res. (2003) 93:776–82. 10.1161/01.RES.0000094746.24774.DC [DOI] [PubMed] [Google Scholar]
- 63.Rosskopf D. Sodium-hydrogen exchange and platelet function. J Thromb Thrombolysis. (1999) 8:15–24. 10.1023/A:1008986329267 [DOI] [PubMed] [Google Scholar]
- 64.Chang HB, Gao X, Nepomuceno R, Hu S, Sun D. Na(+)/H(+) exchanger in the regulation of platelet activation and paradoxical effects of cariporide. Exp Neurol. (2015) 272:11–6. 10.1016/j.expneurol.2014.12.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Aldosari S, Awad M, Harrington EO, Sellke FW, Abid MR. Subcellular reactive oxygen species (ROS) in cardiovascular pathophysiology. Antioxidants (Basel). 7:14. 10.3390/antiox7010014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Seligmann C, Schimmer M, Leitsch T, Bock A, Simsek Y, Tschope C, et al. A thrombocyte-induced myocardial dysfunction in the ischemic and reperfused guinea pig heart is mediated by reactive oxygen species. Free Radic Biol Med. (2000) 29:1244–51. 10.1016/S0891-5849(00)00414-7 [DOI] [PubMed] [Google Scholar]
- 67.Penna C, Bassino E, Alloatti G. Platelet activating factor: the good and the bad in the ischemic/reperfused heart. Exp Biol Med. (2011) 236:390–401. 10.1258/ebm.2011.010316 [DOI] [PubMed] [Google Scholar]
- 68.Davidson SM, Andreadou I, Barile L, Birnbaum Y, Cabrera-Fuentes HA, Cohen MV, et al. Circulating blood cells and extracellular vesicles in acute cardioprotection. Cardiovasc Res. (2019) 115:1156–66. 10.1093/cvr/cvy314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Montrucchio G, Alloatti G, Camussi G. Role of platelet-activating factor in cardiovascular pathophysiology. Physiol Rev. (2000) 80:1669–99. 10.1152/physrev.2000.80.4.1669 [DOI] [PubMed] [Google Scholar]
- 70.Stangl V, Baumann G, Stangl K, Felix SB. Negative inotropic mediators released from the heart after myocardial ischaemia-reperfusion. Cardiovasc Res. (2002) 53:12–30. 10.1016/S0008-6363(01)00420-5 [DOI] [PubMed] [Google Scholar]
- 71.Hidalgo MA, Ojeda F, Eyre P, Labranche TP, Smith C, Hancke JL, et al. Platelet-activating factor increases pH(i) in bovine neutrophils through the PI3K-ERK1/2 pathway. Br J Pharmacol. (2004) 141:311–21. 10.1038/sj.bjp.0705590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Neumann FJ, Marx N, Gawaz M, Brand K, Ott I, Rokitta C, et al. Induction of cytokine expression in leukocytes by binding of thrombin-stimulated platelets. Circulation. (1997) 95:2387–94. [DOI] [PubMed] [Google Scholar]
- 73.Lefer AM, Campbell B, Scalia R, Lefer DJ. Synergism between platelets and neutrophils in provoking cardiac dysfunction after ischemia and reperfusion: role of selectins. Circulation. (1998) 98:1322–8. [DOI] [PubMed] [Google Scholar]
- 74.Habazettl H, Hanusch P, Kupatt C. Effects of endothelium/leukocytes/platelet interaction on myocardial ischemia–reperfusion injury. Z Kardiol. (2000) 89 (Suppl 9):IX/92–5. 10.1007/s003920070038 [DOI] [PubMed] [Google Scholar]
- 75.Sreeramkumar V, Adrover JM, Ballesteros I, Cuartero MI, Rossaint J, Bilbao I, et al. Neutrophils scan for activated platelets to initiate inflammation. Science. (2014) 346:1234–8. 10.1126/science.1256478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Seligmann C, Leitsch T, Kusus M, Bock A, Schimmer M, Simsek Y, et al. PMN/platelets coinfused in guinea pig hearts exposed to low-flow ischemia have no additive cardiodepressive effect. J Vasc Res. (2003) 40:501–8. 10.1159/000074890 [DOI] [PubMed] [Google Scholar]
- 77.Lefer DJ, Flynn DM, Buda AJ. Effects of a monoclonal antibody directed against P-selectin after myocardial ischemia and reperfusion. Am J Physiol. (1996) 270:H88–98. [DOI] [PubMed] [Google Scholar]
- 78.Henn V, Slupsky JR, Grafe M, Anagnostopoulos I, Forster R, Muller-Berghaus G, et al. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature. (1998) 391:591–4. [DOI] [PubMed] [Google Scholar]
- 79.Walsh TG, Poole AW. Do platelets promote cardiac recovery after myocardial infarction: roles beyond occlusive ischemic damage. Am J Physiol Heart Circ Physiol. (2018) 314:H1043–8. 10.1152/ajpheart.00134.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Heindl B, Zahler S, Welsch U, Becker BF. Disparate effects of adhesion and degranulation of platelets on myocardial and coronary function in postischaemic hearts. Cardiovasc Res. (1998) 38:383–94. [DOI] [PubMed] [Google Scholar]
- 81.Yang BC, Virmani R, Nichols WW, Mehta JL. Platelets protect against myocardial dysfunction and injury induced by ischemia and reperfusion in isolated rat hearts. Circ Res. (1993) 72:1181–90. [DOI] [PubMed] [Google Scholar]
- 82.Yang BC, Mehta JL. Platelet-derived adenosine contributes to the cardioprotective effects of platelets against ischemia-reperfusion injury in isolated rat heart. J Cardiovasc Pharmacol. (1994) 24:779–85. [DOI] [PubMed] [Google Scholar]
- 83.Yang BC, Zander DS, Mehta JL. Hypoxia-reoxygenation-induced apoptosis in cultured adult rat myocytes and the protective effect of platelets and transforming growth factor-beta(1). J Pharmacol Exp Ther. (1999) 291:733–8. [PubMed] [Google Scholar]
- 84.Mehta JL, Yang BC, Strates BS, Mehta P. Role of TGF-beta1 in platelet-mediated cardioprotection during ischemia-reperfusion in isolated rat hearts. Growth Factors. (1999) 16:179–90. [DOI] [PubMed] [Google Scholar]
- 85.Walsh TG, Poole AW. Platelets protect cardiomyocytes from ischaemic damage. TH Open. (2017) 1:e24. 10.1055/s-0037-1603928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rossello X, Yellon DM. The RISK pathway and beyond. Basic Res Cardiol. (2017) 113:2. 10.1007/s00395-017-0662-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hadebe N, Cour M, Lecour S. The SAFE pathway for cardioprotection: is this a promising target? Basic Res Cardiol. (2018) 113:9. 10.1007/s00395-018-0670-5: [DOI] [PubMed] [Google Scholar]