

Abstract
A variant (148M) in patatin‐like phospholipase domain‐containing protein 3 (PNPLA3) is a major risk factor for fatty liver disease. Despite its clinical importance, the pathogenic mechanism linking the variant to liver disease remains poorly defined. Previously, we showed that PNPLA3(148M) accumulates to high levels on hepatic lipid droplets (LDs). Here we examined the effect of that accumulation on triglyceride (TG) hydrolysis by adipose triglyceride lipase (ATGL), the major lipase in the liver. As expected, overexpression of ATGL in cultured hepatoma (HuH‐7) cells depleted the cells of LDs, but unexpectedly, co‐expression of PNPLA3(wild type [WT] or 148M) with ATGL inhibited that depletion. The inhibitory effect of PNPLA3 was not caused by the displacement of ATGL from LDs. We tested the hypothesis that PNPLA3 interferes with ATGL activity by interacting with its cofactor, comparative gene identification‐58 (CGI‐58). Evidence supporting such an interaction came from two findings. First, co‐expression of PNPLA3 and CGI‐58 resulted in LD depletion in cultured cells, but expression of PNPLA3 alone did not. Second, PNPLA3 failed to localize to hepatic LDs in liver‐specific Cgi‐58 knockout (KO) mice. Moreover, overexpression of PNPLA3(148M) increased hepatic TG levels in WT, but not in Cgi‐58 KO mice. Thus, the pro‐steatotic effects of PNPLA3 required the presence of CGI‐58. Co‐immunoprecipitation and pulldown experiments in livers of mice and in vitro using purified proteins provided evidence that PNPLA3 and CGI‐58 can interact directly. Conclusion: Taken together, these findings are consistent with a model in which PNPLA3(148M) promotes steatosis by CGI‐58‐dependent inhibition of ATGL on LDs.
Abbreviations
- Ab
antibody
- Ad
adenovirus
- ATGL
adipose triglyceride lipase
- BFP
blue fluorescent protein
- CGI‐58
comparative gene identification‐58
- C‐ter
C‐terminus
- EGFP
enhanced green fluorescent protein
- ER
endoplasmic reticulum
- FA
fatty acid
- GFP
green fluorescent protein
- GST
glutathione S‐transferase
- HSD17B13
17β‐hydroxysteroid dehydrogenase 13
- HSD
high sucrose diet
- KO
knockout
- LD
lipid droplet
- Ls‐Cgi58‐/‐
liver‐specific Cgi58 knockout
- mC
mCherry
- MDH
monodansylpentane
- N‐ter
N‐terminus
- OA
oleic acid
- pAb
polyclonal antibody
- PBS
phosphate‐buffered saline
- PLIN
perilipin
- PNPLA2
patatin‐like domain‐containing protein 2
- PNPLA3
patatin‐like phospholipase domain‐containing protein 3
- PNS
postnuclear supernatant
- RR5
empty adenovirus
- TG
triglyceride
- WT
wild type
Chronic liver disease represents a major public health problem. Decompensated cirrhosis is the fourteenth most common cause of death in adults, and hepatocellular carcinoma (HCC) is the third most common cause of cancer‐related death.1 The main causes of chronic liver diseases are viruses, alcohol, and metabolic abnormalities associated with obesity.2 A common metabolic sequela of obesity is a propensity to accumulate triglyceride (TG) in the liver (hepatic steatosis).3 In some individuals, hepatic steatosis is associated with an inflammatory response (nonalcoholic steatohepatitis) that can progress to fibrosis, cirrhosis, and HCC. We previously identified a missense variant in patatin‐like phospholipase domain‐containing protein 3 (PNPLA3[148M]) that is associated with both liver TG content and circulating levels of alanine aminotransferase.4 Subsequent studies by others have found that PNPLA3(148M) is associated with the full spectrum of fatty liver disease (both nonalcoholic and alcoholic).5, 6, 7
The mechanism by which PNPLA3(148M) promotes fatty liver disease remains poorly defined. In vitro studies using purified recombinant PNPLA3 indicate that the enzyme has TG hydrolase activity and that the 148M substitution reduces that activity by about 80%.8 Expression of PNPLA3(148M) in livers of mice using adenovirus‐mediated or germ‐line transgenesis,9 or by introducing the I148M substitution into the endogenous mouse gene,10 results in an increase in hepatic TG levels that is of similar magnitude to that seen in humans.4 Similarly, a substitution (S47A) in PNPLA3 that abolishes its TG hydrolase activity is associated with hepatic steatosis in mice fed a high sucrose diet (HSD).10 In contrast, neither inactivation11, 12 nor overexpression9 of PNPLA3(wild type [WT]) in the livers of mice cause steatosis. Collectively, these findings are inconsistent with the hypothesis that PNPLA3 causes steatosis due to a simple loss of function or gain of function. Expression of a catalytically defective PNPLA3 protein appears to be required for PNPLA3‐mediated steatosis.10, 11, 12
We showed previously that introduction of the 148M substitution into PNPLA3 in mice is associated with accumulation of PNPLA3 on lipid droplets (LDs) due to reduced turnover of the mutant protein, which evades ubiquitylation and proteasomal degradation.13, 14 It has been suggested that PNPLA3(148M) promotes steatosis by increasing TG synthesis15 or impairing TG secretion from the liver.16 Our laboratory has not found evidence that supports either of these possibilities.8, 14
An alternative hypothesis that we test here is that accumulation of PNPLA3(148M) on LDs interferes with TG hydrolysis by displacing or sequestering a lipase or cofactor. In this study, we investigated the interactions among the major TG hydrolase in the liver, adipose TG lipase (ATGL),17 the cofactor for ATGL, alpha‐beta hydrolase domain containing protein 5 (ABHD5, also called comparative gene identification‐58 [CGI‐58]),18 and PNPLA3 in cultured cells and in mice.
Materials and Methods
Plasmids and Antibodies
All plasmids used in the studies described in this paper are given in Supporting Table S1. Antibodies (Abs) that were used are described in the Supporting Materials and Methods.
Cell Culture and Transfection
QBI‐293A cells (Obiogene, formerly Quantum Biotechnologies, CA) were cultured in complete medium (high glucose Dulbecco's modified Eagle's medium with 5% fetal calf serum [FCS] plus 100 IU/mL penicillin and 100 mg/mL streptomycin). On day 0, cells were plated in 6‐well plates with coverslips (1.5 × 105 cells/well) or in 12‐well plates without coverslips (0.5 × 105 cells/well). On day 1, cells were transfected with target DNA using FuGENE 6 (Promega, Madison, WI). After 5 hours, medium was replaced with fresh medium supplemented with 200 µM oleic acid (OA) bound to bovine serum albumin, and cells were cultured overnight. HuH‐7 cells were cultured the same way except that the concentration of FCS in the medium was 10%. GenJet In Vitro DNA Transfection Reagent (Version II, SignaGen Laboratories, Rockville, MD) was used to transfect HuH‐7 cells.
Immunofluorescence Microscopy and Histochemistry
Cells were rinsed twice with phosphate‐buffered saline (PBS) and fixed with 4% paraformaldehyde (PFA) for 15 minutes. After fixation, cells were rinsed with PBS, incubated with 50 mM NH4Cl for 15 minutes, and then permeabilized for 2 minutes with 0.1% Triton X‐100 in PBS supplemented with fish skin gelatin (0.2%) (Sigma‐Aldrich, St. Louis, MO). Cells were incubated in PBS plus fish skin gelatin (0.2%) overnight at 4°C with the indicated primary Ab, and rinsed three times with PBS before incubation with secondary Abs for 35 minutes at room temperature.
To stain LDs, monodansylpentane (MDH, AutoDOT, Abgent, San Diego, CA) or LipidTOX Green (Invitrogen, Carlsbad, CA) was diluted 1,000 fold in PBS and incubated with cells for 30 minutes. Cells on coverslips were rinsed two to three times with PBS, immersed in a drop of Vectashield antifade mounting medium (H‐1400, Vector Laboratories, Burlingame, CA), and visualized using a confocal microscope (Leica TCS SP5 and Zeiss LSM 880).
For Oil Red O staining, liver samples were fixed in 4% PFA and neutral lipids were visualized using light microscopy (DM2000, Leica) as described.10
For LD morphometry, QBI‐293A cells were cultured in complete medium plus OA (200 µM) without antibiotics and transfected with mCherry (mC), PNPLA3(WT), or 148M using FuGENE 6. Cells were fixed 24 hours after transfection and immunostained with an anti‐PNPLA3 primary Ab and Alexa 555‐conjugated goat antimouse secondary Ab. Sections were visualized using confocal microscopy. Total LD area and the diameters of the 12‐15 largest LDs per cell were measured using ImageJ software. The 10 largest LDs in the cell were selected and the mean diameters (±SD) were compared (≥18 cells/group).
Animals
PNPLA3TgWT/+, PNPLA3Tg148M/+, and Pnpla3148M/Mmice were generated and maintained as described.9, 10 Mice in which the gene‐encoding CGI‐58 was inactivated specifically in the liver (Ls‐Cgi58‐/‐) were kindly provided by Dr. Liqing Yu (University of Maryland, Baltimore, MD).19 All animals were housed in colony cages (≤5 mice/cage) on a 12‐hour light/12‐hour dark cycle and fed a chow diet (Teklad Mouse/Rat Diet 7001) ad libitum. Unless indicated, mice were metabolically synchronized for 3 days before the experiment with a 12‐hour fasting (9 am to 9 pm) and 12‐hour refeeding (9 pm to 9 am) regimen. For dietary challenge studies, mice were fed an HSD (No. 901683, MP Biomedicals; 58.45% sucrose) for 2 weeks. All animal experiments were performed with the approval of the Institutional Animal Care and Research Advisory Committee at the University of Texas Southwestern Medical Center in Dallas, Texas.
Mice were infected with adenovirus (1.25 × 1011 particles) by tail vein injection as described.13 Mice were metabolically synchronized and were sacrificed after the last refeeding cycle to collect the livers.
Subcellular Fractionation and Immunoblotting
Hepatic LDs were isolated from the liver as described except that the postnuclear supernatant (PNS) was centrifuged at 10,000g.10 For immunoblot analysis of the PNS and cytosol fractions, equal proportions of each fraction were loaded onto PAGE gels. For analysis of the proteins on LDs, equal amounts of proteins were loaded, unless otherwise stated. Immunoblot analysis was performed using SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Scientific, Rockford, IL), and signal was detected either by exposure to X‐ray film or by using the LI‐COR Odyssey Imaging System (LI‐COR Biosciences, Lincoln, NE).
Protein Purification and Immunoprecipitation
Epitope‐tagged (deca‐histidine and Flag) fusion proteins of recombinant PNPLA3(WT) and PNPLA3(148M) were purified from Sf9 cells,8, 13 and glutathione S‐transferase (GST)‐CGI‐58 was produced in BL21(DE3) competent E. coli cells as described in the Supporting Materials and Methods.8 Co‐immunoprecipitation studies of PNPLA3, CGI‐58, and ATGL are also described in the Supporting Materials and Methods.
Statistics
Differences among groups were analyzed by one‐way analysis of variance using Turkey's correction or two‐way analysis of variance for multiple comparisons using Sidak correction by GraphPad Prism 7 (GraphPad Software, San Diego, CA). P values less than 0.05 were considered statistically significant.
Results
PNPLA3 does not Localize to the Endoplasmic Reticulum in Cultured Hepatocytes
Previously, we showed that PNPLA3 was recovered in pelleted membranes of cell lysates subjected to ultracentrifugation.13 At the time, we proposed that PNPLA3 is a monotopic protein that is associated with intracellular membranes, most likely the endoplasmic reticulum (ER),13 and traffics to the droplet in a manner that is similar to that described for other so‐called class 1 LD proteins.20 To test this hypothesis, we compared the distribution of PNPLA3 with that of recombinant blue fluorescent protein (BFP), which is targeted to the ER through a C‐terminal (C‐ter) KDEL sequence (BFP‐KDEL).21, 22, 23 We observed no overlap in the distribution of PNPLA3(WT or 148M) and BFP‐KDEL (Fig. 1). These findings are consistent with the notion that PNPLA3 pelleted with cell membranes in previous experiments13 due to its poor solubility and tendency to aggregate. The finding that PNPLA3 is not associated with the ER alters the view that PNPLA3 migrates to LDs through the ER.
Figure 1.
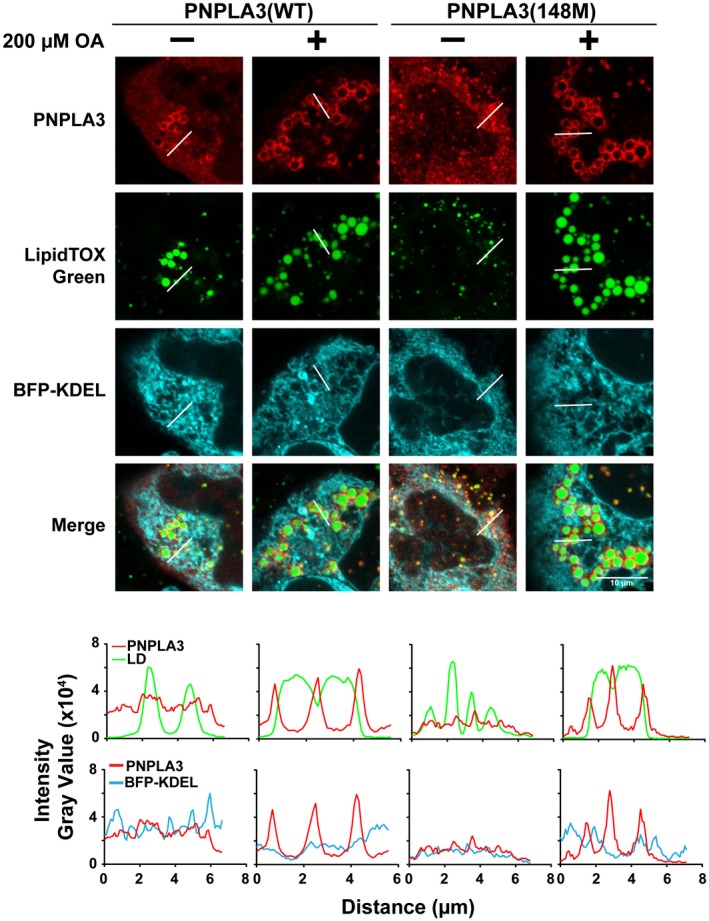
Localization of PNPLA3 in HuH‐7 cells. HuH‐7 cells were cotransfected with BFP‐KDEL and PNPLA3(WT or 148M) in the absence or presence of 200 µM OA for 24 hours. The BFP contains a signal sequence at the N‐ter and an ER retention signal at the C‐ter. Immunofluorescence microscopy was performed using an antihuman PNPLA3 monoclonal antibody (mAb) followed by an Alexa 555‐conjugated goat antimouse polyclonal antibody (pAb) (red). LDs were stained with LipidTOX Green (top). Histograms represent gray value intensity from the merge panel along the indicated line using ImageJ (bottom). The experiment was repeated twice and the results were similar. Scale bar = 10 µm.
PNPLA3 Localizes to the Cytoplasm and LDs in QBI‐293A Cells
To monitor PNPLA3 localization relative to cellular LD content, we expressed PNPLA3(WT and 148M) in cultured human embryonic kidney (QBI‐293A, Fig. 2) and hepatoma cells (HuH‐7, Fig. 2).24 QBI‐293A cells have few LDs when grown without supplemental fatty acids (FAs), but accumulate cytoplasmic TG when OA (200 µM) is added to the medium (Fig. 2A). Immunofluorescence microscopy was used to localize recombinant PNPLA3 in these cells. In cells cultured without OA, both PNPLA3(WT) and PNPLA3(148M) were distributed in a diffuse pattern in the cytosol. Cells supplemented with FAs formed LDs that contained virtually all the PNPLA3 signal (Fig. 2A, left panel). Levels of PNPLA3 protein were comparable in cells expressing PNPLA3(WT) and PNPLA3(148M) (Fig. 2A, right panel), and were not significantly affected by supplementation with FAs.
Figure 2.
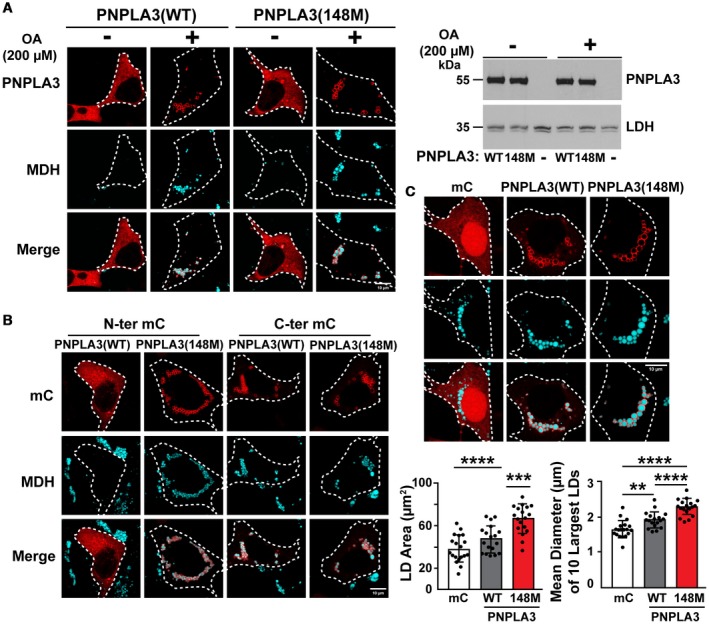
Cellular immunolocalization of PNPLA3(WT and 148M) in QBI‐293A cells. (A) QBI‐293A cells were transfected with the plasmids encoding human PNPLA3(WT or 148M) and then cultured for 24 hours in the presence or absence of 200 µM OA. Immunofluorescence microscopy was performed as described in Fig. 1. LDs were stained using MDH (cyan, left). White dash lines outline the transfected cells. Protein expression was analyzed by immunoblotting using lactate dehydrogenase as a loading control (right). (B) The experiment was repeated using PNPLA3 with a mC epitope tag at the N‐ter or C‐ter in the presence of 200 µM OA. (C) The experiment described in (A) was repeated in the presence of OA with the inclusion of cells expressing mC alone. Sections were analyzed by ImageJ to calculate total LD areas. Diameters of the 10 largest LDs from each cell were measured in 18 or more cells. Tukey's multiple comparisons test; **P < 0.01, ***P < 0.001, ****P < 0.0001. Scale bar = 10 µm. All experiments were repeated twice and yielded similar results. Abbreviation: LDH, lactate dehydrogenase.
Addition of a fluorescent reporter protein to the N‐terminus (N‐ter), but not to the C‐ter, of PNPLA3(WT) altered the subcellular distribution of PNPLA3 in OA‐treated QBI‐293A cells (Fig. 2B and Supporting Fig. S1). Regardless of which reporter protein was used (mC or GFP), N‐ter‐PNPLA3(WT) was distributed more diffusely in the cytosol and the cells had no detectable LDs. In contrast, when the reporter protein was placed at the C‐ter, PNPLA3(WT) localized on LDs. Placement of the reporter did not alter the distribution of PNPLA3(148M) (Fig. 2B and Supporting Fig. S1). Therefore, in subsequent experiments that required labeling of PNPLA3, the protein with the tag at the C‐ter was used, as its distribution resembled that of the native protein (Fig. 2A).
Thus, the addition of a reporter protein at the N‐ter of PNPLA3(WT) appears to alter the conformation of the enzyme such that the TG hydrolase activity8 is unveiled, perhaps by exposing the active site, blocking the binding of an inhibitor, or promoting binding of an activator.
Expression of PNPLA3(148M) is Associated with an Increase in LD Size
We determined the effect of the 148M variant on the size distribution and area of LDs. The mean LD area was about 1.25 times higher in cells expressing PNPLA3(WT) (47.6 ± 12.3 µm2), and 2 times higher in cells expressing PNPLA3(148M) (66.9 ± 13.0 µm2), than in cells transfected with mC alone (38.5 ± 12.4 µm2) (Fig. 2C, lower left panel). In addition, the mean diameter of the 10 largest LDs was higher in cells expressing PNPLA3(148M) compared with PNPLA3(WT) (Fig. 2C, lower right panel). A similar increase in LD size was observed previously in association with PNPLA3(148M) expression in other cell types, including HeLa cells, HuH‐7 cells, and cultured skin carcinoma cells (A‐431).25, 26
In contrast to QBI‐293A cells (Fig. 2), HuH‐7 cells form prominent LDs even in the absence of FA supplementation (Fig. 1). Expression of PNPLA3(WT or 148M) in HuH‐7 cells resulted in diffuse cytoplasmic as well as LD staining. Addition of OA to the medium increased the localization of PNPLA3 on LDs in cells expressing either PNPLA3(WT) or PNPLA3(148M) (Fig. 1).
ATGL(WT), but not ATGL(148M), Hydrolyzes TG and Localizes to the Cytoplasm
The closest paralog of PNPLA3 is PNPLA2. PNPLA2, also called ATGL, is the major TG hydrolase in hepatocytes (as well as adipocytes).17 The isoleucine at residue 148 is conserved between PNPLA3 and ATGL, as is the catalytic serine at amino acid 47.17 To investigate whether PNPLA3 influences the distribution or function of ATGL, we expressed the proteins alone or together in HuH‐7 cells (Fig. 3A‐C).
Figure 3.
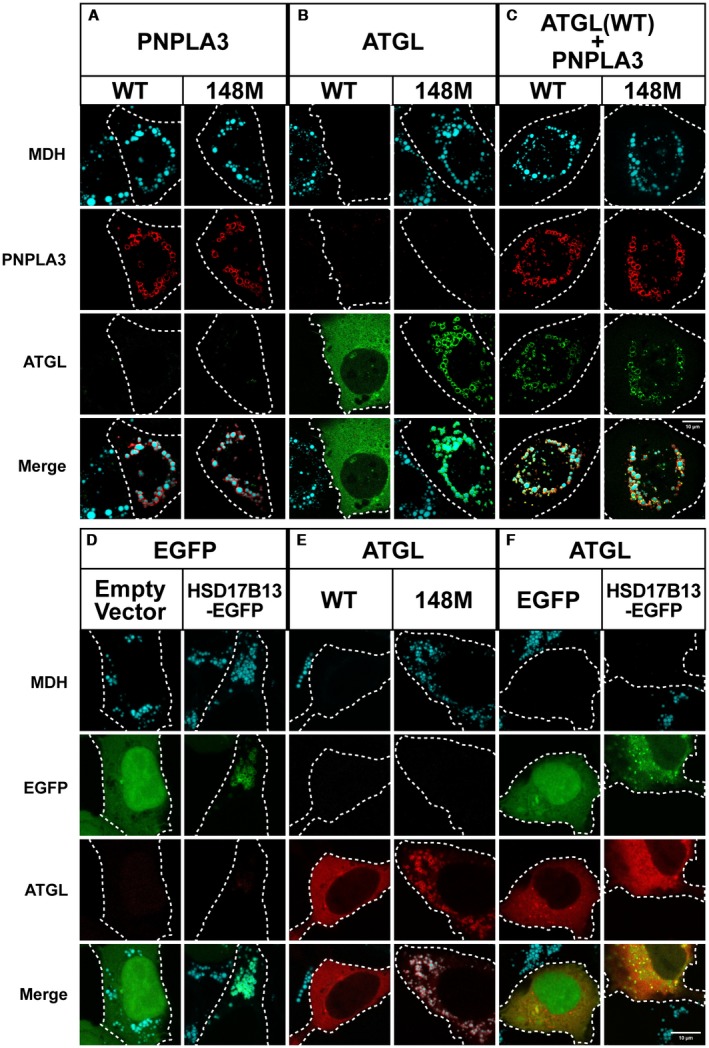
Co‐expression of PNPLA3(WT or 148M) and ATGL in cultured cells. (A‐C) HuH‐7 cells were transfected with PNPLA3(WT or 148M) and GFP‐tagged mouse ATGL and grown in the presence of 200 µM OA as described in Fig. 1. Immunofluorescence was performed as described in Fig. 1. QBI‐293A cells were transfected with enhanced green fluorescent protein (EGFP) and HSD17B13‐EGFP (D), mC‐ATGL(WT or 148M) (E), and ATGL plus EGFP or HSD17B13‐EGFP (F). Cells were transfected and grown in the presence of 200 µM OA as described in Fig. 2. LDs were costained with MDH (cyan). The experiments were repeated 1‐3 times and the results were similar. Scale bar = 10 µm.
Multiple LDs were apparent in OA‐supplemented HuH‐7 cells expressing either PNPLA3(WT) or PNPLA3(148M), and both forms of PNPLA3 localized to LDs (Fig. 3A). In contrast, expression of ATGL(WT) was associated with the diffuse, cytoplasmic staining pattern of the enzyme and no detectable LDs (Fig. 3B),27 regardless of whether the GFP tag was placed at the N‐ter or C‐ter (Supporting Fig. S2A). The N‐ter GFP‐ATGL was used for all subsequent experiments, as expression of ATGL‐GFP (C‐ter) was associated with aggregation of the protein (Supporting Fig. S2A).
Based on these results, ATGL(WT), but not PNPLA3(WT), has TG hydrolase activity when expressed in cells under these conditions.17 Substitution of methionine for the isoleucine at residue 148 of ATGL resulted in a significant increase in LDs, which are decorated with the ATGL(148M) protein (Fig. 3B). Similar results were obtained when the catalytic serine of ATGL was replaced by alanine (Supporting Fig. S2B). Thus, the I148M substitution in ATGL, like in PNPLA3, significantly attenuates the TG hydrolase activity of the enzyme.
Co‐Expression of PNPLA3(WT) or PNPLA3(148M) Inhibits ATGL Activity without Displacing ATGL from LDs
To determine the effect of PNPLA3 in cells expressing ATGL(WT), the two PNPLA family members were co‐expressed in HuH‐7 cells. Unlike cells expressing ATGL(WT) alone, which had no LDs (Fig. 3B, left), LDs were plentiful in cells co‐expressing ATGL(WT) plus either PNPLA3(WT) or PNPLA3(148M) (Fig. 3C). These data indicate that PNPLA3 expression in cells inhibits ATGL‐mediated TG hydrolysis.
Kory et al.28 proposed that lipolysis of LDs decreases the surface area available for protein binding and that the resulting macromolecular crowding causes displacement of proteins from the droplet surface. If this were the case, PNPLA3 would inhibit ATGL activity by displacing ATGL from LDs. We found no evidence to support this scenario; instead, we found that co‐expression of PNPLA3 and ATGL results in colocalization of the two proteins with the MDH‐stained puncta (Fig. 3C).
To determine whether inhibition of ATGL by PNPLA3 is a nonspecific effect caused by overexpression of a LD protein, we co‐expressed ATGL with another LD protein, 17β‐hydroxysteroid dehydrogenase 13 (HSD17B13)29 (Fig. 3F). We found that cells expressing both proteins had no detectable LDs, thus supporting the premise that the inhibitory effect of PNPLA3 expression on ATGL activity is a specific effect of PNPLA3.
Why does expression of both the WT and mutant forms of PNPLA3 inhibit the activity of ATGL? The first hypothesis we tested was that PNPLA3 sequesters CGI‐58, a cofactor of ATGL that significantly augments the TG hydrolase activity of the enzyme.18 As a first step to test this hypothesis, we examined the effect of co‐expressing CGI‐58 and PNPLA3 in cultured cells.
Co‐Expression of CGI‐58 with PNPLA3 Promotes TG Hydrolysis in Cell
We used fluorescence microscopy to assess the size and distribution of LDs in cells co‐expressing CGI‐58 together with a catalytically active (PNPLA3[WT]) and a catalytically inactive (PNPLA3[47A]) form of PNPLA3 (Fig. 4). Expression of CGI‐58 alone was associated with smaller LDs than were seen in cells expressing either the 47A or 148M isoform of PNPLA3 (Fig. 4). We presume that the reduction in LD size is due to CGI‐58 activating endogenous ATGL.
Figure 4.
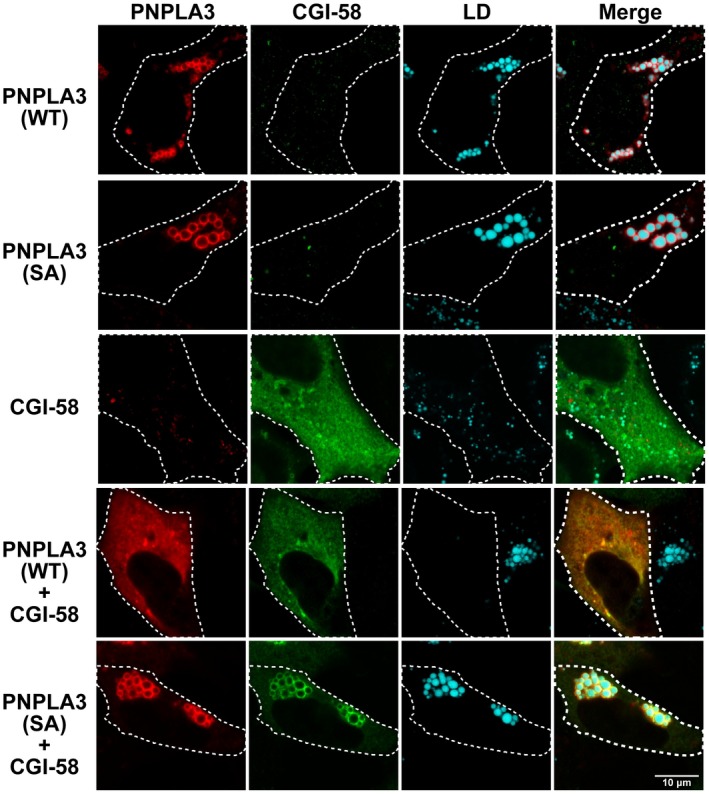
Effect of CGI‐58 on intracellular localization and LD abundance in QBI‐293A cells expressing PNPLA3(WT) and PNPLA3(47A). PNPLA3(WT) and PNPLA3(47A) were expressed in QBI‐293A cells alone or together with CGI‐58, a cofactor for ATGL. Cells were grown in the presence of 200 µM OA for 24 hours. Then, immunofluorescence localization was performed as described in the Materials and Methods using an anti‐PNPLA3 mAb (11C5) and an anti‐myelocytomatosis tag pAb followed by an Alexa 555‐conjugated goat antimouse pAb (red) and an Alexa 488‐conjugated goat antirabbit pAb (green). MDH was used to stain LDs. The white dashed lines indicate the cells that express the protein. Scale bar = 10 µm. The experiment was repeated twice and the results were similar.
In cells expressing both CGI‐58 and PNPLA3(WT), there was further enhancement of LD hydrolysis. It is possible that CGI‐58 activates the enzymatic activity of PNPLA3 in a manner similar to that seen when an epitope tag is attached to the N‐ter. Alternatively, PNPLA3 may assist in positioning CGI‐58 on the droplet so that it can better activate ATGL. To address whether CGI‐58 is activating PNPLA3 directly, we performed the experiment using the catalytically dead form of PNPLA3. When CGI‐58 and PNPLA3(47A) were expressed together, the LDs reappeared. This result raises the possibility that CGI‐58 activates PNPLA3. It is also possible that PNPLA3 alters the propensity of ATGL or CGI‐58 to associate with LDs. Alternatively, expression of PNPLA3 may change the distribution of CGI‐58 and ATGL within LDs of different sizes.
No Effect of PNPLA3 on Detergent‐Mediated Dissociation or Distribution of PNPLA3, ATGL, and CGI‐58 with LDs
To assess the association of PNPLA3, ATGL, and CGI‐58 with LDs in vivo, we isolated LDs from the livers of transgenic animals expressing either PNPLA3(WT) or PNPLA3(148M).9 The LDs were incubated with increasing concentrations of a detergent, n‐dodecyl β‐D‐maltoside (DDM), and the mixture was subjected to centrifugation. The amounts of PNPLA3, ATGL, CGI‐58, and the resident LD protein perilipin 2 (PLIN2) released from LDs were analyzed by immunoblotting (Supporting Fig. S3). As we reported previously, the baseline levels of PNPLA3 were significantly higher in LDs of mice expressing PNPLA3(148M) compared with PNPLA3(WT) (Supporting Fig. S3A, lanes 12 and 6, respectively).10 Although we found that levels of CGI‐58 were also higher in the LDs of mice expressing PNPLA3(148M) (lane 12) compared with PNPLA3(WT) (lane 6), the rates of release of all proteins from LDs were comparable (Supporting Fig. S3A [lanes 1‐5 and 7‐11] and S3B). Thus, the presence of PNPLA3 on droplets does not appear to alter the strength of the association of ATGL or CGI‐58 with LDs.
Next, we examined whether PNPLA3 expression alters the distribution of ATGL or CGI‐58 among LDs of different sizes. To address this question, we used rate‐differential centrifugation to separate LDs based on their size and density in WT and transgenic mice overexpressing human PNPLA3 in the liver (Supporting Fig. S4).30 Because the distribution of PLIN2 among LDs of different sizes was similar, expression levels of other LD proteins were indexed relative to PLIN2 (Supporting Fig. S4C). PNPLA3(148M) was enriched relative to PNPLA3(WT) in all four fractions. The relative amounts of PNPLA3(WT) did not vary much among the four fractions, whereas PNPLA3(148M) was greatly enriched in the fraction containing the largest LDs. In contrast to PNPLA3, only modest differences in the amount or distribution of ATGL were seen among the three strains of mice. Thus, expression of high levels of PNPLA3 had little or no effect on the distribution of ATGL among LDs of different sizes. CGI‐58 was more uniformly distributed among the different fractions than either PNPLA3 or ATGL, and levels tended to be higher in the livers of mice expressing PNPLA3(WT) and PNPLA3(148M) (Supporting Fig. S4C).
To further clarify the functional relationships among PNPLA3, CGI‐58 and ATGL, we examined the expression of PNPLA3 in a strain of mice in which Cgi58 had been inactivated selectively in the liver (Ls‐Cgi58‐/‐ mice), which were obtained from Dr. Liqing Yu (Fig. 5).19
Figure 5.
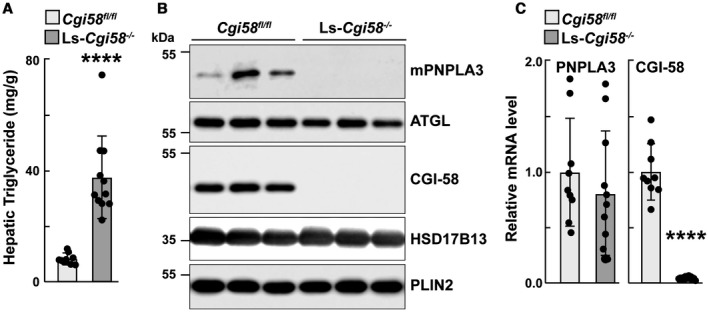
Hepatic TG and LD‐associated protein analysis of Ls‐Cgi‐58‐/‐ mice. Male Cgi‐58flox/flox and Ls‐Cgi‐58‐/‐ mice (n = 4‐6/group, 8‐10 or 11‐13 weeks old) were fed a chow diet and synchronized for 3 days as described in the Materials and Methods. Mice were sacrificed after the last feeding cycle and livers were collected. Lipids were extracted for measurement of hepatic TGs (A), and LDs were prepared for immunoblot analysis as described in the Materials and Methods (B). (C) Total RNA was extracted from the livers of Cgi58fl/fl and Ls‐Cgi58‐/‐ mice, and relative mRNA levels of selected transcripts were quantified by real‐time quantitative PCR. The values were normalized to the levels of mouse cyclophilin A and expressed relative to the levels in chow‐fed Cgi58fl/fl mice, which were arbitrarily set to 1. The data in (A) and (C) were pooled from two independent experiments done under similar conditions.
Liver‐Specific Cgi58‐/‐ Mice have No Detectable PNPLA3 and Reduced ATGL on Hepatic LDs
As reported previously,19 liver TG content was significantly higher in chow‐fed Ls‐Cgi58‐/‐ mice than in mice homozygous for the floxed allele (Cgi58 flox/floxWT [Cgi58fl/fl]) (Fig. 5A). To determine the effect of inactivating Cgi58 on PNPLA3 and ATGL levels, we performed immunoblot analysis on LDs isolated from chow‐fed liver‐specific knockout (KO) mice. No PNPLA3 was detected in the LDs of the KO mice, despite having PNPLA3 mRNA levels that were similar to WT animals (Fig. 5C). In contrast, ATGL levels on LDs were only modestly reduced in the KO mice when compared with the Cgi58fl/fl animals.
PNPLA3 Expression Fails to Augment Hepatic TG Content in Ls‐Cgi58‐/‐ Mice
To determine whether CGI‐58 is required for the recruitment or stabilization of PNPLA3 on LDs, or for the TG‐elevating effects of PNPLA3(148M), we used recombinant adenoviruses (Ads) to overexpress PNPLA3(WT) or PNPLA3(148M) in livers of Cgi58fl/fl and Ls‐Cgi58‐/‐ mice. The levels of mRNAs encoding PNPLA3, CGI‐58, ATGL, and PLIN2 did not differ between the two groups of infected mice, nor were there any differences in body and liver weights or hepatic levels of cholesterol and phospholipids (Supporting Fig. S5). Ls‐Cgi58‐/‐ mice had mean hepatic TG levels that were 4‐fold higher than those of the control animals (38.8 versus 8.28 mg/g tissue, P < 0.0001) (Fig. 6A).19 In Cgi58fl/fl mice, infection with Ad‐PNPLA3(148M) resulted in significantly higher levels of hepatic TG than did infection with Ad‐PNPLA3(WT) (22.89 versus 4.54 mg/g tissue, P = 0.0003) (Fig. 6A, left).13 In contrast with these results, hepatic TG content was not increased by expression of either PNPLA3(WT) or PNPLA3(148M) in Ls‐Cgi58‐/‐ mice (Fig. 6A). The results of the biochemical assays were confirmed by Oil Red O staining of liver sections from the mice (Fig. 6A, right).
Figure 6.
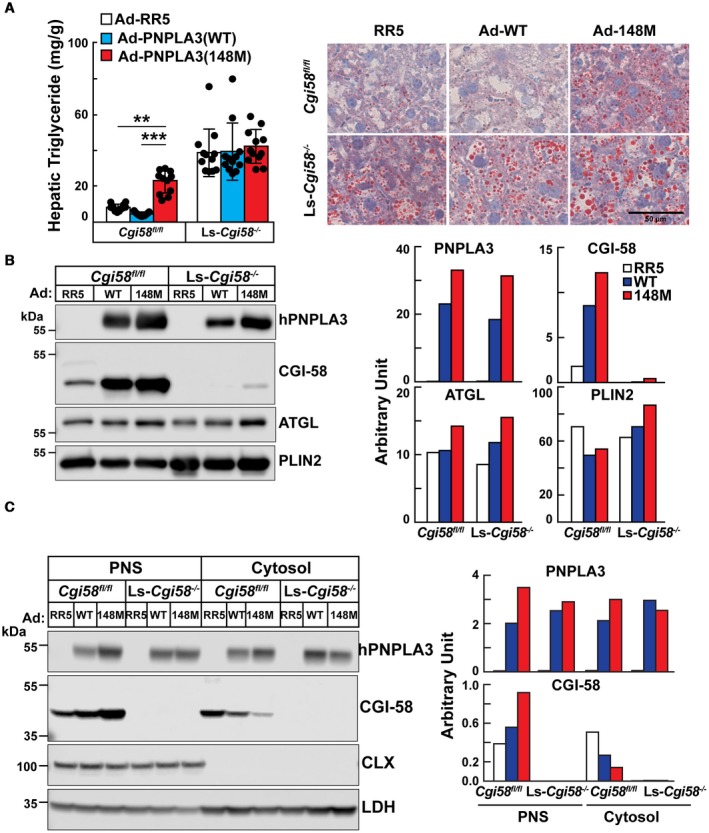
Hepatic lipid (A) and protein analyses (B,C) of Ls‐Cgi58‐/‐ mice infected with RR5, Ad‐PNPLA3(WT), or Ad‐PNPLA3(148M). (A) Chow‐fed Cgi58fl/fl and Ls‐Cgi58‐/‐ mice (n = 4/group, 8‐10 weeks old) were infected with recombinant Ad particles (1.25 × 1011 genome copies) expressing no insert (Vector, RR5) or V5‐tagged versions of human PNPLA3(WT or 148M). Mice were killed 3 days later at the end of the last feeding cycle and hepatic TG levels were measured. Data were pooled from three independent experiments performed under similar conditions (A, left). Oil Red O staining was performed on liver sections at a magnification of ×20. Scale bar = 50 µm (A, right). (B) LDs were isolated from the mice described in (A), and the LD proteins were extracted and pooled. A total of 2 µg of LD protein was subjected to immunoblotting as described in the Materials and Methods. The signals were quantitated using LI‐COR and expressed in arbitrary units. (C) Proteins from the PNS and cytosol from the same mice were pooled and loaded onto the gel in equal proportions before immunoblotting, and the signals were quantitated using LI‐COR. The experiment was repeated twice and the results were similar. Abbreviations: CLX, calnexin; LDH, lactate dehydrogenase.
Next, we assessed the levels of PNPLA3, CGI‐58, and ATGL on LDs of chow‐fed Cgi58fl/fl and Ls‐Cgi58‐/‐ mice. Acute Ad‐mediated expression of PNPLA3, either Ad‐WT or Ad‐148M, in livers of WT mice increased the amount of CGI‐58 associated with LDs when compared with mice expressing an empty Ad (RR5) (Fig. 6B).
To determine whether the increase in LD‐associated CGI‐58 is caused by a redistribution of the cofactor within cells, we compared the amount of CGI‐58 in the PNS, which contains both cytosolic and LD proteins, and in the cytosol, after removal of the LDs (Fig. 6C). Levels of both PNPLA3 and CGI‐58 were greatest in the PNS of mice expressing Ad‐PNPLA3(148M), whereas these mice had the lowest levels of the two proteins in the cytosol (Fig. 6C).
These results are consistent with the notion that PNPLA3 is dependent on CGI‐58 for recruitment and/or stabilization of PNPLA3 on LDs. Moreover, the increase in hepatic TG associated with PNPLA3(148M) requires CGI‐58 expression.
Co‐Immunoprecipitation of Endogenous CGI‐58 with Recombinant PNPLA3 in Mouse Liver
A possible explanation for the absence of PNPLA3 on LDs of Ls‐Cgi58‐/‐ mice (Fig. 5) is that CGI‐58 may promote or stabilize the association of PNPLA3 with LDs. To test this possibility, we immunoprecipitated V5‐tagged PNPLA3 from hepatic LDs of mice expressing Ad‐PNPLA3(WT or 148M) and immunoblotted for CGI‐58. CGI‐58 co‐immunoprecipitated with PNPLA3 in both groups of mice, but not in mice infected with an empty virus (Fig. 7A). ATGL also co‐immunoprecipitated with CGI‐58 under these conditions.
Figure 7.
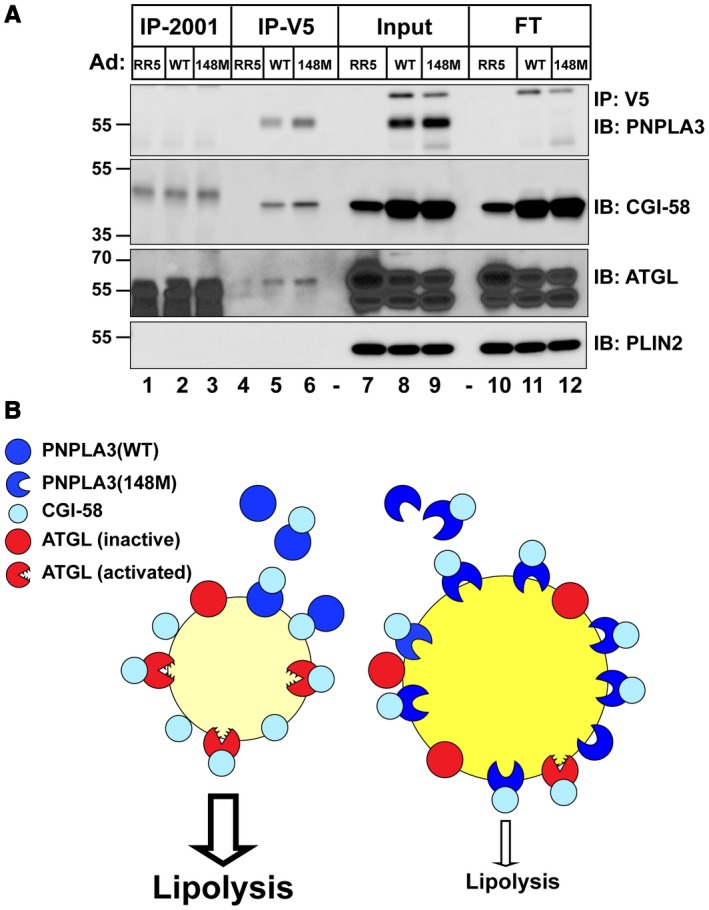
Co‐immunoprecipitation of PNPLA3(WT and 148M) and CGI‐58 in vivo and in vitro. (A) Female WT mice (n = 4/group, 10‐12 weeks old) were infected with Ad‐RR5 or Ad‐PNPLA3‐V5(WT or 148M). After the last refeeding cycle, mice were sacrificed and hepatic LDs were isolated before being incubated with DDM (0.1%) as described in the Materials and Methods. The mixture (3 μg, input) was incubated with anti‐V5 beads at 4°C overnight and then centrifuged at 2,000g. The supernatant (Flowthrough) was collected and the beads were washed before incubation in Laemeli sample buffer (2 times), as described in the Materials and Methods. A total of 1/15 of each fraction was loaded onto the gel. (B) Model of the relationship between PNPLA3(WT) (left) or PNPLA3(148M) (right), CGI‐58, and ATGL and lipolysis of LDs. Abbreviations: FT, Flowthrough; IB, immunoblotting; and IP, immunoprecipitation.
We also examined the physical association of purified, recombinant GST‐tagged CGI‐58 and PNPLA3. GST‐CGI‐58 (70 nM) and PNPLA3 (120 nM) were co‐incubated at 37°C for 1 hour and then precipitated by the addition of glutathione‐linked beads. Proteins were visualized by immunoblot analysis and quantified using LI‐COR. Aliquots of the purified proteins were run on the same gels as the calibrators (Supporting Fig. S6). An estimated 20%‐40% of GST‐CGI‐58 in the incubations was pulled down by the GST beads. PNPLA3 coprecipitated with CGI‐58 (Supporting Fig. S6). The molar ratio of PNPLA3 to CGI‐58 in the precipitates was approximately 1:7 for both PNPLA3(WT) and PNPLA3(148M). Therefore, under the conditions used in this experiment, about 14% of the CGI‐58 molecules that were pulled down were bound to PNPLA3. Although we cannot rule out the possibility that the association of CGI‐58 and PNPLA3 is an artifact of overexpression, our analyses point to a direct interaction and association of these proteins both in vivo and in vitro.
Discussion
The mechanism by which PNPLA3(148M) promotes hepatic steatosis has remained elusive despite extensive studies in vitro, in cultured cells, and in animal models.14 It has been suggested that PNPLA3(148M) promotes steatosis by increasing TG synthesis15 or by impairing TG secretion from hepatocytes,16 but we have not been able to confirm either of these possibilities.8, 14 Here, we tested the hypothesis that accumulation of PNPLA3(148M) on LDs interferes with TG hydrolysis by displacing or sequestering a lipase (specifically ATGL) or its cofactor, CGI‐58. A major finding of this study was that expression of PNPLA3(WT or 148M) inhibited TG hydrolysis by ATGL in cultured human hepatocytes (Fig. 3C) without altering the abundance of ATGL on LDs in these cells. Moreover, we found that PNPLA3 expression in livers of mice did not change the affinity of ATGL for LDs or alter the relative distribution of these proteins among LDs of difference sizes (Supporting Figs. S3 and S4).
Next, we investigated the hypothesis that PNPLA3 perturbs the interaction between ATGL and CGI‐58. The finding that expression of PNPLA3(148M) failed to increase TG levels in the livers of Ls‐Cgi58 ‐/‐ mice (Fig. 6) supported this hypothesis. We found no evidence supporting the notion that PNPLA3(148M) expression displaces CGI‐58 from LDs or results in a mismatch between the abundance of ATGL and CGI‐58 on LDs (Supporting Fig. S4).30
The observation that PNPLA3 can bind directly to CGI‐58 (Fig. 7A and Supporting Fig. S6) raises the possibility that PNPLA3(148M) may sequester CGI‐58 in a manner analogous to that proposed in the widely accepted model of PLIN1/CGI‐58 interaction.31, 32 In this model, PLIN1 plays a key role in the physiological regulation of lipolysis by binding to CGI‐58 and preventing its interaction with ATGL on the droplet surface. Grannerman et al.33 demonstrated that small molecules that blocked the interaction of CGI‐58 with PLIN1 and PLIN5 fully recapitulated the activation of lipolysis and hydrolysis of cellular TG elicited by β‐adrenergic agonists. These data indicate that sequestration of CGI‐58 is sufficient to limit ATGL‐mediated lipolysis. The accumulation of PNPLA3(148M) to high levels in hepatocytes may result in sequestration of CGI‐58 and limit the amount available to activate ATGL.
Three striking findings of the present study cannot readily be explained by the sequestration model and may imply a more complex relationship between PNPLA3 and CGI‐58. First, expression of CGI‐58 together with PNPLA3 in lipid‐supplemented cells resulted in depletion of cellular TG (Fig. 4). This finding is consistent with Chamoun et al.25 and raises the possibility that CGI‐58 acts as a cofactor for PNPLA3, as well as ATGL. However, given that inactivation of PNPLA3 in mice results in specific accumulation of very‐long‐chain polyunsaturated FA‐containing TG,34 but not bulk TG,11, 12 in the liver, and that overexpression of PNPLA3 decreases the same TG species34 without affecting hepatic TG content,9 we think that the depletion of LD in cultured cells is caused by the supraphysiological co‐expression of CGI‐58 and PNPLA3.
The second striking finding was that PNPLA3 was not detected on the LDs of mice that did not express CGI‐58 in the liver (Fig. 5B). Recent studies35, 36 reported that CGI‐58 recruits PNPLA1 to LDs, raising the possibility that CGI‐58 promotes the activity of multiple PNPLA family members by stabilizing their interactions with LDs and access to lipid substrates. This possibility is consistent with Lord et al.,37 who showed that CGI‐58 activates lipolysis independently of ATGL. The mechanism by which CGI‐58 fosters associations of PNPLA proteins with LDs is not clear. CGI‐58 expression may promote (either directly or indirectly) changes in lipid composition or protein architecture that are conducive to association between PNPLA family members and LDs. Alternatively, CGI‐58 may have a more direct effect, binding PNPLA family members and stabilizing their association with LDs. We provide evidence both in vivo and in vitro that PNPLA3 and CGI‐58 can physically associate, but additional studies will be required to further characterize the binding interface and affinity of this association.
Third, we observed that both PNPLA3(WT) and PNPLA3(148M) inhibited ATGL‐mediated lipid hydrolysis in cultured cells (Fig. 3), and both isoforms pulled down CGI‐58 in biochemical assays (Fig. 7 and Supporting Fig. S6). However, only PNPLA3(148M) is associated with hepatic steatosis in mice.9 These apparent discrepancies may reflect differences in the expression levels of the two isoforms in cultured cells and in the livers of mice. In cells, the two proteins are expressed at similar levels (Fig. 2A), whereas in mice, PNPLA3(148M) accumulates to much higher levels on LDs than seen in WT animals.10
Two other observations were made in our studies that alter our conceptions regarding the cell biology of PNPLA3. Previously, our laboratory13 and others26 suggested that PNPLA3 is a monotopic protein that localizes to the ER in lipid‐poor cells and partitions to LD as cellular TG content increases. In the present study, we found no evidence that PNPLA3 or ATGL are associated with the ER (Figs. 1 and 3). Rather, both proteins localize in the cytosol when LDs are sparse and only migrate to preformed LD in cells.27 Exactly how these proteins, which have highly hydrophobic regions, are escorted to LDs remains to be molecularly defined.
Insertion of a fluorescent reporter protein at the N‐terminus of PNPLA3(WT), but not PNPLA3(148M), promoted TG hydrolysis in cells (Fig. 2B and Supporting Fig. S1). This observation suggests that altering the conformation of the N‐ter of PNPLA3 increases lipolytic activity in cells, either by increasing the activity of the enzyme itself or that of another lipase. PNPLA3, like ATGL, may require a coactivator. The fusion of a stable, globular domain at the N‐ter of PNPLA3 may mimic the binding of a coactivator and alter the conformation of the enzyme such that its TG hydrolytic activity is increased, or alternatively that it releases a factor that enhances the activity of another lipase. Experiments are ongoing to explore these different possibilities.
A model of the interactions among PNPLA3, CGI‐58, and ATGL in cells is shown in Fig. 7B. PNPLA3(WT) is synthesized in the cytoplasm in response to carbohydrate feeding and localizes to LDs without any apparent ER association (left). LD localization of PNPLA3 requires CGI‐58, which may chaperone PNPLA3 to droplets or stabilize its interaction with the LD. Once on the LD, PNPLA3(WT) is rapidly degraded. The rapid turnover of the protein limits accumulation of PNPLA3(WT) on LDs (left).14 In contrast to the WT protein, PNPLA3(148M) is poorly ubiquitylated and accumulates on LDs (right).14 We show here that the development of fatty liver due to PNPLA3(148M) is a CGI‐58‐dependent process, and we speculate that the accumulation of PNLA3(148M) sequesters CGI‐58, thus restricting its access to ATGL, or another lipase. Studies to test this hypothesis and further characterize the relationship among PNPLA3, CGI‐58, and ATGL are underway in our laboratory.
Supporting information
Acknowledgments
We thank Dr. Liqing Yu (University of Maryland, Baltimore, MD) for providing the Ls‐Cgi58 KO mice. We thank Drs. Robert Farese and Tobias Walther (Harvard University, Boston, MA) for providing the ssBFP‐KDEL vector and Dr. Orion D. Weiner (University of California at San Francisco, San Francisco, CA) for the pMXs‐CMV‐mC vector. We thank Dorothy Mundy and Katherine Luby‐Phelps in the University of Texas Southwestern Imaging Core for their assistance. Soumik BasuRay provided helpful conversations. We thank Christina Zhao, Liangcai Nie, and Fang Xu for excellent technical assistance.
Supported by the National Institutes of Health (RO1DK090056 and PO1 HL20948) and the Howard Hughes Medical Institute.
Potential conflict of interest: Dr. Cohen consults for Amgen, Pfizer, and Regeneron.
See Editorial on Page 2323
Contributor Information
Jonathan C. Cohen, Email: Jonathan.cohen@utsouthwestern.edu
Helen H. Hobbs, Email: Helen.hobbs@utsouthwestern.edu
References
Author names in bold designate shared co‐first authorship.
- 1. Marcellin P, Kutala BK. Liver diseases: a major, neglected global public health problem requiring urgent actions and large‐scale screening. Liver Int 2018;38(Suppl 1):2‐6. [DOI] [PubMed] [Google Scholar]
- 2. Haga Y, Kanda T, Sasaki R, Nakamura M, Nakamoto S, Yokosuka O. Nonalcoholic fatty liver disease and hepatic cirrhosis: comparison with viral hepatitis‐associated steatosis. World J Gastroenterol 2015;21:12989‐12995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Andronescu CI, Purcarea MR, Babes PA. Nonalcoholic fatty liver disease: epidemiology, pathogenesis and therapeutic implications. J Med Life 2018;11:20‐23. [PMC free article] [PubMed] [Google Scholar]
- 4. Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2008;40:1461‐1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tian C, Stokowski RP, Kershenobich D, Ballinger DG, Hinds DA. Variant in PNPLA3 is associated with alcoholic liver disease. Nat Genet 2010;42:21‐23. [DOI] [PubMed] [Google Scholar]
- 6. Speliotes EK, Butler JL, Palmer CD, Voight BF, Hirschhorn JN. PNPLA3 variants specifically confer increased risk for histologic nonalcoholic fatty liver disease but not metabolic disease. Hepatology 2010;52:904‐912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nischalke HD, Berger C, Luda C, Berg T, Muller T, Grunhage F, et al. The PNPLA3 rs738409 148M/M genotype is a risk factor for liver cancer in alcoholic cirrhosis but shows no or weak association in hepatitis C cirrhosis. PLoS One 2011;6:e27087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huang Y, Cohen JC, Hobbs HH. Expression and characterization of a PNPLA3 protein isoform (I148M) associated with nonalcoholic fatty liver disease. J Biol Chem 2011;286:37085‐37093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li JZ, Huang Y, Karaman R, Ivanova PT, Brown HA, Roddy T, et al. Chronic overexpression of PNPLA3I148M in mouse liver causes hepatic steatosis. J Clin Invest 2012;122:4130‐4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Smagris E, BasuRay S, Li J, Huang Y, Lai KM, Gromada J, et al. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology 2015;61:108‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Basantani MK, Sitnick MT, Cai L, Brenner DS, Gardner NP, Li JZ, et al. Pnpla3/Adiponutrin deficiency in mice does not contribute to fatty liver disease or metabolic syndrome. J Lipid Res 2011;52:318‐329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen W, Chang B, Li L, Chan L. Patatin‐like phospholipase domain‐containing 3/adiponutrin deficiency in mice is not associated with fatty liver disease. Hepatology 2010;52:1134‐1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. He S, McPhaul C, Li JZ, Garuti R, Kinch L, Grishin NV, et al. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J Biol Chem 2010;285:6706‐6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. BasuRay S, Smagris E, Cohen JC, Hobbs HH. The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatology 2017;66:1111‐1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kumari M, Schoiswohl G, Chitraju C, Paar M, Cornaciu I, Rangrez AY, et al. Adiponutrin functions as a nutritionally regulated lysophosphatidic acid acyltransferase. Cell Metab 2012;15:691‐702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Romeo S, Sentinelli F, Dash S, Yeo GS, Savage DB, Leonetti F, et al. Morbid obesity exposes the association between PNPLA3 I148M (rs738409) and indices of hepatic injury in individuals of European descent. Int J Obes (Lond) 2010;34:190‐194. [DOI] [PubMed] [Google Scholar]
- 17. Zimmerman R, Strauss JG, Haemmerle G, Schoiswohl G, Birner‐Gruenberger R, Riederer M, et al. Fat mobolization in adipose tissue is promoted by adipose triglyceride lipase. Science 2004;306:1383‐1386. [DOI] [PubMed] [Google Scholar]
- 18. Lass A, Zimmermann R, Haemmerle G, Riederer M, Schoiswohl G, Schweiger M, et al. Adipose triglyceride lipase‐mediated lipolysis of cellular fat stores is activated by CGI‐58 and defective in Chanarin‐Dorfman Syndrome. Cell Metab 2006;3:309‐319. [DOI] [PubMed] [Google Scholar]
- 19. Guo F, Ma Y, Kadegowda AK, Betters JL, Xie P, Liu G, et al. Deficiency of liver Comparative Gene Identification‐58 causes steatohepatitis and fibrosis in mice. J Lipid Res 2013;54:2109‐2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kory N, Farese RV Jr., Walther TC. Targeting fat: mechanisms of protein localization to lipid droplets. Trends Cell Biol 2016;26:535‐546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang H, Becuwe M, Housden BE, Chitraju C, Porras AJ, Graham MM, et al. Seipin is required for converting nascent to mature lipid droplets. Elife 2016;5:e16582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Munro S, Pelham HR. A C‐terminal signal prevents secretion of luminal ER proteins. Cell 1987;48:899‐907. [DOI] [PubMed] [Google Scholar]
- 23. Dayel MJ, Hom EF, Verkman AS. Diffusion of green fluorescent protein in the aqueous‐phase lumen of endoplasmic reticulum. Biophys J 1999;76:2843‐2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nakabayashi H, Taketa K, Yamane T, Miyazaki M, Miyano K, Sato J. Phenotypical stability of a human hepatoma cell line, HuH‐7, in long‐term culture with chemically defined medium. Gann 1984;75:151‐158. [PubMed] [Google Scholar]
- 25. Chamoun Z, Vacca F, Parton RG, Gruenberg J. PNPLA3/adiponutrin functions in lipid droplet formation. Biol Cell 2013;105:219‐233. [DOI] [PubMed] [Google Scholar]
- 26. Ruhanen H, Perttila J, Holtta‐Vuori M, Zhou Y, Yki‐Jarvinen H, Ikonen E, et al. PNPLA3 mediates hepatocyte triacylglycerol remodeling. J Lipid Res 2014;55:739–‐746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xie X, Langlais P, Zhang X, Heckmann BL, Saarinen AM, Mandarino LJ, et al. Identification of a novel phosphorylation site in adipose triglyceride lipase as a regulator of lipid droplet localization. Am J Physiol Endocrinol Metab 2014;306:E1449‐E1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kory N, Thiam AR, Farese RV Jr., Walther TC. Protein crowding is a determinant of lipid droplet protein composition. Dev Cell 2015;34:351‐363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Su W, Wang Y, Jia X, Wu W, Li L, Tian X, et al. Comparative proteomic study reveals 17beta‐HSD13 as a pathogenic protein in nonalcoholic fatty liver disease. Proc Natl Acad Sci U S A 2014;111:11437‐11442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang S, Wang Y, Cui L, Deng Y, Xu S, Yu J, et al. Morphologically and functionally distinct lipid droplet subpopulations. Sci Rep 2016;6:29539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Subramanian V, Rothenberg A, Gomez C, Cohen AW, Garcia A, Bhattacharyya S, et al. Perilipin A mediates the reversible binding of CGI‐58 to lipid droplets in 3T3‐L1 adipocytes. J Biol Chem 2004;279:42062‐42071. [DOI] [PubMed] [Google Scholar]
- 32. Granneman JG, Moore HP, Krishnamoorthy R, Rathod M. Perilipin controls lipolysis by regulating the interactions of AB‐hydrolase containing 5 (Abhd5) and adipose triglyceride lipase (Atgl). J Biol Chem 2009;284:34538‐34544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rondini EA, Mladenovic‐Lucas L, Roush WR, Halvorsen GT, Green AE, Granneman JG. Novel pharmacological probes reveal ABHD5 as a locus of lipolysis control in white and brown adipocytes. J Pharmacol Exp Ther 2017;363:367‐376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mitsche MA, Hobbs HH, Cohen JC. Patatin‐like phospholipase domain‐containing protein 3 promotes transfer of essential fatty acids from triglycerides to phospholipids in hepatic lipid droplets. J Biol Chem 2018;293:9232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kien B, Grond S, Haemmerle G, Lass A, Eichmann TO, Radner FPW. ABHD5 stimulates PNPLA1‐mediated omega‐O‐acylceramide biosynthesis essential for a functional skin permeability barrier. J Lipid Res 2018;59:2360‐2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ohno Y, Nara A, Nakamichi S, Kihara A. Molecular mechanism of the ichthyosis pathology of Chanarin‐Dorfman syndrome: Stimulation of PNPLA1‐catalyzed omega‐O‐acylceramide production by ABHD5. J Dermatol Sci 2018;92:245‐253. [DOI] [PubMed] [Google Scholar]
- 37. Lord CC, Ferguson D, Thomas G, Brown AL, Schugar RC, Burrows A, et al. Regulation of hepatic triacylglycerol metabolism by CGI‐58 does not require ATGL co‐activation. Cell Rep 2016;16:939‐949. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials