

Abstract
Background
Levodopa‐induced dyskinesias are an often debilitating side effect of levodopa therapy in Parkinson's disease. Although up to 90% of individuals with PD develop this side effect, uniformly effective and well‐tolerated antidyskinetic treatment remains a significant unmet need. The pathognomonic loss of striatal dopamine in PD results in dysregulation and disinhibition of striatal CaV1.3 calcium channels, leading to synaptopathology that appears to be involved in levodopa‐induced dyskinesias. Although there are clinically available drugs that can inhibit CaV1.3 channels, they are not adequately potent and have only partial and transient impact on levodopa‐induced dyskinesias.
Methods
To provide unequivocal target validation, free of pharmacological limitations, we developed a CaV1.3 shRNA to provide high‐potency, target‐selective, mRNA‐level silencing of striatal CaV1.3 channels and examined its ability to impact levodopa‐induced dyskinesias in severely parkinsonian rats.
Results
We demonstrate that vector‐mediated silencing of striatal CaV1.3 expression in severely parkinsonian rats prior to the introduction of levodopa can uniformly and completely prevent induction of levodopa‐induced dyskinesias, and this antidyskinetic benefit persists long term and with high‐dose levodopa. In addition, this approach is capable of ameliorating preexisting severe levodopa‐induced dyskinesias. Importantly, motoric responses to low‐dose levodopa remained intact in the presence of striatal CaV1.3 silencing, indicating preservation of levodopa benefit without dyskinesia liability.
Discussion
The current data provide some of the most profound antidyskinetic benefit reported to date and suggest that genetic silencing of striatal CaV1.3 channels has the potential to transform treatment of individuals with PD by allowing maintenance of motor benefit of levodopa in the absence of the debilitating levodopa‐induced dyskinesia side effect. © 2019 The Authors. Movement Disorders published by Wiley Periodicals, Inc. on behalf of International Parkinson and Movement Disorder Society.
Keywords: adeno‐associated virus (AAV), CaV1.3 channels, dyskinesia, levodopa, striatum
Symptomatic treatment of individuals with Parkinson's disease (PD) includes dopamine (DA) replacement therapies, with the DA precursor, levodopa, being the gold standard. Long‐term levodopa therapy is associated with motor complications including levodopa‐induced dyskinesias (LID) that often negatively impact the quality of life of those afflicted. Although there are varying accounts of the occurrence, LIDs are estimated to occur in roughly 50% of PD patients after approximately 3‐5 years of treatment, with the incidence escalating to approximately 90% after 10‐15 years.1, 2 Maintaining motor benefits of therapy while avoiding this often debilitating side effect remains an unmet clinical need.
Central to the biology of LIDs are changes in synaptic plasticity associated with striatal medium spiny neurons (MSNs), critical targets of convergent cortical glutamate and nigrostriatal DA input. MSNs, which constitute approximately 95% of striatal neurons, express both CaV1.3 and CaV1.2 voltage‐gated L‐type calcium channels. In PD and animal models of PD, striatal DA depletion results in loss of dendritic spines on MSNs,3, 4, 5, 6, 7, 8, 9, 10 an aberrant feature accompanied by secondary loss of glutamate synapses from corticostriatal projections.3, 5, 6, 7, 10, 11 Introduction of levodopa in this environment results in restoration of dendritic spines12 and reestablishment of glutamate input, but in an aberrant pattern of apparent “miswiring.”5, 8, 10, 13
Retraction of dendritic spines on MSNs associated with loss of striatal DA has been linked to dysregulation of intraspinous CaV1.3 channels.3 Blockade of these channels with CaV1.2/1.3 channel antagonists (ie, nimodipine, isradipine) prevents spine retraction despite severe loss of DA in the parkinsonian striatum.9, 14 As such, it has been hypothesized that preventing initial spine loss and associated synaptopathology in PD and models of PD may prevent abnormal rewiring of glutamate inputs and diminish liability for LID despite loss of DA. Indeed, we9 and others14 have shown that CaV1.2/1.3 channel antagonists can prevent induction of LID produced by low‐dose levodopa (6 mg/kg14) and high‐dose levodopa (12.5 mg/kg9); however, this effect is partial and lost over time, and this paradigm is incapable of reversing established LID.14 The transient nature of the antidyskinetic effect of currently available CaV1.3 antagonists is speculated to be because of pharmacologic limitations of these drugs, including lack of specificity and potency for CaV1.3 channels.15, 16
To provide the first unequivocal proof‐of‐principle evidence, devoid of pharmacological limitations, on the ability of CaV1.3 silencing to provide meaningful and lasting functional protection against LID, we developed a recombinant adeno‐associated virus (rAAV)‐mediated short hairpin RNA (shRNA) to provide continuous, high‐potency, and target‐selective mRNA‐level silencing of striatal CaV1.3 channels. We present here functional data demonstrating that delivery of rAAV‐CaV1.3‐shRNA to the DA‐depleted striatum of unilaterally parkinsonian rats prior to the introduction of levodopa provides robust and lasting prevention of LIDs and, to a lesser extent, also can reverse LID in parkinsonian rats with established severe LID behavior.
Methods
Experimental Subjects
All procedures were performed on adult male Sprague‐Dawley (SD) rats (250 g; Envigo RMS Inc., Indianapolis, IN) in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care International guidelines and following approval of the Michigan State University Institutional Animal Care and Use Committee.
Vector Design and Production
Portions of the CaV1.3 gene (Cacna1d) were cloned from rat cDNA and sequenced. shRNA sequences (CaV1.3 [5′‐GAAGAGGCGCGGCCAAGAC‐3′] or scrambled [5′‐CAACAAGATGAAGAGCACC‐3′]) were designed using standard algorithms as described previously17 and compared against available rat genome data to ensure specificity. Particular attention was paid to ensure that the shRNA contained negligible overlap with CaV1.2. Then shRNA was cloned into a rAAV genome under control of the H1 promoter. The same genome contained green fluorescent protein (GFP) under control of the hybrid chicken b‐actin/cytomegalovirus promoter as a transduction marker.17
The rAAV was packaged as described previously18 into AAV9 capsids using triple transfection in HEK293T cells. Viral capsids were purified using an iodixanol step gradient and concentrated using buffer exchange. Virus titers were determined using dot‐blot and normalized to 1 × 1013 vector genomes (vg)/mL.19
Dyskinesia Rating
LIDs refer here to abnormal involuntary behaviors including dystonia, hyperkinesia, and/or stereotypies noted in the presence of levodopa in parkinsonian rats. Injections were administered to allow assessment of LID behaviors for 1 minute every 30 minutes beginning 20 minutes after injection and continued up to 170 or 200 minutes. Rats were randomized and rated by the same blinded investigator throughout the experiment. Each rat was given a total LID severity score for each point assessed based on a rating system developed with a clinical movement disorders specialist (R.K.20) as previously detailed.20, 21
LID Prevention Studies
All stereotaxic surgeries (prevention and reversibility studies) were performed as previously detailed.22 To test the hypothesis that shRNA‐mediated silencing of striatal CaV1.3 channels prior to levodopa would prevent the development and provide sustained amelioration of LIDs, rats received unilateral injections of either rAAV‐CaV1.3‐shRNA or the control rAAV‐Scrambled (Scr)‐shRNA (1.0 × 1013 vg/mL) into 2 dorsolateral sites within the left striatum (AP0.0, ML+3.0, DV‐5.2; and AP+1.6, ML+2.7, DV‐4.9). Vector surgery preceded by 1 week 6‐hydroxydopamine (6‐OHDA) neurotoxin surgery used to induce unilateral parkinsonism (Fig. 1A). Parkinsonism was induced using stereotaxic injection of 6‐OHDA into substantia nigra (SN) and medial forebrain bundle per our usual protocol.10, 23 Vector delivery prior to 6‐OHDA was used to minimize spine loss associated with CaV1.3 dysregulation, which occurs secondary to striatal DA depletion. However, because AAV is retrogradely transported to nigral DA neurons in which CaV1.3 silencing could interfere with 6‐OHDA‐induced cell death,24 timing for these proof‐of‐principle experiments was systematically worked out in pilot feasibility studies. Three weeks following 6‐OHDA (4 weeks postvector surgery), rats began receiving daily levodopa injections given at escalating doses ranging from low (6 mg/kg) to moderate (9 mg/kg) to high (12 mg/kg) to what is referred to here as “extreme” (18 mg/kg); see Figure 1A. Each dose was given daily (Monday through Friday) for 2 weeks with a constant dose of carbidopa (12 mg/kg), a peripheral decarboxylase inhibitor. LID behaviors were rated on days 1, 6, and 10 of each dose.
Figure 1.
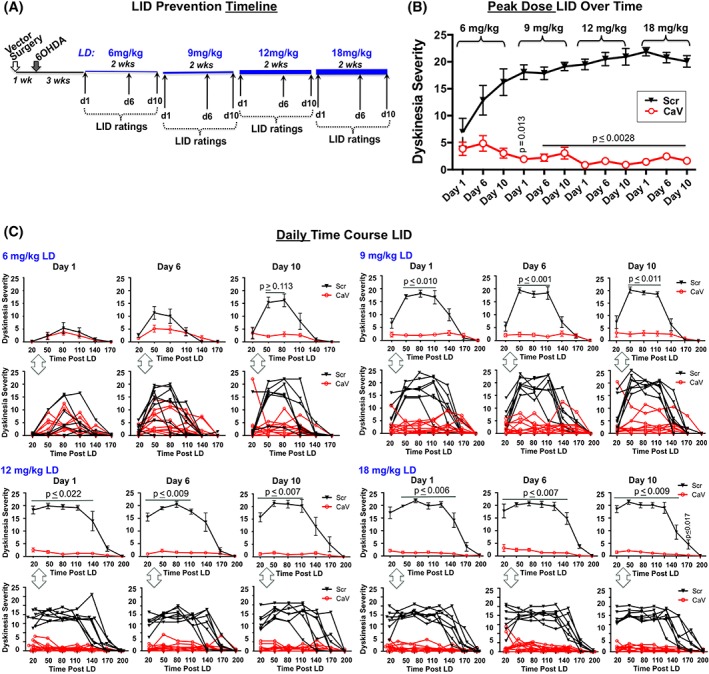
LID prevention study. (A) Treatment timeline. (B) Peak dose LID severity (80 minutes postlevodopa) across time and doses. (C) Daily time course (20‐170/200 minutes postlevodopa). For each dose and day, the top graphs reflect mean ± SEM; bottom graphs show individual subject responses over time. Statistics: Friedman and Kruskal‐Wallis tests with Dunn's multiple‐comparison post hoc as shown in graphs. Scr, rAAV‐Scrambled‐shRNA (n = 7); CaV, rAAV‐CaV1.3‐shRNA (n = 10). [Color figure can be viewed at wileyonlinelibrary.com]
LID Reversibility Studies
To test the hypothesis that in rats with established LID behaviors, ameliorating dysfunctional calcium signaling in an environment of established “miswiring” would lessen LIDs, rats were first rendered unilaterally parkinsonian per referenes 10 and 23. As depicted in Figure 2A, 3 weeks after 6‐OHDA rats began receiving daily high‐dose levodopa (12 mg/kg, Monday through Friday; levodopa:carbidopa 1:1). After 3 weeks of treatment, all rats exhibiting stable, high levels of LIDs were assigned to either a rAAV‐CaV1.3‐shRNA or control rAAV‐Scr‐shRNA group. Groups were assigned to ensure the average peak‐dose LID severity between groups was not different. Vector surgeries were identical to those for the LID prevention study.
Figure 2.
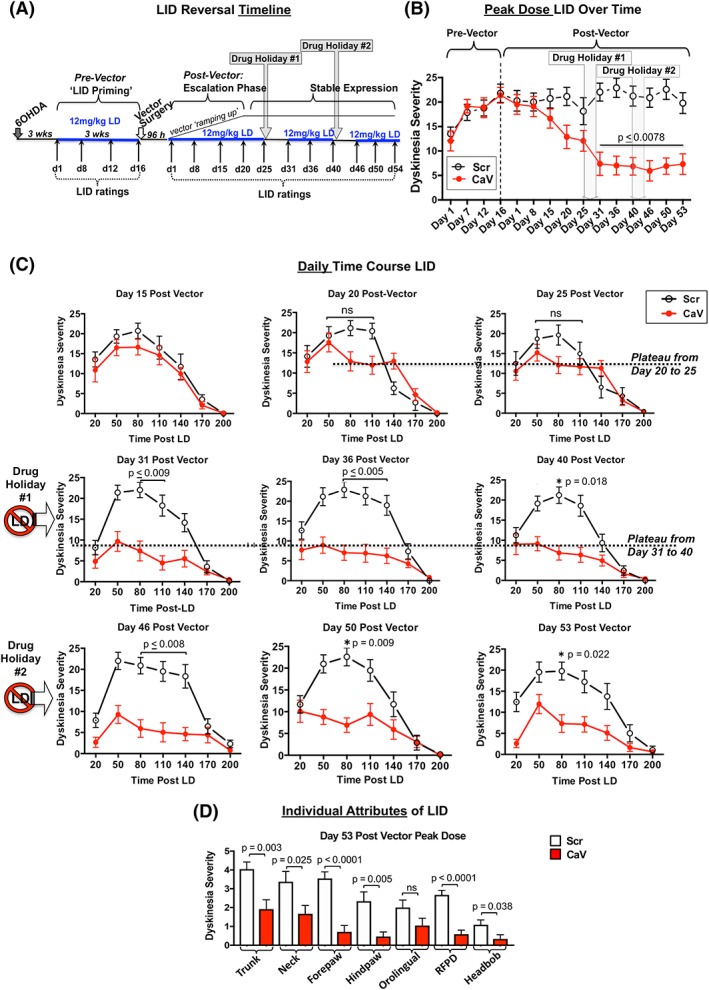
LID reversibility study. (A) Treatment timeline. (B) Peak‐dose LID severity (80 minutes postlevodopa) across time. (C) Daily time course of LID behavior ranging from 20 to 200 minutes postlevodopa on each rating day. (D) Individual attributes of LID. As seen in this depiction of peak LID behaviors on day 53 postvector, the impact of CaV1.3 channel silencing appears to uniformly reduce all aspects of LID and is not selective on any particular attribute. As shown in the timeline, (A) all rats received daily high‐dose (12 mg/kg) levodopa at all times indicated. Data represent the mean ± SEM. Statistics: Kruskal‐Wallis tests with Dunn's multiple‐comparison post hoc as shown in graphs. LD, levodopa; Scr, rAAV‐Scrambled‐shRNA (n = 12); CaV, rAAV‐CaV1.3‐shRNA (n = 11). [Color figure can be viewed at wileyonlinelibrary.com]
Figure 3.
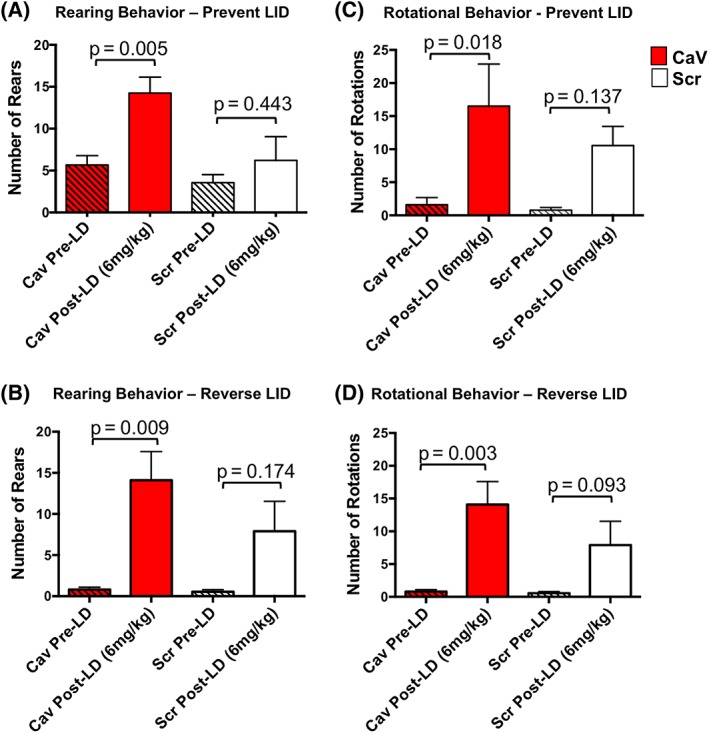
Motor response to low‐dose (6 mg/kg) levodopa. Data represent total number of rears (A, B) or rotations (C, D) over a 5‐minute test period, prior to or beginning 50 minutes after levodopa. Behavioral responses were examined in all subjects in the LID prevention study (‘prevent LID’; A, C) and the LID reversibility study (‘reverse LID’; B, D). Statistics: 2‐way ANOVA with post hoc Sidak's multiple‐comparisons test as shown in the graphs; additional comparisons are provided in the Results section. Pre‐LD, prelevodopa; post‐LD, postlevodopa; Scr, rAAV‐Scrambled‐shRNA; CaV, rAAV‐CaV1.3‐shRNA. [Color figure can be viewed at wileyonlinelibrary.com]
Cylinder Motor Test
A modified cylinder task was used to examine motor response in the absence of drug (prelevodopa [pre‐LD]) and in response to low‐dose levodopa (6 mg/kg; 12 mg/kg benserazide) in all rats in both prevention and reversibility studies. Rats were placed in a clear plexiglass cylinder (16 cm in diameter, 25 cm in height) and videotaped for 5 minutes prior to and again 50 mins postlevodopa injection. The number of 360° rotations and rears were quantified by a blinded investigator.
Euthanasia
One day after the final LID behavioral rating, rats were administered a final dose of levodopa (12 mg/kg), LID‐rated, and videotaped 50 minutes postinjection and subsequently euthanized per our usual protocol.10, 25
TH, GFP, and NeuN Immunohistochemistry
All postmortem analyses were done by blinded investigators. To examine nigral lesion status, region of vector transduction, and lack of striatal neuron toxicity, individual series (1 in 6) of sections (40‐μm thickness) were processed for tyrosine hydroxylase (TH; Millipore‐AB152b rabbit anti‐TH, 1:4000), GFP (Millipore‐Ab290 rabbit anti‐GFP, 1:20,000), or neuronal nuclei protein (NeuN; pan‐neuronal marker of mature neurons; Millipore‐Mab377, 1:1000) immunochemistry (IHC) per our previously reported methods (TH,23 GFP,18 NeuN26).
The degree of nigral DA neuron depletion in each animal was confirmed by total enumeration of TH‐positive neurons.27 Nigral TH neuron loss of ≥95% in the lesioned hemisphere compared with the unlesioned hemisphere was used as a final inclusion criterion. This magnitude of SN DA neuron depletion is required in this model to produce reliable LIDs.9, 21, 23, 28, 29
The striatal volume of vector transduction in GFP‐immunostained sections was determined with the Cavalieri estimator using StereoInvestigator software (MicroBrightField, Williston, VT). Briefly, contours were defined for both the striatum and striatal region of GFP immunoreactivity. The outlines of each structure were traced at 1× in approximately 6 coronal sections along the entire rostral‐caudal extent of the striatum.
Prior to undertaking our functional studies, a small cohort of rats received either rAAV‐CaV1.3‐shRNA (n = 4) or rAAV‐Scr‐shRNA (n = 4, as described above) to ensure the absence of nonspecific shRNA or GFP‐associated toxicity.30, 31 The number of NeuN+ cells was quantified in the GFP+ region of striatum employing stereological techniques according to our previously reported method.26
CaV Knockdown Analyses
To determine degree and specificity of knockdown of the CaV1.3 transcript in the presence of our vectors, we employed tissue from animals in the LID prevention study. The relative abundance of CaV1.3 and CaV1.2 transcripts was quantified in the dorsolateral striatum using commercially available RNAscope in situ hybridization (ISH) (ACD, Newark, CA) probes generated against CaV1.3 (cacna1d, NM_017298.1, nucleotides 5401‐6474) and CaV1.2 (Cacna1c, NM_012517.2, nucleotides 5183‐6142). To estimate degree of CaV1.3 protein knockdown, dual‐label CaV1.3 (Santa Cruz SC‐515679, L‐type‐Ca2+CPα1D [CACNA1D] mouse anti‐cav, 1:600) plus GFP IHC, and confocal microscopy was employed in a random subset of rats in which transcript was measured (n = 4 rAAV‐CaV1.3‐shRNA, n = 4 rAAV‐Scr‐shRNA).
For RNAscope ISH, 3 images in the GFP+ dorsolateral vector‐injected or noninjected striatum were acquired on an OlympusBX51 light microscope at 20×. For dual‐label IHC, 2 images in the vector‐injected or noninjected striatum in the same general striatal regions as the ISH micrographs were acquired at 20× on a Nikon ElipseTi confocal using NIS Elements. The CaV immunoreactive ISH signal, which is sensitive enough to detect single transcripts, or CaV1.3 immunostaining analyses were performed using ImageJ software (NIH) using the threshold function. All microscope and camera settings were identical for all images within a given transcript or antibody. Data are represented as mean area or percent total area above the threshold. CaV1.3 transcript silencing is expressed as relative level of transcript in the rAAV‐CaV‐shRNA versus rAAV‐Scr‐shRNA striatum.
Statistical Analyses
All LID behavioral data were analyzed with nonparametric statistics (Freidman [FM] or Kruskal‐Wallis [H] with Dunn's multiple‐comparisons test). Percent nigral lesion comparisons was analyzed with a nonparametric Mann‐Whitney test. CaV1.3 mRNA expression in the rAAV‐CaV‐shRNA versus rAAV‐Scr‐shRNA striatum was compared using an unpaired, 2‐tailed t test. Statistical evaluation of CaV1.2 mRNA expression in the injected and noninjected striata of rAAV‐CaV‐shRNA and rAAV‐Scr‐shRNA was done using 1‐way ANOVA with Tukey's multiple‐comparisons post hoc test. Nonparametric Spearman correlation test was used for correlation of LID severity with CaV1.3 levels. We10 and others32 have previously reported that in the outbred SD rat strain there is a small subset (15%‐20%) of subjects that despite having an equal degree of striatal DA depletion and levodopa treatment remains resistant to LID. We characterize these rats as LID negative and having a peak LID severity of ≤3.0. In the LID prevention study we identified 2 statistical outliers in the rAAV‐Scr‐shRNA group, verified with the ROUT method (robust regression and outlier removal test; 3‐step test automated within Prism; suggested Q coefficient detects outliers with false discovery rate <1%) and having a cumulative mean (across all 4 doses of levodopa) of 0.5 ± 0.3 and 2.5 ± 0.5; these 2 rats were omitted from final LID analyses. In addition, 1 rat in the AAV‐CaV1.3‐shRNA group in the LID prevention study was not adequately lesioned (ie, 21.6%) and was excluded from final behavioral analyses. Final subject numbers for the LID prevention study were: rAAV‐CaV1.3‐shRNA, n = 10; rAAV‐Scr‐shRNA, n = 7; for the LID reversibility study were: rAAV‐CaV1.3‐shRNA, n = 11; rAAV‐Scr‐shRNA, n = 12. All analyses were performed with Prism GraphPad.v7 for MacOSX.
Results
Striatal rAAV‐CaV1.3‐shRNA Potently Prevents Induction of LID
Levodopa was administered using a dose‐escalation paradigm beginning 4 weeks postvector (3 weeks post‐6OHDA; Fig. 1A). As demonstrated in Figure 1B, rats receiving the control rAAV‐Scr‐shRNA displayed a typical escalation in LID severity over time that remained stable and severe with increasing doses of levodopa (Scr [black line]: day 1, 6 mg/kg versus day 6, 10‐12 and 10‐18 mg/kg; FM, 39.63; P = 0.0002; Dunn's post hoc P ≤ 0.0326; Fig. 1B). In contrast, rats treated with rAAV‐CaV1.3‐shRNA showed significant suppression of the development of LIDs compared with rAAV‐Scr‐shRNA rats (red lines, CaV; Fig. 1B,C). This notable prevention of LID escalation continued to persist as a near‐complete absence of dyskinetic behavior in the rAAV‐CaV‐shRNA rats, which showed a significant difference from control rAAV‐Scr‐shRNA rats beginning on day 1 of 9 mg/kg levodopa (H, 159.6; P < 0.0001; Dunn's post hoc: Scr vs CaV, 50 minutes; P = 0.0027; Scr vs CaV, 80 minutes; P = 0.0010; Scr vs CaV, 110 minutes; P = 0.0109). This potent antidyskinetic effect was noted in all rAAV‐CaV‐shRNA parkinsonian rats despite continuing elevation in dose of levodopa over 2 months (Fig. 1C, lower graphs illustrate uniform individual subject responses; detailed statistics provided for each day/dose in figure legends).
Quantification of TH‐positive SN DA neuron loss revealed equivalent ≥95% depletion in both CaV and Scr groups (CaV, 98.6% ± 0.5%; Scr, 98.3% ± 0.5%; TH‐positive cell loss compared with intact side; mean ± SEM; P = 0.7642 Mann‐Whitney U test).
Striatal rAAV‐CaV1.3‐shRNA Partially Reverses Established, Severe LID
To examine whether striatal CaV1.3 silencing would impact expression of established LIDs, parkinsonian rats received chronic high‐dose levodopa (12 mg/kg) for 3 weeks prior to receiving an intrastriatal injection of either rAAV‐CaV1.3‐shRNA or control rAAV‐Scr‐shRNA vector (Fig. 2A). Groups were balanced to ensure that the average peak‐dose LID severity between the groups did not differ (Mann‐Whitney, 2‐tailed, P = 0.6631; U = 53.50; mean rAAV‐CaV1.3, 22.8 ± 0.7; rAAV‐Scr, 23.2 ± 1.3; median rAAV‐CaV1.3, 23; rAAV‐Scr, 24).
As depicted in Figure 2B,C, rAAV‐mediated CaV1.3 silencing in parkinsonian rats with established high‐level LIDs can result in progressive and significant amelioration of existing LIDs. Specifically, the ability of CaV1.3 silencing to reverse peak‐dose LIDs showed a nonsignificant trend of decreasing LID severity in the rAAV‐CaV1.3‐shRNA group (Fig. 2B,C) compared with the rAAV‐Scr‐shRNA group beginning on day 20 postvector. This trend continued to be nonsignificant through day 25. As such, we examined whether a short, 5‐day withdrawal of levodopa (ie, “drug holiday”), once popular for improving patient response to levodopa including potentially decreasing LIDs,33, 34 would provide an environment in which vector‐mediated reduction in aberrant calcium signaling might translate into reduction of LIDs.
As presented in Figure 2B,C on day 31 postvector, the first day immediately following the 1‐week levodopa‐free drug holiday, LID severity in rAAV‐CaV1.3‐shRNA rats was significantly reduced compared with that seen in the control rAAV‐Scr‐shRNA group that predictably maintained a sustained high level of LID severity from the prevector through the entire postvector time frame (day 31, F = 135.7, P < 0.0001; post hoc Dunn's test P = 0.0009; Fig. 2B; F = 97.2, P < 0.0001; post hoc Dunn's test P ≤ 0.0094; Fig. 2C). The benefit from this first drug holiday for rAAV‐CaV1.3 rats was sustained over the next 2 weeks (Fig. 2B,C, day 31 through day 40); however, there was no further diminution of LIDs with continued daily high‐dose levodopa. Given this plateau of response together with the apparent benefit of the first drug holiday, we employed a second 1‐week drug holiday to determine whether further enhancement of LID reversal was possible. However, there was no further dampening of LID severity for the rAAV‐CaV1.3 vector group after a second 1‐week drug holiday when levodopa was maintained at this high dose of 12 mg/kg (day 40 vs day 46, P > 0.999).
Quantification of TH‐positive SN neuron loss in the lesioned hemisphere of all rats revealed nearly identical levels of unilateral lesioning between CaV and Scr groups (CaV, 99.1% ± 0.3%; Scr, 98.4% ± 0.6%; cell loss compared with the intact side, P = 0.9394 Mann‐Whitney).
Striatal CaV1.3 Silencing Does Not Interfere With Levodopa Motor Response
Quantification of exploratory rearing and contralateral rotational behavior using a modified cylinder test revealed no significant difference in either behavior under baseline drug‐free (pre‐LD) conditions (2‐way ANOVA with post hoc Sidak's multiple‐comparisons test; rearing, P = 0.8503 for LID prevention, P = 0.9999 for LID reversibility; rotations, P = 0.9984 for LID prevention, P = 0.9999 for LID reversibility). However, following low‐dose levodopa (6 mg/kg), rAAV‐CaV1.3‐shRNA‐ but not control rAAV‐Scr‐shRNA‐treated rats showed a significant increase in both exploratory rearing and rotational behavior compared with baseline levels (Fig. 3 contains detailed statistics), suggesting that knockdown of CaV1.3 channels in the DA‐depleted striatum may enhance motor response to levodopa. Additional evidence demonstrating that motor effects of levodopa are readily apparent and not impaired by gene‐level CaV1.3 silencing can be seen in the accompanying videos (Supplemental Fig. 1).
mRNA Knockdown, Specificity of Knockdown, and Transduction Spread
ISH analyses using probes generated against CaV1.3 (Cacna1d) and CaV1.2 (Cacna1c) mRNA revealed that our rAAV‐CaV1.3‐shRNA vector resulted in an average 84.77% reduction in CaV1.3 mRNA in the GFP‐transduced region of the striatum compared with that seen with rAAV‐Scr‐shRNA (Fig. 4A,C). A similar degree of CaV1.3 protein knockdown was found (87.3%; Fig. 4Cii). The range of mRNA silencing was 43.6% to 99.1% (Fig. 4D). However, there was no significant correlation of degree of CaV1.3 silencing with final LID severity scores (Spearman correlation, r = 0.0886, 2‐tailed P = 0.4116), suggesting that even partial silencing of overactive CaV1.3 channels in the parkinsonian striatum is capable of completely preventing LID induction. Importantly, in contrast to an 84.77% knockdown of CaV1.3 mRNA, we detected no significant change in CaV1.2 mRNA (Fig. 4Ciii), demonstrating the specificity of our genetic approach.
Figure 4.
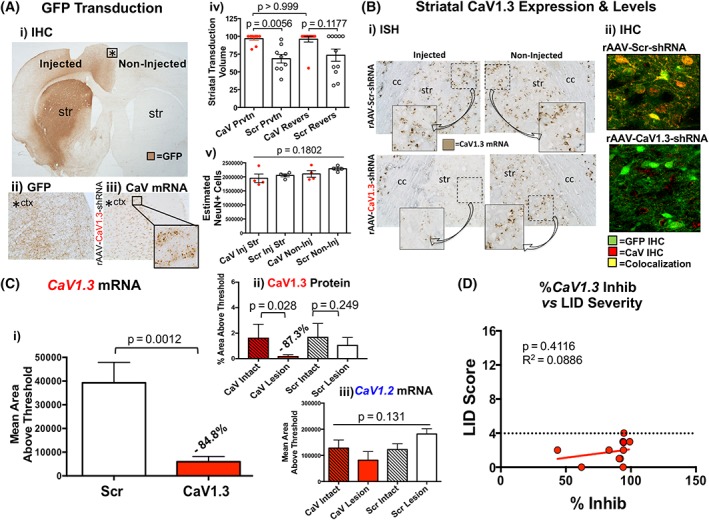
CaV silencing and vector distribution. (Ai) Representative section with GFP immunohistochemistry (IHC) demonstrating the striatal targeting and spread of vector. (Aii) Higher‐magnification image of that seen in Ai showing that the GFP that is expressed in the cortex is expressed exclusively in neuritic fibers. (Aiii) CaV1.3 ISH in the same region as Aii, demonstrating that despite neuritic GFP expression in this region in a subject injected with rAAV‐CaV1.3‐shRNA, there is no impact on cellular CaV1.3 mRNA. (Aiv) Striatal transduction volume. (Av) Estimate of NeuN‐positive cells within the striatum of vector‐ and noninjected striatum. Statistics: 1‐way ANOVA with post hoc Tukey's test. (B) CaV1.3 mRNA expression and protein levels. (Bi) RNAscope in situ hybridization (ISH) showing relative levels of striatal CaV1.3 mRNA expression. (Bii) Dual‐label immunohistochemistry (IHC) for GFP and CaV1.3. (C) Densitometric analysis of CaV1.3 ISH and IHC. (Ci) CaV1.3 ISH, (Cii) CaV1.3 IHC, and (Ciii) CaV1.2 ISH using ImageJ software in the region of the striatum stained positive for GFP IHC in rAAV‐CaV1.3‐shRNA rats compared with rAAV‐Scr‐shRNA rats; (i) CaV1.3: unpaired t test; (ii) Mann‐Whitney U; (iii) CaV1.2: 1‐way ANOVA. (D) Nonparametric Spearman correlation analysis of LID severity score for day 10 18 mg/kg versus % inhibition of CaV1.3 mRNA. The dashed line indicates the maximal LID observed in the rAAV‐CaV1.3‐shRNA rats shown in this graph. Str, striatum. [Color figure can be viewed at wileyonlinelibrary.com]
There was no difference in the volume of striatal transduction in rAAV‐CaV1.3‐shRNA rats between the LID prevention and LID reversibility studies, nor between these 2 studies in rats injected with rAAV‐Scr‐shRNA (Kruskal‐Wallis, post hoc Dunn's multiple comparisons: P > 0.999; Fig. 4Aii). Although not of functional significance, there was less volume of the striatum transduced from the inert control rAAV‐Scr‐shRNA vector compared with the rAAV‐CaV1.3‐shRNA in rats in the LID prevention study (P = 0.0056; Fig. 4Aii). We found no evidence of toxicity related to our vectors, as demonstrated by equal numbers of NeuN‐positive cells in intact versus vector‐injected striatum regardless of the vector (ANOVA P = 0.182, F = 1.920; Fig. 4Aiii).
As depicted in Figure 4A, GFP expression was prominent in the target region of the striatum, but could also be seen as a result of retrograde transduction to striatal input areas including various cortical regions. In some but not all animals sparse and highly variable patterns of GFP+ fibers were also noted in the globus pallidus, medial and/or lateral geniculate nuclei, and thalamic and/or hypothalamic regions. Despite the variable presence of extrastriatal GFP‐immunoreactive fibers, little GFP was observed in cell bodies (eg, Fig. 4Aii). Visual examination of CaV1.3 ISH in these extrastriatal regions confirmed that despite some transduction, there was no apparent reduction in cellular CaV1.3 mRNA levels (Fig. 4Aiii). Our findings are in agreement with the relatively low retrograde transduction efficacy seen with wild‐type AAV capsids.35, 36 Although involvement of CaV1.3 silencing in extrastriatal regions cannot definitively be ruled out as a contributing factor to the LID prevention/amelioration in the current studies, the highly variable nature of its expression in such regions, in the face of uniform LID reduction suggests that extrastriatal CaV1.3 silencing is not the principal mechanism of LID suppression.
Discussion
The current genetic‐based studies, developed to provide high‐potency striatal CaV1.3 channel silencing, were derived from a set of exploratory preclinical studies aimed at demonstrating the novel application of common US Food and Drug Administration‐approved antihypertensive dihydropyridine (DHP) drugs for a new therapeutic application in PD, specifically prevention and/or reversal of LIDs.9, 14 Indeed, although initial studies provided evidence that pharmacological antagonism of CaV1.3 channels could dampen LID in parkinsonian rats, the efficacy of these DHP drugs (ie, isradipine and nimodipine) was partial and transient.
Supported by existing data, which includes evidence that CaV1.3 channels are incompletely inhibited even by high concentrations of current DHP drugs,15, 37 we hypothesized that if other methods to block CaV1.3 channels were available, this target would provide an optimal anti‐LID response. The current study has confirmed this to be true. Using the approach of continuous, high‐potency, target‐specific genetic silencing of striatal CaV1.3 calcium channels with rAAV‐CaV1.3‐shRNA, we have demonstrated that mRNA‐level silencing can provide potent, uniform, and long‐term (>2 months) prevention of LIDs, even with extreme doses of daily levodopa. In addition, this approach was capable of reversing preexisting severe LIDs, which was not possible with pharmacological CaV silencing.14 Importantly, motor benefit from levodopa was maintained with the knockdown of CaV1.3. We contend that these findings provide some of the strongest preclinical data to date demonstrating the amelioration of LIDs without compromise of motor benefit.
LID Reversibility and Clinical Relevance
It is perhaps most compelling, and surprising, based on the presumed mechanism of LID prevention being the prevention of PD‐associated spine changes, that the approach of CaV1.3 silencing employed in these studies could significantly reverse severe LID behavior in parkinsonian rats. Although we observed that this method was capable of significant reversal of severe LID behavior associated with high‐dose levodopa, the effect was nonetheless incomplete. In addition, our experimental design in these proof‐of‐principle studies included a short‐term withdrawal of levodopa that coincided with a significant reduction of LIDs in rAAV‐CaV1.3‐shRNA rats when high‐dose (12 mg/kg) treatment was reintroduced, which contrasted with maintenance of severe LIDs in rAAV‐Scr‐shRNA control rats. It is unclear whether levodopa withdrawal at this early point after vector surgery (ie, days 26‐30 postvector) was necessary for this significant amelioration of LIDs. Because there was no additional benefit with a second drug withdrawal, it could be suggested that short‐term drug withdrawal in the presence of CaV1.3 silencing may not enhance or be necessary for LID amelioration. The mechanism(s) by which reduction in CaV1.3 expression can significantly dampen existing severe LIDs, whether or not a short drug withdrawal is indicated, remains to be determined.
Although beyond the scope of this report to detail such potential mechanisms, it is clear that there are adaptations in MSNs unrelated to dendritic spine density that may impact LID in the presence of CaV1.3 silencing. Specifically, these channels are linked directly to signaling cascades that not only impact, for example, long‐term alterations in synaptic strength and short‐term dendritic excitability, but also impact synaptic function through selective signaling to the nucleus altering transcriptional activity (eg, references 3, 38, 39, 40). The loss of DA tone (specifically D2 receptor tone) after DA depletion disinhibits CaV1.3 channels, leading to structural (eg, spine retraction, synaptic pruning) and functional adaptations.3 Accordingly, it is reasonable to suggest that silencing dysfunctional CaV1.3 channel activity in the parkinsonian striatum may ameliorate LIDs through functional adaptations distinct from altered dendritic spine density per se.
Despite the current studies demonstrating that the antidyskinetic potency of CaV1.3 channel silencing is most robust in a prevention paradigm, the ability to reverse LIDs holds more immediate clinical relevance. Specifically, it is unlikely that any gene therapy approach prior to induction of LIDs would be considered despite the current statistics of its occurrence in up to 90% of patients over the lifetime of the disease.1, 2 Accordingly, we posit that future studies are warranted to extend an understanding of the capacity, limitations, and mechanism(s) of striatal CaV1.3 silencing to reverse a behavioral phenomenon that is often considered steadfast. Such studies should be directed not only at understanding mechanisms of action but also the necessity of drug withdrawal and the relationship of levodopa dose and/or LID severity with the ability to maximally reverse this aberrant behavior. Indeed, it could be anticipated that if gene therapy were undertaken when LID severity and/or levodopa dose were low (eg, early in the disease), complete reversal and stable suppression of LIDs could be achieved. This idea is supported by our LID prevention studies demonstrating that once expression of the CaV1.3 channel and LIDs are maximally suppressed, the underlying mechanism(s) allowing for this near‐complete amelioration is maintained despite the escalation of levodopa doses to very high levels.
Beyond LID Prevention and Future Directions
Although we interpret our motor behavior findings with due prudence, they appear to suggest that knockdown of CaV1.3 channels in the DA‐depleted striatum does not impede and may benefit motor response to levodopa. Indeed, when a “subtherapeutic” dose of levodopa was administered to our severely parkinsonian rats, only those with CaV1.3 silencing showed a significant motor response. The undeniable maintenance of the motor effect of levodopa is clearly evident in the videos of the rAAV‐CaV1.3‐shRNA‐expressing rats (Supplemental Fig. 1). Although our current findings in the rat model of PD are highly encouraging, the approach of high potency, target‐selective silencing of CaV1.3 channels on LIDs and motor response to DA agonists needs to be validated in other models of PD including nonhuman primates.
A common debate about preclinical models of LID is that because rats require significant DA depletion and because in rodent and NHP models LIDs develop more rapidly than in patients, these models do not reflect PD. However, it is often underappreciated that regardless of species, near‐complete loss of striatal DA innervation is generally required for LIDs to manifest (eg, references 10, 32, 41, 42, 43). Indeed, as reported by Kordower and colleagues,44 near‐complete loss of striatal DA innervation occurs during the first 3‐5 years following a PD diagnosis, a time frame that correlates well with onset of LIDs in patients.1, 2 Future studies aimed at understanding the mechanisms of this therapy, especially in the reversal paradigm, and whether the direct versus indirect pathway of MSN silencing has similar efficacy, as well as its impact on neurophysiological and molecular indices of LIDs, are warranted to allow a more complete understanding of the safety and efficacy of this approach in PD.
Summary
Gene therapy‐mediated silencing of striatal CaV1.3 expression provides some of the most profound antidyskinetic benefit reported to date. The spatial control achieved with a gene therapy approach importantly circumvents potential off‐target side effects, whose relevance to DHP drugs could include cardiovascular, learning and memory, and neuroendocrine effects.45, 46, 47, 48 If the current findings can be translated into a clinical application with a similar magnitude, this would provide a much‐needed breakthrough in the treatment of individuals with PD and has the potential to allow the most powerful antiparkinsonian therapy identified (ie, levodopa) to work unabated through the duration of the disease.
Author contributions
Designed research: K.S.C., F.P.M., T.J.C., C.E.S.; acquisition and analysis of data: K.S.C., J.A.S., T.J.C., I.M.S., N.J.C., N.M.M., C.E.S., F.P.M.; manuscript preparation: principally K.S.C., F.P.M., and T.J.C., with significant contributions by all authors.
Financial Disclosures (preceding 12 months)
K.S.C.: NINDS: NS090107, NS098079, NS110398; Michael J. Fox Foundation Target Advancement Program grant; NIH grant reviewer honorarium.
J.A.S.: NINDS: NS090107; Michael J. Fox Foundation Target Advancement Program grant (KSC PI).
N.J.C.: none.
I.M.S.: Parkinson Foundation PDF‐FBS‐1634; St. Mary's Foundation
N.M.M.: graduate stipend.
C.E.S.: NINDS: NS090107, NS098079; grant reviewer honorarium from Michael J. Fox Foundation, NIH, Weston Brain Institute.
T.J.C.: NINDS: NS110398, NS094460.
F.P.M.: NINDS: NS090107, NS098079, NS110398; grant reviewer honorarium from Michael J. Fox Foundation, NIH, Parkinson Foundation.
Supporting information
SUPPLEMENT (VIDEO) FIG. 1 In the first representative parkinsonian rat, which received the rAAV‐Scr‐shRNA vector, 50 minutes after a 12‐mg/kg dose of levodopa, there is an abundance of tight rotational behavior intermixed with LID behavioral profiles including orolingual behaviors (ie, licking and chewing, tongue protrusion), severe hindlimb extension/dystonia, forelimb dystonia and hyperkinetic movements, and severe trunk and neck dystonia. In contrast, as shown in 2 representative rats that had received the rAAV‐CaV1.3‐shRNA vector, at this same dose and a time 50 minutes postlevodopa, there is no evidence of the prototypical LID behaviors readily displayed in parkinsonian rats treated with our control vector. In the first rAAV‐CaV1.3‐shRNA rat, this parkinsonian subject displays motor activation that includes continuous sniffing and exploring, left and right forepaw tapping, digging at the litter, and 1 incident of a wet‐dog shake. It is evident that this rat has a unilateral nigral lesion, in that it walks only clockwise around the perimeter of the cage, but does not display typical tight rotational behavior seen in the rAAV‐Scr‐shRNA rats. The second rAAV‐CaV1.3‐shRNA rat shows sniffing and exploring, mild tapping and pushing at the litter, and again walking only clockwise in the cage. These motor activation behaviors start at approximately 20 minutes and continue until approximately 140‐170 minutes after the levodopa injection, a time course similar to that seen for LID behaviors.
note: As presented in the Methods section, in the outbred SD rat strain there is a small subset (15%‐20%) of these rats that despite having an equal degree of striatal DA depletion and levodopa treatment remain resistant to LID expression. Their behavior is very different from the LID‐negative rAAV‐CaV1.3‐shRNA rats that show what could be suggested to be enhanced motor efficacy to levodopa. Specifically, the 15%‐20% of control (non‐vector injected) rats that remain LID negative generally show mild activation including sniffing and exploring, most likely in response to the injection per se, which lasts approximately 5‐10 minutes, after which they fall asleep in the corner of the cage for the remainder of the rating period.
Acknowledgments
We acknowledge the excellent technical assistance of Nathan Kuhn, BS, Brian F. Daley, BS, and Christopher J. Kemp, MS. We also acknowledge the excellent videographic efforts of Madison R. Collier, BS. We dedicate this article to D. Secrest, who has just begun her struggles with PD.
Relevant conflicts of interest/financial disclosures: The authors have no conflicts to report.
Funding agencies: This study was supported by the National Institute of Neurological Disorders and Stroke NS090107 (to K.S.C., F.P.M., C.E.S.), NS098079 (to F.P.M., K.S.C.), and NS110398 (K.S.C., F.P.M., T.J.C.), and the Parkinson Disease Foundation International Research Grants Program, now the Parkinson Foundation (to K.S.C., F.P.M.).
References
- 1. Huot P, Johnston TH, Koprich JB, Fox SH, Brotchie JM. The pharmacology of L‐DOPA‐induced dyskinesia in Parkinson's disease. Pharmacol Rev 2013;65(1):171–222. [DOI] [PubMed] [Google Scholar]
- 2. Hauser RA, Pahwa R, Tanner CM, et al. ADS‐5102 (Amantadine) Extended‐Release Capsules for Levodopa‐Induced Dyskinesia in Parkinson's Disease (EASE LID 2 Study): Interim Results of an Open‐Label Safety Study. J Parkinsons Dis 2017;7(3):511–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Day M, Wang Z, Ding J, et al. Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nat Neurosci 2006;9(2):251–259. [DOI] [PubMed] [Google Scholar]
- 4. Fieblinger T, Cenci MA. Zooming in on the small: the plasticity of striatal dendritic spines in L‐DOPA‐induced dyskinesia. Mov Disord 2015;30(4):484–493. [DOI] [PubMed] [Google Scholar]
- 5. Fieblinger T, Graves SM, Sebel LE, et al. Cell type‐specific plasticity of striatal projection neurons in parkinsonism and L‐DOPA‐induced dyskinesia. Nat Commun 2014;5:5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ingham CA, Hood SH, Arbuthnott GW. Spine density on neostriatal neurones changes with 6‐hydroxydopamine lesions and with age. Brain Res 1989;503(2):334–338. [DOI] [PubMed] [Google Scholar]
- 7. McNeill TH, Brown SA, Rafols JA, Shoulson I. Atrophy of medium spiny I striatal dendrites in advanced Parkinson's disease. Brain Res 1988;455(1):148–152. [DOI] [PubMed] [Google Scholar]
- 8. Nishijima H, Suzuki S, Kon T, et al. Morphologic changes of dendritic spines of striatal neurons in the levodopa‐induced dyskinesia model. Mov disord 2014;29(3):336–343. [DOI] [PubMed] [Google Scholar]
- 9. Soderstrom KE, O'Malley JA, Levine ND, Sortwell CE, Collier TJ, Steece‐Collier K. Impact of dendritic spine preservation in medium spiny neurons on dopamine graft efficacy and the expression of dyskinesias in parkinsonian rats. Eur J Neurosci 2010;31(3):478–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang Y, Meredith GE, Mendoza‐Elias N, Rademacher DJ, Tseng KY, Steece‐Collier K. Aberrant restoration of spines and their synapses in L‐DOPA‐induced dyskinesia: involvement of corticostriatal but not thalamostriatal synapses. J Neurosci 2013;33(28):11655–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ingham CA, Hood SH, Taggart P, Arbuthnott GW. Plasticity of synapses in the rat neostriatum after unilateral lesion of the nigrostriatal dopaminergic pathway. J Neurosci 1998;18(12):4732–4743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bastide MF, Meissner WG, Picconi B, et al. Pathophysiology of L‐dopa‐induced motor and non‐motor complications in Parkinson's disease. Prog Neurobiol 2015;132:96–168. [DOI] [PubMed] [Google Scholar]
- 13. Suarez LM, Solis O, Carames JM, et al. L‐DOPA treatment selectively restores spine density in dopamine receptor D2‐expressing projection neurons in dyskinetic mice. Biol Psychiatry 2014;75(9):711–722. [DOI] [PubMed] [Google Scholar]
- 14. Schuster S, Doudnikoff E, Rylander D, et al. Antagonizing L‐type Ca2+ channel reduces development of abnormal involuntary movement in the rat model of L‐3,4‐dihydroxyphenylalanine‐induced dyskinesia. Biol psychiatry 2009;65(6):518–526. [DOI] [PubMed] [Google Scholar]
- 15. Kang S, Cooper G, Dunne SF, Luan CH, Surmeier DJ, Silverman RB. Structure‐activity relationship of N,N'‐disubstituted pyrimidinetriones as Ca(V)1.3 calcium channel‐selective antagonists for Parkinson's disease. J Med Chem 2013;56(11):4786–4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xu W, Lipscombe D. Neuronal Ca(V)1.3alpha(1) L‐type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J Neurosci 2001;21(16):5944–5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Benskey MJ, Sellnow RC, Sandoval IM, Sortwell CE, Lipton JW, Manfredsson FP. Silencing Alpha Synuclein in Mature Nigral Neurons Results in Rapid Neuroinflammation and Subsequent Toxicity. Front Mol Neurosci 2018;11:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Benskey MJ, Sandoval IM, Manfredsson FP. Continuous Collection of Adeno‐Associated Virus from Producer Cell Medium Significantly Increases Total Viral Yield. Hum Gene Ther Methods 2016;27(1):32–45. [DOI] [PubMed] [Google Scholar]
- 19. Benskey MJ, Manfredsson FP. Intraparenchymal Stereotaxic Delivery of rAAV and Special Considerations in Vector Handling. Methods Mol Biol 2016;1382:199–215. [DOI] [PubMed] [Google Scholar]
- 20. Steece‐Collier K, Collier TJ, Danielson PD, Kurlan R, Yurek DM, Sladek JR, Jr . Embryonic mesencephalic grafts increase levodopa‐induced forelimb hyperkinesia in parkinsonian rats. Mov Disord 2003;18(12):1442–1454. [DOI] [PubMed] [Google Scholar]
- 21. Maries E, Kordower JH, Chu Y, et al. Focal not widespread grafts induce novel dyskinetic behavior in parkinsonian rats. Neurobiol Dis 2006;21(1):165–180. [DOI] [PubMed] [Google Scholar]
- 22. Burger C, Nguyen FN, Deng J, Mandel RJ. Systemic mannitol‐induced hyperosmolality amplifies rAAV2‐mediated striatal transduction to a greater extent than local co‐infusion. Mol Ther 2005;11(2):327–331. [DOI] [PubMed] [Google Scholar]
- 23. Collier TJ, O'Malley J, Rademacher DJ, et al. Interrogating the aged striatum: robust survival of grafted dopamine neurons in aging rats produces inferior behavioral recovery and evidence of impaired integration. Neurobiol Dis 2015;77:191–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ilijic E, Guzman JN, Surmeier DJ. The L‐type channel antagonist isradipine is neuroprotective in a mouse model of Parkinson's disease. Neurobiol Dis 2011;43(2):364–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Levine ND, Rademacher DJ, Collier TJ, et al. Advances in thin tissue Golgi‐Cox impregnation: fast, reliable methods for multi‐assay analyses in rodent and non‐human primate brain. J Neurosci Methods 2013;213(2):214–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Paumier KL, Sortwell CE, Madhavan L, et al. Chronic amitriptyline treatment attenuates nigrostriatal degeneration and significantly alters trophic support in a rat model of parkinsonism. Neuropsychopharmacology 2015;40(4):874–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gombash SE, Manfredsson FP, Mandel RJ, et al. Neuroprotective potential of pleiotrophin overexpression in the striatonigral pathway compared with overexpression in both the striatonigral and nigrostriatal pathways. Gene Ther 2014;21(7):682–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee CS, Cenci MA, Schulzer M, Bjorklund A. Embryonic ventral mesencephalic grafts improve levodopa‐induced dyskinesia in a rat model of Parkinson's disease. Brain 2000;123(Pt 7):1365–1379. [DOI] [PubMed] [Google Scholar]
- 29. Putterman DB, Munhall AC, Kozell LB, Belknap JK, Johnson SW. Evaluation of levodopa dose and magnitude of dopamine depletion as risk factors for levodopa‐induced dyskinesia in a rat model of Parkinson's disease. J Pharmacol Exp Ther 2007;323(1):277–284. [DOI] [PubMed] [Google Scholar]
- 30. McBride JL, Boudreau RL, Harper SQ, et al. Artificial miRNAs mitigate shRNA‐mediated toxicity in the brain: implications for the therapeutic development of RNAi. Proc Natl Acad Sci U S A 2008;105(15):5868–5873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Samaranch L, Sebastian WS, Kells AP, et al. AAV9‐mediated expression of a non‐self protein in nonhuman primate central nervous system triggers widespread neuroinflammation driven by antigen‐presenting cell transduction. Mol Ther 2014;22(2):329–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Konradi C, Westin JE, Carta M, et al. Transcriptome analysis in a rat model of L‐DOPA‐induced dyskinesia. Neurobiol Dis 2004;17(2):219–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Goetz CG, Tanner CM, Klawans HL. Drug holiday in the management of Parkinson disease. Clin Neuropharmacol 1982;5(4):351–364. [DOI] [PubMed] [Google Scholar]
- 34. Koller WC, Weiner WJ, Perlik S, Nausieda PA, Goetz CG, Klawans HL. Complications of chronic levodopa therapy: long‐term efficacy of drug holiday. Neurology 1981;31(4):473–476. [DOI] [PubMed] [Google Scholar]
- 35. Kanaan NM, Sellnow RC, Boye SL, et al. Rationally Engineered AAV Capsids Improve Transduction and Volumetric Spread in the CNS. Mol Ther Nucleic Acids 2017;8:184–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tervo DG, Hwang BY, Viswanathan S, et al. A Designer AAV Variant Permits Efficient Retrograde Access to Projection Neurons. Neuron 2016;92(2):372–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kang S, Cooper G, Dunne SF, Luan CH, James Surmeier D, Silverman RB. Antagonism of L‐type Ca2+ channels CaV1.3 and CaV1.2 by 1,4‐dihydropyrimidines and 4H‐pyrans as dihydropyridine mimics. Bioorg Med Chem 2013;21(14):4365–4373. [DOI] [PubMed] [Google Scholar]
- 38. Olson PA, Tkatch T, Hernandez‐Lopez S, et al. G‐protein‐coupled receptor modulation of striatal CaV1.3 L‐type Ca2+ channels is dependent on a Shank‐binding domain. J Neurosci 2005;25(5):1050–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Surmeier DJ, Ding J, Day M, Wang Z, Shen W. D1 and D2 dopamine‐receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci 2007;30(5):228–235. [DOI] [PubMed] [Google Scholar]
- 40. Wang X, Marks CR, Perfitt TL, et al. A novel mechanism for Ca(2+)/calmodulin‐dependent protein kinase II targeting to L‐type Ca(2+) channels that initiates long‐range signaling to the nucleus. J Biol Chem 2017;292(42):17324–17336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cotzias GC, Papavasiliou PS, Gellene R. L‐dopa in parkinson's syndrome. N Engl J Med 1969;281(5):272. [DOI] [PubMed] [Google Scholar]
- 42. Cenci MA, Crossman AR. Animal models of l‐dopa‐induced dyskinesia in Parkinson's disease. Mov Disord 2018;33(6):889–899. [DOI] [PubMed] [Google Scholar]
- 43. Morin N, Jourdain VA, Di Paolo T. Modeling dyskinesia in animal models of Parkinson disease. Exp Neurol 2014;256:105–116. [DOI] [PubMed] [Google Scholar]
- 44. Kordower JH, Olanow CW, Dodiya HB, et al. Disease duration and the integrity of the nigrostriatal system in Parkinson's disease. Brain 2013;136(Pt 8):2419–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Clark NC, Nagano N, Kuenzi FM, et al. Neurological phenotype and synaptic function in mice lacking the CaV1.3 alpha subunit of neuronal L‐type voltage‐dependent Ca2+ channels. Neuroscience 2003;120(2):435–442. [DOI] [PubMed] [Google Scholar]
- 46. Kang S, Cooper G, Dunne SF, et al. CaV1.3‐selective L‐type calcium channel antagonists as potential new therapeutics for Parkinson's disease. Nat Commun 2012;3:1146. [DOI] [PubMed] [Google Scholar]
- 47. Berger SM, Bartsch D. The role of L‐type voltage‐gated calcium channels Cav1.2 and Cav1.3 in normal and pathological brain function. Cell Tissue Res 2014;357(2):463–476. [DOI] [PubMed] [Google Scholar]
- 48. Ortner NJ, Striessnig J. L‐type calcium channels as drug targets in CNS disorders. Channels (Austin). 2016;10(1):7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENT (VIDEO) FIG. 1 In the first representative parkinsonian rat, which received the rAAV‐Scr‐shRNA vector, 50 minutes after a 12‐mg/kg dose of levodopa, there is an abundance of tight rotational behavior intermixed with LID behavioral profiles including orolingual behaviors (ie, licking and chewing, tongue protrusion), severe hindlimb extension/dystonia, forelimb dystonia and hyperkinetic movements, and severe trunk and neck dystonia. In contrast, as shown in 2 representative rats that had received the rAAV‐CaV1.3‐shRNA vector, at this same dose and a time 50 minutes postlevodopa, there is no evidence of the prototypical LID behaviors readily displayed in parkinsonian rats treated with our control vector. In the first rAAV‐CaV1.3‐shRNA rat, this parkinsonian subject displays motor activation that includes continuous sniffing and exploring, left and right forepaw tapping, digging at the litter, and 1 incident of a wet‐dog shake. It is evident that this rat has a unilateral nigral lesion, in that it walks only clockwise around the perimeter of the cage, but does not display typical tight rotational behavior seen in the rAAV‐Scr‐shRNA rats. The second rAAV‐CaV1.3‐shRNA rat shows sniffing and exploring, mild tapping and pushing at the litter, and again walking only clockwise in the cage. These motor activation behaviors start at approximately 20 minutes and continue until approximately 140‐170 minutes after the levodopa injection, a time course similar to that seen for LID behaviors.
note: As presented in the Methods section, in the outbred SD rat strain there is a small subset (15%‐20%) of these rats that despite having an equal degree of striatal DA depletion and levodopa treatment remain resistant to LID expression. Their behavior is very different from the LID‐negative rAAV‐CaV1.3‐shRNA rats that show what could be suggested to be enhanced motor efficacy to levodopa. Specifically, the 15%‐20% of control (non‐vector injected) rats that remain LID negative generally show mild activation including sniffing and exploring, most likely in response to the injection per se, which lasts approximately 5‐10 minutes, after which they fall asleep in the corner of the cage for the remainder of the rating period.