

Abstract
Objective
To investigate the causal role of cardiometabolic risk factors in osteoarthritis (OA) using associated genetic variants.
Methods
We studied 27,691 adults from the Malmö Diet and Cancer Study (MDCS) and replicated novel findings among 376,435 adults from the UK Biobank. Trait‐specific polygenic risk scores for low‐density lipoprotein (LDL) and high‐density lipoprotein (HDL) cholesterol levels, triglyceride levels, body mass index (BMI), fasting plasma glucose (FPG) levels, and systolic blood pressure (BP) were used to test the associations of genetically predicted elevations in each trait with incident OA diagnosis (n = 3,559), OA joint replacement (n = 2,780), or both (total OA; n = 4,226) in Mendelian randomization (MR) analyses in the MDCS, and with self‐reported and/or hospital‐diagnosed OA (n = 65,213) in the UK Biobank. Multivariable MR, MR‐Egger, and weighted median MR were used to adjust for potential pleiotropic biases.
Results
In the MDCS, genetically predicted elevation in LDL cholesterol level was associated with a lower risk of OA diagnosis (odds ratio [OR] 0.83 [95% confidence interval (95% CI) 0.73–0.95] per 1SD increase) and total OA (OR 0.87 [95% CI 0.78–0.98]), which was supported by multivariable MR for OA diagnosis (OR 0.84 [95% CI 0.75–0.95]) and total OA (0.87 [95% CI 0.78–0.97]), and by conventional 2‐sample MR for OA diagnosis (OR 0.86 [95% CI 0.75–0.98]). MR‐Egger indicated no pleiotropic bias. Genetically predicted elevation in BMI was associated with an increased risk of OA diagnosis (OR 1.65 [95% CI 1.14–2.41]), while MR‐Egger indicated pleiotropic bias and a larger association with OA diagnosis (OR 3.25 [1.26–8.39]), OA joint replacement (OR 3.81 [95% CI 1.39–10.4]), and total OA (OR 3.41 [95% CI 1.43–8.15]). No associations were observed between genetically predicted HDL cholesterol level, triglyceride level, FPG level, or systolic BP and OA outcomes. The associations with LDL cholesterol levels were replicated in the UK Biobank (OR 0.95 [95% CI 0.93–0.98]).
Conclusion
Our MR study provides evidence of a causal role of lower LDL cholesterol level and higher BMI in OA.
INTRODUCTION
It has been hypothesized that there are pathophysiologic links between cardiometabolic disease and osteoarthritis (OA) 1, 2. Several studies have previously shown that cardiometabolic risk factors and diseases are associated with an increased risk of OA 2, 3. Potential mechanisms could relate to systemic processes, including cholesterol metabolism and associated inflammatory processes 2, 4. OA encompasses changes in articular, bone, and cartilage structures 5, and the current clinical focus is on the modification of mechanical loading as a causal factor, treatment of psychosocial factors, or replacement of intraarticular cartilage 6. Yet studies of generalized OA suggest a potential role of systemic processes, which has raised the hypothesis that lipid metabolism disorders 7, 8 could be involved in pathogenic mechanisms of OA 9. Supporting evidence for this hypothesis includes the shared mesenchymal origin of adipocytes and articular cells 10, 11, in vitro studies showing that higher lipid levels in the synovial fluid can induce arthritic changes 12, and experimental mouse models of atherosclerosis showing arthritic changes with a high cholesterol diet 13, 14. Epidemiologic studies have provided further support for the hypothesis, indicating that cardiovascular disease and OA commonly co‐occur 15, share similar risk factors 16, 17, and are both associated with higher mortality 18.
Observational studies, however, suffer from biases, mainly due to reverse causation and confounding, making it difficult to infer a causal role between cardiometabolic traits and OA. Since genetic markers are distributed randomly at conception, they can be used to infer causal relationships between these traits and OA. This analysis has previously been performed in a Mendelian randomization (MR) study using a genetic variant in the FTO locus associated with adiposity measured as body mass index (BMI), and more recently in the UK Biobank using polygenic risk scores 19. Those studies have provided evidence supporting a causal role of higher BMI in increasing the risk of OA 20. Similarly, genetic variants associated with other cardiometabolic traits can be leveraged for deciphering their causal role in OA. However, for genetic variants to be used in MR studies they must be reliably associated with the exposure of interest, should exert their effect on the outcome solely through the exposure, and should not associate with other factors that affect the outcome. Those assumptions are violated when variants exhibit horizontal pleiotropy, meaning that variants have effects on disease outside of their effect on the exposure, which leads to bias in MR studies. Since combinations of many genetic variants are needed to power most MR studies, there is an increased risk of violations through horizontal pleiotropy. Several methods have been developed to correct for such bias in the 2‐sample MR setting using exposure and outcome summary statistics for genetic associations obtained from independent samples.
In light of the conflicting evidence, we aimed to use previously identified cardiometabolic genetic variants, and an MR approach, to test the hypothesis that each of the 6 cardiometabolic traits, including low‐density lipoprotein (LDL) cholesterol, high‐density lipoprotein (HDL) cholesterol, triglycerides, BMI, fasting plasma glucose (FPG), and systolic blood pressure (BP), plays a causal role in the pathogenesis of OA.
MATERIALS AND METHODS
Study population
The Malmö Diet and Cancer Study (MDCS) cohort includes nearly 30,000 adults ages 45–64 years recruited from the population between 1991 and 1996. Baseline data obtained with subject consent included anthropometrics, body composition, cardiovascular physiologic measures, socioeconomic and lifestyle factors, and medical history 21. The data on the MDCS population were linked to clinical registries using the civil registration numbers that uniquely identify all inhabitants of Sweden. The MDCS was approved by the Regional Ethics Committee at Lund University (LU 51‐90), and the final data provided to the research team was anonymized. We also used the UK Biobank, a large cohort of ~500,000 adults ages 40–69 years, to replicate novel findings. We excluded samples with nonwhite ancestry, sex mismatches, excess heterozygosity, missingness, and second‐degree relatives 22. Analyses in the UK Biobank were conducted via application 7089 via a protocol approved by the Partners HealthCare Institutional Review Board.
Baseline assessments of subjects in the MDCS
BP was measured using a mercury‐column sphygmomanometer after 10 minutes of rest in the supine position. A balance‐beam scale was used to measure weight (in kg) with subjects wearing light clothing and no shoes. A fixed stadiometer was used to measure height (in cm). BMI was measured as weight in kilograms divided by height in meters squared. Fasting serum lipids and fasting blood glucose were measured from blood samples obtained after an overnight fast. Fasting blood glucose was converted to FPG by multiplying the values by 1.13. Samples were analyzed by routine standard methods at the Department of Clinical Chemistry, Malmö University Hospital (Malmö, Sweden). Fasting blood measurements were only available in the MDCS Cardiovascular Cohort (n = 5,675). Apolipoprotein B (Apo B) and Apo A‐I were measured in nonfasting plasma samples from all subjects in the MDCS in a blinded manner (with regard to case or control status) by Quest Diagnostics, using an immunonephelometry assay run on a Siemens BN II system. The interassay variability was <4.0% for both Apo A‐I and Apo B.
Genetic data
In the MDCS, a matrix‐assisted laser desorption ionization–time‐of‐flight mass spectrometer (Sequenom MassArray) was used to genotype DNA samples using Sequenom reagents and protocols. Proxy single‐nucleotide polymorphisms (SNPs) were identified using SNAP version 2.2.2 when commercial primers were not available. SNPs that failed Sequenom genotyping were genotyped individually using TaqMan or KASPar allele discrimination on an ABI 7900HT (Applied Biosystems, Life Technologies), according to the manufacturer's instructions. Individuals with <60% of SNPs successfully genotyped, and SNPs with a genotype success rate of <90% or deviation from Bonferroni‐corrected Hardy‐Weinberg equilibrium in each set of SNPs of the specific traits were excluded. Using these exclusion criteria, genetic data were available for 26,435 individuals. Genotyped SNPs were selected based on previous genome‐wide association studies (GWAS) and included 31 SNPs for BMI 23, 29 for systolic BP 24, 25, 26, 52 for LDL cholesterol 27, 28, 41 for HDL cholesterol 27, 26 for triglycerides 27, and 15 for FPG 29. Trait‐specific MDCS‐weighted polygenic risk scores were created using Plink software (version 1.07).
In the UK Biobank, we extracted data on 185 lipid‐associated SNPs, including 76 independent LDL cholesterol–associated SNPs, defined as SNPs showing associations with LDL cholesterol level with a P value less than 5 × 10−8, from the latest Global Lipids Genetics Consortium GWAS 28. Additionally, gene‐specific instruments for HMGCR, PCSK9, LDLR, and NPC1L1 were constructed by using independent LDL cholesterol–associated SNPs in each locus, as previously described by Ference et al 30, 31, to estimate the causal effect of LDL cholesterol level through each of these genes. Gene‐specific genotype data were not available in the MDCS.
Cardiovascular and OA‐related outcomes
Cardiovascular disease cases were defined as fatal or nonfatal myocardial infarction (MI) or stroke, or death due to ischemic heart disease, and identified using the Swedish Hospital Discharge Register, the Swedish Cause of Death Register, and the Stroke Register of Malmö. MI cases were defined using codes 410 and I21 (in the International Classification of Diseases, Ninth Revision [ICD‐9] and ICD‐10, respectively), and stroke cases were defined using codes 430, 431, 434, and 436 of the ICD‐9, and codes I60, I61, I63, and I64 of the ICD‐10.
Data from national registries provided by the Swedish Board on Health and Welfare (Swedish National Discharge Registers) and by Statistics Sweden were used to define the following 3 main study outcomes: incident OA diagnosis (a clinical record of either knee or hip OA), incident OA joint replacement (a record of either a hip or knee joint arthroplasty), and total OA (either a diagnosis or joint replacement). Incident outcome was defined as a new case occurring at any point during the follow‐up period after the baseline screening date (1996) to the end of 2014, which means there was a nearly 20‐year mean follow‐up period for the study participants. The case definitions for the outcomes were based on ICD coding systems that covered the time period from the 1970s to the current version (ICD‐10). For OA arthroplasty the codes were 8423, 8424, 8428, 8433, NGB09, NGB19, NGB29, NGB39, and NGB49 (for knee arthroplasty) and 8410, 8411, NFB29, NFB39, and NFB49 (for hip arthroplasty). Additionally, participants with both joint replacements and a record of fractures (any cause) were excluded from the OA outcomes sample, to ensure that the indication was likely to be related to OA. In the MDCS, based on ICD‐10 codes, a total of 3,559 individuals had an OA diagnosis, 2,780 had an OA joint replacement, 2,113 had both an OA diagnosis and an OA joint replacement, and 4,226 were included in the category “total OA” (1,446 who had an OA diagnosis only, 667 who had a joint replacement only, and 2,113 who had both an OA diagnosis and a joint replacement).
In the UK Biobank the OA definition was based on self‐reported medical history and/or ICD‐9 and ICD‐10 codes in hospital‐based medical records. A total of 65,213 individuals had either self‐reported or medical records–based OA, 36,128 had self‐reported OA, 43,744 had medical records–based OA, and 14,659 had both.
Statistical analysis
In this study, we performed several different MR analyses. In the MDCS, we performed 1‐sample conventional and multivariable MR using SNP exposure and SNP outcome data from the MDCS, and 2‐sample conventional and multivariable MR using SNP exposure data from publicly available GWAS databases and SNP outcome data from the MDCS 32, 33. For significant associations, we continued with further methods to control for pleiotropy. We used 2‐sample MR‐Egger analyses and estimation of the Egger intercept, which reflects total horizontal pleiotropy 34, and 2‐sample weighted median MR which can provide accurate estimates given that at least 50% of the variants are valid instruments 35. For LDL cholesterol, we performed replication analyses in the UK Biobank using 2‐sample conventional MR, and correcting for pleiotropy by multivariable MR, MR‐Egger, and weighted median MR. In the MDCS, prevalent OA cases were excluded and controls were defined as all individuals who did not develop OA during the follow‐up period. In the UK Biobank, cases included both prevalent and incident OA and controls were defined as all individuals without OA.
In order to achieve normality of distribution and comparability between the traits studied, values for all of the traits (LDL cholesterol, HDL cholesterol, triglycerides, BMI, systolic BP, and FPG) were natural log–transformed and converted to Z scores in the MDCS. Linear regression was used to study the association between the weighted polygenic risk scores and their respective traits. To estimate the unconfounded causal effect of the trait on incident cases, we performed a conventional 1‐sample MR analysis using a 2‐stage regression. The predicted fitted values from the linear regression of the traits by their respective polygenic risk scores were used as the predictor variables for OA outcomes in logistic regression models adjusted for age and sex.
The polygenic risk scores included pleiotropic SNPs, which may have biased our results. Excluding these SNPs would have largely weakened the polygenic risk scores as instrumental variables; therefore, we used a previously described and applied inverse‐variance–weighted multivariable MR approach 36. In this approach, the SNP outcome β coefficients obtained from logistic regression of 168 independent SNPs (r2 < 0.2) on incident OA were included as outcome variables in a multivariable model with SNP–LDL cholesterol, SNP–HDL cholesterol, SNP–triglycerides, SNP–BMI, SNP–FPG, and SNP–systolic BP β coefficients obtained from the linear regression of the same SNPs on each of these cardiometabolic traits in the MDCS. This multivariable model was weighted by the inverse‐variance of the SNP–outcome association and the intercept was fixed to 0. The multivariable MR model corrects for pleiotropic bias across the traits studied and not for potential pleiotropy with other traits not included in the model 32, 37. To correct for possible bias from total or cardiovascular mortality as competing risks, we used the Fine and Gray proportional subdistribution hazard model and age as the underlying time variable in sensitivity analyses.
We followed up significant findings using summary level data for the same set of SNPs in the most recent publicly available GWAS data, except for BP 28, 38, 39, which was not publicly available, to perform 2‐sample MR analyses using inverse‐variance–weighted MR or conventional MR. This is a regression of SNP–outcome β coefficients on each instrumental SNP–exposure β coefficient weighted by the inverse‐variance of the SNP–outcome associations with the intercept fixed to 0 33. To detect and correct for known and unknown pleiotropic bias, multivariable MR and MR‐Egger were performed. MR‐Egger is similar to the inverse‐variance–weighted MR, but the intercept is left unconstrained and represents the average unbalanced pleiotropy by each instrument. It provides valid estimates even if all SNPs are invalid instruments. However, it is limited by the untestable “instrument strength independent of direct effect” assumption that the distribution of the pleiotropic effects of the SNPs on the outcome are independent of their associations with the exposure 34. We additionally performed weighted median MR which provides a causal estimate given that at least 50% of the variants are valid instruments 35.
Finally, we performed 2‐sample conventional MR association analysis between a 76‐SNP LDL cholesterol GWAS‐weighted 28 polygenic risk score and OA in the UK Biobank including 65,213 OA cases and 311,222 controls. Restricted conventional MR analyses were also performed using a 16‐SNP LDL cholesterol–specific GWAS‐weighted polygenic risk score, defined as LDL cholesterol–associated SNPs that remained after excluding those associated with HDL cholesterol and/or triglycerides, using a P value of 0.05. Then, we performed sensitivity MR analyses including MR‐Egger and weighted median MR using the 76 LDL cholesterol–associated SNPs, and multivariable MR using 185 LDL cholesterol–, HDL cholesterol–, and/or triglyceride‐associated SNPs 28. Finally, we performed conventional MR analyses of the association between LDL cholesterol and OA using GWAS‐weighted gene‐specific LDL cholesterol polygenic risk scores in each of the HMGCR, PCSK9, LDLR, and NPC1L1 genes.
Stata SE 13.1 and R (version 3.3) were used for statistical analysis. For the 2‐sample MR analyses, we used the “MendelianRandomization” package in R 40.
RESULTS
Incident OA end points in the MDCS
Of the total sample of 27,691 MDCS participants at baseline, by the end of the mean follow‐up time of 17.4 years, 19,350 (70%) were alive, 8,091 (29.2%) had died, and 250 (0.9%) had emigrated. During the follow‐up period, 3,559 (12.9%) had an OA diagnosis, 2,780 (10%) had an OA joint replacement, and 4,226 (15.3%) were included in the category “total OA” (Table 1). The variances explained by polygenic risk scores were 7.3% for LDL cholesterol, 5.7% for HDL cholesterol, 4.3% for triglycerides, 0.8% for BMI, 2.3% for FPG, and 0.5% for systolic BP in the MDCS.
Table 1.
Baseline characteristics of the subjects in the Malmö Diet and Cancer Studya
| OA diagnosis (n = 3,559) | OA joint replacement (n = 2,780) | Total OA (n = 4,226)b | Non‐OA (n = 23,465)c | |
|---|---|---|---|---|
| Age, years (n = 27,691) | 58.5 ± 7.3d | 60.2 ± 7.3d | 59.2 ± 7.4d | 57.8 ± 7.6 |
| Men, no. (%) (n = 10,916) | 1,183 (33.2)d | 875 (31.5)d | 1,371 (32.4)d | 9,545 (40.7) |
| Women, no. (%) (n= 16,775) | 2,376 (66.8)d | 1,905 (68.5)d | 2,855 (67.6)d | 13,920 (59.3) |
| BMI (n = 27,649) | 27.3 ± 4.3d | 26.9 ± 4.3d | 27.0 ± 4.3d | 25.5 ± 3.8 |
| LDL cholesterol, mmoles/liter (n = 5,137) | 4.2 ± 1.0 | 4.3 ± 1.0 | 4.2 ± 1.0 | 4.2 ± 1.0 |
| Apo A, mg/dl (n = 27,022) | 158 ± 28 | 159 ± 28 | 158 ± 28 | 156 ± 28 |
| Apo B, mg/dl (n = 27,018) | 107 ± 25 | 108 ± 25 | 108 ± 26 | 107 ± 26 |
| Systolic BP, mm Hg (n = 27,648) | 141 ± 19 | 143 ± 20 | 142 ± 20 | 141 ± 20 |
| Diastolic BP, mm Hg (n = 27,646) | 86 ± 10 | 86 ± 10 | 86 ± 10 | 86 ± 10 |
| Never smoked, no. (%) (n = 10,461) | 1,466 (41.2)d | 1,168 (42.0)d | 1,736 (41.1)d | 8,725 (37.2) |
| Former smoker, no. (%) (n = 9,375) | 1,276 (35.9)d | 958 (34.5)d | 1,471 (34.8)d | 7,904 (33.7) |
| Current smoker, no. (%) (n = 7,843) | 815 (22.9)d | 654 (23.5)d | 1,017 (24.1)d | 6,826 (29.1) |
| Myocardial infarction, no. (%) (n = 538) | 59 (1.7) | 47 (1.7) | 73 (1.7) | 465 (2.0) |
| Cardiovascular disease, no. (%) (n = 837) | 79 (2.2)d | 64 (2.3)d | 101 (2.4)d | 736 (3.1) |
| Diabetes, no. (%) (n = 1,209) | 144 (4.0) | 120 (4.3) | 186 (4.4) | 1,023 (4.4) |
Except where indicated otherwise, values are the mean ± SD. Of the total cohort, 39.4% were men and 60.6% were women. BMI = body mass index; LDL = low‐density lipoprotein; Apo A = apolipoprotein A; BP = blood pressure.
Includes participants with incident osteoarthritis (OA) diagnosis only (n = 1,446), OA joint replacement only (n = 667), or both (n = 2,113).
Participants without incident OA diagnosis or OA joint replacement.
P < 0.05 versus the rest of the population, after adjustment for age and sex.
Conventional and multivariable 1‐sample MR in the MDCS
The associations of elevations in cardiometabolic traits predicted by polygenic risk score with OA outcomes in the MDCS, determined using conventional MR, are shown in Figure 1. Genetically predicted elevations in LDL cholesterol level were associated with a lower risk of OA diagnosis, with an odds ratio (OR) of 0.83 (95% confidence interval [95% CI] 0.73–0.95) and total OA (OR 0.87 [95% CI 0.78–0.98]) but not with OA joint replacement (OR 0.94 [95% CI 0.81–1.08]). Genetically predicted elevations in BMI were associated with an increased risk of OA diagnosis (OR 1.65 [95% CI 1.14–2.41]) and with a trend toward an increased risk of total OA (OR 1.42 [95% CI 1.00–2.02]) but not with OA joint replacement (OR 1.31 [95% CI 0.86–2.00]). Genetically predicted elevations in systolic BP were associated with a reduced risk of OA diagnosis (OR 0.54 [95% CI 0.35–0.83]), OA joint replacement (OR 0.43 [95% CI 0.26–0.70]), and total OA (OR 0.58 [95% CI 0.39–0.88]).
Figure 1.
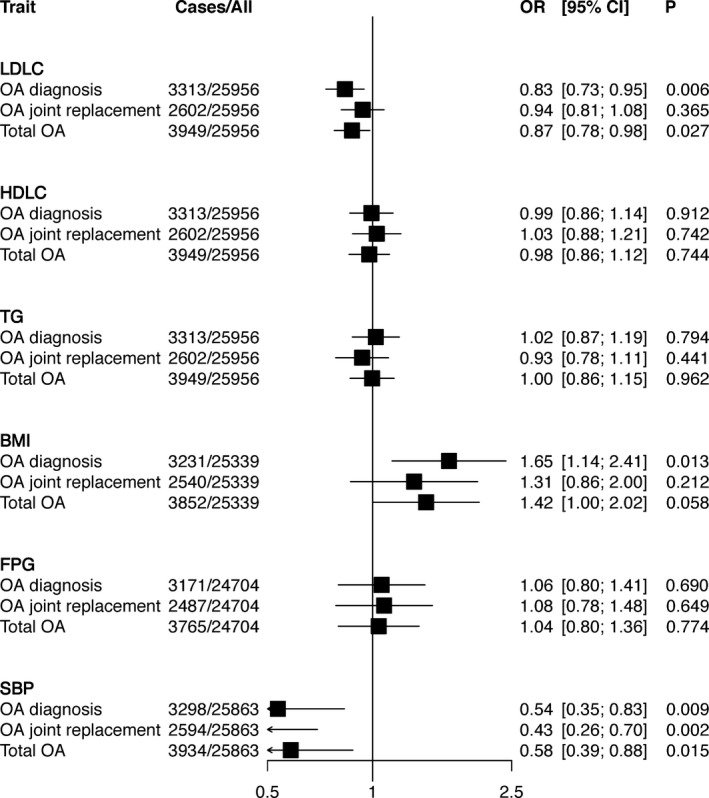
One‐sample conventional Mendelian randomization analyses of genetically predicted elevations in cardiometabolic traits and osteoarthritis (OA) outcomes in the Malmö Diet and Cancer Study (MDCS). The odds ratios (ORs) and 95% confidence intervals (95% CIs) for OA outcomes per genetically predicted 1SD increase in levels of cardiometabolic traits determined using respective polygenic risk scores are shown. Fitted values predicted by the polygenic risk score for each trait were used as predictors of incident OA outcomes in the MDCS in a 2‐stage least squares regression analysis. Genetically predicted elevation in low‐density lipoprotein cholesterol (LDLC) level was associated with a lower risk of OA diagnosis and total OA, genetically predicted elevation in body mass index (BMI) was associated with a higher risk of OA diagnosis, and genetically predicted elevation in systolic blood pressure (SBP) was associated with a lower risk of all OA outcomes. High‐density lipoprotein cholesterol (HDLC) level, triglycerides (TG), and fasting plasma glucose (FPG) were not associated with OA outcomes.
Multivariable MR adjusted for pleiotropy across the 6 cardiometabolic traits supported the observed associations of BMI and LDL cholesterol level with OA (for example, for LDL cholesterol level, the OR for OA diagnosis was 0.84 [95% CI 0.75–0.95] and the OR for total OA was 0.87 [95% CI 0.78–0.97]). However, the association between genetically predicted elevation in systolic BP and reduced risk of OA outcomes was lost when adjusting for pleiotropy (see Supplementary Figure 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40812/abstract). Sensitivity analyses that accounted for competing risks showed that the association between LDL cholesterol level and OA diagnosis was not likely to be biased by total mortality (subdistribution hazard ratio [SHR] 0.83 [95% CI 0.74–0.94]) or cardiovascular mortality (SHR 0.84 [95% CI 0.74–0.94]) as competing risks (Supplementary Table 1, online at http://onlinelibrary.wiley.com/doi/10.1002/art.40812/abstract).
Two‐sample multivariable MR, MR‐Egger, and weighted median MR analysis to correct for pleiotropy using SNP–exposure associations from publicly available GWAS databases for SNP–outcome associations in the MDCS
Using published GWAS summary level data for cardiometabolic traits, we performed 2‐sample MR analyses of the association of LDL cholesterol level and BMI with OA outcomes in the MDCS. Conventional MR indicated an inverse association between LDL cholesterol level and OA diagnosis (OR 0.86 [95% CI 0.75–0.98]). Multivariable MR adjusting for estimates of HDL cholesterol level, triglycerides, BMI, and FPG indicated a lower risk of OA diagnosis in subjects with genetically predicted elevation in LDL cholesterol level (OR 0.85 [95% CI 0.73–0.98). Similar analyses indicated no association with OA joint replacement. The MR‐Egger intercept indicated no pleiotropic bias (P‐intercept = 0.51), and the MR‐Egger estimate was consistent with conventional and multivariable MR, yet not significant (P = 0.41). Weighted median MR indicated an inverse association between LDL cholesterol level and total OA (OR 0.82 [95% CI 0.68–0.99]) and a tendency toward an inverse association between LDL cholesterol level and OA diagnosis (OR 0.83 [95% CI 0.68–1.02]) (Table 2).
Table 2.
Two‐sample MR for association of LDL cholesterol level and BMI with OA end points in the Malmö Diet and Cancer Studya
| MR method and OA end point | LDL cholesterol | BMI | ||
|---|---|---|---|---|
| OR (95% CI) | P | OR (95% CI) | P | |
| Conventional MRb | ||||
| OA diagnosis | 0.86 (0.75–0.98) | 0.029 | 1.41 (0.96–2.08) | 0.089 |
| OA joint replacement | 1.01 (0.85–1.20) | 0.910 | 1.19 (0.77–1.82) | 0.442 |
| Total OA | 0.93 (0.81–1.06) | 0.281 | 1.24 (0.86–1.79) | 0.266 |
| Multivariable MRc | ||||
| OA diagnosis | 0.85 (0.73–0.98) | 0.022 | 1.39 (1.05–1.86) | 0.025 |
| OA joint replacement | 1.02 (0.87–1.19) | 0.853 | 1.35 (0.98–1.86) | 0.069 |
| Total OA | 0.91 (0.80–1.04) | 0.178 | 1.32 (1.01–1.72) | 0.048 |
| MR‐Eggerc | ||||
| OA diagnosis | 0.91 (0.73–1.13) | 0.405 | 3.25 (1.26–8.39) | 0.015 |
| OA joint replacement | 0.97 (0.73–1.28) | 0.807 | 3.81 (1.39–10.4) | 0.009 |
| Total OA | 0.94 (0.75–1.18) | 0.583 | 3.41 (1.43–8.15) | 0.006 |
| Egger interceptd | ||||
| OA diagnosis | 1.00 (0.98–1.01) | 0.505 | 0.97 (0.93–1.00) | 0.062 |
| OA joint replacement | 1.00 (0.99–1.02) | 0.689 | 0.96 (0.92–0.99) | 0.014 |
| Total OA | 1.00 (0.98–1.01) | 0.882 | 0.96 (0.93–0.99) | 0.013 |
| Weighted median MRe | ||||
| OA diagnosis | 0.83 (0.68–1.02) | 0.078 | 1.39 (0.87–2.24) | 0.172 |
| OA joint replacement | 0.90 (0.72–1.13) | 0.354 | 2.01 (1.19–3.39) | 0.009 |
| Total OA | 0.82 (0.68–0.99) | 0.045 | 2.07 (1.33–3.21) | 0.001 |
LDL = low‐density lipoprotein; BMI = body mass index; OA = osteoarthritis; OR = odds ratio; 95% CI = 95% confidence interval.
Inverse‐variance–weighted Mendelian randomization (MR) provides estimates without correcting for pleiotropy 33.
Inverse‐variance–weighted multivariable MR provides estimates after correcting for pleiotropy with other cardiometabolic traits 32.
Egger intercept reflects total horizontal pleiotropy 34.
Weighted median MR provides accurate estimates given that at least 50% of variants are valid instruments 35.
Conventional MR analyses showed a tendency toward an increased risk of OA diagnosis (but not other OA outcomes) in subjects with genetically predicted elevation in BMI (OR 1.41 [95% CI 0.96–2.08]). Multivariable MR showed a significantly increased risk of OA diagnosis and total OA in subjects with elevated BMI (OR 1.39 [95% CI 1.05–1.86] and OR 1.32 [95% CI 1.01–1.72], respectively). The MR‐Egger intercept, however, indicated that the BMI SNPs likely exhibited unbalanced pleiotropy in their association with all 3 OA outcomes (P‐intercept = 0.06 for OA diagnosis and 0.01 for both OA joint replacement and total OA). The MR‐Egger estimates suggested an ~3.3–3.8 fold increased risk of OA diagnosis (OR 3.25 [95% CI 1.26–8.39]), OA joint replacement (OR 3.81 [95% CI 1.39–10.4]), and total OA (OR 3.41 [95% CI 1.43–8.15]) per 1SD increase in BMI. Higher BMI was associated with both OA joint replacement and total OA using weighted median MR (OR 2.01 [95% CI 1.19–3.39] and OR 2.07 [95% CI 1.33–3.21], respectively) (Table 2).
Two‐sample MR analyses in the UK Biobank to replicate the association between LDL cholesterol level and OA
Finally, we performed 2‐sample MR analyses in the UK Biobank. A 76‐SNP LDL cholesterol instrument was used in more than 65,213 OA cases and 311,222 controls in the UK Biobank. Using the 76‐SNP GWAS‐weighted polygenic risk score, each 1SD increase in LDL cholesterol level was associated with a lower risk of OA (OR 0.95 [95% CI 0.93–0.98]). Restricting the polygenic risk score to 16 LDL cholesterol–specific SNPs indicated a larger magnitude of decreased risk of OA (OR 0.86 [95% CI 0.79–0.95]). Gene‐specific LDL cholesterol polygenic risk score analyses showed an inverse association between the LDLR gene–specific polygenic risk score and OA (OR 0.92 [95% CI 0.88–0.97]). While the HMGCR gene–specific polygenic risk score showed a tendency toward lower OA risk (OR 0.95 [95% CI 0.90–1.01]), the NPC1L1 gene–specific polygenic risk score suggested an increased risk of OA (OR 1.12 [95% CI 0.98–1.27]). Finally, sensitivity 2‐sample MR‐Egger, multivariable MR, and weighted median MR analyses with SNP data from public GWAS databases indicated inverse associations between LDL cholesterol level and OA, with ORs ranging from 0.91 to 0.95 (Figure 2), and the MR‐Egger intercept did not suggest evidence of pleiotropy. Similar results were obtained when self‐reported and medical records–based OA were analyzed separately (Supplementary Figures 2 and 3, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40812/abstract).
Figure 2.
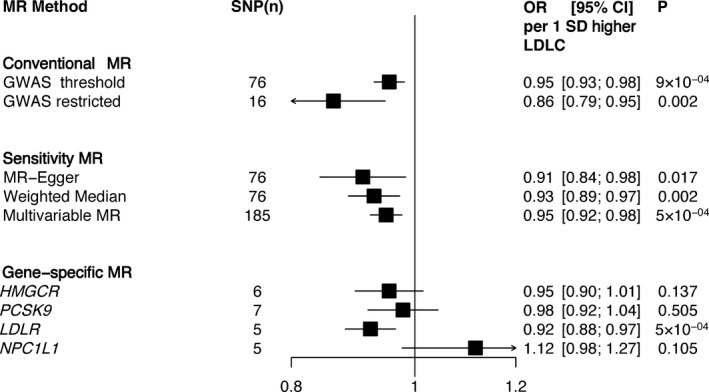
Association between genetic predisposition for elevated low‐density lipoprotein cholesterol (LDLC) and osteoarthritis (OA) in the UK Biobank. Odds ratios (ORs) and 95% confidence intervals (95% CIs) for OA using 2‐sample Mendelian randomization (MR) analyses in the UK Biobank are shown. Conventional MR estimates were obtained by analyzing polygenic risk scores for LDL cholesterol created from single‐nucleotide polymorphisms (SNPs) identified in genome‐wide association studies (GWAS) and gene‐specific SNPs related to OA in the UK Biobank weighted by SNP–LDL cholesterol associations in the Global Lipids Genetics Consortium 28. In the conventional MR analysis, for “GWAS threshold,” the polygenic risk score for LDL cholesterol was created using SNPs that were previously associated with LDL cholesterol at the GWAS significance level (P < 5 × 10−8) in the Global Lipids Genetics Consortium 28. For “GWAS restricted,” the polygenic risk score for LDL cholesterol was created using SNPs that were previously associated with LDL cholesterol at the GWAS significance level (P < 5 × 10−8) and were not associated with either HDL cholesterol level or triglycerides (P > 0.05) in the Global Lipids Genetics Consortium 28. Two‐sample sensitivity MR analyses were performed using MR‐Egger 34, weighted median MR 35, and multivariable MR 32 using summary SNP exposure data from the Global Lipids Genetic Consortium 28 and summary SNP outcome data from the UK Biobank. Genes for the gene‐specific analyses (HMGCR,PCSK9,LDLR, and NPC1L1) were mainly selected due to the fact that they encode for LDL cholesterol–lowering targets. Gene‐specific analyses indicated a lower risk of OA by LDLR‐mediated higher LDL cholesterol level. A similar trend was observed with the HMGCR instrument, although it did not reach statistical significance. MR indicated that a genetically predicted elevation in LDL cholesterol level decreases the risk of OA.
DISCUSSION
Our study has 3 separate key findings. First, our results suggest an inverse causal role between LDL cholesterol level and OA in large Swedish and British cohorts, which was not explained after adjustment for the genetically predicted levels of other cardiometabolic traits. Second, our findings support previously presented evidence of a direct causal role between BMI and OA 19, 20. Third, there was a lack of association between genetically predicted levels of other cardiometabolic factors and OA.
Our findings regarding LDL cholesterol are in contrast to those of previous observational studies, which have suggested elevated serum cholesterol levels as a risk factor for OA 1, 17, 41. Previously, it has been hypothesized that lipid accumulation in the cartilage might trigger OA development 1, and a high cholesterol diet has been shown to induce arthritic changes in experimental mouse models of atherosclerosis 13, 14. However, our present findings provide an alternative and opposite hypothesis that higher LDL cholesterol levels may be associated with a lower risk of OA.
The key implication of our findings relates to the potential role of LDL cholesterol–lowering drugs in the pathogenesis of OA. Statins are key and widespread drugs for lowering LDL cholesterol levels in cardiovascular disease management, and observational studies have provided conflicting results on the relationship between statins and OA. While some epidemiologic studies have indicated that statin use is associated with a lower risk 42, 43, others have observed an increased risk 44, 45, or a lack of association 46, 47. The key limitations of the previous studies are that they, with few exceptions, have either been small or have used OA joint replacement as the outcome. Further, observational associations are prone to biases due to confounding and reverse causation, which can be bypassed by using genetic variants as instruments.
Our study is, to the best of our knowledge, the first MR study to examine causal associations of genetically predicted LDL cholesterol level with incident OA end points. The association between lower LDL cholesterol level and OA identified using the LDL cholesterol polygenic risk score and LDLR gene–specific polygenic risk score, and the tendency toward an association between lower LDL cholesterol level through the HMGCR gene (encoding statin target) and higher OA risk contradicts most previous evidence and suggests that statins may increase the risk of OA. The implication is that future research needs to specifically investigate the effects of statins on OA with radiographic definitions to demonstrate pathologic progression or nonprogression.
Our findings also support the notion of direct causality between BMI and OA 19, 20. The higher prevalence of the metabolic syndrome among patients with OA and the lower age at onset of OA among individuals with the metabolic syndrome has led to the hypothesis that OA could be part of a systemic metabolic disorder characterized by obesity, dyslipidemia (based on HDL cholesterol and/or triglyceride levels), dysglycemia, and hypertension 16, 48, 49. The only component of the metabolic syndrome for which a causal link to OA was indicated in our study was BMI. The effect estimates using the different MR methods were notably different. While the conventional MR analysis in the MDCS indicated an ~65% increased risk of OA diagnosis with each genetically predicted 1SD increase in BMI, the MR‐Egger analysis using GWAS summary data indicated that the BMI genetic instrument exhibited pleiotropic bias and thus this analysis, which corrected for pleiotropy, may have provided a more accurate estimate. It showed that each genetically predicted 1SD increase in BMI increased the risk of all OA outcomes, with ORs ranging from 3.25 to 3.81.
Previous studies have suggested that hyperglycemia or diabetes status is associated with an increased risk of OA 1, 50, but we did not observe an association between FPG and OA in our study. This result is consistent with recent observations in the UK Biobank, where no association was found between genetic risk for type 2 diabetes and OA 19. It is possible that hyperglycemia may still have a causal but weak association with OA, as lack of evidence could be a consequence of the rather weak instrument for FPG and insufficient statistical power. Regardless, it is intriguing that earlier MR studies have shown clear evidence that a genetically predicted decrease in LDL cholesterol level increases the risk of type 2 diabetes 30, 51, similar to what was observed in our study of OA.
In the initial conventional MR analyses, we observed an inverse association between genetically predicted elevation in systolic BP and a lower risk of OA, which lost significance in multivariable MR analyses. The genetic instrument for systolic BP was the weakest, explaining only 0.5% of the variance in baseline systolic BP, which also means that a causal association with OA cannot be excluded pending stronger genetic instruments for BP.
The main strength of our study is the use of different MR analyses to investigate the causal role of cardiometabolic traits in OA. We used both 1‐sample and 2‐sample strategies that account for biases mainly due to pleiotropy. Importantly, we were able to replicate the rather controversial finding that lower LDL cholesterol level increases the risk of OA, in the large UK Biobank using up‐to‐date summary level genetic data for lipid traits. However, the MR estimate for OA using the 76‐SNP GWAS‐weighted LDL cholesterol polygenic risk score was smaller in the UK Biobank than the MR estimate using the 52‐SNP LDL cholesterol polygenic risk score in the MDCS. This could be due to misclassification in the self‐reported OA phenotype in the UK Biobank.
Nevertheless, we must acknowledge some limitations in our study. First, we cannot confirm negative associations and thus we cannot rule out possible weaker causal associations of HDL cholesterol, triglycerides, FPG, and systolic BP with OA outcomes, since the genetic instruments and the sample size used might be underpowered to detect them. Second, we used 2 different OA end points, one based on clinical diagnosis and the other on the surgical intervention of arthroplasty. Clinical diagnosis provides the phenotype for the presentation of symptomatic OA, whatever the joint location, whereas joint arthroplasty provides the phenotype for intervention for either severe large joint pain or radiographic joint destruction 52. This approach enabled the ascertainment of the most symptomatic OA cases, yet with the caveat that the study‐defined phenotype is heterogeneous and prone to other nonrandom factors that influence indications for joint replacement 53. Third, we did not correct for multiple testing. However, considering a Bonferroni corrected P value of 0.0028, the LDL cholesterol–OA association findings (P = 0.004 from multivariable MR) can be considered borderline significant, and replication in the UK Biobank further strengthened this observation. In addition, prior evidence of a causal role of BMI in OA motivated the analyses of BMI as a positive control. Fourth, it is important to note that our results are likely relevant to hip and knee OA, as they are the most prevalent diagnoses in our cases, and thus may not necessarily be true for OA in other joints, such as the hand. Finally, misclassification of OA outcomes in our samples may have biased our estimates toward the null and could at least partially explain some of the null findings.
In conclusion, our study suggests a causal association between lower LDL cholesterol level and an increased risk of OA. This evidence challenges the current perspectives from epidemiologic studies and indicates that future investigations need to focus on the mechanisms linking lower LDL cholesterol level with OA pathogenesis to potentially identify therapeutic targets, and on investigating how the widespread use of LDL cholesterol–lowering drugs may impact the pathogenesis of OA and related outcomes.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Drs. Hindy and Orho‐Melander had full access to all of the data and take responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Hindy, Åkesson, Melander, Kadam, Orho‐Melander.
Acquisition of data
Hindy, Melander, Aragam, Haas, Nilsson, Kadam, Orho‐Melander.
Analysis and interpretation of data
Hindy, Åkesson, Melander, Nilsson, Kadam, Orho‐Melander.
Supporting information
ACKNOWLEDGMENTS
We thank the participants of the Malmö Diet and Cancer Study and the UK Biobank.
Drs. Hindy and Orho‐Melander's work was supported by the Swedish Research Council. Dr. Orho‐Melander's work was supported by the European Research Council (consolidator grant 649021) and the Region Skåne.
Drs. Kadam and Orho‐Melander contributed equally to this work.
Dr. Åkesson has received consulting fees, speaking fees, and/or honoraria from Amgen, UCB, and Renapharma (less than $10,000 each). Dr. Nilsson has received consulting fees, speaking fees, and/or honoraria from AstraZeneca, Merck, Novo Nordisk, and Getz Pharma (less than $10,000 each). No other disclosures relevant to this article were reported.
References
- 1. Zhuo Q, Yang W, Chen J, Wang Y. Metabolic syndrome meets osteoarthritis. Nat Rev Rheumatol 2012;8:729–37. [DOI] [PubMed] [Google Scholar]
- 2. Tall AR, Yvan‐Charvet L. Cholesterol, inflammation and innate immunity. Nat Rev Immunol 2015;15:104–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kadam UT, Holmberg A, Blagojevic M, Nilsson PM, Akesson K. Risk factors for cardiovascular disease and future osteoarthritis‐related arthroplasty: a population‐based cohort study in men and women from Malmo, Sweden. Scand J Rheumatol 2011;40:478–85. [DOI] [PubMed] [Google Scholar]
- 4. Aspden RM, Scheven BA, Hutchison JD. Osteoarthritis as a systemic disorder including stromal cell differentiation and lipid metabolism. Lancet 2001;357:1118–20. [DOI] [PubMed] [Google Scholar]
- 5. Brandt KD, Doherty M, Lohmander S. Osteoarthritis. New York: Oxford University Press; 2003. [Google Scholar]
- 6. Zhang W, Moskowitz RW, Nuki G, Abramson S, Altman RD, Arden N, et al. OARSI recommendations for the management of hip and knee osteoarthritis, part I: critical appraisal of existing treatment guidelines and systematic review of current research evidence. Osteoarthritis Cartilage 2007;15:981–1000. [DOI] [PubMed] [Google Scholar]
- 7. Thijssen E, van Caam A, van der Kraan PM. Obesity and osteoarthritis, more than just wear and tear: pivotal roles for inflamed adipose tissue and dyslipidaemia in obesity‐induced osteoarthritis. Rheumatology (Oxford) 2015;54:588–600. [DOI] [PubMed] [Google Scholar]
- 8. Dahaghin S, Bierma‐Zeinstra SM, Koes BW, Hazes JM, Pols HA. Do metabolic factors add to the effect of overweight on hand osteoarthritis? The Rotterdam Study. Ann Rheum Dis 2007;66:916–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Conaghan PG, Vanharanta H, Dieppe PA. Is progressive osteoarthritis an atheromatous vascular disease? Ann Rheum Dis 2005;64:1539–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, et al. Multilineage potential of adult human mesenchymal stem cells. Science 1999;284:143–7. [DOI] [PubMed] [Google Scholar]
- 11. Diascro DD Jr, Vogel RL, Johnson TE, Witherup KM, Pitzenberger SM, Rutledge SJ, et al. High fatty acid content in rabbit serum is responsible for the differentiation of osteoblasts into adipocyte‐like cells. J Bone Miner Res 1998;13:96–106. [DOI] [PubMed] [Google Scholar]
- 12. Lippiello L, Walsh T, Fienhold M. The association of lipid abnormalities with tissue pathology in human osteoarthritic articular cartilage. Metabolism 1991;40:571–6. [DOI] [PubMed] [Google Scholar]
- 13. De Munter W, Blom AB, Helsen MM, Walgreen B, van der Kraan PM, Joosten LA, et al. Cholesterol accumulation caused by low density lipoprotein receptor deficiency or a cholesterol‐rich diet results in ectopic bone formation during experimental osteoarthritis. Arthritis Res Ther 2013;15:R178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gierman LM, Kuhnast S, Koudijs A, Pieterman EJ, Kloppenburg M, van Osch GJ, et al. Osteoarthritis development is induced by increased dietary cholesterol and can be inhibited by atorvastatin in APOE*3Leiden.CETP mice: a translational model for atherosclerosis. Ann Rheum Dis 2014;73:921–7. [DOI] [PubMed] [Google Scholar]
- 15. Kadam UT, Jordan K, Croft PR. Clinical comorbidity in patients with osteoarthritis: a case‐control study of general practice consulters in England and Wales. Ann Rheum Dis 2004;63:408–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Singh G, Miller JD, Lee FH, Pettitt D, Russell MW. Prevalence of cardiovascular disease risk factors among US adults with self‐reported osteoarthritis: data from the Third National Health and Nutrition Examination Survey. Am J Manag Care 2002;8 Suppl:S383–91. [PubMed] [Google Scholar]
- 17. Sturmer T, Sun Y, Sauerland S, Zeissig I, Gunther KP, Puhl W, et al. Serum cholesterol and osteoarthritis: the baseline examination of the Ulm Osteoarthritis Study. J Rheumatol 1998;25:1827–32. [PubMed] [Google Scholar]
- 18. Nuesch E, Dieppe P, Reichenbach S, Williams S, Iff S, Juni P. All cause and disease specific mortality in patients with knee or hip osteoarthritis: population based cohort study. BMJ 2011;342:d1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zengini E, Hatzikotoulas K, Tachmazidou I, Steinberg J, Hartwig FP, Southam L, et al. Genome‐wide analyses using UK Biobank data provide insights into the genetic architecture of osteoarthritis. Nat Genet 2018;50:549–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Panoutsopoulou K, Metrustry S, Doherty SA, Laslett LL, Maciewicz RA, Hart DJ, et al. The effect of FTO variation on increased osteoarthritis risk is mediated through body mass index: a Mendelian randomisation study. Ann Rheum Dis 2014;73:2082–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Manjer J, Carlsson S, Elmstahl S, Gullberg B, Janzon L, Lindstrom M, et al. The Malmo Diet and Cancer Study: representativity, cancer incidence and mortality in participants and non‐participants. Eur J Cancer Prev 2001;10:489–99. [DOI] [PubMed] [Google Scholar]
- 22. Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, et al. Genome‐wide genetic data on ~500,000 UK Biobank participants. bioRxiv. 2017.
- 23. Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet 2010;42:937–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. International Consortium for Blood Pressure Genome‐Wide Association Studies , Ehret GB, Munroe PB, Rice KM, Bochud M, Johnson AD, et al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature 2011;478:103–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Newton‐Cheh C, Johnson T, Gateva V, Tobin MD, Bochud M, Coin L, et al. Genome‐wide association study identifies eight loci associated with blood pressure. Nat Genet 2009;41:666–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wain LV, Verwoert GC, O'Reilly PF, Shi G, Johnson T, Johnson AD, et al. Genome‐wide association study identifies six new loci influencing pulse pressure and mean arterial pressure. Nat Genet 2011;43:1005–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010;466:707–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, et al. Discovery and refinement of loci associated with lipid levels. Nat Genet 2013;45:1274–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dupuis J, Langenberg C, Prokopenko I, Saxena R, Soranzo N, Jackson AU, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet 2010;42:105–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ference BA, Robinson JG, Brook RD, Catapano AL, Chapman MJ, Neff DR, et al. Variation in PCSK9 and HMGCR and risk of cardiovascular disease and diabetes. N Engl J Med 2016;375:2144–53. [DOI] [PubMed] [Google Scholar]
- 31. Ference BA, Majeed F, Penumetcha R, Flack JM, Brook RD. Effect of naturally random allocation to lower low‐density lipoprotein cholesterol on the risk of coronary heart disease mediated by polymorphisms in NPC1L1, HMGCR, or both: a 2 × 2 factorial Mendelian randomization study. J Am Coll Cardiol 2015;65:1552–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Burgess S, Thompson SG. Multivariable Mendelian randomization: the use of pleiotropic genetic variants to estimate causal effects. Am J Epidemiol 2015;181:251–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol 2013;37:658–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol 2015;44:512–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol 2016;40:304–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Do R, Willer CJ, Schmidt EM, Sengupta S, Gao C, Peloso GM, et al. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat Genet 2013;45:1345–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Burgess S, Dudbridge F, Thompson SG. Re: “multivariable Mendelian randomization: the use of pleiotropic genetic variants to estimate causal effects” [letter]. Am J Epidemiol 2015;181:290–1. [DOI] [PubMed] [Google Scholar]
- 38. Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 2015;518:197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Manning AK, Hivert MF, Scott RA, Grimsby JL, Bouatia‐Naji N, Chen H, et al. A genome‐wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet 2012;44:659–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yavorska OO, Burgess S. MendelianRandomization: an R package for performing Mendelian randomization analyses using summarized data. Int J Epidemiol 2017;46:1734–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hart DJ, Doyle DV, Spector TD. Association between metabolic factors and knee osteoarthritis in women: the Chingford Study. J Rheumatol 1995;22:1118–23. [PubMed] [Google Scholar]
- 42. Kadam UT, Blagojevic M, Belcher J. Statin use and clinical osteoarthritis in the general population: a longitudinal study. J Gen Intern Med 2013;28:943–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Clockaerts S, Van Osch GJ, Bastiaansen‐Jenniskens YM, Verhaar JA , Van Glabbeek F, Van Meurs JB, et al. Statin use is associated with reduced incidence and progression of knee osteoarthritis in the Rotterdam study. Ann Rheum Dis 2012;71:642–7. [DOI] [PubMed] [Google Scholar]
- 44. Beattie MS, Lane NE, Hung YY, Nevitt MC. Association of statin use and development and progression of hip osteoarthritis in elderly women. J Rheumatol 2005;32:106–10. [PubMed] [Google Scholar]
- 45. Mansi IA, Mortensen EM, Pugh MJ, Wegner M, Frei CR. Incidence of musculoskeletal and neoplastic diseases in patients on statin therapy: results of a retrospective cohort analysis. Am J Med Sci 2013;345:343–8. [DOI] [PubMed] [Google Scholar]
- 46. Riddle DL, Moxley G, Dumenci L. Associations between statin use and changes in pain, function and structural progression: a longitudinal study of persons with knee osteoarthritis. Ann Rheum Dis 2013;72:196–203. [DOI] [PubMed] [Google Scholar]
- 47. Michaelsson K, Lohmander LS, Turkiewicz A, Wolk A, Nilsson P, Englund M. Association between statin use and consultation or surgery for osteoarthritis of the hip or knee: a pooled analysis of four cohort studies. Osteoarthritis Cartilage 2017;25:1804–13. [DOI] [PubMed] [Google Scholar]
- 48. Puenpatom RA, Victor TW. Increased prevalence of metabolic syndrome in individuals with osteoarthritis: an analysis of NHANES III data. Postgrad Med 2009;121:9–20. [DOI] [PubMed] [Google Scholar]
- 49. Engström G, Gerhardsson de Verdier M, Rollof J, Nilsson PM, Lohmander LS. C‐reactive protein, metabolic syndrome and incidence of severe hip and knee osteoarthritis: a population‐based cohort study. Osteoarthritis Cartilage 2009;17:168–73. [DOI] [PubMed] [Google Scholar]
- 50. Schett G, Kleyer A, Perricone C, Sahinbegovic E, Iagnocco A, Zwerina J , et al. Diabetes is an independent predictor for severe osteoarthritis: results from a longitudinal cohort study. Diabetes Care 2013;36:403–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. White J, Swerdlow DI, Preiss D, Fairhurst‐Hunter Z, Keating BJ, Asselbergs FW, et al. Association of lipid fractions with risks for coronary artery disease and diabetes. JAMA Cardiol 2016;1:692–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hernandez‐Vaquero D, Fernandez‐Carreira JM. Relationship between radiological grading and clinical status in knee osteoarthritis: a multicentric study. BMC Musculoskelet Disord 2012;13:194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ibrahim SA, Siminoff LA, Burant CJ, Kwoh CK. Understanding ethnic differences in the utilization of joint replacement for osteoarthritis: the role of patient‐level factors. Med Care 2002;40 Suppl:I44–51. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials