

Abstract
The integration of enzymes with synthetic materials allows efficient electrocatalysis and production of solar fuels. Here, we couple formate dehydrogenase (FDH) from Desulfovibrio vulgaris Hildenborough (DvH) to metal oxides for catalytic CO2 reduction and report an in‐depth study of the resulting enzyme–material interface. Protein film voltammetry (PFV) demonstrates the stable binding of FDH on metal‐oxide electrodes and reveals the reversible and selective reduction of CO2 to formate. Quartz crystal microbalance (QCM) and attenuated total reflection infrared (ATR‐IR) spectroscopy confirm a high binding affinity for FDH to the TiO2 surface. Adsorption of FDH on dye‐sensitized TiO2 allows for visible‐light‐driven CO2 reduction to formate in the absence of a soluble redox mediator with a turnover frequency (TOF) of 11±1 s−1. The strong coupling of the enzyme to the semiconductor gives rise to a new benchmark in the selective photoreduction of aqueous CO2 to formate.
Keywords: artificial photosynthesis, carbon dioxide fixation, formate dehydrogenase, interfaces, photocatalysis
Electrocatalytic‐ and solar‐driven fuel synthesis from the greenhouse gas CO2 is a desirable approach to simultaneously produce sustainable energy carriers and combat increasing atmospheric CO2 levels.1 Formate is a stable intermediate in the reduction of CO2 and can be used as liquid energy carrier in fuel cells, as a hydrogen storage material, or feedstock for the synthesis of fine chemicals.2 Metals and synthetic molecular systems have been widely studied as electrocatalysts for CO2 reduction to formate, but largely lack the required efficiency, selectivity or affordability to enable carbon capture and utilization technologies.3, 4
There is avid research into both biological and artificial CO2 fixation. Semi‐artificial photosynthesis provides a common stage for these contrasting approaches as components from synthetic and biological origin can be combined in hybrid model systems.5 To date, enzyme‐based visible‐light‐driven CO2 reduction to formate relies on diffusional mediators, such as methyl viologen (MV2+) and nicotinamide adenine dinucleotide (NAD+).6, 7 Mediated processes are inefficient as they consume energy, are kinetically slow, and cause short‐circuit reactions. MV2+ is toxic to microorganisms,8 and NAD+ is prohibitively expensive for fuel production.6
In this work, we selected wild‐type formate dehydrogenase (FDH) from Desulfovibrio vulgaris Hildenborough (DvH) as it has previously displayed robustness and high activity for the oxidation of formate in solution assays,10, 11 and the electrochemical reduction of CO2.12 Initially, protein film voltammetry (PFV) was employed to study the interfacial electron transfer between FDH and porous indium‐doped tin oxide (ITO) and TiO2 electrodes in the absence of a mediator. Immobilization and loading of FDH on TiO2 were then investigated using a quartz crystal microbalance (QCM) and attenuated total reflection infrared (ATR‐IR) spectroscopy. FDH was finally coupled directly to dye‐sensitized TiO2 nanoparticles for the selective photocatalytic reduction of CO2 to formate in a diffusional mediator‐free colloidal system (Figure 1).
Figure 1.
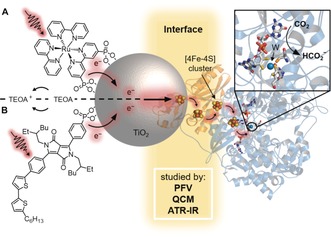
Schematic CO2 conversion with a dye–semiconductor–FDH photocatalyst system. Photoexcited electrons from the dye, RuP in (A) or DPP in (B), are transferred via the conduction band (CB) of TiO2 across the enzyme–material interface through the intraprotein [4Fe–4S] relays to the W‐active site of FDH for the reduction of CO2 to formate. The oxidized dye is regenerated by triethanolamine (TEOA). A protein structure homologous to DvH FDH is shown.9
The electrocatalytic activity of FDH on metal‐oxide electrodes was studied by PFV on mesoporous ITO (mesoITO) and TiO2 (mesoTiO2) electrodes with a film thickness of approximately 2.5 μm (Supporting Information, Figure S1).13 FDH (21.5 μm) was activated by incubation with the reducing agent dl‐dithiothreitol (DTT, 50 mm)9 and the resulting solution (2 μL) was drop‐cast on the electrode surface. The FDH‐modified electrode was placed in an electrolyte solution containing CO2/NaHCO3 and KCl at pH 6.5 under a CO2 atmosphere.
Figure 2 A shows the electrochemically reversible interconversion of CO2 and formate by FDH immobilized on a conductive mesoITO electrode (mesoITO|FDH). The onset potential for both CO2 reduction and formate oxidation was observed close to the thermodynamic potential (E 0′=−0.36 V vs. standard hydrogen electrode (SHE), pH 6.5),14 demonstrating that interfacial electron transfer by the [4Fe–4S] relays and catalysis at the W‐active site are highly efficient.15 Similar electrochemically reversible characteristics have previously only been reported for FDHs from Escherichia coli and Syntrophobacter fumaroxidans on graphite electrodes.14, 16, 17
Figure 2.
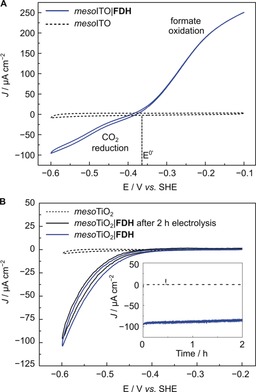
PFV (ν=5 mV s−1) showing A) reversible reduction of CO2 to formate by mesoITO|FDH (blue trace) and B) CO2 reduction by mesoTiO2|FDH before (blue) and after 2 h CPE (black). Inset: CPE at −0.6 V vs. SHE. Conditions for A and B: 43 pmol FDH (amount drop‐cast), 100 mm CO2/NaHCO3, 50 mm KCl, 20 mm formate (only present in A), 1 atm CO2, pH 6.5, 25 °C, Pt counter electrode. Dashed traces show control experiments of FDH‐free electrodes.
When FDH was immobilized on a semiconducting mesoTiO2 electrode (mesoTiO2|FDH), a similar onset potential for CO2 reduction (−0.4 V vs. SHE) was observed and the current density reached −100 μA cm−2 at −0.6 V vs. SHE (Figure 2 B). Formate oxidation could not be observed for mesoTiO2|FDH electrodes as TiO2 behaves as an insulator at the required potentials. Controlled‐potential electrolysis (CPE) at −0.6 V vs. SHE for 2 h produced formate with a Faradaic efficiency of (92±5)% (Figure 2 B, inset). Comparison of PFV scans before and after CPE showed that approximately 90 % of the initial FDH activity remains after 2 h, demonstrating the excellent stability of the immobilized enzyme.
The interaction of FDH and TiO2 was quantitatively investigated with a previously described QCM cell.18, 19 Upon flowing an FDH‐containing solution over a planarTiO2‐covered quartz chip (12 nm in 100 mm TEOA), the surface of TiO2 reached saturation after 1 h, resulting in approximately 3.5 pmol cm−2 of adsorbed FDH (planarTiO2|FDH, Figure 3 A). The strength of the enzyme–TiO2 interaction was probed by exposing the planarTiO2|FDH electrode to buffer solutions with different ionic strengths. Rinsing the QCM cell with an enzyme‐free solution for 1 h desorbed only 6 % of the preloaded FDH. Upon increasing the KCl concentration to 0.5–3.0 m KCl, 70–60 % of FDH remained adsorbed on the TiO2 surface. The finding that 60 % FDH remained adsorbed on TiO2 after multiple rinsing steps with high KCl concentrations suggests a contribution from chemisorption to the attachment of the enzyme.20, 21 Amino‐acid residues exposed on the FDH surface are likely involved in binding. For example, aspartic and glutamic acid have previously been suggested to form a strong interaction with TiO2.22, 23
Figure 3.
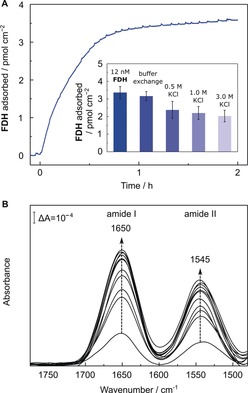
A) QCM analysis of the adsorption process of FDH on a planarTiO2‐coated quartz chip. Conditions: 12 nm FDH, 100 mm TEOA, open circuit potential of −0.1 to 0.0 V vs. SHE, pH 6.5, 25 °C, N2 atmosphere, circulation (0.141 mL min−1). Inset: Desorption of FDH by replacing the solution with fresh solution (100 mm TEOA) and subsequent increase of the ionic strength (each condition was held for 1 h). Error bars correspond to standard deviation (N=3). B) ATR‐IR absorbance spectra of the amide‐band region of FDH during the adsorption process over time onto a planarTiO2‐coated Si prism (100 nm thickness). Arrows indicate successively recorded spectra of every 1.5 min up to 7.5 min and subsequently every 30 min. Conditions: 1.0 μm FDH, 100 mm TEOA, total volume: 150 μL, open circuit potential, pH 6.5, 25 °C.
The adsorption of FDH was also probed by surface‐selective ATR‐IR spectroscopy using a Si prism coated with a planar or a mesoTiO2 layer (100 or 400 nm thickness, respectively). After the addition of FDH to the buffer solution covering the planarTiO2 (Figure 3 B) or mesoTiO2 (Supporting Information, Figure S2) coated prism, the two characteristic amide I and amide II bands of the protein backbone structure were detected at 1650 cm−1 and 1545 cm−1, respectively.24 The protein adsorption was monitored in situ over 2 h of incubation time and no (in the case of planarTiO2) or slight (in the case of mesoTiO2) changes to the band features in the amide‐band region were observed, suggesting a mainly retained backbone structure of FDH on the surface of TiO2. During the adsorption process, amide I and amide II band intensities showed an increase over time (Figure 3 B). The majority of FDH remained adsorbed on the surface of planarTiO2 (Supporting Information, Figure S3) upon increasing the ionic strength of the buffer, which agrees with the QCM experiments (Figure 3 A, inset) and supports a stronger than purely electrostatic interaction between FDH and TiO2.
After establishing the strong interface between FDH and TiO2, visible‐light‐driven CO2 reduction to formate was investigated with FDH immobilized on dye‐sensitized TiO2 nanoparticles (dye|TiO2|FDH, Figures 1 and 4). The colloidal system was self‐assembled by adding FDH (pre‐activated with DTT) to a suspension of TiO2 nanoparticles containing TEOA and a phosphonate group‐bearing dye, either a ruthenium tris‐2,2′‐bipyridine complex (RuP) or a diketopyrrolopyrrole (DPP) at pH 6.5 and 25 °C under N2 atmosphere (to protect the enzyme from aerobic damage). Both dyes are known to adsorb onto TiO2 via their phosphonate‐anchoring groups and DPP provides a precious‐metal‐free alternative to RuP.25 CO2 was introduced to the solution via the addition of NaHCO3. Upon UV‐filtered irradiation, the photoexcited dye injects electrons into the conduction band (CB) of TiO2 (E CB(TiO2)=−0.67 V vs. SHE at pH 6.5),25 whereupon the electrons are conveyed to the catalytic W‐center of FDH to drive CO2 reduction. The oxidized dye is regenerated by the sacrificial electron donor (Figure 1).26
Figure 4.
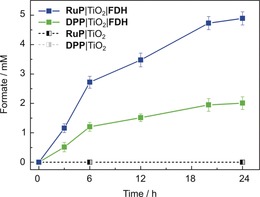
Photocatalytic CO2 reduction to formate with FDH in a colloidal dye‐sensitized TiO2 system. Conditions: 12 nm FDH, 10 mm DTT, 0.83 mg mL−1 TiO2, 16.7 μm dye (RuP or DPP), 100 mm TEOA, 100 mm NaHCO3, pH 6.5, 25 °C, total volume: 1.0 mL, assembled in an anaerobic glove box, UV‐filtered simulated solar‐light irradiation: 100 mW cm−2, AM 1.5G, λ>420 nm. Error bars correspond to standard deviation (N=3). Dashed traces show control experiments in the absence of FDH.
The dye|TiO2|FDH systems showed stable formate production for approximately 6 h (Figure 4). The formation of gaseous or dissolved side‐products was not detected by gas chromatography, ion chromatography, and 1H NMR spectroscopy. The activity of RuP|TiO2|FDH was not limited by the amount of dye or the light intensity (Supporting Information, Figures S4 and S5). A solution assay monitoring the activity of FDH by UV/Vis spectroscopy (via formate oxidation in presence of 2 mm MV2+) showed that approximately 36±7 % FDH remained active after 24 h of photocatalysis (Supporting Information, Figure S6), suggesting that inactivation of FDH is likely the main reason for activity loss. The addition of MV2+ as a soluble redox mediator to RuP|TiO2|FDH showed that not all FDH present in the system is accessible to direct electron transfer across the enzyme–material interface (Supporting Information, Figure S7). Control experiments demonstrated that all components are essential for formate production (Supporting Information, Figures S8 and S9) and support oxidative quenching and “through‐particle” electron transfer as depicted in Figure 1 (Supporting Information, Figures S10 and S11).26 Isotopic‐labeling studies confirmed that formate was produced from CO2 (Supporting Information, Figure S12).
For photocatalytic experiments, an enzyme loading of approximately 0.03 pmol cm−2 was calculated assuming that all FDH is adsorbed on TiO2 with a surface area of 50 m2 g−1. Saturation of the TiO2 surface with FDH in the QCM experiment was only observed when two orders of magnitude higher amounts of FDH were adsorbed (Figure 3 A). As QCM and ATR‐IR spectroscopy indicate stronger than purely electrostatic interactions, close‐to‐quantitative adsorption of FDH on the TiO2 nanoparticle in the colloidal system is likely. A turnover frequency (TOF) of 11±1.0 and 5±0.6 s−1 (based on CO2 conversion after 6 h) and approximately 4.9±0.2 and 2.0±0.2 μmol formate (after 24 h) were observed from CO2 using RuP and DPP‐sensitized TiO2, respectively (Figure 4). The results of all photocatalysis experiments are presented in Tables S1 and S2 in the Supporting Information.
Table 1 shows a comparison of state‐of‐the‐art catalysts (enzymatic and synthetic) in combination with dye‐sensitized TiO2 nanoparticles without diffusional mediators for CO2 reduction and H2 evolution. Previous studies showed that enzymes outperform the synthetic systems in terms of TOF.30 Among the compared systems, the presented RuP|TiO2|FDH system exhibits the highest TOF for CO2 reduction. The DPP|TiO2|FDH system shows that comparable activities can also be achieved in an entirely precious‐metal‐free system. In semi‐artificial systems, rapid electron transfer from TiO2 to the enzyme was previously found to be essential for efficient catalysis,22, 31 suggesting that the strong interfacial interaction plays an important role for the high activity and stability of dye|TiO2|FDH. Previously reported photocatalyst systems employing NAD+‐dependent FDHs for CO2 reduction to formate rely on soluble redox mediators and only produced TOFs in the range of 10–20 h−1.32
Table 1.
Comparison of TOFs for dye‐sensitized TiO2 systems with enzymatic and synthetic catalysts for CO2 reduction and H2 evolution.
| reaction | dye | catalyst | TOF [h−1] | ref. |
|---|---|---|---|---|
| CO2 → HCO2 − | RuP | DvH FDH [a] | 4.0×104 | this work |
| DPP | DvH FDH [a] | 1.8×104 | this work | |
| CO2 → CO | RuP | Ch CODH I[b] | 5.4×102 | 27 |
| dye[c] | Re [d] | 8.6 | 28 | |
| H+ → H2 | RuP | Db [NiFeSe]‐H2ase[e] | 1.8×105 | 22 |
| DPP | Db [NiFeSe]‐H2ase[e] | 8.7×103 | 25 | |
| CNx [f] | Db [NiFeSe]‐H2ase[e] | 2.8×104 | 23 | |
| RuP | Ni [g] | 3.2×102 | 29 |
[a] W‐FDH from DvH. [b] Carbon monoxide dehydrogenase (CODH) I from Carboxydothermus hydrogenoformans (Ch). [c] (E)‐2‐cyano‐3‐(5′‐(5′′‐(p‐(diphenylamino)phenyl)thiophen‐2′′‐yl)thiophen‐2′‐yl)‐acrylic acid. [d] Synthetic rhenium catalyst (Re) in N,N‐dimethyl formamide (DMF) and water. [e] [NiFeSe]‐hydrogenase from Desulfomicrobium baculatum (Db). [f] Polyheptazine carbon nitride polymer melon (CNx). [g] Synthetic nickel(II) bis(diphosphine) catalyst (Ni).
In summary, FDH immobilized on metal‐oxide electrodes is established as a reversible electrocatalyst for the selective conversion of CO2 to formate. The porous metal‐oxide scaffolds allow for high FDH loading and consequently high current densities, which makes the protein‐modified electrodes not only a relevant model system for CO2 utilization, but also for formate oxidation in formate fuel cells. An excellent interface between TiO2 and FDH is confirmed by QCM analysis and ATR‐IR spectroscopy. The direct (diffusional mediator‐free) electron transfer across the enzyme–metal‐oxide interface is exploited for visible‐light‐driven CO2 reduction to formate. These results underline the importance of characterizing the interactions at the enzyme–material interface and future improvements in performance may arise from more controlled immobilization and more efficient electron transfer with the directly wired FDH.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by an ERC Consolidator Grant “MatEnSAP” (682833), the Royal Society (NF160054, Newton fellowship), the Christian Doppler Research Association and OMV Group, SFRH/BD/116515/2016, grant PTDC/BIA‐MIC/2723/2014 and R&D units UID/Multi/04551/2013 (Green‐IT) and LISBOA‐01‐0145‐FEDER‐007660 (Most‐Micro) cofunded by FCT/MCTES and FEDER funds through COMPETE2020/POCI and the European Union's Horizon 2020 research and innovation programme (GA 810856). We thank Prof. J. Hirst, Dr. A. Eisenschmidt, Dr. S. Roy, A. Wagner, and D. Antón García for fruitful discussions.
M. Miller, W. E. Robinson, A. R. Oliveira, N. Heidary, N. Kornienko, J. Warnan, I. A. C. Pereira, E. Reisner, Angew. Chem. Int. Ed. 2019, 58, 4601.
Contributor Information
Melanie Miller, http://www‐reisner.ch.cam.ac.uk/.
Prof. Erwin Reisner, Email: reisner@ch.cam.ac.uk.
References
- 1. Nocera D. G., Acc. Chem. Res. 2017, 50, 616–619. [DOI] [PubMed] [Google Scholar]
- 2. Boddien A., Mellmann D., Gärtner F., Jackstell R., Junge H., Dyson P. J., Laurenczy G., Ludwig R., Beller M., Science 2011, 333, 1733–1736. [DOI] [PubMed] [Google Scholar]
- 3. Gao S., Lin Y., Jiao X., Sun Y., Luo Q., Zhang W., Li D., Yang J., Xie Y., Nature 2016, 529, 68–71. [DOI] [PubMed] [Google Scholar]
- 4. Dalle K. E., Warnan J., Leung J. J., Reuillard B., Karmel I. S., Reisner E., Chem. Rev. 2019, in print (DOI: 10.1021/acs.chemrev.8b00392). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kornienko N., Zhang J. Z., Sakimoto K. K., Yang P., Reisner E., Nat. Nanotechnol. 2018, 13, 890–899. [DOI] [PubMed] [Google Scholar]
- 6. Kim J., Lee S. H., Tieves F., Choi D. S., Hollmann F., Paul C. E., Park C. B., Angew. Chem. Int. Ed. 2018, 57, 13825–13828; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 14021–14024. [Google Scholar]
- 7. Parkinson B. A., Weaver P. F., Nature 1984, 309, 148–149. [Google Scholar]
- 8. Rowe S. F., Le Gall G., Ainsworth E. V., Davies J. A., Lockwood C. W. J., Shi L., Elliston A., Roberts I. N., Waldron K. W., Richardson D. J., et al., ACS Catal. 2017, 7, 7558–7566. [Google Scholar]
- 9. Raaijmakers H., Macieira S., Dias J. M., Teixeira S., Bursakov S., Huber R., Moura J. J. G., Moura I., Romão M. J., Structure 2002, 10, 1261–1272. [DOI] [PubMed] [Google Scholar]
- 10. da Silva S. M., Pimentel C., Valente F. M. A., Rodrigues-Pousada C., Pereira I. A. C., J. Bacteriol. 2011, 193, 2909–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. da Silva S. M., Voordouw J., Leitão C., Martins M., Voordouw G., Pereira I. A. C., Microbiology 2013, 159, 1760–1769. [DOI] [PubMed] [Google Scholar]
- 12. Sokol K. P., Robinson W. E., Oliveira A. R., Warnan J., Nowaczyk M. M., Ruff A., Pereira I. A. C., Reisner E., J. Am. Chem. Soc., 2018, 140, 16418–16422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kato M., Cardona T., Rutherford A. W., Reisner E., J. Am. Chem. Soc. 2012, 134, 8332–8335. [DOI] [PubMed] [Google Scholar]
- 14. Reda T., Plugge C. M., Abram N. J., Hirst J., Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 10654–10658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Armstrong F. A., Hirst J., Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 14049–14054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bassegoda A., Madden C., Wakerley D. W., Reisner E., Hirst J., J. Am. Chem. Soc. 2014, 136, 15473–15476. [DOI] [PubMed] [Google Scholar]
- 17. Robinson W. E., Bassegoda A., Reisner E., Hirst J., J. Am. Chem. Soc. 2017, 139, 9927–9936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kornienko N., Heidary N., Cibin G., Reisner E., Chem. Sci. 2018, 9, 5322–5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nam D. H., Zhang J. Z., Andrei V., Kornienko N., Heidary N., Wagner A., Nakanishi K., Sokol K. P., Slater B., Zebger I., et al., Angew. Chem. Int. Ed. 2018, 57, 10595–10599; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 10755–10759. [Google Scholar]
- 20. Frasca S., von Graberg T., Feng J.-J., Thomas A., Smarsly B. M., Weidinger I. M., Scheller F. W., Hildebrandt P., Wollenberger U., ChemCatChem 2010, 2, 839–845. [Google Scholar]
- 21. Ly H. K., Marti M. A., Martin D. F., Alvarez-Paggi D., Meister W., Kranich A., Weidinger I. M., Hildebrandt P., Murgida D. H., ChemPhysChem 2010, 11, 1225–1235. [DOI] [PubMed] [Google Scholar]
- 22. Reisner E., Powell D. J., Cavazza C., Fontecilla-Camps J. C., Armstrong F. A., J. Am. Chem. Soc. 2009, 131, 18457–18466. [DOI] [PubMed] [Google Scholar]
- 23. Caputo C. A., Wang L., Beranek R., Reisner E., Chem. Sci. 2015, 6, 5690–5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Barth A., Biochim. Biophys. Acta Bioenerg. 2007, 1767, 1073–1101. [DOI] [PubMed] [Google Scholar]
- 25. Warnan J., Willkomm J., Ng J. N., Godin R., Prantl S., Durrant J. R., Reisner E., Chem. Sci. 2017, 8, 3070–3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Willkomm J., Orchard K. L., Reynal A., Pastor E., Durrant J. R., Reisner E., Chem. Soc. Rev. 2016, 45, 9–23. [DOI] [PubMed] [Google Scholar]
- 27. Woolerton T. W., Sheard S., Pierce E., Ragsdale S. W., Armstrong F. A., Energy Environ. Sci. 2011, 4, 2393–2399. [Google Scholar]
- 28. Lee J.-S., Won D.-I., Jung W.-J., Son H.-J., Pac C., Kang S. O., Angew. Chem. Int. Ed. 2017, 56, 976–980; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 996–1000. [Google Scholar]
- 29. Gross M. A., Reynal A., Durrant J. R., Reisner E., J. Am. Chem. Soc. 2014, 136, 356–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wombwell C., Caputo C. A., Reisner E., Acc. Chem. Res. 2015, 48, 2858–2865. [DOI] [PubMed] [Google Scholar]
- 31. Woolerton T. W., Sheard S., Reisner E., Pierce E., Ragsdale S. W., Armstrong F. A., J. Am. Chem. Soc. 2010, 132, 2132–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee S. H., Choi D. S., Kuk S. K., Park C. B., Angew. Chem. Int. Ed. 2018, 57, 7958–7985; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 8086–8116. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary