

Abstract
Aim:
We report the prevalence and effect of genetic variability on pharmacokinetic parameters of isoniazid and rifampicin.
Materials & methods:
Genotypes for SLCO1B1, NAT2, PXR, ABCB1 and UGT1A genes were determined using a TaqMan® Genotyping OpenArray™. Nonlinear mixed-effects models were used to describe drug pharmacokinetics.
Results:
Among 172 patients, 18, 43 and 34% were classified as rapid, intermediate and slow NAT2 acetylators, respectively. Of the 58 patients contributing drug concentrations, rapid and intermediate acetylators had 2.3- and 1.6-times faster isoniazid clearance than slow acetylators. No association was observed between rifampicin pharmacokinetics and SLCO1B1, ABCB1, UGT1A or PXR genotypes.
Conclusion:
Clinical relevance of the effects of genetic variation on isoniazid concentrations and low first-line tuberculosis drug exposures observed require further investigation.
Keywords: : isoniazid, pharmacogenetics, pharmacokinetics, pyrazinamide, rifampicin, tuberculosis
Tuberculosis ranks alongside HIV as a leading cause of death globally, affecting more than 10.4 million people in 2016 [1]. Despite efforts to introduce new drugs and shorter drug regimens for drug-susceptible tuberculosis, the standard first-line treatment for drug-susceptible tuberculosis has not changed over 50 years [2]. Rifampicin, isoniazid and pyrazinamide are critical components of the current standard tuberculosis treatment regimens. Several studies are being conducted to optimize current drug regimens using appropriate dosing strategies and dose optimization of drugs such as rifampicin [3–5]. Studies using the hollow fiber model of tuberculosis suggest that microbiologic failure and acquired drug resistance are primarily driven by low drug concentrations that result from pharmacokinetic variability independent of adherence to treatment [6]. Rifampicin in particular exhibits concentration dependant activity [4], and low isoniazid and pyrazinamide drug levels have been linked to poor treatment outcomes [7–10]. High between-subject pharmacokinetic variability for rifampicin, isoniazid and pyrazinamide has been reported in prior studies [11–15]. Several possible reasons could explain the variability, some of these include drug formulation, quality of tablets, patient weight, age, sex, treatment adherence patterns and comorbidities such as HIV [13–16]. Furthermore, pharmacogenetic variability in genes coding for drug-metabolizing and transporter enzymes has been associated with variable drug concentrations and pharmacokinetics (PK) [12,17–19].
Rifampicin is metabolized by hepatic esterases and biliary excretion of rifampicin occurs after hepatocellular uptake, which is mediated primarily by organic anion-transporting polypeptide 1B1, coded by the SLCO1B1 gene [20,21]. Rifampicin metabolism may be mediated by Phase I and II pathways [22]. Rifampicin is also a substrate of the drug efflux pump P-glycoprotein, coded by the ABCB1 gene, and it activates the Pregnane X receptor (PXR/NRI12), which regulates the expression of multiple metabolic enzymes and drug transporters including UGT enzymes coded by the UGT1A genes [22]. These genes are reported as highly polymorphic, which suggests that genetic variability may affect the PK of rifampicin and other drugs [12,23–26].
Isoniazid is metabolized by the N-acetyltransferases coded for by the NAT2 genes, which determine individual acetylator status [27]. The rate of elimination of isoniazid has been shown to be trimodally distributed in accordance with NAT2 metabolic activity [28].
Previous studies in African populations report changes to rifampicin PK and reduced drug concentrations due to genetic variability in the SLCO1B1 gene, which includes rs4149032, rs4149056 and rs11045819 [12,17,29]. Variable isoniazid plasma concentrations due to NAT2 genotype and acetylator status affecting therapeutic outcomes and adverse events have been well documented [28,30]. No data is currently available on the effect of genetic variation on pyrazinamide PK [19].
Rifampicin, isoniazid and pyrazinamide concentrations are reported as low in African patients, resulting in worse treatment outcomes [12,16,17,29,31,32]. African populations have high levels of host genetic diversity resulting in differences in tuberculosis disease susceptibility [33]. Furthermore, genetic diversity in drug-metabolizing and transport enzymes are shown to result in lower tuberculosis drug concentrations and variations in response to standard first-line tuberculosis drugs [12,17,34]. However, data on the pharmacogenetic determinants of antituberculosis drug exposure among tuberculosis-endemic African populations remain limited.
We describe here the pharmacokinetic profiles of rifampicin, isoniazid and pyrazinamide in South African patients with drug-susceptible tuberculosis and determine the prevalence and effect of pharmacogenetic variability in genes coding for relevant drug-metabolizing enzymes on pharmacokinetic parameters of rifampicin and isoniazid. Moxifloxacin PK and pharmacogenetic associations in the study has been described previously [18,35].
Materials & methods
A prospective pharmacokinetic substudy was conducted within the ongoing Improving Retreatment Success open-label randomized controlled trial (NCT02114684) from October 2013 to June 2017, in KwaZulu-Natal, Durban, South Africa. Details on study objectives, design and inclusion or exclusion criteria are available at ClinicalTrials.gov (https://clinicaltrials.gov/ct2/show/NCT02114684).
Patients in the intervention arm of the study that provided informed consent to be included in the pharmacokinetic substudy had blood samples collected for pharmacokinetic analysis at predefined time points. Baseline whole blood samples were collected in all patients consenting to storage of samples for later genetic testing. All participants recruited to the study were >18 years of age, had a confirmed history of tuberculosis within the last 3 years and had been diagnosed with sputum smear positive, rifampicin sensitive, mycobacterium tuberculosis based on microscopy and GeneXpert technology. Only those with no predefined laboratory or clinical abnormalities were included, regardless of HIV status.
Drug regimens
Patients randomized to the intervention arm of the study received daily moxifloxacin 400 mg (Avelox®, Bayer Healthcare, Leverkusen, Germany), and weight-adjusted rifampicin, isoniazid, and, only during the 2-month intensive phase, pyrazinamide. Patients below 55 kg received 450 mg of rifampicin, 225 mg of isoniazid and 1500 mg of pyrazinamide, while those above 55 kg received 1/3 more of each drug. Drugs were not administered as fixed-drug combinations except rifampicin/isoniazid (Rifinah®).
Follow-up
Patients were followed-up for 24 months, and clinical and safety monitoring was done bimonthly for the first 6 months, or as clinically indicated. Adherence to tuberculosis treatment was measured using pill count data. The HIV coinfected patients received standard first-line antiretroviral therapy (ART) containing efavirenz, emtricitabine, and tenofovir. Treatment and prophylaxis for opportunistic infections and concomitant treatment used was recorded on case report forms.
Pharmacokinetic sampling
Blood samples were collected prior to drug dose and at 2.5, 6 and 24 h postobserved dose at 1 and/or 2 months (during the intensive phase of tuberculosis treatment) and at 6 months (during the continuation phase of tuberculosis treatment). The month, at which PK sampling was conducted before and after an observed dose, was defined as an occasion. Blood, collected in EDTA tubes, was centrifuged at 3000 rpm, placed on ice, sent within 1 h of collection to the CAPRISA laboratory to be stored at -80°C. Drug concentrations were quantified in clinical plasma samples at the KwaZulu-Natal Research Institute for Tuberculosis and HIV (KRITH) pharmacology laboratory using validated HPLC–MS/MS. The bioanalytical method was developed and validated according to US FDA guidelines (2011) [22]. Plasma sample preparation included a protein precipitation with acetonitrile and subsequent dilution with water. Analytes were chromatographically separated using a Zorbax C18, 3.5 μm, 50 × 2.1 mm column and detected using the ABI Sciex 5500 QTrap mass spectrometer operated in positive mode. The following transitions were used; precursor ion → product ion (all in units of m/z): rifampicin: 823.3 → 791.4 and 823.3 → 95; pyrazinamide: 124.1 → 54 and 124.1→79; isoniazid: 137.9 → 120.8 and 137.9 → 93. The two internal standards used were ciprofloxacin: 331.6 → 231.0 and 331.6 → 288.1 for isoniazid and pyrazinamide and rifaximin: 786.2 → 754.3 and 786.2 → 95.0 for rifampicin. Isoniazid, pyrazinamide and rifampicin were analyzed isocratically with acetonitrile/water/0.1% formic acid mobile phase (at 22% acetronile for pyrazinamide and isoniazid and 55% acetronile for rifampicin). The injection volume was 2 μl and a total analytical run time of 5 min. The method was validated over the concentration range of 20–10,000 ng/ml for isoniazid, 150–75,000 ng/ml for pyrazinamide and 40–20,000 ng/ml for rifampicin. Overall precision, based on quality control samples evaluated at a low, medium and high concentrations, during the validation and analysis of samples ranged from ranged from 4.3 to 6.7%, 5.4 to 10.1% and 5.5 to 6.5%, and accuracy ranged from 103.9 to 109.5%, 96 to 98.5% and 92.9 to 96.4% for isoniazid, pyrazinamide and rifampicin, respectively. Calculated carry over at the LLOQ was 2.8, 1.8 and 2.3% for isoniazid, pyrazinamide and rifampicin, respectively. The LC–MS/MS system was interfaced with a DELL® Windows®7 computer running Analyst® software version 1.6.2, used for chromatographic data acquisition, peak integration and quantification of analytes.
Pharmacogenetic sampling
A total of 25 SNPs, four within NAT2, four within SLCO1B1, three in the PXR/NRI, six within ABCB1 and eight within UGT1A were selected for analysis in this study based on previous evidence of functional significance on drug response using the PharmGKB database and relevant literature [12,17,23,25,29,36,37]. The SNPs selected included; rs2306283, rs4149032, rs4149056, rs4149015 for SLCO1B1, rs1799931, rs1801280, rs1801279 and rs1799930 for NAT2, rs2307424, rs2472677, rs1523130 for PXR or NR1, rs10276036, rs1128503, rs2032582, rs1045642, rs2235033 and rs2235013 for ABCB1; and rs4148323, rs2003569, rs3755319, rs11692021, rs2070959, rs28900377, rs1983023 and rs8175347 for UGT1A.
Genotypes were determined for each of the SNPs using a TaqMan® Genotyping OpenArray™ (Thermo Fisher Scientific, MA, USA). For rs8175347, due to the limited ability of TaqMan® probes to reliably distinguish all genotypes, a high resolution melt analysis was used to determine genotype [38]. The TaqMan® Genotyping OpenArray™ was performed as follows: genomic DNA was extracted from whole blood for each sample using the Qiagen QIAamp MiniKit (Germany, Cat. No. 51306), according to manufacturer instructions. Approximately 50 μg/ml of DNA and 2 X TaqMan Genotyping OpenArray Master Mix were used for the assay. Plates were loaded using the Accufill liquid handling robot and run on the Applied Biosystems™ QuantStudio™ 12K Flex Real-Time PCR instrument according to Applied Biosystems™ user guide. Analysis was performed using the TaqMan® Genotyper Software version 3.1. The genotypes for UGT1A1*28 (rs8175347) were determined using genomic DNA as previously described [38] with minor modifications. The real-time PCR conditions were changed as follows: 95°C for 5 min, followed by 45 cycles of 95°C for 10 s, 67°C for 10 s and 76°C for 10 s, and finally 76°C for 1 min.
Acetylator status based on NAT2 genotypes were determined using a four-SNP genotyping panel including rs1799931, rs1801280, rs1801279 and rs1799930 [39]. Samples homozygous common for all SNPs were classified as rapid acetylator phenotype, samples heterozygous for any one of the SNPs were classified as intermediate acetylator phenotype and samples homozygous variant for one or more SNPs or heterozygous for two or more SNPs were classified as slow acetylator phenotype.
Pharmacokinetic modeling
Nonlinear mixed-effects modeling was employed to interpret the concentration-time data using the software NONMEM 7.3 [40], and the algorithm First-Order Conditional Estimation with eta-epsilon interaction (FOCE-I). Pirana, Perl-speaks-NONMEM, and Xpose4 were used to aid the modeling process and prepare model diagnostics [41]. The modeling approach is summarized below. Several structural models were tested: one- and two-compartment disposition kinetics with first-order elimination and several approaches for absorption: first-order, with or without a lag time or a chain transit compartments [42]. Since very little information was available in the absorption phase, we used literature values as priors (with 30% uncertainty) to improve parameter estimation and stabilize the model [43]. Parameter estimates reported by Wilkins et al., Denti et al. and Chirehwa et al. were used as priors for the pharmacokinetic model for isoniazid, rifampicin and pyrazinamide, respectively [13,44,45]. The statistical model assumed log-normal distribution for the between-subject and -occasion random effects, and a combined additive and proportional structure for the residual unexplained variability, with the additive component of the error bound to be at least 20% of the LLOQ. Concentrations below the formal LLOQ of the assay were available for analysis, while the values below the limit of detection were imputed to LLOQ/10 and handled with the M6 method suggested in Beal [46]. Rifampicin, isoniazid and pyrazinamide concentrations were below LLOQ in 43, 27 and 3% of the samples, and below the limit of detection in 18, 11 and 1%. Most of these values were observed in the predose concentrations.
Allometric scaling with either total bodyweight (WT) or fat-free mass (FFM) was applied to all clearance (CL and Q) and volume of distribution (Vc and Vp) parameters, as advocated by Anderson and Holford [47]. Equations 1 and 2 describes how body size is related to oral clearance and volume of distribution, respectively.
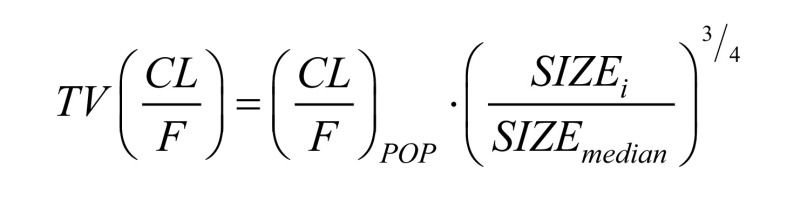 |
(1) |
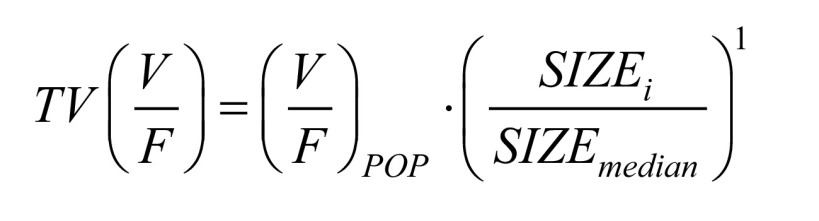 |
(2) |
The SIZEi denotes individual values of the two size descriptors (WT or FFM). The SIZEmedian is the median body size in the cohort. The (CL/F)POP is the typical oral clearance for an individual whose body size is equal to the median body size. The (V/F)POP is the typical volume of distribution for an individual whose body size is equal to the median body size in the cohort.
The effect of covariates on pharmacokinetic parameters was tested and included in the model based on significant decreases (p < 0.05) in the objective function value (OFV) and physiological plausibility. The effect of the following covariates was evaluated: antiretroviral therapy, treatment phase, genetic polymorphisms on rifampicin and isoniazid, and serum creatinine.
Results
We included 172 South African tuberculosis patients: 119 (69.2%) male, 170 (98.8%) of Black African ethnicity and 127 (73.8%) HIV coinfected (Table 1). Rifampicin, isoniazid and pyrazinamide concentration-time data were available for 58 of 172 patients. Of these: median weight, fat-free mass and age were 56.9 kg (interquartile range [IQR]: 51.1–65.2), 46.8 kg (IQR: 42.5–50.3) and 37 years (IQR: 31–42), respectively. Total of 41 (70.7%) patients were male, 42 (72.4%) HIV coinfected, with 40/42 (95%) on efavirenz-based ART (Table 1).
Table 1. . Baseline data.
| Variable | N = 172 (all participants) | N = 58 (participants in PK substudy with drug concentrations) |
|---|---|---|
| Age (years), median (IQR) | 35 (30–41) | 37 (31–42) |
| Male, n (%) | 119 (69.2) | 41 (70.7) |
| Race, n (%) – Black African ethnicity/Caucasian/colored‡ | 170 (98.8)/1 (0.6)/1 (0.6) | 56 (96.6)/1 (1.7)/1 (1.7) |
| Weight (kg), median (IQR) | 55.7 (50.3–62.1) | 56.9 (51.1–65.2) |
| Fat-free mass (kg) | – | 46.8 (42.5–50.3) |
| BMI(kg/m2), median (IQR) | 19.7 (18.3–22.5) | 19.6 (18.0–23.3) |
| HIV status, n (%) – positive/negative | 127 (73.8)/45 (26.2) | 42 (72.4)/16 (27.6) |
| ART, n (%)† : | ||
| – Efavirenz + emtricitabine + tenofovir | 117 (95.1) | 40 (95.2) |
| – Lopinavir/ritonavir + lamivudine + tenofovir | 2 (1.6) | 2 (4.8) |
| – Lopinavir/ritonavir + lamivudine + zidovudine | 2 (1.6) | – |
| – Lopinavir/ritonavir + emtricitabine + tenofovir | 1 (0.8) | – |
| – Efavirenz/lamivudine/zidovudine | 1 (0.8) | – |
| – CD4+ count (cells/mm3), median (IQR)†,§ | 241.0 (129.0–407.0) | 277.0 (139.0–384.0) |
| – Viral load (log10 copies/ml)†,¶ | 3.7 (1.3–5.0) | 3.3 (1.3–4.2) |
†Only for HIV + patients.
‡Mixed race.
§10/172, 4/58 missing data.
¶12/172, 5/58 missing data.
IQR: Interquartile range; PK: Pharmacokinetics.
Genotype & allele frequency of UGT1A, ABCB1, SLCO1B1, NAT2, NR1I
The genotypic frequencies for variants within UGT1A and ABCB1, SLCO1B1, NAT2, NR1I are presented in Table 2 for all patients (n = 172) included in the study and the subset of 58 patients with rifampicin, isoniazid and pyrazinamide drug level data. Allelic frequencies for all SNPs examined are presented in Table 3. Total of 18% of patients were classified as rapid acetylators, and 34 and 43% as slow and intermediate acetylators, respectively.
Table 2. . Genotype frequency for UGT1A, ABCB1, SLCO1B1, NAT2 and PXR (NR1I) SNPs.
| UGT1A | ABCB1 | SLCO1B1 | NAT2 | PXR/NR1I | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP | Genotype | n = 172 | n = 58 | SNP | Genotype | n = 172 | n = 58 | SNP | Genotype | n = 172 | n = 58 | SNP | Genotype | n = 172 | n = 58 | SNP | Genotype | n = 172 | n = 58 |
| rs11692021 234591205T>C | CC | 0.04 | 0.05 | TT | 0.74 | 0.74 | rs2306283 21329738A>G |
AA | 0.03 | 0.02 | rs1799931 18258370G>A |
GG | 0.00 | 0.00 | AA | 0.00 | 0.00 | ||
| CT | 0.18 | 0.22 | rs10276036 87180198C>T |
CT | 0.23 | 0.24 | AG | 0.19 | 0.25 | AG | 0.03 | 0.00 | rs2307424 161202605G>A |
AG | 0.10 | 0.10 | |||
| TT | 0.78 | 0.73 | CC | 0.03 | 0.02 | GG | 0.78 | 0.73 | AA | 0.97 | 1.00 | GG | 0.90 | 0.90 | |||||
| rs4148323 | GG | 1.00 | 1.00 | AA | 0.01 | 0.00 | rs4149032 21317791C>T |
TT | 0.51 | 0.42 | rs1801280 18257854T>C |
CC | 0.09 | 0.16 | TT | 0.10 | 0.20 | ||
| 234669144G>A | GA | 0.00 | 0.00 | rs1128503 | AG | 0.14 | 0.16 | TC | 0.41 | 0.47 | CT | 0.44 | 0.40 | rs2472677 119518417C>T |
CT | 0.50 | 0.40 | ||
| AA | 0.00 | 0.00 | 87179601A>G | GG | 0.85 | 0.84 | CC | 0.08 | 0.11 | TT | 0.47 | 0.44 | CC | 0.40 | 0.40 | ||||
| rs2003569 234667937G>A | AA | 0.17 | 0.09 | AA | 0.00 | 0.00 | rs4149056 21331549T>C |
TT | 1.00 | 1.00 | rs1801279 18257704G>A |
GG | 0.00 | 0.00 | TT | 0.90 | 0.90 | ||
| AG | 0.38 | 0.40 | rs2032582 | CA | 0.02 | 0.03 | CT | 0.00 | 0.00 | AG | 0.09 | 0.11 | rs1523130 119499507T>C |
CT | 0.10 | 0.10 | |||
| GG | 0.45 | 0.51 | 87160618A>C | CC | 0.98 | 0.97 | CC | 0.00 | 0.00 | AA | 0.91 | 0.89 | CC | 0.00 | 0.00 | ||||
| rs3755319 | AA | 0.06 | 0.05 | AA | 0.02 | 0.00 | rs4149015 21283322G>A |
GG | 1.00 | 1.00 | rs1799930 18258103G>A |
GG | 0.08 | 0.07 | |||||
| 234667582A>C | AC | 0.32 | 0.41 | rs1045642 | AG | 0.14 | 0.19 | GA | 0.00 | 0.00 | AG | 0.25 | 0.33 | ||||||
| CC | 0.62 | 0.54 | 87138645A>G | GG | 0.84 | 0.81 | AA | 0.00 | 0.00 | AA | 0.67 | 0.60 | |||||||
| rs2070959 234602191A>G | AA | 0.78 | 0.74 | TT | 0.29 | 0.37 | |||||||||||||
| AG | 0.18 | 0.21 | rs2235013 | CT | 0.47 | 0.43 | |||||||||||||
| GG | 0.04 | 0.05 | 87178626C>T | CC | 0.24 | 0.20 | |||||||||||||
| rs28900377 234652257C>T | TT | 0.01 | 0.00 | AA | 0.24 | 0.20 | |||||||||||||
| CT | 0.06 | 0.09 | rs2235033 | AG | 0.76 | 0.80 | |||||||||||||
| CC | 0.93 | 0.91 | 87179143A>G | GG | 0.00 | 0.00 | |||||||||||||
| rs1983023 | TT | 0.06 | 0.07 | ||||||||||||||||
| 234637022T>C | CT | 0.39 | 0.36 | ||||||||||||||||
| CC | 0.55 | 0.57 | |||||||||||||||||
| rs8175347 TA TA [5][6][7][8] | TA5/6 – UGT1A1*36/*1 | 0.11 | 0.10 | ||||||||||||||||
| TA5/7 – UGT1A1*36/*28 | 0.01 | 0.00 | |||||||||||||||||
| TA5/8 – UGT1A1*36/*37 | 0.01 | 0.00 | |||||||||||||||||
| TA6/6 – UGT1A1*1/*1 | 0.48 | 0.50 | |||||||||||||||||
| TA6/7 – UGT1A1*1/*28 | 0.16 | 0.12 | |||||||||||||||||
| TA6/8 – UGT1A1*1/*37 | 0.02 | 0.02 | |||||||||||||||||
| TA7/7 – UGT1A1*28/*28 | 0.08 | 0.12 | |||||||||||||||||
| TA7/8 – UGT1A1*28/*37 | 0.13 | 0.14 | |||||||||||||||||
The genotypes represented for ABCB1, SLCO1B1, NAT2 and NR1I represent the forward orientation while the gene is transcribed in the reverse direction. Genomic placements are relative to the human reference genome GRCh37.
Table 3. . Allele frequency for UGT1A, ABCB1, SLCO1B1, NAT2 and NR1I SNPs.
| UGT1A | ABCB1 | SLCO1B1 | NAT2 | NR1I | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP | Allele | Frequency | SNP | Allele | Frequency | SNP | Allele | Frequency | SNP | Allele | Frequency | SNP | Allele | Frequency |
| rs11692021 | C | 0.13 | rs10276036 | T | 0.85 | rs2306283 | A | 0.10 | rs1799931 | G | 0.00 | rs2307424 | A | 0.00 |
| T | 0.87 | C | 0.15 | G | 0.90 | A | 1.00 | G | 1.00 | |||||
| rs4148323 | G | 1.00 | rs1128503 | A | 0.08 | rs4149032 | T | 0.70 | rs1801280 | C | 0.30 | rs2472677 | T | 0.40 |
| A | 0.00 | G | 0.92 | C | 0.30 | T | 0.70 | C | 0.60 | |||||
| rs2003569 | A | 0.36 | rs2032582 | A | 0.01 | rs4149056 | T | 1.00 | rs1801279 | G | 0.00 | rs1523130 | T | 1.00 |
| G | 0.64 | C | 0.99 | C | 0.00 | A | 1.00 | C | 0.00 | |||||
| rs3755319 | A | 0.22 | rs1045642 | A | 0.06 | rs4149015 | G | 1.00 | rs1799930 | G | 0.20 | |||
| C | 0.78 | G | 0.94 | A | 0.00 | A | 0.80 | |||||||
| rs2070959 | A | 0.87 | rs2235013 | T | 0.53 | |||||||||
| G | 0.13 | C | 0.47 | |||||||||||
| rs28900377 | T | 0.04 | rs2235033 | A | 0.50 | |||||||||
| C | 0.96 | G | 0.50 | |||||||||||
| rs1983023 | T | 0.25 | ||||||||||||
| C | 0.75 | |||||||||||||
| rs8175347 TA [5][6][7][8] | TA5 – UGT1A1*36 | 0.07 | ||||||||||||
| TA6 – UGT1A1*1 | 0.63 | |||||||||||||
| TA7 – UGT1A1*28 | 0.23 | |||||||||||||
| TA8 – UGT1A1*37 | 0.07 | |||||||||||||
The genotypes represented for ABCB1, SLCO1B1, NAT2 and NR1I represent the forward orientation while the gene is transcribed in the reverse direction.
Linkage disequilibrium (LD) was tested using LDlink 2.0. online software [49]. The SNPs from UGT1A (rs11692021 and rs2070959) were found to be in perfect LD (D’ = 1, r2 = 1). In addition, ABCB1 SNPs (rs2235033 and rs2235013) were found to be in perfect LD (D’ = 1, r2 = 1). We also observed a set of SNPs within the SLCO1B1 gene (rs2306283 and rs4149032) that were found in strong linkage (D’ = 0.91, r2 = 0.51).
PK of first-line tuberculosis drugs
Concentration data were available for a total of 574, 573 and 328 sampling points for rifampicin, isoniazid and pyrazinamide, respectively.
The final pharmacokinetic model for rifampicin was a one-compartment disposition model with first-order elimination and a chain of transit compartments describing the delayed onset of absorption. The final parameter estimates together with 95% empirical CI based on a nonparametric bootstrap are shown in Table 4 and a visual predictive check shown in Figure 1, shows that the models suitably fits the data. Parameter estimates from a previous study [44] were used as priors for absorption rate constant and mean transit time. The FFM was a better size descriptor for allometric scaling compared with total bodyweight. Between subject variability (BSV) was supported on clearance, and between occasion variability (BOV) was supported on clearance and bioavailability. The model did not detect the effect of any of the genetic polymorphisms tested for SLCO1B1, UGT, ABCB1 and NR1I, or weight adjusted daily dose on rifampicin pharmacokinetic parameters.
Table 4. . Population parameter estimates of rifampicin, isoniazid and pyrazinamide pharmacokinetics.
| Parameter description | Typical value (95% CI)† | Random variability (95% CI) |
|---|---|---|
| Rifampicin | ||
| Clearance (l/h)‡ | 22.8 (20.5–25.1) | BSV: 5.5% (0.1–12.3) BOV: 21.3% (11.6–27.9) |
| Volume of central compartment (l)‡ | 77.4 (68.5–85.8) | |
| Bioavailability | 1 FIXED | BOV: 43.4% (33.5–51.1) |
| Ka – absorption rate (1/h)§ | 1.57 (1.25–1.96) PRIOR: 1.04 |
|
| Mean transit time (h)§ | 0.53 (0.46–0.59) PRIOR: 0.62 |
|
| Number of transit compartments§ | 34.6 (33.3–35.8) PRIOR: 34.5 |
|
| Proportional error (%) | 37.4% (32.4–41.1) | |
| Additive error (mg/l) | 0.008 FIXED | |
| Isoniazid | ||
| Clearance (l/h): | ||
| – Fast‡ | 40.5 (35.5; 45.4) | BSV: 26.3% (14.2–33.8) BOV: 13% (3.8–18.8) |
| – Intermediate‡ | 28.4 (24.6–33.1) | |
| – Slow‡ | 17.4 (15.2–20.9) | |
| Volume of central compartment (l)‡ | 73.4 (66.7–83.9) | |
| Inter-compartmental clearance (l/h)‡ | 1.1 (0.7; 1.5) | |
| Volume of peripheral compartment (l)‡ | 19.8 (15.0–27.9) | |
| Bioavailability | 1 FIXED | BOV: 27.4% (19.3–40.3) |
| Absorption lag time (h), prior§ | 0.13 (0.12–0.15) PRIOR: 0.18 |
|
| Ka – absorption rate (1/h), prior§ | 3.9 (3.1– 4.3) PRIOR: 1.85 |
BOV: 23.5% (0.9–97.5) |
| Scaling factor for variability in bioavailability for unobserved doses (-fold)¶ | 3.9 (2.7– 5.0) | |
| Proportional error (%) | 27.8 (20.8– 31.9) | |
| Additive error (mg/l) | 0.004 FIXED | |
| Pyrazinamide | ||
| Clearance (l/h)‡ | 5.41 (4.91– 5.81) | BSV: 19.1% (16.4– 26.0) |
| Volume of central compartment (l)‡ | 62.8 (57.6– 66.3) | BSV: 12.3% (0.1–14.9) |
| Bioavailability | 1 FIXED | BSV: 30.6% (20.8–34.6) BOV: 14.4% (7.8–19.3) |
| Absorption lag Time (h), prior§ | 0.47 (0.42–0.74) PRIOR: 0.49 |
BOV: 74.6 (49.3–84.7) |
| Ka - Absorption rate (1/h), prior§ | 1.26 (1.15–1.83) PRIOR: 3.51 |
BOV: 13.9% (0.1–14.0) |
| Scaling factor for BOV in bioavailability for unobserved doses (-fold)¶ | 2.93 (2.13–5.61) | |
| Proportional Error (%) | 7.78 (6.0–10.9) | |
| Additive error (mg/l) | 1.17 (0.68–1.45) | |
†Obtained with a non-parametric bootstrap (n = 300).
‡All clearance and volume parameters have been allometrically scaled with fat-free mass, and the typical values reported here refer to the typical patient, with fat-free mass of 47 kg.
§These parameters were estimated using a prior.
¶These scaling factors modulate the size of the between-occasion variability in bioavailability for the unobserved doses.
BOV: Between occasion variability; BSV: Between subject variability.
Figure 1. . Visual predictive check for rifampicin, pyrazinamide and isoniazid.

Visual predictive check for pharmacokinetics of rifampicin (A), pyrazinamide (B) and isoniazid stratified by NAT2 acetylator status (C). The dashed and solid lines are the 5th percentile, median and 95th percentile of the observed concentrations, while the shaded regions represent the corresponding 95% CIs for the same percentiles. The subplot in each panel or stratum shows the same VPC with a logarithmic transformation applied to the y-axis.
VPC: Visual predictive check.
For isoniazid, the concentration time data were best described by a two-compartment disposition with first-order elimination and first-order absorption process with a lag time. Priors were included on the absorption rate constant and lag time using previously reported parameter estimates [13]. Compared with total bodyweight, FFM was best size descriptor for allometric scaling of all clearance and volume parameters. Clearance of isoniazid was dependent on NAT2 acetylator status: (ΔOFV = 41, 2 degrees of freedom, p-value < 0.001) among fast and intermediate acetylators clearance was 2.3- and 1.6-times that of slow acetylators, respectively. The final parameter estimates together with 95% CIs are shown in Table 4 and a visual predictive check is displayed in Figure 1, showing how the model suitably describes the difference in concentrations among the acetylator groups.
Pyrazinamide concentrations were best described by using a one-compartment model with first-order absorption and elimination. Priors were introduced on absorption parameters (rate constant ka and absorption lag) based on parameter estimates of a previous pharmacokinetic model [45] as there was insufficient data to characterize the process. Parameter estimates from the final model are presented in Table 4, and a visual predictive check showing good fit of the model to the data is provided in Figure 1. Between-subject variability was retained in the final model on clearance, volume and bioavailability, and between-occasion variability was supported on absorption lag, absorption rate constant and bioavailability.
For both isoniazid and pyrazinamide, the between-occasion variability for bioavailability was found to be 3.9- and 2.9-times higher for the data following dose administration not supervised at the clinic, in other words, the predose concentrations on the pharmacokinetic visit day. This was included in the model to account for the uncertainty around the precise information about dose intake and other factors that may affect the absorption, such as food intake with the dose. The effect of scaling the between-occasion variability for bioavailability on data following unsupervised dose was not supported by the model describing the PK of rifampicin. The additive error for rifampicin and isoniazid was fixed to 20% of the limit of detection because the estimate from the model was very close to 0 and poorly determined. This conservative choice had little effect on the model fit, but it was included to ensure that the model does not treat the lower concentrations as more precise than they realistically are.
Model derived individual values of exposure area under the curve (AUC and Cmax) for rifampicin isoniazid, and pyrazinamide are presented in Figure 2. The median (IQR) AUC and Cmax for rifampicin, isoniazid and pyrazinamide were 24.1 h·mg/l (19.2–34.8) and 4.70 mg/l (3.93–5.98), 10.0 h·mg/l (7.07–17.8) and 2.89 mg/l (2.43–3.29), 350 h·mg/l (279–450) and 28.0 mg/l (22.0–34.1), respectively.
Figure 2. . Model-predicted individual exposures (AUC0–24 and Cmax).

Model-predicted individual exposures (AUC0–24 and Cmax) of rifampicin (A), pyrazinamide (B) and isoniazid (C). For isoniazid, the values are stratified by NAT2 acetylator status. Each point represents the average AUC or Cmax per individual.
AUC: Area under the curve.
Discussion
We determined the prevalence of genetic variability in selected SLCO1B1, NAT2, PXR, UGT1A and ABCB1 SNPs in a cohort of South African tuberculosis patients, the majority being of Black African ethnicity. Our data add to the current evidence of genetic variants having pharmacogenetic relevance among Africans. We describe the PK of first-line tuberculosis drugs and the effects of genetic variation on rifampicin and isoniazid pharmacokinetic parameters. As hypothesized, NAT2 acetylator status was significantly associated with isoniazid clearance, resulting in higher clearance and lower drug exposures in rapid and intermediate acetylators. None of the genotypes tested showed an effect on rifampicin pharmacokinetic parameters. Model derived typical concentrations of first-line tuberculosis drugs included in the drug regimens were low overall, when compared with the commonly cited therapeutic ranges [50,51] but in keeping with drug levels reported in some other African cohorts [10,12,17,32].
There have been several conflicting reports from studies investigating the effects of genetic variation in the SLCO1B1 gene on rifampicin PK. Studies from South Africa and Uganda [12,17,29] found significant effects of the SLCO1B1 rs4149032 and rs11045819 SNP, while studies in another African population and a study in South India [52,53] found no effects of these SNPs on rifampicin pharmacokinetic parameters. The frequency of the rs4149032 SNP in our study was similar to reports from previous African studies. The frequency of the rs4149032 CC genotype was 10% in our study, similar to other African cohorts (12–14%) [12,53] and lower than a study in India reporting 23% frequency [52]. There may be several plausible reasons for the differences in effects of the SNPs studied on rifampicin PK; such as disparities in prevalence of the SNP variation, use of tag SNPs that may not have the same LD patterns within different populations, the complexity of competing metabolic processes and small sample sizes studied. Furthermore, South Africa has a highly diverse population and race or ethnic mixture may contribute to the differences in genetic effects.
The significant effects of isoniazid acetylator status on clearance of isoniazid are in keeping with several studies investigating the impact on isoniazid efficacy based on adequate drug concentrations or risk of toxicity. Similar to findings from other studies in Africa and Asia, the majority of our patients were slow or intermediate acetylators (>80%) based on NAT2 genotype with less than 10–20% of population tested having a rapid acetylator status. Isoniazid concentrations were low overall in our patients with >60% of the patients having intermediate or rapid acetylator status resulting in higher clearance with no significant toxicity requiring drug discontinuation reported. Some studies suggest that isoniazid dose adjustment based on acetylator status improves treatment outcomes and lowers risk of serious adverse events and need for drug discontinuation [28,30].
The clinical relevance of the low drug concentrations reported in our study are unclear and require further investigation. There have been conflicting results from studies investigating the effects of the low drug concentrations on clinical outcomes, with some studies showing a significant association between drug concentrations and sputum culture conversion rates or other clinical outcomes, while others did not [51]. Lack of association could be due to the limitations of these studies using retrospective design, small sample size, single 2-h concentrations as a proxy for drug exposure and differing treatment outcome measures (sputum culture conversion definitions or culture methods). One study using a classification and regression tree analysis found new AUC and Cmax cutoffs that predict >91% of the treatment outcomes [9]. The Cmax cutoffs were higher than currently used cutoffs for isoniazid (8.8 vs 3 mg/l) and pyrazinamide (58.3 vs 20 mg/l) but lower for rifampicin (6.6 vs 8 mg/l) and found an association between Cmax of pyrazinamide and 2-month sputum conversion. Other studies found, for patients with isoniazid Cmax >4.6 mg/l higher isoniazid exposures were associated with improved rates of culture conversion [10] and rifampicin Cmax >8.2 mg/l associated with improved treatment outcomes [54]. Although these and other currently cited reference ranges and targets need further validation and clinically validated targets need to be better defined, it is clear that adequate concentrations of drugs in plasma and sites of action are needed for improved outcomes and to prevent emergent resistance to existing antituberculosis drugs.
We acknowledge the limitations of our study. This includes a relatively small sample of patients with drug concentration data and the relatively sparse sampling schedule. However, nonlinear mixed-effects modeling used in this study has been shown to analyze sparse data well. The lack of significant associations observed between SNPs studied and pharmacokinetic parameters for rifampicin may be due to inadequate numbers of patients in the groups for each of the genotypes studied. We included a limited number of SNPs in our study based on previous literature that showed association between these SNPs and drug response, toxicity or disease susceptibility. Other relevant SNPs or rare variants that were not included in this study may significantly impact rifampicin or isoniazid pharmacokinetic parameters. Genotype frequencies in our population may differ from other populations, and our findings may therefore be limited to similar African populations.
In conclusion, we found a high prevalence of genetic variation in our population. We found no significant association between SNPs tested and rifampicin pharmacokinetic parameters. Drug concentrations were low overall and isoniazid fast and intermediate acetylator genotype resulted in higher clearance and lower concentrations of isoniazid in the majority of patients. Clinical relevance of the effects of genetic variation on first-line tuberculosis drug concentrations, low drug concentrations and the need for isoniazid dose adjustment based on acetylator status requires further investigation.
Summary points.
We report the prevalence and effect of genetic polymorphisms in relevant drug-metabolizing and transporter enzymes on pharmacokinetic parameters of isoniazid and rifampicin, in a cohort of South African tuberculosis patients.
We found a high prevalence of genetic variation in our population.
Among 172 patients (∼70% HIV coinfected), 18, 43 and 34% were classified as rapid, intermediate and slow NAT2 acetylators, respectively.
Among the 58 patients contributing drug concentrations, rapid and intermediate acetylators had 2.3- and 1.6-times faster isoniazid clearance than slow acetylators.
No association was observed between rifampicin pharmacokinetics and SLCO1B1, ABCB1, UGT1A or PXR genotypes.
The median (interquartile range) area under the concentration–time curve for rifampicin, isoniazid and pyrazinamide was 24.1 h·mg/l (19.2–34.8), 10.0 h·mg/l (7.07–17.8), 350 h·mg/l (279–450), respectively.
Drug concentrations were low overall compared with cited reference ranges.
The NAT2 genotype was significantly associated with isoniazid clearance.
In contrast to previous studies in African subjects, but in keeping with more recent reports, we did not detect any effect of SLCO1B1 on rifampicin pharmacokinetic parameters.
Clinical relevance of the effects of genetic variation on first-line tuberculosis drug concentrations, low drug concentrations, and the need for isoniazid dose adjustment based on acetylator status requires further investigation.
Acknowledgements
The authors gratefully acknowledge the study participants fortheir participation in the IMPRESS pharmacokinetic study; study nurses, counsellors, clinicians, the study co-ordinator Ms Dhineshree Govender and the IMPRESS study team for their work in the pharmacokinetic study. Special acknowledgement to Mrs Magashree Govender and the pharmacy team for support on the study. We thank the following laboratories and laboratory staff; CAPRISA laboratory (Mrs Natasha Samsunder, and lab staff), KwaZulu-Natal Research Institute for Tuberculosis and HIV (KRITH) Pharmacology Core (Dr John Adamson and Ms Katya Govender) and Global Clinical & Viral Laboratories, for laboratory support. Mr Day Munatsi and Mrs Precious Radebe for data management on the study. Dr Jane Bredenkamp (Forest Molecular Genetics Programme, University of Pretoria) for the TaqMan® OpenArray genotyping assay analysis.
The Division of Clinical Pharmacology at the University of CapeTown acknowledges Novartis Pharma for their support of the development of pharmacometric skills in Africa.
Footnotes
Author contributions
The study concept and design were devised by A Naidoo, N Padayatchi, K Naidoo, H McIlleron and V Ramsuran. The manuscript was drafted by A Naidoo, V Ramsuran, P Denti, M Chirehwa. Statistical analysis (NLME modelling and data analysis) was conducted by P Denti and M Chirehwa with general statistical support from N Yende-Zuma. Pharmacokinetic analysis was by J Adamson and K Govender. All authors participated in acquisition, analysis, or interpretation of data.
All authors had access to data, commented on drafts, and approvedthe final report. The corresponding author had final responsibility for the decision to submit for publication.
Financial & competing interests disclosure
This publication was made possible by grant number 5R24TW008863from the Office of Global AIDS Coordinator and the US Department of Health and Human Services, National Institutes of Health (NIH OAR and NIH OWAR – grant number 5R24TW008863). Research reported in this publication was supported by the European & Developing Countries Clinical Trials Partnership (EDCTP) (TA.2011.40200.044), the South African Medical Research Council CAPRISA HIV-TB Pathogenesis and Treatment Research Unit the South African Medical Research Council under a Self-Initiated Research Grant. Study drug was donated by Bayer Pharmaceuticals. A Naidoo was the recipient of the University of KwaZulu-Natal College of Health Sciences Scholarship.The sponsor of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report.
H McIlleron is supported in part by theWellcome Trust (206379/Z/17/Z). K Naidoo and N Padayatchi were supported by the Columbia University – South Africa Fogarty AIDS260 International Training and Research Program (AITRP, grant number D43TW000231). V Ramsuran was supported in part by a research Flagship grant from the South African Medical Research Council (MRC-RFA-UFSP-01-2013/UKZN HIVEPI) and by the Sub-Saharan African Network for TB/HIV Research Excellence (SANTHE) – a DELTAS Africa Initiative programme (grant number DEL-15-006).
N Padayatchi is the principal studyinvestigator, on the Improving Retreatment Success Trial (IMPRESS) (NCT02114684). Bayer Pharmaceuticals donated the study drug (moxifloxacin) used during the trial. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Disclaimer
The contents of the manuscript are solely the responsibility ofthe authors and do not necessarily represent the official views of the US government, EDCTP, Wellcome Trust, Fogarty International Center, National Institutes of Health or the Medical Research Council.
References
- 1.WHO. Global Tuberculosis Report 2017.http://www.who.int/tb/publications/global_report/en/ [Google Scholar]
- 2.South African Department of Health. Pretoria: National Department of Health; 2014. South African National Tuberculosis Management Guidelines.www.health-e.org.za/2014/06/10/guidelines-national-tuberculosis-management-guidelines/ [Google Scholar]
- 3.Boeree MJ, Diacon AH, Dawson R, et al. A dose-ranging trial to optimize the dose of rifampin in the treatment of tuberculosis. Am. J. Respir. Crit. Care Med. 2015;191:1058–1065. doi: 10.1164/rccm.201407-1264OC. [DOI] [PubMed] [Google Scholar]
- 4.Boeree MJ, Heinrich N, Aarnoutse R, et al. High-dose rifampicin, moxifloxacin, and SQ109 for treating tuberculosis: a multi-arm, multi-stage randomised controlled trial. Lancet Infect. Dis. 2017;17:39–49. doi: 10.1016/S1473-3099(16)30274-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Ingen J, Aarnoutse RE, Donald PR, et al. Why do we use 600 mg of rifampicin in tuberculosis treatment? Clin. Infect. Dis. 2011;52:e194–e199. doi: 10.1093/cid/cir184. [DOI] [PubMed] [Google Scholar]
- 6.Srivastava S, Pasipanodya JG, Meek C, et al. Multidrug-resistant tuberculosis not due to noncompliance but to between-patient pharmacokinetic variability. J. Infect. Dis. 2011;204:1951–9. doi: 10.1093/infdis/jir658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chideya S, Winston CA, Peloquin CA, et al. Isoniazid, rifampin, ethambutol, and pyrazinamide pharmacokinetics and treatment outcomes among a predominantly HIV-infected cohort of adults with tuberculosis from Botswana. Clin. Infect. Dis. 2009;48:1685–94. doi: 10.1086/599040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mah A, Kharrat H, Ahmed R, et al. Serum drug concentrations of INH and RMP predict 2-month sputum culture results in tuberculosis patients. Int. J. Tuberc. Lung Dis. 2015;19:210–215. doi: 10.5588/ijtld.14.0405. [DOI] [PubMed] [Google Scholar]
- 9.Pasipanodya JG, McIlleron H, Burger A, et al. Serum drug concentrations predictive of pulmonary tuberculosis outcomes. J. Infect. Dis. 2013;208:1464–1473. doi: 10.1093/infdis/jit352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rockwood N, Pasipanodya JG, Denti P, et al. Concentration-dependent antagonism and culture conversion in pulmonary tuberculosis. Clin. Infect. Dis. 2017;64:1350–1359. doi: 10.1093/cid/cix158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilkins JJ, Langdon G, McIlleron H, et al. Variability in the population pharmacokinetics of pyrazinamide in South African tuberculosis patients. Eur. J. Clin. Pharmacol. 2006;62:727–35. doi: 10.1007/s00228-006-0141-z. [DOI] [PubMed] [Google Scholar]
- 12.Chigutsa E, Visser ME, Swart EC, et al. The SLCO1B1 rs4149032 polymorphism is highly prevalent in South Africans and is associated with reduced rifampin concentrations: dosing implications. Antimicrob. Agents Chemother. 2011;55:4122–4127. doi: 10.1128/AAC.01833-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilkins JJ, Langdon G, McIlleron H, et al. Variability in the population pharmacokinetics of isoniazid in South African tuberculosis patients. Br. J. Clin. Pharmacol. 2011;72:51–62. doi: 10.1111/j.1365-2125.2011.03940.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pasipanodya JG, Srivastava S, Gumbo T. Meta-analysis of clinical studies supports the pharmacokinetic variability hypothesis for acquired drug resistance and failure of antituberculosis therapy. Clin. Infect. Dis. 2012;55:169–177. doi: 10.1093/cid/cis353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Devaleenal Daniel B, Ramachandran G, Swaminathan S. The challenges of pharmacokinetic variability of first-line anti-TB drugs. Expert Rev. Clin. Pharmacol. 2017;10:47–58. doi: 10.1080/17512433.2017.1246179. [DOI] [PubMed] [Google Scholar]
- 16.McIlleron H, Rustomjee R, Vahedi M, et al. Reduced antituberculosis drug concentrations in HIV-infected patients who are men or have low weight: implications for international dosing guidelines. Antimicrob. Agents Chemother. 2012;56:3232–3238. doi: 10.1128/AAC.05526-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gengiah TN, Botha JH, Soowamber D, et al. Low rifampicin concentrations in tuberculosis patients with HIV infection. J. Infect. Dev Ctries. 2014;8:987–93. doi: 10.3855/jidc.4696. [DOI] [PubMed] [Google Scholar]
- 18.Naidoo A, Ramsuran V, Chirehwa M, et al. Effect of genetic variation in UGT1A and ABCB1 on moxifloxacin pharmacokinetics in South African patients with tuberculosis. Pharmacogenomics. 2018;19:17–29. doi: 10.2217/pgs-2017-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramachandran G, Swaminathan S. Role of pharmacogenomics in the treatment of tuberculosis: a review. Pharmgenomics Pers. Med. 2012;5:89–98. doi: 10.2147/PGPM.S15454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim RB. Organic anion-transporting polypeptide (OATP) transporter family and drug disposition. Eur. J. Clin. Invest. 2003;33(Suppl. 2):1–5. doi: 10.1046/j.1365-2362.33.s2.5.x. [DOI] [PubMed] [Google Scholar]
- 21.Roth M, Obaidat A, Hagenbuch B. OATPs, OATs and OCTs: the organic anion and cation transporters of the SLCO and SLC22A gene superfamilies. Br. J. Clin. Pharmacol. 2012;165:1260–1287. doi: 10.1111/j.1476-5381.2011.01724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen J, Raymond K. Roles of rifampicin in drug–drug interactions: underlying molecular mechanisms involving the nuclear pregnane X receptor. Ann. Clin. Microbiol. Antimicrob. 2006;5:3. doi: 10.1186/1476-0711-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barbarino JM, Haidar CE, Klein TE, et al. PharmGKB summary: very important pharmacogene information for UGT1A1 . Pharmacogenet. Genomics. 2014;24:177–183. doi: 10.1097/FPC.0000000000000024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hariparsad N, Chu X, Yabut J, et al. Identification of pregnane-X receptor target genes and coactivator and corepressor binding to promoter elements in human hepatocytes. Nucleic Acids Res. 2009;37:1160–1173. doi: 10.1093/nar/gkn1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hodges LM, Markova SM, Chinn LW, et al. Very important pharmacogene summary: ABCB1 (MDR1, P-glycoprotein) Pharmacogenet. Genomics. 2011;21:152–161. doi: 10.1097/FPC.0b013e3283385a1c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kliewer SA, Goodwin B, Willson TM. The nuclear pregnane x receptor: a key regulator of xenobiotic metabolism. Endocr. Rev. 2002;23:687–702. doi: 10.1210/er.2001-0038. [DOI] [PubMed] [Google Scholar]
- 27.Ellard GA, Gammon PT. Pharmacokinetics of isoniazid metabolism in man. J. Pharmacokinet. Biopharm. 1976;4:83–113. doi: 10.1007/BF01086149. [DOI] [PubMed] [Google Scholar]
- 28.Azuma J, Ohno M, Kubota R, et al. NAT2 genotype guided regimen reduces isoniazid-induced liver injury and early treatment failure in the 6-month four-drug standard treatment of tuberculosis: a randomized controlled trial for pharmacogenetics-based therapy. Eur. J. Clin. Pharmacol. 2013;69:1091–1101. doi: 10.1007/s00228-012-1429-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weiner M, Peloquin C, Burman W, et al. Effects of tuberculosis, race, and human gene SLCO1B1 polymorphisms on rifampin concentrations. Antimicrob. Agents Chemother. 2010;54:4192–4200. doi: 10.1128/AAC.00353-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jung JA, Kim TE, Lee H, et al. A proposal for an individualized pharmacogenetic-guided isoniazid dosage regimen for patients with tuberculosis. Drug Des. Devel. Ther. 2015;9:5433–5438. doi: 10.2147/DDDT.S87131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Denti P, Jeremiah K, Chigutsa E, et al. Pharmacokinetics of isoniazid, pyrazinamide, and ethambutol in newly diagnosed pulmonary TB Patients in Tanzania. PLoS ONE. 2015;10 doi: 10.1371/journal.pone.0141002. e0141002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tostmann A, Mtabho CM, Semvua HH, et al. Pharmacokinetics of first-line tuberculosis drugs in Tanzanian patients. Antimicrob. Agents Chemother. 2013;57:3208–3213. doi: 10.1128/AAC.02599-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mboowa G. Genetics of Sub-Saharan African human population: implications for HIV/AIDS, tuberculosis, and malaria. Int. J. Evol. Biol. 2014;2014 doi: 10.1155/2014/108291. 108291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McIlleron H, Willemse M, Werely CJ, et al. Isoniazid plasma concentrations in a cohort of South African children with tuberculosis: implications for international pediatric dosing guidelines. Clin. Infect. Dis. 2009;48:1547–1553. doi: 10.1086/598192. [DOI] [PubMed] [Google Scholar]
- 35.Naidoo A, Chirehwa M, McIlleron H, et al. Effect of rifampicin and efavirenz on moxifloxacin concentrations when co-administered in patients with drug-susceptible TB. J. Antimicrob. Chemother. 2017;72:1441–1449. doi: 10.1093/jac/dkx004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McDonagh EM, Boukouvala S. PharmGKB summary: very important pharmacogene information for N-acetyltransferase 2. Pharmacogenet. Genomics. 2014;24(8):409–425. doi: 10.1097/FPC.0000000000000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whirl-Carrillo M, McDonagh EM, Hebert JM, et al. Pharmacogenomics knowledge for personalized medicine. Clin. Pharmacol. Ther. 2012;92:414–417. doi: 10.1038/clpt.2012.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Farrar JS, Palais RA, Wittwer CT. Snapback primer genotyping of the Gilbert syndrome UGT1A1 (TA)(n) promoter polymorphism by high-resolution melting. Clin. Chem. 2011;57:1303–1310. doi: 10.1373/clinchem.2011.166306. [DOI] [PubMed] [Google Scholar]
- 39.Hein DW, Doll MA. Accuracy of various human NAT2 SNP genotyping panels to infer rapid, intermediate and slow acetylator phenotypes. Pharmacogenomics. 2012;13:31–41. doi: 10.2217/pgs.11.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beal S, Sheiner L, Boeckmann A, et al. MD, USA: ICON Development Solutions; 2009. NONMEM users guides (1989-2009) [Google Scholar]
- 41.Keizer RJ, Karlsson MO, Hooker A. Modeling and simulation workbench for NONMEM: tutorial on Pirana, PsN, and Xpose. CPT Pharmacometrics Syst. Pharmacol. 2013;2:e50. doi: 10.1038/psp.2013.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Savic RM, Jonker DM, Kerbusch T, et al. Implementation of a transit compartment model for describing drug absorption in pharmacokinetic studies. J. Pharmacokinet. Pharmacodyn. 2007;34:711–726. doi: 10.1007/s10928-007-9066-0. [DOI] [PubMed] [Google Scholar]
- 43.Gisleskog PO, Karlsson MO, Beal SL. Use of prior information to stabilize a population data analysis. J. Pharmacokinet. Pharmacodyn. 2002;29:473–505. doi: 10.1023/a:1022972420004. [DOI] [PubMed] [Google Scholar]
- 44.Denti P, Smythe W, Simonsson US, et al. The 3rd International Workshop on Clinical Pharmacology of TB Drugs. Boston, MA, USA: 11 September 2010. A population pharmacokinetic model for rifampicin auto-induction. Presented at: [Google Scholar]
- 45.Chirehwa MT, McIlleron H, Rustomjee R, et al. Pharmacokinetics of pyrazinamide and optimal dosing regimens for drug-sensitive and -resistant tuberculosis. Antimicrob. Agents Chemother. 2017:61. doi: 10.1128/AAC.00490-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beal SL. Ways to fit a PK model with some data below the quantification limit. J. Pharmacokinet. Pharmacody. 2001;28:481–504. doi: 10.1023/a:1012299115260. [DOI] [PubMed] [Google Scholar]
- 47.Anderson BJ, Holford NH. Mechanism-based concepts of size and maturity in pharmacokinetics. Annu. Rev. Pharmacol. Toxicol. 2008;48:303–332. doi: 10.1146/annurev.pharmtox.48.113006.094708. [DOI] [PubMed] [Google Scholar]
- 48.Kendall EA, Shrestha S, Cohen T, et al. Priority-setting for novel drug regimens to treat tuberculosis: an epidemiologic model. PLoS Med. 2017;14 doi: 10.1371/journal.pmed.1002202. e1002202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Machiela MJ, Chanock SJ. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics. 2015;31:3555–3557. doi: 10.1093/bioinformatics/btv402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Egelund EF, Alsultan A, Peloquin CA. Optimizing the clinical pharmacology of tuberculosis medications. Clin. Pharmacol. Ther. 2015;98(4):387–393. doi: 10.1002/cpt.180. [DOI] [PubMed] [Google Scholar]
- 51.Sekaggya-Wiltshire C, Lamorde M, Kiragga AN, et al. The utility of pharmacokinetic studies for the evaluation of exposure–response relationships for standard dose anti-tuberculosis drugs. Tuberculosis. 2018;108:77–82. doi: 10.1016/j.tube.2017.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ramesh K, Hemanth Kumar AK, Kannan T, et al. SLCO1B1 gene polymorphisms do not influence plasma rifampicin concentrations in a south Indian population. Int. J. Tuberc. Lung Dis. 2016;20:1231–1235. doi: 10.5588/ijtld.15.1007. [DOI] [PubMed] [Google Scholar]
- 53.Sloan DJ, McCallum AD, Schipani A, et al. Genetic determinants of the pharmacokinetic variability of rifampin in Malawian adults with pulmonary tuberculosis. Antimicrob. Agents Chemother. 2017;61 doi: 10.1128/AAC.00210-17. e00210-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chigutsa E, Pasipanodya JG, Visser ME, et al. Impact of nonlinear interactions of pharmacokinetics and MICs on sputum bacillary kill rates as a marker of sterilizing effect in tuberculosis. Antimicrob. Agents Chemother. 2015;59:38–45. doi: 10.1128/AAC.03931-14. [DOI] [PMC free article] [PubMed] [Google Scholar]