

Abstract
Many drug–drug interactions (DDIs) are based on alterations of the plasma concentrations of a victim drug due to another drug causing inhibition and/or induction of the metabolism or transporter‐mediated disposition of the victim drug. In the worst case, such interactions cause more than tenfold increases or decreases in victim drug exposure, with potentially life‐threatening consequences. There has been tremendous progress in the predictability and modeling of DDIs. Accordingly, the combination of modeling approaches and clinical studies is the current mainstay in evaluation of the pharmacokinetic DDI risks of drugs. In this paper, we focus on the methodology of clinical studies on DDIs involving drug metabolism or transport. We specifically present considerations related to general DDI study designs, recommended enzyme and transporter index substrates and inhibitors, pharmacogenetic perspectives, index drug cocktails, endogenous substrates, limited sampling strategies, physiologically‐based pharmacokinetic modeling, complex DDIs, methodological pitfalls, and interpretation of DDI information.
Unintentional and mismanaged drug–drug interactions (DDIs) are a common reason for preventable adverse events.1 As the population is aging and polypharmacotherapy is becoming progressively more common, there is an increased likelihood of DDIs that can inadvertently lead to exaggeration of adverse effects or—in some cases—loss of drug efficacy. As these kind of events cannot be prevented without recognizing the need to adjust medications according to DDI risks, there is a need for carefully planned preclinical and clinical DDI studies during drug development, and typically also after marketing approval, as well as for modeling studies, databases, and clinical decision support systems that can be easily implemented and used to improve clinical decision making.
In the past, extreme safety concerns caused by DDIs have led to multiple market withdrawals, such as those of mibefradil, terfenadine, cisapride, and cerivastatin in the late 1990s and early 2000s. Due to such unfortunate incidents and the rapid accumulation of scientific knowledge that has improved the understanding of DDI mechanisms and awareness of DDI risks, regulatory agencies have frequently updated their guidances on drug interaction studies. For example, the last clinical drug interaction studies guidance by the US Food and Drug Administration (FDA) was published in 2017 and that by the European Medicines Agency (EMA) is currently being revised.2, 3 Even though these guidelines are directed for studies performed for drugs under development, their concepts can be applied to drugs on the market as well.
The above developments have led to marked advances in the conduct of DDI studies during drug development. As a result, the number of drug withdrawals due to DDIs has dramatically decreased and detailed knowledge on mechanisms, clinical relevance, and management of DDIs mediated by inhibition or induction of cytochrome P450 (CYPs) enzymes, some other enzymes, and key transporters are, in most cases, available already at the time of marketing approval. For example, among the 34 drugs approved by the FDA in 2017, 5 had been identified as sensitive substrates of CYP3A or organic anion‐transporting polypeptide (OATP) 1B1, and 3 had been considered as strong inhibitors of CYP3A, OATP1B1, or breast cancer resistance protein (BCRP), whereas no strong inducers had been identified.4
A major proportion of harmful drug interactions is based on alterations of the plasma concentrations of the victim drug due to the perpetrator drug causing a change in the metabolism or transporter‐mediated disposition of the victim drug. Inhibition of drug metabolism or transporter‐dependent elimination in most cases leads to elevated concentrations of the victim drug, whereas induction increases metabolic elimination, decreasing the concentrations of the victim. In the worst case, such interactions can lead to several hundred‐fold variations in drug exposure.5, 6 During the past decade, several review articles have been published focusing on various specific aspects related to clinical DDI studies.7, 8, 9, 10 In this paper, we present an overview of the basic methodology of clinical DDI studies that can be used when investigating a specific drug as a victim or perpetrator of pharmacokinetic DDIs mediated by inhibition or induction of drug‐metabolizing enzymes and/or transporters, with an attempt to pinpoint specific considerations that we have found important on the basis of our own experience in clinical DDI studies. As much of the methodology described in regulatory guidance is focused on studies carried out during drug development, we extend the review beyond the regulatory guidance, highlighting certain specific questions related to complex DDIs, pharmacogenetics, methodological pitfalls, and interpretation of DDI information.
DESIGN OF CLINICAL DDI STUDIES
General considerations of clinical DDI study design
With regard to pharmacokinetic DDIs, the study hypotheses and objectives essentially define the most suitable study design. Based on prior information, an evaluation of interaction risk is usually carried out continuously during drug development (Figure 1 ). When the drug enters clinical development, this evaluation is typically based on in vitro, animal, and in silico data, but the evaluation is rapidly strengthened by accumulating clinical data. If the drug is in later stages of clinical development or already on the market, DDI studies may be triggered by retrospective analyses of population data, case reports, or additional in vitro data. Consequently, the drug of interest can be evaluated either as a substrate (victim drug), or as an inhibitor or inducer (perpetrator drug) of drug‐metabolizing enzymes or transporters. For example, a retrospective epidemiologic analysis of the risk of rhabdomyolysis in cerivastatin users with concomitant clopidogrel published in 201211 triggered an investigation that resulted in identification of clopidogrel as an inhibitor of CYP2C8.12
Figure 1.
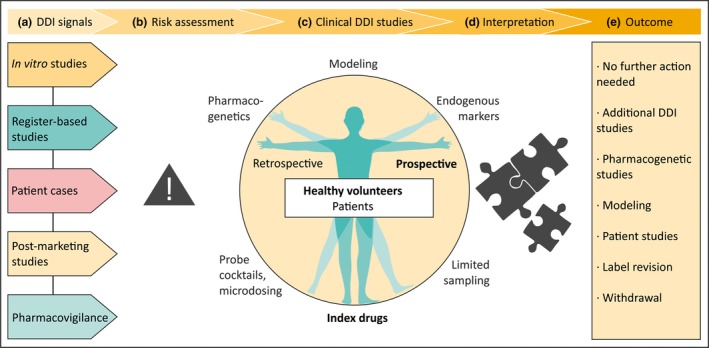
Investigation of drug–drug interactions (DDIs). (a) Signals of a potential DDI may arise from a variety of sources. (b) Signals need careful assessment before further action is taken. (c) The golden standard for clinical DDI studies is a prospective crossover study with index drugs, usually performed in healthy volunteers. (d) The interpretation of the obtained results determines the (e) implications of the study.
There are multiple design issues and requirements that need to be carefully considered already when preparing a clinical DDI study protocol (Table 1 ). The golden standard for clinical DDI studies is a prospective crossover study, usually performed in healthy volunteers. The crossover design reduces the effect of interindividual variability because individuals act as their own controls. Furthermore, interindividual variability is reduced in healthy subjects who have normal kidney and liver function, are not using concomitant medication, and are devoid of other interfering factors (e.g., smoking). In a two‐way pharmacokinetic crossover DDI study in healthy volunteers, sample sizes as low as 10–12 individuals are often sufficient to demonstrate clinically significant interaction, although a 2–3 times larger sample may be needed to demonstrate lack of DDI. In a crossover study, the washout period between study phases should be long enough to allow the drugs, metabolites, and their effects to be completely eliminated before the next phase, even when their elimination is impaired by strong inhibitors. When drug toxicity or adverse effects are an issue, DDI studies can be performed in patient populations, but they typically require a larger sample size due to greater variability caused by various individual factors, particularly if a parallel group design is used.
Table 1.
List of recommended considerations and requirements of interventional DDI studies and examples of potential pitfalls in DDI studies and their interpretation
| Typical requirements of a DDI study | Potential pitfalls of DDI studies |
|---|---|
| General design issues | |
| Healthy volunteers if no safety concerns | Risky drugs given to healthy subjects |
| Patients if safety concerns or clinical focus | Bias and confounding in observational patient studies |
| Crossover design | Parallel group design may produce bias |
| Sufficient washout to eliminate carry‐over effects | Insufficient washout (e.g., a slowly eliminated metabolite still present) |
| Placebo control and blinding (e.g., in case of pharmacodynamic end points) | |
| Dietary control/restrictions as necessary | |
| Appropriate sample collection, storage and analytical method to cover > 80–90% of the AUC of the victim drug and metabolites | Inaccurate or insensitive analytical method, degradation of analytes during storage or analysis |
| Monitoring of perpetrator pharmacokinetics (compliance, quantification of exposure, presence after washout) | Perpetrator exposure not documented |
| Pharmacodynamic assessment | Pharmacodynamic assessments neglected |
| DNA samples | |
| Biomarker samples in selected cases | |
| Necessary prior knowledge considered in design | Deficiencies in preclinical and early clinical data (e.g., in mass‐balance studies) |
| Safety issues | |
| Strict exclusion criteria (e.g., contraindications, pregnancy) | Careless exclusion criteria leading to risk of adverse effects |
| History, clinical examination, laboratory tests, and genotyping as necessary | |
| Safety monitoring and sufficient follow‐up | Insufficient follow‐up (residual drug effects) |
| Precautions and interventions to avoid adverse effects, even in the worst‐case DDI scenario | Rescue interventions not prearranged |
| Blood sampling should generally not exceed the volume of blood donation | |
| Perpetrator (inhibitor/inducer) | |
| Selectivity and strength of index inhibitor (> 5‐fold increase in AUC possible) |
Suboptimal strength Nonselective inhibitor |
| Clinically relevant (high) dose depending on tolerability | Too low dose, leading to weak inhibition |
| Dosing to reach and maintain steady‐state, including time‐dependent inhibition and induction |
Clinically atypical dosing Duration of dosing too short/long to document maximal DDI |
| Victim substrate | |
| Sensitivity of index substrate | Lack of sensitivity (particularly if not considered in interpretation) |
| Documented selectivity of index substrate | Lack of selectivity (particularly if not considered in interpretation) |
| High first‐pass and short half‐life preferable | Long half‐life victim with a short‐acting or “presystemic” inhibitor |
| Monitoring a specific metabolic ratio can be useful | |
| Dose, expecting the worst‐case scenario | Too high dose |
| Staggered dosing | Victim drug administered too soon (or too long) after perpetrator to document maximal DDI |
AUC, area under plasma concentration‐time curve; DDI, drug–drug interaction.
If inhibition or induction caused by a drug is investigated, a probe or index substrate for a specific pharmacokinetic pathway is typically used as the victim drug (Table 2 ). Particularly in drug development, such studies are first focused on the enzyme toward which the investigated drug is expected to cause the strongest inhibition on the basis of in vitro inhibition data. If the DDI potential of a drug as an inhibitor is being studied, it should be administered preferably with multiple doses reaching and maintaining concentrations approximating steadystate at the highest clinically used doses, so that the study could reveal the worst‐case scenario (Figure 2 ). In addition, our recommendation is to use dose staggering, rather than simultaneous administration, to allow maximum inhibitor concentrations at the site of inhibition (e.g., an interval of 1 hour between the inhibitor and victim drug administration) to reveal the full extent of interaction, particularly if the absorption of the inhibitor can be delayed or if the victim is rapidly eliminated and has extensive first‐pass metabolism. On the other hand, to evaluate induction effects, a sufficient multiple dosing period to reach near full induction should be used. However, when both induction and inhibition are possible, a delay between perpetrator and victim drug administration of, for example, 12 hours can be considered to ensure full induction effect of the perpetrator. For example, the prototypical CYP inducer rifampin13 inhibits OATP1B1/1B3 immediately after dosing in vivo,14 but this effect is no longer present after 12 hours.15
Table 2.
Characteristics of possible index substrates of CYP enzymes
| Enzyme | Substrate | Sensitivitya | Other relevant enzymes/transporters | F | t1/2 (h) | Remarks | E/Fb |
|---|---|---|---|---|---|---|---|
| CYP1A2 | Agomelatine | ++++ | 0.05 | 1–2 | Limited availability in some countries | ||
| Caffeined | ++++ | NAT, XO | 1.00 | 3–7 | E/F | ||
| Melatonin | ++++ | CYP1A1c | 0.03 | 0.9 | |||
| Theophylline | ++ | CYP3A4, CYP2E1 | 0.96 | 9 | Adverse effects at high concentrations | E | |
| Tizanidined | ++++ | 0.34 | 2.5 | Adverse effects at high concentrations | F | ||
| CYP2B6 | Bupropiond | + | 11β‐HSD1c | 0.9 | 11 | Hydroxybupropion to bupropion AUC ratio is a selective and more sensitive marker of CYP2B6 than bupropion AUC | E |
| (S)‐Ketamine | ++ | CYP3A4 | 0.08 | 2–6 | Parenteral formulation can be given orally | ||
| Efavirenz |
N/A (++) |
CYP2A6 | N/A | 52–76 | Limited DDI data | E | |
| CYP2C8 | Amodiaquine | N/A | N/A | 5 |
Limited DDI data Limited availability Metabolite t1/2 > 100 hours |
E | |
| Daprodustat | ++++ | N/A | 1 |
Limited DDI data Limited availability, currently in phase III |
|||
| Dasabuvird | ++++ | CYP3A4 | 0.70 | 5.5–6 | |||
| Repaglinided | +++ | CYP3A4, OATP1B1 | 0.56 | 0.8 | Adverse effects at high concentrations | E/F | |
| CYP2C9 | (S)‐Warfarin |
++ (+++) |
0.93 (racemic) | 21–43 | Bleeding risk | E/F | |
| Flurbiprofen | ++ | 0.92 | 5.5 |
4′‐Hydroxyflurbiprofen/flurbiprofen ratio is a sensitive marker of CYP2C9 Limited availability of appropriate formulation |
|||
| Fluvastatind |
+ (++) |
OATP1B1 | 0.29 | 0.7 | |||
| Tolbutamided |
++ (+++) |
0.8–0.9 | 5.9 |
Limited availability Adverse effects at high concentrations |
E/F | ||
| CYP2C19 | Lansoprazole |
++ (+++) |
CYP3A4 | 0.81 | 0.9 | Delayed absorption | F |
| Omeprazoled |
+++ (++++) |
CYP3A4 | 0.53 | 0.7 |
Delayed absorption Inhibitor of CYP2C19 |
E/F | |
| Pantoprazoled |
N/A (+++) |
0.77 | 1.0 |
Delayed absorption Breath test established |
|||
| Rabeprazole |
++ (+++) |
CYP3A4 | 0.52 | 1.5 | Delayed absorption | ||
| CYP2D6 | Desipramine |
+++ (+++) |
CYP3A4 | 0.38 | 28 | Limited availability in some countries | E/F |
| Dextromethorphand |
++++ (++++) |
CYP3A4 | N/A | 3.4 | Dextrorphan/dextromethorphan ratio used as the index | F | |
| Metoprolol |
++ (+++) |
CYP3A4 | 0.38 | 3.2 | E | ||
| Nebivolol |
+++ (++++) |
CYP2C19 | N/A | 11 | F | ||
| Tolterodine |
++++ (++++) |
CYP3A4 | 0.26 (NMs) | 2.3 (NMs) | |||
| CYP3A4 | Buspirone | ++++ | 0.04 | 2.4 | Sensitive to intestinal CYP3A4 inhibition | ||
| Midazolamd | ++++ | 0.44 | 1.9 |
i.v. formulation available to assess hepatic CYP3A4 Adverse effects at high concentrations |
E/F | ||
| Sildenafil | ++++ | CYP2C9 | 0.38 | 2.4 | |||
| Simvastatin (lactone)d | ++++ | < 0.05 | 2–3 |
Simvastatin acid substrate of OATP1B1 Sensitive to intestinal CYP3A4 inhibition |
|||
| Triazolam | ++++ | 0.44 | 2.9 | Adverse effects at high concentrations | F |
The table contains all the US Food and Drug Administration (FDA) and European Medicines Agency (EMA)‐recommended index substrates and some selected alternatives based on potential advantages in sensitivity, selectivity, and/or relative safety. The data are compiled primarily from the UW Metabolism and Transport Drug Interaction Database (Copyright University of Washington 1999–2019. Accessed: January−February 2019), secondarily from drug labels.
AUC, area under plasma concentration‐time curve; CYP, cytochrome P450; E/F, European Medicines Agency/US Food and Drug Administration; F, bioavailability; HSD‐1, hydroxysteroid dehydrogenase type 1; NAT, N‐acetyl transferase; NMs, normal metabolizers; N/A, not available; OATP, organic anion‐transporting polypeptide; t1/2, elimination half‐life; XO, xanthine oxidase.
Indication of sensitivity is based on data from a trial with an EMA/FDA‐recommended index inhibitor of the affected CYP. In some cases, a second sensitivity value is given within brackets, based on pharmacogenetic data. Classification: ++++ a maximal > 10‐fold increase in AUC, +++ a maximal 5–10‐fold increase, ++ a maximal two to fivefold increase, + a maximal 1.25−2‐fold increase. Values for maximal fold increases in AUC are given in Table S1 .
Indicates if the drug is recommended as a clinical CYP probe substrate by the EMA (E) and/or the FDA (F) (2–3).
Based on in vitro data only.
Our recommended substrate.
Figure 2.
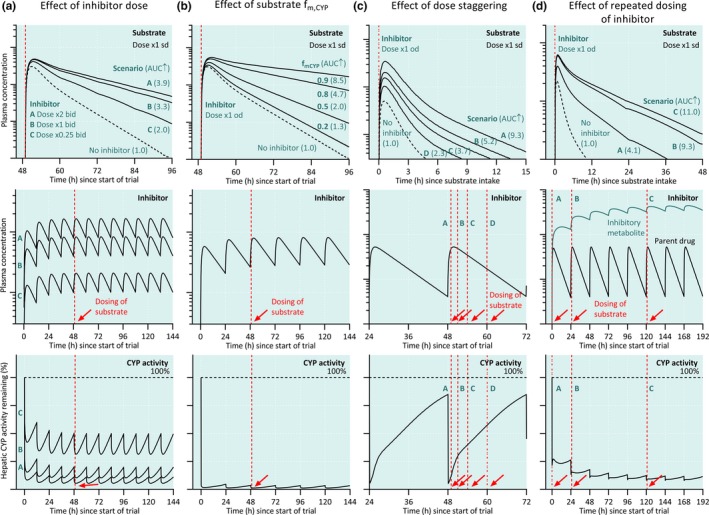
Design of drug–drug interaction (DDI) studies. (a) Effect of varying inhibitor dose (scenarios A‐C) on the plasma concentrations of a substrate drug. A sufficient dose of the perpetrator drug is necessary to reach strong inhibition and to detect and accurately quantify a potential DDI. (b) Effect of a cytochrome P450 (CYP) inhibitor on the plasma concentrations of a substrate with varying fractions metabolized by the affected CYP (fm,CYP). In combination with a CYP inhibitor, a substrate drug with a low fm,CYP will give a smaller DDI than a sensitive substrate drug with a high fm,CYP. (c) Effect of dose staggering (scenarios A‐D) on DDI magnitude. Strongest DDI can be detected when the victim drug is administered shortly after the inhibitor drug. (d) Effect of repeated dosing of perpetrator whose metabolite inhibits the CYP enzyme (scenarios A‐C) on DDI magnitude. To study the worst‐case scenario, the perpetrator should be dosed to steadystate and victim drug given at the time of the peak concentrations of the perpetrator. AUC, area under plasma concentration‐time curve; od, once daily; sd, single dose.
When the drug is investigated as a victim, probe or index inhibitors can be utilized to reveal the contributions of specific pathways to the drug's pharmacokinetics (i.e., to document the clinical relevance of the key enzymes and transporters involved; Table 3 ). In this situation, a low single dose of the victim drug is usually sufficient. If a strong interaction is expected, even subtherapeutic victim drug doses may be necessary for ethical reasons to ensure a large enough safety margin in drug exposure. Such a dosing scheme is adequately informative provided that the results can be extrapolated to steady‐state conditions at clinically relevant doses (i.e., the victim drug displays linear dose‐dependent and time‐dependent pharmacokinetics). On the other hand, when the effect of a strong enzyme/transporter inducer is investigated, a relatively high victim drug dose may be necessary to allow quantification of its concentrations. Monitoring of pharmacodynamic effects may be required for safety and can be useful when evaluating the clinical significance of the DDI. Pharmacodynamic end points are particularly useful in cases where the victim drug has active metabolites, where organ specific disposition of the drug is anticipated to be altered (e.g., entry to the central nervous system via blood–brain barrier), or where concomitant administration of the perpetrator and victim is anticipated to occur in clinical practice. If subjective pharmacodynamic measurements are used, double‐blinding of the treatment phases is crucial.
Table 3.
Characteristics of possible index inhibitors of CYP enzymes
| Enzyme | Inhibitor | Dose used in DDI trialsa | Strengthb | Also inhibits | t1/2 (h) | Remarks | E/Fc |
|---|---|---|---|---|---|---|---|
| CYP1A2 | Ciprofloxacine | 500 mg b.i.d. | +++ | CYP3A4 | 3.3 | Some safety concerns | |
| Enoxacin | 400 mg b.i.d. | +++ | 3.3 |
Some safety concerns Limited availability |
E | ||
| Fluvoxaminee | 25 mg b.i.d. to 100 mg q.d. | ++++ | CYP2C19, CYP2D6, CYP3A4 | 15 | Strong CYP2C19 inhibitor | F | |
| CYP2B6 | Ticlopidinee | 250 mg b.i.d. | + | CYP2C19 | 98 |
Strong CYP2B6 inhibitor based on substrate metabolic ratio Strong CYP2C19 inhibitor MBI Limited availability Bleeding risk |
E |
| CYP2C8 | Clopidogrel | 75 mg q.d. (first dose 300 mg) | +++ | CYP2B6, CYP2C19 | 6 |
Moderate CYP2C8 inhibitor with 75 mg daily MBI (glucuronide) Bleeding risk |
F |
| Gemfibrozile | 600 mg b.i.d. | ++++ | OATP1B1, OAT3 | 1.1 | MBI (glucuronide) | E/F | |
| CYP2C9 | Fluconazolee | 200 mg q.d. | ++ | CYP2C19, CYP3A4 | 32 | Strong CYP2C19 inhibitor | E/F |
| CYP2C19 | Fluconazolee | 200 mg q.d. | +++ | CYP2C9, CYP3A4 | 32 | ||
| Fluoxetine | 60 mg q.d. | ++ | CYP2D6 | 53 |
MBI Strong CYP2D6 inhibitor t½ of norfluoxetine 4–16 days |
||
| Fluvoxaminee | 25 mg b.i.d. to 100 mg q.d. | +++ | CYP1A2, CYP2D6, CYP3A4 | 15 | Strong CYP1A2 inhibitor | F | |
| Omeprazolee | 20–40 mg q.d. | ++ | 0.7 |
Strength based on nonsensitive probe substrates MBI |
E | ||
| CYP2D6 | Bupropion | 150–300 mg q.d. | +++ | 11 | |||
| Fluoxetine | 60 mg q.d. | ++++ | CYP2C19 | 53 |
Strong CYP2C19 inhibitor t1/2 of norfluoxetine 4–16 days |
E/F | |
| Mirabegron | 50 mg q.d. | ++ | 50 | F | |||
| Paroxetinee | 20 mg q.d. | ++++ | 17 | MBI | E/F | ||
| Quinidine | 100 mg q.d., 100–600 mg s.d. | ++++ | P‐gp | 6.2 | Prolongs QT‐interval | E | |
| Terbinafine | 250 mg q.d. | +++ | CYP1A2 | 200–400 | |||
| CYP3A4 | Clarithromycin | 250–500 mg b.i.d. | +++ | CYP2C19, P‐gp | 3.3 | MBI | E/F |
| Cobicistate | 150 mg q.d. | ++++ | CYP2D6, P‐gp | 5.2 | MBI | ||
| Erythromycin | 500 mg t.i.d. | +++ | P‐gpd | 1.6 | MBI | F | |
| Fluconazole | 200 mg q.d. | ++ | CYP2C19, CYP2C9 | 32 | Strong CYP2C19 inhibitor | F | |
| Itraconazolee | 100–200 mg q.d. | ++++ | CYP2J2,d P‐gp | 21 | E/F | ||
| Ketoconazole | 200 mg b.i.d. to 400 mg q.d. | ++++ | CYPC19, P‐gp | 3.3 | Limited availability | E | |
| Posaconazole | 300–400 mg b.i.d. | +++ | 31 | ||||
| Ritonavire | 100–200 mg b.i.d. | ++++ | P‐gp | 4 |
MBI Induces CYP enzymes |
E | |
| Verapamil | 80 mg t.i.d. to 240 mg q.d. | ++ | P‐gp | 4 | MBI | F |
The table contains all the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) recommended index inhibitors and some selected alternatives based on potential advantages in sensitivity, selectivity, and/or relative safety. The data are compiled primarily from the UW Metabolism and Transport Drug Interaction Database (Copyright University of Washington 1999–2019. Accessed: January−February 2019), secondarily from drug labels.
CYP, cytochrome P450; DDI, drug–drug interaction; E/F, European Medicines Agency/US Food and Drug Administration; F, bioavailability; MBI, mechanism‐based inhibition; OAT, organic anion transporter; P‐gp, P‐glycoprotein; s.d., single dose; t1/2, elimination half‐life.
Typical dose used in clinical DDI trials. Cocktail studies not included.
Indication of strength is based on a trial with an index substrate of the affected CYP. Cocktail studies not included. Classification: ++++ a maximal > 10‐fold increase in index substrate AUC, +++ a maximal 5–10‐fold increase, ++ a maximal two to fivefold increase, + a maximal 1.25−2‐fold increase. Values for maximal fold increases in AUC are given in Table S2 .
Indicates if the drug is recommended as a CYP probe inhibitor by EMA (E) and/or FDA (F) (2–3).
Erythromycin: based on in vitro data only. Itraconazole: based on interaction with astemizole.
Our recommended inhibitor.
Properties of suitable index substrates of drug‐metabolizing enzymes
The most important property of an index substrate for DDI studies is sensitivity for the specific pharmacokinetic pathway investigated (Table 2 ). In the context of drug‐metabolizing enzymes, the fraction metabolized by the enzyme should optimally be > 80%, so that an over fivefold increase in the area under plasma concentration‐time curve (AUC) can occur when the enzyme is completely inhibited (Figure 2 ). Furthermore, other disposition mechanisms of the index substrate should be known in detail, especially in cases where the perpetrator has the potential to cause interactions via other mechanisms. The properties of the substrate should also be suitable for thorough characterization of its pharmacokinetic parameters in a clinical study. Most importantly, the elimination half‐life (t1/2) of the probe should be sufficiently short to avoid unnecessarily long sample collection (needed to cover 80–90% of its total AUC) and washout period. From this point of view, efavirenz, warfarin, and desipramine are far from ideal index substrates. Particularly if it is important to quantify a transient effect on enzyme activity, an index drug with extensive first‐pass metabolism and short t1/2 should be preferred. An optimal index substrate exhibits linear pharmacokinetics and has itself no effects on the pharmacokinetics of other drugs. Tolerability and lack of toxicity are also key issues, and the safety margin should be large enough to cover changes of several folds in exposure. Other desirable qualities for an index substrate include ease of quantification and availability of metabolite standards, as well as commercial availability at suitable doses. If equally good substrates for the specific purposes are available, the choice should be made on the basis of the clinical relevance of the potential combinations.
There are several sensitive and selective index substrates for some CYP enzymes (e.g., CYP1A2, CYP2D6, and CYP3A), whereas sensitive index substrates for some CYPs (CYP2A6, CYP2B6, CYP2C9, and CYP2J2) are currently not available (Table 2 ). In some cases, sensitive index substrates are available, but they are also substrates for a transporter protein. For example, more than 80% of repaglinide and simvastatin are metabolized by CYP2C8 and CYP3A, respectively, making them sensitive index substrates of these enzymes, but both are also fairly sensitive substrates of OATP1B1/1B3.16 The usefulness of both these drugs as index substrates has been documented extensively, and their nonselectivity can thus be taken into account when interpreting the results of the study. For non‐CYP enzymes, knowledge is still evolving, and useful probe substrates are known only for few of them (Table 4 ). Lastly, if a sufficiently sensitive index substrate is not otherwise available or feasible, an alternative can be to use a specific reaction (i.e., a metabolic ratio as an index of enzyme activity; see also Limited sampling strategies below).
Table 4.
Examples of DDIs involving non‐CYP drug‐metabolizing enzymes
| Enzyme | Victim | Perpetrator (inhibitor) | Consequence |
|---|---|---|---|
| Acylpeptide hydrolase | Valproic acid glucuronide | Carbapenems, e.g., meropenem | Decreased serum valproic acid levels92 |
| COMT | Levodopa | Entacapone | AUC 1.5‐fold93 |
| DPD | 5‐fluorouracil | Sorivudine | Fluoropyrimidine toxicity94 |
| Monoamine oxidases (A and B) | Dopamine | Moclobemide | Increased dopamine effects95 |
| Thymidine phosphorylase | Trifluridine | Tipiracil | AUC 37‐fold96 |
| UGT1A1 | SN‐38 (irinotecan active metabolite) | Lopinavir‐ritonavir | AUC 3‐fold97 |
| UGT1A9 | Dapagliflozin | Mefenamic acid | AUC 1.5‐fold98 |
| UGT2B7 | Lamotrigine | Valproic acid | AUC up to 2‐fold99 |
| Xanthine oxidase | 6‐mercaptopurine | Allopurinol | AUC 5‐fold100 |
AUC, area under plasma concentration‐time curve; COMT, catechol‐O‐methyltransferase; CYP, cytochrome P450; DDI, drug–drug interaction; DPD, dihydropyrimidine dehydrogenase; UGT, uridine 5′‐diphosphate glucuronosyltransferase.
Properties of suitable index inhibitors and inducers of drug‐metabolizing enzymes
In addition to safety, the two most important characteristics of index inhibitors are strength of inhibition and selectivity. The FDA and EMA guidances define a strong index inhibitor as a drug that increases the AUC of a sensitive index substrate of a given metabolic pathway more than or equal to fivefold,2, 3 which equals at least an 80% inhibition of clearance. Strong inhibition typically requires a repeated dosing of sufficient inhibitor doses for about five inhibitor half‐lives before the victim drug is administered, followed by inhibitor dosing until victim AUC is fully characterized (Tables 1 and 3 , Figure 2 ). Strong inhibition increases the sensitivity to detect interaction effects, especially in cases where the victim drug is not solely dependent on the single elimination pathway. An optimal index inhibitor is selective (i.e., only inhibits one enzyme), so that direct mechanistic conclusions can be drawn from the result with reasonable confidence. On the other hand, if the index inhibitor inhibits several enzymes and/or transporters, the observed effects on victim drug pharmacokinetics cannot be attributed to a single pathway without further studies. Lastly, a long t1/2 of the inhibitor makes it impractical in terms of the required washout period. For example, the t1/2 of 4–6 days of parent fluoxetine and 4–16 days of its metabolite norfluoxetine make fluoxetine a suboptimal index inhibitor of CYP2D6 in crossover studies. There are fairly good inhibitors for most CYP enzymes, but adequately documented strong and selective inhibitors for CYP2A6, CYP2B6, and CYP2C9 are missing (Table 3 ). For instance, high doses of fluconazole result in strong inhibition of CYP2C9, but they also cause a strong inhibition of CYP3A4 and CYP2C19. Likewise, suitable strong inhibitors for non‐CYP enzymes are mainly lacking; for example, the strongest identified uridine 5′‐diphosphate glucuronosyltransferase (UGT)‐mediated DDIs are limited to a two to threefold increase in AUC (Table 4 ). Therefore, studies with moderate, weak, and nonselective inhibitors are also needed in particular if concomitant clinical use is likely to occur frequently.
Unlike inhibition, induction of drug‐metabolizing enzymes and transporters is usually highly nonselective, with few exceptions, such as induction of CYP1A2 by polycyclic aromatic hydrocarbon compounds.17 Of note, the inducibility of CYP2D6 by known drugs is low or negligible, even though its activity can be increased by certain conditions, such as pregnancy. Rifampin is the prototypical inducer that is most commonly used in clinical DDI studies because of its safety compared with other strong inducers, such as carbamazepine and phenytoin, and the strength of its inducing effect.13 Like other compounds causing pregnane X receptor‐mediated induction, rifampin causes a particularly strong induction of intestinal and hepatic CYP3A but also induces many other drug‐metabolizing enzymes and certain transporters. Thus, it can be used to test the sensitivity of a drug to strong enzyme induction. It should be noted that the CYP3A inducing effect of rifampin is so strong that it can dramatically increase the fraction of the drug metabolized by CYP3A, so that even a drug that is normally mainly eliminated via noninducible or weakly inducible pathways becomes dependent on CYP3A4.
Specific considerations for transporter DDI studies
Current evidence indicates that of the numerous transporters, at least BCRP, OATP1B1, and P‐glycoprotein (P‐gp) mediate clinically relevant DDIs. In addition, multidrug and toxin extrusion protein (MATE)1, MATE2‐K, organic anion transporter (OAT)1, OAT3, OATP1B3, OATP2B1, organic cation transporter (OCT)1, and OCT2 can be involved in DDIs.18 Accordingly, drug transporters are routinely investigated already during preclinical drug development, providing a wealth of information regarding the properties of an investigational drug as a substrate and inhibitor of different transporters.19 With regard to clinical DDI studies, the FDA guidance instructs that the need of clinical transporter DDI studies of transporter substrates identified in in vitro studies should be considered on the basis of the drug's site of action, route of elimination, likeliness of concomitant use, and safety issues.2 For example, in the cases of P‐gp and BCRP‐mediated DDIs, most clinically relevant DDIs (aliskiren, dabigatran, digoxin, and fexofenadine as P‐gp substrates, and rosuvastatin as a BCRP substrate) are based on inhibition of the absorption limiting effects of these transporters in the small intestine or, in some cases, biliary or renal excretion.20, 21, 22 In addition, based on limited evidence, P‐gp–mediated DDIs may also occur at blood–tissue barriers, such as the blood–brain barrier, warranting DDI studies if the site of pharmacological or toxic effects of a P‐gp substrate drug is behind such a barrier. Moreover, for drugs targeting the liver or eliminated via the liver, current guidance states that DDI studies focusing on OATP1B1 or OATP1B3 can be warranted, and for drugs eliminated via renal excretion, studies on OAT1 or 3, OCT2, or MATE‐mediated DDIs can be needed.2 Recent studies support the roles of OCT1 and OATP2B1 in mediating hepatic and intestinal DDIs, indicating that they also need to be considered.19 Furthermore, if the drug is suspected to be a transporter inhibitor on the basis of in vitro data, clinical DDI studies could be conducted with likely concomitant substrate drugs that could be subject to a clinically relevant DDI, regardless of the suspected perpetrator's own route of elimination.
DDI studies focusing on transporters have some challenges compared with drug‐metabolizing enzymes. First, the lack of specific index substrates and inhibitors makes extrapolation of the results of DDI studies with other drugs challenging (Table 5 , Table S3 ). For example, dabigatran etexilate could be used as a fairly specific P‐gp substrate because its intestinal absorption is limited by P‐gp, but dabigatran elimination is not dependent on renal P‐gp and its sensitivity to P‐gp inhibition at clinically used doses is only modest, so that even the strongest P‐gp inhibitors have increased its AUC less than threefold.23 Similarly, recognized substrates of BCRP (e.g., rosuvastatin), OAT1/3 (e.g., adefovir, ganciclovir, and famotidine), and MATE or OCT2 transporters (e.g., dofetilide and metformin) have been at most moderately sensitive to inhibition of the respective transporters.24 In the cases of OATP1B1 and 1B3, there are some moderately sensitive substrates, such as simvastatin (acid) and pitavastatin, but it is unclear if their AUC could be increased more than fivefold by inhibition of a single transporter only.
Table 5.
Examples of transporter probe substrates and inhibitors for clinical DDI studies
| Transporter | Substrates | Inhibitors |
|---|---|---|
| BCRP | Rosuvastatin | Cyclosporine |
| Sulfasalazine | Eltrombopag | |
| OATP1B1 or OATP1B3 | Atorvastatin | Cyclosporine |
| Pitavastatin | Rifampin (single dose) | |
| Pravastatin | ||
| Repaglinide | ||
| Rosuvastatin | ||
| Simvastatin (acid) | ||
| OATP2B1 | Celiprolol | Apple, orange and grapefruit juices (intestine) |
| OCT1 | Sumatriptan | N/A |
| Metformin | ||
| OCT2 | Metformin | Cimetidine |
| OAT1 | Adefovir | Pyrimethamine |
| Probenecid | ||
| OAT3 | Benzylpenicillin | Probenecid |
| P‐gp | Aliskiren | Itraconazole |
| Dabigatran etexilate | Verapamil | |
| Digoxin | ||
| Fexofenadine |
The data are compiled primarily from the University of Washington Metabolism and Transport Drug Interaction Database (Copyright University of Washington 1999–2019. Accessed: February 2019). More information regarding the properties of substrates and inhibitors are given in Table S3 .
BCRP, breast cancer resistance protein; DDI, drug–drug interaction; N/A, not available; OAT, organic anion transporter; OATP, organic anion‐transporting polypeptide; OCT, organic cation transporter; P‐gp, P‐glycoprotein.
Well‐established transporter inhibitors are typically either nonselective or cause only limited extent of inhibition. Although probenecid can be used as a relatively selective inhibitor of OATs 1 and 3, P‐gp inhibitors, such as clarithromycin, itraconazole, quinidine, and verapamil, also have inhibitory effects on CYP enzymes, and cyclosporine acts as a multitransporter inhibitor of BCRP, P‐gp, and OATPs, and also inhibits CYP3A4. The OATP inhibitor rifampin also acts as a strong inducer of drug‐metabolizing enzymes. In the case of rifampin, the inducing effect can be largely avoided by using only a single rifampin dose, but this is primarily a relevant dosing scheme only in mechanistic DDI studies. In addition to the general lack of selectivity of transporter inhibitors, the strength of inhibition of many inhibitors may be limited, as it is unclear if any of the inhibitors have a sufficiently strong inhibitory effect on a single transporter to cause a more than fivefold increase in the AUC of a victim drug.
Second, modulation of transporter activity can result in changes in tissue distribution depending on the localization of the transporter, without significant changes in plasma concentrations. Therefore, solely determining the plasma exposure of the victim drug might not be a valid end point for a transporter DDI study. In such cases, it can be useful to measure the pharmacodynamic effect of the victim drug to find out if the perpetrator caused any clinically relevant changes in drug concentration at the site of action. A good example is metformin, which is not metabolized but whose absorption from the intestine, hepatic disposition, and renal elimination is subject to active transport. A DDI study design for metformin has been proposed, where the end points consist of systemic pharmacokinetics, renal clearance, and antihyperglycemic effect.25 Another possibility to study tissue distribution of drugs is provided by nuclear imaging techniques, such as positron emission tomography and single‐photon emission computed tomography, where the radiolabeled drug of interest is administered and noninvasively measured.26
Pharmacogenetics
Our recommendation is to routinely collect DNA samples from participants of DDI studies and to genotype for well‐characterized genetic variants of enzymes and transporters involved in pharmacokinetics of investigated drugs. For example, in studies with index inhibitors or substrates of enzymes and transporters that exhibit common genetic variation, genotype information can be important in interpretation of the data, as genotype can markedly influence the extent of the DDI.27 Genotype information may also be valuable in detecting causes for pharmacokinetic variability or outliers. In some cases, pharmacogenetic studies can also complement or replace DDI studies. They can be particularly useful if no selective or strong inhibitor is available, which is common with drug transporters and non‐CYP enzymes. If the enzyme/transporter exhibits considerable genetic variability resulting in altered activity, especially loss‐of‐function, individuals with different genotypes can be studied in a genotype panel study to obtain clear mechanistic understanding of the importance of the specific pathway for the pharmacokinetics of the investigational drug. This type of study is only possible in cases where functionally relevant variants are known with suitable population frequencies, allowing recruitment of a sufficient number of subjects. For example, a common single‐nucleotide variant in SLCO1B1 (c.521T>C, rs4149056), encoding the hepatic uptake transporter OATP1B1, results in decreased transport activity.16 Thus, pharmacokinetics in heterozygous and homozygous variant allele carriers can be compared with that in reference allele carriers, to inform about the relevance of OATP1B1 in the disposition of the investigational drug, and its sensitivity to inhibition of OATP1B1. Of drug transporters and drug‐metabolizing enzymes, at least ABCG2, CES1, CYP2C9, CYP2C19, CYP2D6, CYP3A4, CYP3A5, DPYD, NAT2, SLC22A1, SLCO1B1, TPMT, UGT1A1, UGT1A3, UGT2B10, and UGT2B17 show significant genetic variability.28, 29, 30 Variants in these genes could be used to define subject groups in a genotype panel study and genotyping for these genes should be considered if they are likely to be involved in the pharmacokinetics of the victim or perpetrator of a DDI study.
Index drug cocktails and microdosing
In the cocktail approach, more than one index substrate is administered simultaneously to measure activities of several drug‐metabolizing enzymes or transporters. In the context of DDI studies, this allows a drug to be studied as a perpetrator for several pharmacokinetic pathways in the same clinical study. A cocktail needs to be validated so that its components do not interact with each other, usually by administering each substrate separately and as a part of the cocktail.31 Recent advances in this area have occurred in the utilization of very low doses of the substrate drugs (i.e., microdosing) and in the development of drug cocktails to assess drug transporter function.32 The use of microdosing is beneficial in terms of safety and reduced likelihood of interactions between substrates. A potential problem is that it may be difficult to extrapolate the findings to clinically relevant doses if the substrate displays nonlinear pharmacokinetics.
Several phenotyping cocktails, including index substrates for different CYP enzymes, have been developed over the last 20 years.31 Most of the proposed cocktails include index drugs for up to six CYP enzymes that have most commonly been CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and/or CYP3A4. The individual substrates are usually relatively selective for a single enzyme so that mechanistic conclusions can be drawn from the results. One of the challenges related to a few of the earlier cocktails is that some of the suggested index substrates are no longer commercially available in some countries. Two of the more recent cocktails called the Basel and Geneva cocktails, including substrates for CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2D6, and CYP3A4 (Geneva cocktail also including fexofenadine as an index for P‐gp), have been specifically validated for DDI studies.33, 34, 35
One of the biggest challenges in development of drug transporter cocktails is lack of selective index substrates. The first published cocktail approach for transporters included digoxin for P‐gp, furosemide for OAT1 and OAT3, metformin for OCT2, MATE1, and MATE2‐K, and rosuvastatin for OATP1B1, OATP1B3, and BCRP.36 The initial study showed mutual interaction resulting in higher rosuvastatin peak plasma concentration (Cmax) and AUC when administered as a part of the cocktail than individually. In further studies, the doses of furosemide and metformin were reduced, and a cocktail consisting of 0.25 mg digoxin, 1 mg furosemide, 10 mg metformin, and 10 mg rosuvastatin was shown to be devoid of mutual interactions.37, 38 The sensitivity of the cocktail as well as the potential for mutual interactions in a situation where the exposure to one of the components is increased due to an inhibitor being studied, remains to be determined. Recently, a transporter cocktail utilizing microdosing was evaluated with known inhibitors in DDI studies.39 The microdose cocktail consisted of 10 μg midazolam (CYP3A4), 375 μg dabigatran etexilate (P‐gp), 10 μg pitavastatin (OATP1B), 25 μg rosuvastatin (BRCP, OATP, and P‐gp), and 50 μg atorvastatin (OATP, BCRP, P‐gp, and CYP3A4). In the DDI studies, the cocktail produced similar results as the index drugs had produced in previous studies, and only dabigatran etexilate showed marked differences to clinical doses with an approximately twofold lower dose‐normalized oral exposure and higher magnitude of interactions when microdosing was used.
Endogenous substrates
Instead of using index substrate drugs to assess the perpetrator's impact on a specific pharmacokinetic pathway, an endogenous compound as a measure of activity of this pathway could be a useful alternative. The current status of endogenous biomarkers for DDI studies, their limitations, as well as strategies to investigate novel biomarkers for this purpose has been reviewed in detail recently.24, 40, 41, 42 The main advantage of this approach is that it does not require the additional intervention of administering the index substrate. This could be especially useful during early clinical drug development, or when the perpetrator cannot be administered to healthy volunteers due to safety concerns. The endogenous biomarker needs to be validated to fit this purpose, it should be selective for the enzyme or transporter under investigation, and it should not be altered by disease states or dietary factors. Well‐validated endogenous biomarkers that could function as an alternative to formal DDI studies are, however, still lacking. For example, 4β‐hydroxycholesterol, 6β‐hydroxycortisol, and 6β‐hydroxycortisone could be used as sensitive biomarkers of CYP3A activity, unconjugated bilirubin as a nonselective marker of UGT1A1, and coproporphyrin as a marker of OATP1B1/1B3, but they are only suitable in initial characterization of DDI risks.41
Limited sampling strategies and population pharmacokinetic modeling
In a stand‐alone DDI study, the pharmacokinetic profile of the victim drug should be fully characterized with a suitably frequent and long sampling schedule, so that the extrapolated part of the total AUC is < 20%.2 In contrast to studies in healthy volunteers, rich sampling strategy is usually not feasible in large patient populations. However, population pharmacokinetic analyses can be useful to assess the clinical impact of concomitant medications on the pharmacokinetics of the drug of interest despite sparse or limited sampling. The strength of such studies is that DDIs can be investigated in the clinically relevant patient group using typical doses on long‐term basis and that the effects of frequently coprescribed drugs can be studied irrespective of prior knowledge on relevant DDI mechanisms. To make the best use of population pharmacokinetic data, the study protocol, including sampling strategy, should be carefully designed before commencing data collection. In particular, accurate documentation of the dosage and timing of drug administration, as well as sampling times, is essential. This is especially important for drugs with variable absorption rate and short t1/2. Ideally, also perpetrator drug exposure should be documented by using available samples. The methodology and design of DDI studies using population pharmacokinetic analyses is discussed in a recent commentary by the International Society of Pharmacometrics Working Group.43 For example, a simulation case study with methotrexate and trastuzumab has shown that, with careful design, population pharmacokinetic studies can be a viable alternative to DDI studies.44
Another instance where limited sampling can be used is with index substrate drugs whose validated metrics of enzyme activity is based on only few or single sample time points, typically by calculating a metabolic ratio from the parent drug and its metabolite, whose formation is selective for the enzyme in question. The interpretation of such indices should be done cautiously particularly if the fate of the metabolite is subject to changes by perpetrators. In an ideal case of a fully specific index ratio, a decrease of 80% in a metabolic ratio indicates a maximal increase of fivefold in the AUC of a sensitive substrate drug. Furthermore, limited sampling can be subject to large variability due to, for example, delayed drug absorption, especially if the metric is based on the parent drug concentration only. For example, limited sampling strategies for midazolam and warfarin have shown poor correlation with full pharmacokinetic profiling.45, 46 On the other hand, determination of a metabolic ratio from a single time point can be a more useful metric that accurately reflects the systemic clearance of the drug. A good example of such a metric is caffeine, which is selectively metabolized by CYP1A2 to paraxanthine. The paraxanthine/caffeine ratio from a single plasma sample, taken 4–6 hours after caffeine intake, can be used to assess CYP1A2 activity.47 Such single time point metabolic ratios have been tested in index drug cocktails as well.34, 48 In cocktail approaches, however, a single sampling time represents a compromise between the different substrates and might not be optimal for each index substrate.
One approach to limit blood sample volumes and to make blood sampling more feasible is to use capillary blood microsamples and dried blood as the sample matrix.34 The main advantage of this approach is its low invasiveness and the flexibility it potentially offers, such as applicability in low resource environments or self‐sampling by the study participants. However, the measurement of whole‐blood concentrations in this matrix needs to be validated against samples collected by venipuncture, and whole blood concentrations of drugs often differ from those in plasma/serum. Use of dried blood samples can be very useful in large‐scale studies, such as phase III trials or in studies in vulnerable populations, such as children, typically in conjunction with limited sampling strategies, but its usefulness for formal DDI studies in healthy volunteers is limited.
Physiologically‐based pharmacokinetic modeling
In the past 2 decades, major advances have been made in the modeling methods used for prediction of DDIs on the basis of in vitro CYP inhibition data.49, 50 In particular, physiologically‐based pharmacokinetic (PBPK) modeling, a dynamic approach incorporating changes in drug concentrations over time, has evolved and is becoming increasingly used even for regulatory decisions.8, 51, 52 PBPK models simulate concentration‐time profiles of drugs in plasma and/or an organ of interest, and allow for simultaneous modeling of multiple drug disposition processes, providing a range of opportunities.53, 54 The reliability of PBPK models increases with accumulating clinical data and information on the disposition/elimination pathways of the drugs and their metabolites. In the ideal case, the model can be cross‐verified using data from multiple clinical studies concerning both the perpetrator and the victim drug.
PBPK DDI modeling is encouraged by regulatory agencies,2, 3 and the pharmaceutical industry is increasingly using PBPK modeling to predict DDIs and related dosing decisions as replacement for clinical studies.8 Examples of submissions where clinical DDI studies have been waived on the basis of PBPK DDI simulations include drugs such as cobimetinib, lesinurad, and olaparib.55 In regulatory submissions, PBPK DDI modeling has been applied to (i) predict DDIs with perpetrator/victim drugs other than those used in clinical studies, (ii) predict DDIs in various subpopulations (e.g., specific patient populations or populations with genetically different CYP activities), (iii) predict the effects of altered dosing regimens/repeated dosing of perpetrator/victim drug, (iv) predict the effects of pH modulation on drug solubility/absorption, and (v) predict the effects of a perpetrator drug on single drug enzymes/transporters.55 In addition, PBPK DDI modeling has been applied to support the design of clinical DDI studies, and mechanistically explain clinically observed DDIs.
There are many examples of successful PBPK DDI models, in particular in cases where the interaction is based on simple reversible CYP inhibition. In contrast, modeling of the effects of combined time‐dependent inhibition and reversible inhibition, or time‐dependent inhibition and induction are not as established.56, 57 Moreover, modeling of DDIs affecting non‐CYP drug‐metabolizing enzymes and drug transporters may be difficult due to lack of selective in vivo data, and uncertainties concerning the expression levels and turnover characteristics of transporters and enzymes, as well as scaling of drug concentration levels at the relevant cellular or subcellular sites.55, 57, 58 A good example highlighting the challenges with modeling of transporter‐DDIs, is related to OATP1B1‐mediated influx of drugs to hepatocytes. The measured in vitro OATP1B1 Ki values of clarithromycin, clopidogrel acyl‐β‐D glucuronide, cyclosporine, and rifampin do not seem to be low enough to recover DDIs observed in clinical studies, which has forced modelers to adjust OATP1B1 inhibition constant values in PBPK models against measured values.59, 60, 61 A possible explanation of this disconnect from physiological principles is time‐dependent inhibition of OATP1B1 by these inhibitors and interplay between drug transporters and drug‐metabolizing enzymes.62 It is obvious that the biochemical and physiological processes related to transporter‐mediated DDIs are not fully understood, which poses one of the biggest challenges to development of PBPK modeling and in vitro–in vivo predictions in general.
Overall, PBPK modeling is a useful tool for DDI research to be used in concert with clinical DDI studies. However, the current limitations of PBPK modeling in complex situations, where multiple mechanisms and DDIs are involved, should be taken into account. On the whole, there has been a lack of consistency in model development and quality assessment practices, demonstrating a need for best‐practice guidelines, as well as for harmonization of guidelines between regulatory agencies.49, 55 At present, however, PBPK models can be highly valuable in interpretation and extrapolation of clinical DDI studies to different clinical situations, as well as in planning and design of further clinical studies.
Complex Ddis
Although DDIs based on a single well‐defined pharmacokinetic mechanism can already be predicted and extrapolated to different clinical situations with great confidence, it has become increasingly recognized that pharmacokinetic interaction mechanisms are often complex.50, 63, 64 Complex DDIs can be difficult to anticipate and control, and can pose a big challenge during early drug development. From the drug development perspective, as well as from the clinical point of view, some of the most problematic complex mechanisms are those involving autoinhibition or induction of the drug's own metabolism, the possibility of several simultaneous mechanisms, inhibition, or induction by major drug metabolites and time‐dependent inhibition.64 Although comprehensive preclinical data and modeling approaches are fundamental for understanding such DDIs, clinical DDI studies are eventually indispensable to understand their clinical relevance.
For example, on the basis of modern preclinical research methods, autoinduction and autoinhibition can be suspected early, but only the first clinical pharmacokinetic studies reveal if they are clinically relevant. In the worst case, autoinduction can be so strong that adequate drug exposure cannot be obtained despite extreme doses and the drug development program fails. On the other hand, both autoinduction and autoinhibition can result in dose‐dependent and time‐dependent changes in the relative importance of different disposition pathways and thereby alter the sensitivity of the drug to inhibitory perpetrators. For example, the tyrosine kinase inhibitor imatinib is a mechanism‐based inhibitor of CYP3A465 that can increase the plasma concentrations of CYP3A4 substrates, such as simvastatin. As CYP3A4 plays an important role in the metabolism of imatinib, the CYP3A4 inhibitory effect can lead to a complicated increase in the significance of the alternative elimination pathway (CYP2C8), and an increased risk of increased imatinib levels due to CYP2C8 inhibitors during multiple imatinib dosing.66
An even more complex drug is the kinase inhibitor midostaurin (Figure 3 ), which is a substrate, time‐dependent inhibitor and inducer of CYP3A4.67, 68, 69 Based on mechanistic information, it is conceivable that midostaurin could cause a temporary net (auto)inhibition of CYP3A4, which gradually changes to net (auto)induction. On the basis of a short study with midazolam as a victim drug, the effect of midostaurin on CYP3A4 could be interpreted as negligible.67 However, a DDI study carried out at two early time points may not have revealed a temporary inhibition in the beginning of treatment and leaves unclear if midostaurin acts as an inducer decreasing concentrations of CYP3A4 substrates in long‐term treatment. In addition, the sensitivity of single‐dose midostaurin to CYP3A4 inhibition may not have revealed its full sensitivity to CYP3A4 inhibition at steady state (i.e., after autoinduction has increased the role of CYP3A4 in its metabolism). Deciphering this kind of dose‐dependent and time‐dependent complexity requires clinical DDI studies where the drug is the perpetrator, as well as studies where the drug is the victim. Moreover, clinical DDI studies at different time points during the course of treatment are needed, as PBPK models are not reliable in this area, particularly because there is paucity of clinical data on the dose and time dependency of DDIs involving drugs with nonlinear pharmacokinetics, and because enzyme‐specificity of induction is not thoroughly understood.
Figure 3.
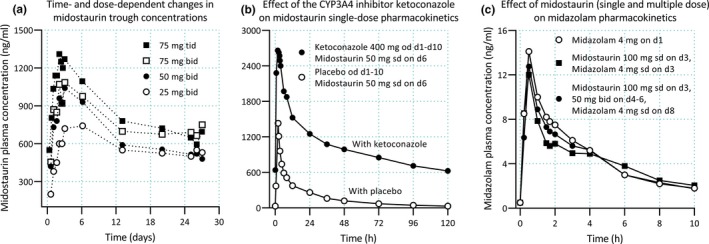
Pharmacokinetics and drug–drug interactions of midostaurin. (a) Midostaurin exhibits time‐dependent pharmacokinetics, with increasing trough plasma concentrations during the first 3–6 days of therapy, followed by a 60–70% decline in concentrations until a steady state is reached.68, 69 Autoinduction of cytochrome P450 (CYP)3A4 by midostaurin and its two major metabolites is thought to be involved in the time‐dependent pharmacokinetics of midostaurin.69 (b) Midostaurin is a sensitive CYP3A4 substrate, and ketoconazole has increased its SD area under plasma concentration‐time curve (AUC) by more than tenfold.67 (c) Although both midostaurin and its two major metabolites are inducers and mechanism‐based inhibitors of CYP3A4 in vitro, the drug has had no effect on midazolam pharmacokinetics in healthy subjects, following either a single midostaurin dose (100 mg) or 2 days after the last dose of multiple midostaurin doses (50 mg twice daily for 3 days).67 Clinical data shown are from refs. 67 and 68. d, day; od, once daily; sd, single dose; tid, three times daily.
Examples of DDIs involving multiple mechanisms simultaneously (i.e., DDIs where a multipathway substrate is given simultaneously with an inhibitor and/or inducer of the same pathways), include the gemfibrozil‐repaglinide (OATP1B1/1B3 and CYP2C8 inhibition), cyclosporine‐rosuvastatin (OATP1B and BCRP inhibition), sofosbuvir/velpatasvir/voxilaprevir‐rosuvastatin (inhibition of OATP1B1/1B3 and BCRP), and many ritonavir‐victim drug (P‐gp and CYP3A4 inhibition and CYP induction) interactions.70, 71, 72, 73 The key to building an understanding of this kind of DDI risk is to first learn with enzyme and transporter selective preclinical and clinical studies, which specific enzymes and transporters are involved in the drug's metabolism and disposition and which are inhibited or induced by the drug. Thereafter, relevant DDIs with potential for multiple mechanisms simultaneously can be studied clinically using standard methods. If additional mechanistic understanding is needed, studies with multiple perpetrators or different inhibitor dosing or timing experiments may be useful.
What comes to other types of complex DDIs, those caused by inhibitory or inducing major metabolites and mechanism‐based inhibition typically require carefully performed in vitro studies to initially identify the risks. The contribution of metabolites as perpetrators has only been realized during the past decade. It has, for example, been estimated that the clinically observed CYP2C9 inhibition by amiodarone, CYP2D6 inhibition by bupropion, and CYP3A4 inhibition by sertraline could only be explained accurately when considering their inhibitory metabolites.74 Of note, even glucuronide metabolites can act as mechanism‐based inhibitors of CYP enzymes, capable of causing strong DDIs.75 Such DDIs can usually be studied using standard clinical DDI study methodology, although in the case of mechanism‐based inhibition and in the case of metabolites that have a very long t1/2, the time‐dependency of the DDI risk may warrant long‐term administration or other more focused studies. In addition, the growing number of fixed‐dose combination products among newly marketed drugs (especially antiviral agents) represents a unique challenge of understanding the contribution of each drug component to the overall clinical effect when the combination is evaluated as a perpetrator.
From a clinical point of view, involvement of multiple perpetrator drugs simultaneously, and the influence of genetic variants or disease states on the risk of DDIs76 often necessitate specific DDI studies. For example, concomitant treatment with a CYP2C8 inhibitor (e.g., gemfibrozil) and a CYP3A4 inhibitor (e.g., itraconazole) can lead to an extremely strong effect on the pharmacokinetics of drugs that are substrates for both these enzymes, such as repaglinide and loperamide.71, 77 In case of genetic variants, poor metabolizer individuals are typically insensitive to inhibition of the genetically deficient enzyme, such as inhibition of CYP2C19 by omeprazole or inhibition of CYP2D6 by paroxetine,78, 79 but they may be particularly susceptible to DDIs mediated by inhibition of an alternative pathway, such as CYP3A4. Genetic variants leading to increased activity usually have an opposite effect. On the other hand, genetic variants can also influence the concentrations of the perpetrator drug, such as the CYP2C19 substrate voriconazole, thereby modifying the extent of inhibition/induction.80 Moreover, for example, fluvoxamine inhibited the oral clearance of theophylline by 62% in healthy subjects but only by 12% in patients with severe cirrhosis, indicating that patients with severe cirrhosis are not susceptible to this DDI.81 The conduct of clinical studies on these types of complex DDIs is based on the characteristics of the victim drug (i.e., which enzymes and transporters are relevant to the drug). Such studies typically require a larger clinical study, which includes an adequate number of subjects in each genotype or disease group, or a multiphase crossover study, where different perpetrators and perpetrator combinations can be compared. The study designs are relatively straightforward, but the recruitment of a sufficient number of individuals with a rare genotype or rare disease may be challenging.
Pitfalls and Interpretation of Ddi Studies
Pitfalls
There are multiple potential pitfalls in the design and conduct of DDI studies that can lead to inaccurate results, ethical problems, false alerts, or, in the worst case, failure to identify relevant DDI risks (Table 1 ). Particularly, deficiencies in preclinical data or lack of understanding of the importance of specific pharmacokinetic pathways have led to failures to identify clinically hazardous DDIs. The past cases that have led to market withdrawals, such as those of cerivastatin, cisapride, and terfenadine as victims and sorivudine as a perpetrator, have been caused mainly by failure to understand basic mechanistic issues, together with dose‐dependent toxicity of the victim drugs.82 In the following, we are not presenting an exhaustive review of all possible pitfalls but instead describe three case examples of pitfalls that can lead to inappropriate conclusions of the DDI risk.
In the late 1990s, the importance of CYP2C8 in drug metabolism was not understood. Eventually, the gemfibrozil‐cerivastatin interaction revealed the potential significance of the enzyme after a series of in vitro and clinical studies that were needed to understand the mechanisms of this DDI.83 Initially, cerivastatin was brought to the market with arguments of a favorable interaction potential due to metabolism by both CYP3A4 and CYP2C8. However, it was shortly thereafter withdrawn from the market in 2001 owing to multiple cases of rhabdomyolysis. In about half of the cases, the patients had been using gemfibrozil and cerivastatin concomitantly, and, sadly, many of the cases had a fatal outcome. Soon, gemfibrozil was shown to increase the AUC of cerivastatin five to sixfold and to greatly reduce the formation of its CYP2C8‐dependent M‐23 metabolite, whereas strong CYP3A4 inhibition by itraconazole had only a minor effect on cerivastatin exposure.84, 85 At first, gemfibrozil was identified as a rather modest and nonselective inhibitor of CYP2C8 in vitro, and it was only later discovered that the main metabolite of gemfibrozil, gemfibrozil 1‐O‐β‐glucuronide, is a strong mechanism‐based inhibitor of CYP2C8.86 This case concretely illustrated that it is difficult to anticipate DDIs without systematic studies and thorough understanding of the DDI characteristics of both the victim and perpetrator drugs.
The cyclooxygenase‐2 selective nonsteroidal anti‐inflammatory drug rofecoxib was initially considered to be a modest inhibitor of CYP1A2. This information in the labeling was mainly based on a DDI study using theophylline as a CYP1A2 index substrate, where rofecoxib 25 mg once daily increased the AUC of theophylline only 1.5‐fold.87 However, a DDI study with a similar design using tizanidine as a probe substrate resulted in a 13.6‐fold increase in its AUC, indicating strong CYP1A2 inhibition by rofecoxib.88 The reason for this apparent discrepancy is theophylline's lack of sensitivity as an in vivo index substrate of CYP1A2, partly due to its high bioavailability and lack of first‐pass metabolism. At the time, in vitro data regarding the effect of rofecoxib on CYP1A2 were also lacking, and it was only later shown that rofecoxib is a potent, mechanism‐based inhibitor of CYP1A2.89
As demonstrated in Figure 2 , the inhibitor dose and timing of the substrate in relation to inhibitor administration can greatly affect the magnitude of the observed interaction. An example of this is provided by examining drug interaction studies involving the intestinal CYP3A inhibitor grapefruit juice and the CYP3A substrate lovastatin. In the study by Rogers et al.,90 grapefruit juice was administered for 4 days in the morning and 40 mg lovastatin was administered in the evening of day 3. Grapefruit juice increased the AUC of lovastatin 1.9‐fold and that of lovastatin acid 1.6‐fold only. It is notable that the exact dosing interval between the perpetrator and victim is not given in the paper. The results are in contrast with an earlier study by Kantola et al.,91 where grapefruit juice increased the AUC values of lovastatin and lovastatin acid by 15‐fold and 5‐fold, respectively. In this study, grapefruit juice was administered thrice daily for 2 days, and lovastatin was administered simultaneously with grapefruit juice on day 3, followed by additional grapefruit juice doses 0.5 and 1.5 hours after lovastatin. Even though this amount of grapefruit juice intake is not likely to occur frequently in real life, the results represent a potential worst‐case scenario of the grapefruit juice‐lovastatin interaction. This example also makes evident how important it is to consider the study design both when planning and interpreting the results of a DDI study.
During the past 2 decades, DDI guidelines have been frequently updated by the regulatory authorities, and most serious pitfalls can now be avoided by following the guidelines. However, knowledge on non‐CYP enzyme‐mediated metabolism and transporters still continues to emerge, and is going to further improve DDI study methodology and the predictability of DDIs. Yet, it is still difficult to evaluate the quantitative roles of all relevant disposition pathways of a drug based on in vitro data. Consequently, a potentially dangerous pitfall is to use PBPK modeling in situations where its reliability without additional clinical data is questionable (e.g., in the case of complex DDIs, as outlined above).
Interpretation and extrapolation of findings
It is usually fairly straightforward to interpret the findings of standard DDI studies with index inhibitors or index substrates and to classify the tested drug's sensitivity to inhibition of a specific pathway or its strength as an inhibitor or inducer of a transporter or enzyme. Key issues to consider when interpreting DDI data are the strength and selectivity of the index perpetrator at the given dose and the sensitivity of the index substrate, as well as the general design of the study, including the dose and time relationships (Figure 2 ). For example, if the index drug is not sensitive enough, even full inhibition of a single metabolic pathway does not produce an over fivefold AUC change that would classify the perpetrator drug as a strong inhibitor.
When it comes to interpretation of complex situations and extrapolation of the findings with other scenarios, PBPK modeling can be highly valuable. Yet, if multiple mechanisms are involved in the DDI, such as involvement of both transporters and metabolism, or simultaneous induction and inhibition, extrapolation of the findings with other dosing scenarios or different drugs should be done with caution. In cases where a single mechanism (e.g., CYP3A4 inhibition) is crucial, extrapolation of the findings with other scenarios can be made with more confidence. It should be emphasized, however, that a very sensitive but nonselective index substrate due to combined roles of metabolism and transport can be superior to a poorly sensitive index substrate when the purpose is to detect a DDI risk. On the other hand, when the clinical significance of a DDI is evaluated, also complex issues, such as multiple simultaneous mechanisms, time‐dependency and dose‐dependency of transporter/enzyme inhibition and induction, as well as time‐dependent and dose‐dependent pharmacokinetics of the victim drug, often need to be considered in interpretation and extrapolation of the findings. As highlighted by the midostaurin and imatinib cases above, the sensitivity of a victim drug to DDIs may be altered by time‐dependent changes in its own pharmacokinetics. Moreover, the magnitude of DDIs occurring during the absorption and first‐pass metabolism of the victim may depend on the dosing interval between the perpetrator and victim, which should be considered in the interpretation (e.g., using PBPK modeling).
Conclusion
Evaluation of the DDI potential of drugs under development and on the market is a crucial issue for drug safety. Advances in in vitro methodology and modeling methods have greatly advanced our understanding of molecular mechanisms of DDIs and the ways to predict and interpret them. Clinical DDI studies remain, however, an integral part of the process of evaluating the DDI risks. Moreover, clinical DDI studies have revealed previously unknown DDIs and shed light on their mechanisms, which have then been confirmed by, for example, in vitro studies through reverse translation. As clinical DDI studies may not cover every single permutation of various parameters affecting the outcome, mechanistically focused clinical studies in concert with in silico models have great synergy in providing comprehensive understanding of DDIs and their clinical relevance. In conclusion, there is no optimal way to study DDIs, but the design of every study needs to be based on careful evaluation of the available data, to ensure safety and maximal usefulness of the study.
Funding
This work has been financially supported by grants from the Academy of Finland (Grant decision 278123, 2014), European Research Council (ERC; Grant agreement 725249), Sigrid Juselius Foundation and by State funding for university‐level health research, Finland.
Conflict of Interest
The authors declared no competing interests for this work.
Supporting information
Table S1. Characteristics of possible index substrates of cytochrome P450 enzymes. The table contains all the FDA and the EMA‐recommended index substrates and some selected alternatives based on potential advantages in sensitivity, selectivity, and/or relative safety.
Table S2. Characteristics of possible index inhibitors of cytochrome P450 enzymes. The table contains all the FDA and the EMA‐recommended index inhibitors and some selected alternatives based on potential advantages in sensitivity, selectivity, and/or relative safety.
Table S3. Characteristics of transporter substrates and inhibitors.
References
- 1. Dechanont, S. , Maphanta, S. , Butthum, B. & Kongkaew, C. Hospital admissions/visits associated with drug‐drug interactions: a systematic review and meta‐analysis. Pharmacoepidemiol. Drug Saf. 23, 489–497 (2014). [DOI] [PubMed] [Google Scholar]
- 2. US Food and Drug Administration, Center for Drug Evaluation and Research . Clinical Drug Interaction Studies — Study Design, Data Analysis, and Clinical Implications Guidance for Industry <https://www.fda.gov/downloads/drugs/guidances/ucm292362.pdf> (2017). Accessed February 15, 2019.
- 3. European Medicines Agency . Guideline on the investigation of drug interactions <https://www.ema.europa.eu/documents/scientific-guideline/guideline-investigation-drug-interactions_en.pdf> (2012). Accessed February 15, 2019.
- 4. Yu, J. , Petrie, I.D. , Levy, R.H. & Ragueneau‐Majlessi, I. Mechanisms and clinical significance of pharmacokinetic‐based drug‐drug interactions with drugs approved by the U.S. Food and Drug Administration in 2017. Drug Metab. Dispos. 47, 135–144 (2019). [DOI] [PubMed] [Google Scholar]
- 5. Backman, J.T. , Kivistö, K.T. , Olkkola, K.T. & Neuvonen, P.J. The area under the plasma concentration‐time curve for oral midazolam is 400‐fold larger during treatment with itraconazole than with rifampicin. Eur. J. Clin. Pharmacol. 54, 53–58 (1998). [DOI] [PubMed] [Google Scholar]
- 6. de Jong, J. et al Effect of CYP3A perpetrators on ibrutinib exposure in healthy participants. Pharmacol. Res. Perspect. 3, e00156 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fuhr, U. , Hsin, C.H. , Li, X. , Jabrane, W. & Sörgel, F. Assessment of pharmacokinetic drug‐drug interactions in humans: in vivo probe substrates for drug metabolism and drug transport revisited. Annu. Rev. Pharmacol. Toxicol. 59, 507–536 (2019). [DOI] [PubMed] [Google Scholar]
- 8. Grimstein, M. et al Physiologically based pharmacokinetic modeling in regulatory science: an update from the U.S. Food and Drug Administration's Office of Clinical Pharmacology. J. Pharm. Sci. 108, 21–25 (2019). [DOI] [PubMed] [Google Scholar]
- 9. Prueksaritanont, T. et al Drug‐drug interaction studies: regulatory guidance and an industry perspective. AAPS J. 15, 629–645 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rekic, D. et al Clinical drug‐drug interaction evaluations to inform drug use and enable drug access. J. Pharm. Sci. 106, 2214–2218 (2017). [DOI] [PubMed] [Google Scholar]
- 11. Floyd, J.S. et al A screening study of drug‐drug interactions in cerivastatin users: an adverse effect of clopidogrel. Clin. Pharmacol. Ther. 91, 896–904 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tornio, A. et al Glucuronidation converts clopidogrel to a strong time‐dependent inhibitor of CYP2C8: a phase II metabolite as a perpetrator of drug‐drug interactions. Clin. Pharmacol. Ther. 96, 498–507 (2014). [DOI] [PubMed] [Google Scholar]
- 13. Niemi, M. , Backman, J.T. , Fromm, M.F. , Neuvonen, P.J. & Kivistö, K.T. Pharmacokinetic interactions with rifampicin: clinical relevance. Clin. Pharmacokinet. 42, 819–850 (2003). [DOI] [PubMed] [Google Scholar]
- 14. Backman, J.T. , Luurila, H. , Neuvonen, M. & Neuvonen, P.J. Rifampin markedly decreases and gemfibrozil increases the plasma concentrations of atorvastatin and its metabolites. Clin. Pharmacol. Ther. 78, 154–167 (2005). [DOI] [PubMed] [Google Scholar]
- 15. Lau, Y.Y. , Huang, Y. , Frassetto, L. & Benet, L.Z. Effect of OATP1B transporter inhibition on the pharmacokinetics of atorvastatin in healthy volunteers. Clin. Pharmacol. Ther. 81, 194–204 (2007). [DOI] [PubMed] [Google Scholar]
- 16. Niemi, M. , Pasanen, M.K. & Neuvonen, P.J. Organic anion transporting polypeptide 1B1: a genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol. Rev. 63, 157–181 (2011). [DOI] [PubMed] [Google Scholar]
- 17. Hukkanen, J. Induction of cytochrome P450 enzymes: a view on human in vivo findings. Expert Rev. Clin. Pharmacol. 5, 569–585 (2012). [DOI] [PubMed] [Google Scholar]
- 18. Zamek‐Gliszczynski, M.J. et al Transporters in drug development: 2018 ITC recommendations for transporters of emerging clinical importance. Clin. Pharmacol. Ther. 104, 890–899 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. International Transporter Consortium , Giacomini, K.M. , Huang, S.M. et al Membrane transporters in drug development. Nat. Rev. Drug Discov. 9, 215–236 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Elsby, R. , Martin, P. , Surry, D. , Sharma, P. & Fenner, K. Solitary inhibition of the breast cancer resistance protein efflux transporter results in a clinically significant drug‐drug interaction with rosuvastatin by causing up to a 2‐fold increase in statin exposure. Drug Metab. Dispos. 44, 398–408 (2016). [DOI] [PubMed] [Google Scholar]
- 21. Lund, M. , Petersen, T.S. & Dalhoff, K.P. Clinical implications of P‐glycoprotein modulation in drug‐drug interactions. Drugs 77, 859–883 (2017). [DOI] [PubMed] [Google Scholar]
- 22. Jalava, K.M. , Partanen, J. & Neuvonen, P.J. Itraconazole decreases renal clearance of digoxin. Ther. Drug Monit. 19, 609–613 (1997). [DOI] [PubMed] [Google Scholar]
- 23. US Food and Drug Administration, Center for Drug Evaluation and Research . Clinical Pharmacology Biopharmaceutics Review(s): Pradaxa <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2010/022512Orig1s000ClinPharmR_Corrrected%203.11.2011.pdf> (2010). Accessed February 19, 2019.
- 24. Chu, X. et al Clinical probes and endogenous biomarkers as substrates for transporter drug‐drug interaction evaluation: perspectives from the international transporter consortium. Clin. Pharmacol. Ther. 104, 836–864 (2018). [DOI] [PubMed] [Google Scholar]
- 25. Zamek‐Gliszczynski, M.J. et al ITC commentary on metformin clinical drug‐drug interaction study design that enables an efficacy‐ and safety‐based dose adjustment decision. Clin. Pharmacol. Ther. 104, 781–784 (2018). [DOI] [PubMed] [Google Scholar]
- 26. Guo, Y. et al Advancing predictions of tissue and intracellular drug concentrations using in vitro, imaging and physiologically based pharmacokinetic modeling approaches. Clin. Pharmacol. Ther. 104, 865–889 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kajosaari, L.I. , Niemi, M. , Neuvonen, M. , Laitila, J. , Neuvonen, P.J. & Backman, J.T. Cyclosporine markedly raises the plasma concentrations of repaglinide. Clin. Pharmacol. Ther. 78, 388–399 (2005). [DOI] [PubMed] [Google Scholar]
- 28. Tornio, A. & Backman, J.T. Cytochrome P450 in pharmacogenetics: an update. Adv. Pharmacol. 83, 3–32 (2018). [DOI] [PubMed] [Google Scholar]
- 29. Yee, S.W. et al Influence of transporter polymorphisms on drug disposition and response: a perspective from the international transporter consortium. Clin. Pharmacol. Ther. 104, 803–817 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bank, P.C.D. et al Comparison of the guidelines of the clinical pharmacogenetics implementation consortium and the Dutch pharmacogenetics working group. Clin. Pharmacol. Ther. 103, 599–618 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fuhr, U. , Jetter, A. & Kirchheiner, J. Appropriate phenotyping procedures for drug metabolizing enzymes and transporters in humans and their simultaneous use in the “cocktail” approach. Clin. Pharmacol. Ther. 81, 270–283 (2007). [DOI] [PubMed] [Google Scholar]
- 32. Mikus, G . Probes and cocktails for drug‐drug interaction evaluation: the future is microdosing? Clin. Pharmacol. Ther. 105, 1335–1337 (2019). [DOI] [PubMed] [Google Scholar]
- 33. Bosilkovska, M. et al Geneva cocktail for cytochrome p450 and P‐glycoprotein activity assessment using dried blood spots. Clin. Pharmacol. Ther. 96, 349–359 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bosilkovska, M. et al Evaluation of mutual drug‐drug interaction within Geneva cocktail for cytochrome P450 phenotyping using innovative dried blood sampling method. Basic Clin. Pharmacol. Toxicol. 119, 284–290 (2016). [DOI] [PubMed] [Google Scholar]
- 35. Derungs, A. , Donzelli, M. , Berger, B. , Noppen, C. , Krahenbuhl, S. & Haschke, M. Effects of cytochrome P450 inhibition and induction on the phenotyping metrics of the Basel cocktail: a randomized crossover study. Clin. Pharmacokinet. 55, 79–91 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stopfer, P. et al Pharmacokinetic evaluation of a drug transporter cocktail consisting of digoxin, furosemide, metformin, and rosuvastatin. Clin. Pharmacol. Ther. 100, 259–267 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stopfer, P. et al Effects of metformin and furosemide on rosuvastatin pharmacokinetics in healthy volunteers: implications for their use as probe drugs in a transporter cocktail. Eur. J. Drug Metab. Pharmacokinet. 43, 69–80 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Stopfer, P. et al Optimization of a drug transporter probe cocktail: potential screening tool for transporter‐mediated drug‐drug interactions. Br. J. Clin. Pharmacol. 84, 1941–1949 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Prueksaritanont, T. et al Validation of a microdose probe drug cocktail for clinical drug interaction assessments for drug transporters and CYP3A. Clin. Pharmacol. Ther. 101, 519–530 (2017). [DOI] [PubMed] [Google Scholar]
- 40. Chu, X. , Chan, G.H. & Evers, R. Identification of endogenous biomarkers to predict the propensity of drug candidates to cause hepatic or renal transporter‐mediated drug‐drug interactions. J. Pharm. Sci. 106, 2357–2367 (2017). [DOI] [PubMed] [Google Scholar]
- 41. Mariappan, T.T. , Shen, H. & Marathe, P. Endogenous biomarkers to assess drug‐drug interactions by drug transporters and enzymes. Curr. Drug Metab. 18, 757–768 (2017). [DOI] [PubMed] [Google Scholar]
- 42. Müller, F. , Sharma, A. , König, J. & Fromm, M.F. Biomarkers for in vivo assessment of transporter function. Pharmacol. Rev. 70, 246–277 (2018). [DOI] [PubMed] [Google Scholar]
- 43. Bonate, P.L. et al Methods and strategies for assessing uncontrolled drug‐drug interactions in population pharmacokinetic analyses: results from the International Society of Pharmacometrics (ISOP) Working Group. J. Pharmacokinet. Pharmacodyn. 43, 123–135 (2016). [DOI] [PubMed] [Google Scholar]
- 44. Wang, D.D. et al The utility of a population approach in drug‐drug interaction assessments: a simulation evaluation. J. Clin. Pharmacol. 57, 1268–1278 (2017). [DOI] [PubMed] [Google Scholar]
- 45. Masters, J.C. , Harano, D.M. , Greenberg, H.E. , Tsunoda, S.M. , Jang, I.J. & Ma, J.D. Limited sampling strategy of partial area under the concentration‐time curves to estimate midazolam systemic clearance for cytochrome P450 3A phenotyping. Ther. Drug Monit. 37, 84–89 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chang, A.T. et al S‐Warfarin limited sampling models to estimate area under the concentration versus time curve for cytochrome P450 2C9 baseline activity and after induction. Ther. Drug Monit. 38, 383–387 (2016). [DOI] [PubMed] [Google Scholar]
- 47. Fuhr, U. & Rost, K.L. Simple and reliable CYP1A2 phenotyping by the paraxanthine/caffeine ratio in plasma and in saliva. Pharmacogenetics 4, 109–116 (1994). [DOI] [PubMed] [Google Scholar]
- 48. de Andrés, F. , Terán, S. , Bovera, M. , Fariñas, H. , Terán, E. & LLerena, A. Multiplex phenotyping for systems medicine: a one‐point optimized practical sampling strategy for simultaneous estimation of CYP1A2, CYP2C9, CYP2C19, and CYP2D6 activities using a cocktail approach. OMICS 20, 88–96 (2016). [DOI] [PubMed] [Google Scholar]
- 49. Sager, J.E. , Yu, J. , Ragueneau‐Majlessi, I. & Isoherranen, N. Physiologically based pharmacokinetic (PBPK) modeling and simulation approaches: a systematic review of published models, applications, and model verification. Drug Metab. Dispos. 43, 1823–1837 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yoshida, K. et al In vitro‐in vivo extrapolation of metabolism‐ and transporter‐mediated drug‐drug interactions‐overview of basic prediction methods. J. Pharm. Sci. 106, 2209–2213 (2017). [DOI] [PubMed] [Google Scholar]
- 51. European Medicines Agency . Guideline on the qualification and reporting of physiologically based pharmacokinetic (PBPK) modelling and simulation <http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2016/07/WC500211315.pdf> (2016). Accessed February 15, 2019.
- 52. US Food and Drug Administration, Center for Drug Evaluation and Research . Physiologically based pharmacokinetic analyses — format and content guidance for industry <https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM531207.pdf> (2018). Accessed February 15, 2019.
- 53. Yeo, K.R. , Jamei, M. & Rostami‐Hodjegan, A. Predicting drug‐drug interactions: application of physiologically based pharmacokinetic models under a systems biology approach. Expert Rev. Clin. Pharmacol. 6, 143–157 (2013). [DOI] [PubMed] [Google Scholar]
- 54. Huang, S.M. & Rowland, M. The role of physiologically based pharmacokinetic modeling in regulatory review. Clin. Pharmacol. Ther. 91, 542–549 (2012). [DOI] [PubMed] [Google Scholar]
- 55. Shebley, M. et al Physiologically based pharmacokinetic model qualification and reporting procedures for regulatory submissions: a consortium perspective. Clin. Pharmacol. Ther. 104, 88–110 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jones, H.M. et al Physiologically based pharmacokinetic modeling in drug discovery and development: a pharmaceutical industry perspective. Clin. Pharmacol. Ther. 97, 247–262 (2015). [DOI] [PubMed] [Google Scholar]
- 57. Wagner, C. et al Application of physiologically based pharmacokinetic (PBPK) modeling to support dose selection: report of an FDA public workshop on PBPK. CPT Pharmacometrics Syst. Pharmacol. 4, 226–230 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pan, Y. et al The application of physiologically based pharmacokinetic modeling to predict the role of drug transporters: scientific and regulatory perspectives. J. Clin. Pharmacol. 56 (suppl. 7), S122–S131 (2016). [DOI] [PubMed] [Google Scholar]
- 59. Shebley, M. , Fu, W. , Badri, P. , Bow, D. & Fischer, V. Physiologically based pharmacokinetic modeling suggests limited drug‐drug interaction between clopidogrel and dasabuvir. Clin. Pharmacol. Ther. 102, 679–687 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Varma, M.V. , Lai, Y. , Feng, B. , Litchfield, J. , Goosen, T.C. & Bergman, A. Physiologically based modeling of pravastatin transporter‐mediated hepatobiliary disposition and drug‐drug interactions. Pharm. Res. 29, 2860–2873 (2012). [DOI] [PubMed] [Google Scholar]
- 61. Yoshikado, T. et al Quantitative analyses of hepatic OATP‐mediated interactions between statins and inhibitors using PBPK modeling with a parameter optimization method. Clin. Pharmacol. Ther. 100, 513–523 (2016). [DOI] [PubMed] [Google Scholar]
- 62. Shitara, Y. & Sugiyama, Y. Preincubation‐dependent and long‐lasting inhibition of organic anion transporting polypeptide (OATP) and its impact on drug‐drug interactions. Pharmacol. Ther. 177, 67–80 (2017). [DOI] [PubMed] [Google Scholar]
- 63. VandenBrink, B.M. & Isoherranen, N. The role of metabolites in predicting drug‐drug interactions: focus on irreversible cytochrome P450 inhibition. Curr. Opin. Drug Discov. Devel. 13, 66–77 (2010). [PMC free article] [PubMed] [Google Scholar]
- 64. Varma, M.V. , Pang, K.S. , Isoherranen, N. & Zhao, P. Dealing with the complex drug‐drug interactions: towards mechanistic models. Biopharm. Drug Dispos. 36, 71–92 (2015). [DOI] [PubMed] [Google Scholar]
- 65. Filppula, A.M. , Laitila, J. , Neuvonen, P.J. & Backman, J.T. Potent mechanism‐based inhibition of CYP3A4 by imatinib explains its liability to interact with CYP3A4 substrates. Br. J. Pharmacol. 165, 2787–2798 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Filppula, A.M. , Neuvonen, M. , Laitila, J. , Neuvonen, P.J. & Backman, J.T. Autoinhibition of CYP3A4 leads to important role of CYP2C8 in imatinib metabolism: variability in CYP2C8 activity may alter plasma concentrations and response. Drug Metab. Dispos. 41, 50–59 (2013). [DOI] [PubMed] [Google Scholar]
- 67. Dutreix, C. , Munarini, F. , Lorenzo, S. , Roesel, J. & Wang, Y. Investigation into CYP3A4‐mediated drug‐drug interactions on midostaurin in healthy volunteers. Cancer Chemother. Pharmacol. 72, 1223–1234 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wang, Y. , Yin, O.Q. , Graf, P. , Kisicki, J.C. & Schran, H. Dose‐ and time‐dependent pharmacokinetics of midostaurin in patients with diabetes mellitus. J. Clin. Pharmacol. 48, 763–775 (2008). [DOI] [PubMed] [Google Scholar]
- 69. Yin, O.Q. , Wang, Y. & Schran, H. A mechanism‐based population pharmacokinetic model for characterizing time‐dependent pharmacokinetics of midostaurin and its metabolites in human subjects. Clin. Pharmacokinet. 47, 807–816 (2008). [DOI] [PubMed] [Google Scholar]
- 70. Simonson, S.G. et al Rosuvastatin pharmacokinetics in heart transplant recipients administered an antirejection regimen including cyclosporine. Clin. Pharmacol. Ther. 76, 167–177 (2004). [DOI] [PubMed] [Google Scholar]
- 71. Niemi, M. , Backman, J.T. , Neuvonen, M. & Neuvonen, P.J. Effects of gemfibrozil, itraconazole, and their combination on the pharmacokinetics and pharmacodynamics of repaglinide: potentially hazardous interaction between gemfibrozil and repaglinide. Diabetologia 46, 347–351 (2003). [DOI] [PubMed] [Google Scholar]
- 72. US Food and Drug Administration, Center for Drug Evaluation and Research . Clinical Pharmacology Biopharmaceutics Review(s): Vosevi <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/209195Orig1s000ClinPharmR.pdf> (2017). Accessed February 18, 2019.
- 73. Foisy, M.M. , Yakiwchuk, E.M. & Hughes, C.A. Induction effects of ritonavir: implications for drug interactions. Ann. Pharmacother. 42, 1048–1059 (2008). [DOI] [PubMed] [Google Scholar]
- 74. Yeung, C.K. , Fujioka, Y. , Hachad, H. , Levy, R.H. & Isoherranen, N. Are circulating metabolites important in drug‐drug interactions?: Quantitative analysis of risk prediction and inhibitory potency. Clin. Pharmacol. Ther. 89, 105–113 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Backman, J.T. , Filppula, A.M. , Niemi, M. & Neuvonen, P.J. Role of cytochrome P450 2C8 in drug metabolism and interactions. Pharmacol. Rev. 68, 168–241 (2016). [DOI] [PubMed] [Google Scholar]
- 76. Storelli, F. , Samer, C. , Reny, J.L. , Desmeules, J. & Daali, Y. Complex drug‐drug‐gene‐disease interactions involving cytochromes P450: systematic review of published case reports and clinical perspectives. Clin. Pharmacokinet. 57, 1267–1293 (2018). [DOI] [PubMed] [Google Scholar]
- 77. Niemi, M. , Tornio, A. , Pasanen, M.K. , Fredrikson, H. , Neuvonen, P.J. & Backman, J.T. Itraconazole, gemfibrozil and their combination markedly raise the plasma concentrations of loperamide. Eur. J. Clin. Pharmacol. 62, 463–472 (2006). [DOI] [PubMed] [Google Scholar]
- 78. Brøsen, K. , Hansen, J.G. , Nielsen, K.K. , Sindrup, S.H. & Gram, L.F. Inhibition by paroxetine of desipramine metabolism in extensive but not in poor metabolizers of sparteine. Eur. J. Clin. Pharmacol. 44, 349–355 (1993). [DOI] [PubMed] [Google Scholar]
- 79. Yu, K.S. et al Effect of omeprazole on the pharmacokinetics of moclobemide according to the genetic polymorphism of CYP2C19. Clin. Pharmacol. Ther. 69, 266–273 (2001). [DOI] [PubMed] [Google Scholar]
- 80. Weiss, J. et al CYP2C19 genotype is a major factor contributing to the highly variable pharmacokinetics of voriconazole. J. Clin. Pharmacol. 49, 196–204 (2009). [DOI] [PubMed] [Google Scholar]
- 81. Orlando, R. , Padrini, R. , Perazzi, M. , De Martin, S. , Piccoli, P. & Palatini, P. Liver dysfunction markedly decreases the inhibition of cytochrome P450 1A2‐mediated theophylline metabolism by fluvoxamine. Clin. Pharmacol. Ther. 79, 489–499 (2006). [DOI] [PubMed] [Google Scholar]
- 82. Onakpoya, I.J. , Heneghan, C.J. & Aronson, J.K. Delays in the post‐marketing withdrawal of drugs to which deaths have been attributed: a systematic investigation and analysis. BMC Med. 13, 26 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Tornio, A. , Neuvonen, P.J. , Niemi, M. & Backman, J.T. Role of gemfibrozil as an inhibitor of CYP2C8 and membrane transporters. Expert Opin. Drug Metab. Toxicol. 13, 83–95 (2017). [DOI] [PubMed] [Google Scholar]
- 84. Backman, J.T. , Kyrklund, C. , Neuvonen, M. & Neuvonen, P.J. Gemfibrozil greatly increases plasma concentrations of cerivastatin. Clin. Pharmacol. Ther. 72, 685–691 (2002). [DOI] [PubMed] [Google Scholar]
- 85. Kantola, T. , Kivistö, K.T. & Neuvonen, P.J. Effect of itraconazole on cerivastatin pharmacokinetics. Eur. J. Clin. Pharmacol. 54, 851–855 (1999). [DOI] [PubMed] [Google Scholar]
- 86. Ogilvie, B.W. et al Glucuronidation converts gemfibrozil to a potent, metabolism‐dependent inhibitor of CYP2C8: implications for drug‐drug interactions. Drug Metab. Dispos. 34, 191–197 (2006). [DOI] [PubMed] [Google Scholar]
- 87. Bachmann, K. et al An evaluation of the dose‐dependent inhibition of CYP1A2 by rofecoxib using theophylline as a CYP1A2 probe. J. Clin. Pharmacol. 43, 1082–1090 (2003). [DOI] [PubMed] [Google Scholar]
- 88. Backman, J.T. , Karjalainen, M.J. , Neuvonen, M. , Laitila, J. & Neuvonen, P.J. Rofecoxib is a potent inhibitor of cytochrome P450 1A2: studies with tizanidine and caffeine in healthy subjects. Br. J. Clin. Pharmacol. 62, 345–357 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Karjalainen, M.J. , Neuvonen, P.J. & Backman, J.T. Rofecoxib is a potent, metabolism‐dependent inhibitor of CYP1A2: implications for in vitro prediction of drug interactions. Drug Metab. Dispos. 34, 2091–2096 (2006). [DOI] [PubMed] [Google Scholar]
- 90. Rogers, J.D. et al Grapefruit juice has minimal effects on plasma concentrations of lovastatin‐derived 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase inhibitors. Clin. Pharmacol. Ther. 66, 358–366 (1999). [DOI] [PubMed] [Google Scholar]
- 91. Kantola, T. , Kivistö, K.T. & Neuvonen, P.J. Grapefruit juice greatly increases serum concentrations of lovastatin and lovastatin acid. Clin. Pharmacol. Ther. 63, 397–402 (1998). [DOI] [PubMed] [Google Scholar]
- 92. Suzuki, E. et al Identification of valproic acid glucuronide hydrolase as a key enzyme for the interaction of valproic acid with carbapenem antibiotics. Drug Metab. Dispos. 38, 1538–1544 (2010). [DOI] [PubMed] [Google Scholar]
- 93. Myllylä, V.V. , Sotaniemi, K.A. , Illi, A. , Suominen, K. & Keränen, T. Effect of entacapone, a COMT inhibitor, on the pharmacokinetics of levodopa and on cardiovascular responses in patients with Parkinson's disease. Eur. J. Clin. Pharmacol. 45, 419–423 (1993). [DOI] [PubMed] [Google Scholar]
- 94. Diasio, R.B. Sorivudine and 5‐fluorouracil; a clinically significant drug‐drug interaction due to inhibition of dihydropyrimidine dehydrogenase. Br. J. Clin. Pharmacol. 46, 1–4 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Grana, E. , Barbieri, A. , Zonta, F. & Lucchelli, A. Potentiating actions of tranylcypromine and moclobemide on the sympathomimetic activity of dopamine. Pharmacol. Res. 24, 337–343 (1991). [DOI] [PubMed] [Google Scholar]
- 96. Cleary, J.M. , Rosen, L.S. , Yoshida, K. , Rasco, D. , Shapiro, G.I. & Sun, W. A phase 1 study of the pharmacokinetics of nucleoside analog trifluridine and thymidine phosphorylase inhibitor tipiracil (components of TAS‐102) vs trifluridine alone. Invest. New Drugs 35, 189–197 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Corona, G. et al Lopinavir‐ritonavir dramatically affects the pharmacokinetics of irinotecan in HIV patients with Kaposi's sarcoma. Clin. Pharmacol. Ther. 83, 601–606 (2008). [DOI] [PubMed] [Google Scholar]
- 98. Kasichayanula, S. , Liu, X. , Griffen, S.C. , Lacreta, F.P. & Boulton, D.W. Effects of rifampin and mefenamic acid on the pharmacokinetics and pharmacodynamics of dapagliflozin. Diabetes Obes. Metab. 15, 280–283 (2013). [DOI] [PubMed] [Google Scholar]
- 99. Gidal, B.E. , Sheth, R. , Parnell, J. , Maloney, K. & Sale, M. Evaluation of VPA dose and concentration effects on lamotrigine pharmacokinetics: implications for conversion to lamotrigine monotherapy. Epilepsy Res. 57, 85–93 (2003). [DOI] [PubMed] [Google Scholar]
- 100. Zimm, S. , Collins, J.M. , O'Neill, D. , Chabner, B.A. & Poplack, D.G. Inhibition of first‐pass metabolism in cancer chemotherapy: interaction of 6‐mercaptopurine and allopurinol. Clin. Pharmacol. Ther. 34, 810–817 (1983). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Characteristics of possible index substrates of cytochrome P450 enzymes. The table contains all the FDA and the EMA‐recommended index substrates and some selected alternatives based on potential advantages in sensitivity, selectivity, and/or relative safety.
Table S2. Characteristics of possible index inhibitors of cytochrome P450 enzymes. The table contains all the FDA and the EMA‐recommended index inhibitors and some selected alternatives based on potential advantages in sensitivity, selectivity, and/or relative safety.
Table S3. Characteristics of transporter substrates and inhibitors.