

Abstract
Specific metabolic underpinnings of androgen receptor (AR)-driven growth in prostate adenocarcinoma (PCa) are largely undefined, hindering the development of strategies to leverage the metabolic dependencies of this disease when hormonal manipulations fail. Here we show that the mitochondrial pyruvate carrier (MPC), a critical metabolic conduit linking cytosolic and mitochondrial metabolism, is transcriptionally regulated by AR. Experimental MPC inhibition restricts proliferation and metabolic outputs of the citric acid cycle (TCA) including lipogenesis and oxidative phosphorylation in AR-driven PCa models. Mechanistically, metabolic disruption resulting from MPC inhibition activates the eIF2α/ATF4 integrated stress response (ISR). ISR signaling prevents cell cycle progression while coordinating salvage efforts, chiefly enhanced glutamine assimilation into the TCA, to regain metabolic homeostasis. We confirm that MPC function is operant in PCa tumors in-vivo using isotopomeric metabolic flux analysis. In turn, we apply a clinically viable small molecule targeting the MPC, MSDC0160, to pre-clinical PCa models and find that MPC inhibition suppresses tumor growth in hormone-responsive and castrate-resistant conditions. Collectively, our findings characterize the MPC as a tractable therapeutic target in AR-driven prostate tumors.
Introduction
Metabolic reprogramming, a recognized hallmark of cancer, is inextricably linked to mitogenic cell signaling pathways1. To fuel proliferation, tumor cells constitutively import nutrients and engage biosynthetic pathways to generate nucleotides, lipids, proteins, and other macromolecules required for cell division2. While the absolute biosynthetic requirements for cellular proliferation are relatively conserved3, many interacting factors dictate how these requirements are ultimately met. Tissue of origin, microenvironment, host factors, and oncogenic driver mutations can all impact tumor metabolism4. It follows that many widely-studied oncogenes (e.g. MYC, KRAS) drive specific metabolic alterations and dependencies while promoting tumor growth1. Likewise, accumulating evidence demonstrates hormone and hormone-related nuclear receptors directly regulate metabolic pathways to supply the biosynthetic demands of proliferation5,6. In prostate adenocarcinoma (PCa), androgen receptor (AR) is a hormone-responsive nuclear receptor transcription factor that coordinates anabolic processes to enable tumor proliferation through transcriptional regulation of metabolic pathways7. AR is widely recognized as the primary molecular driver of PCa progression, but a detailed understanding of the metabolic programs it coordinates in PCa is currently limited.
Locally advanced and metastatic PCa is typically managed with agents that disrupt AR and its signaling axis by inhibiting androgen production or directly antagonizing AR itself8. However, through a variety of resistance mechanisms9, AR signaling is reactivated and drives disease progression in a castrate-resistant manner that is ultimately lethal. Though castrate-resistant PCa remains largely dependent on AR signaling10, the multitude of castration-resistance mechanisms in PCa underscore the difficulty of directly targeting AR in this setting. While substantial clinical efforts have focused on preventing AR action at the level of its transcriptional activity, identifying and disrupting downstream metabolic components of AR-driven proliferation may enable novel and complimentary approaches for the treatment of AR-driven castrate-resistant PCa.
To this end, we implemented a bioinformatic screen to identify AR-regulated genes driving metabolic processes in PCa. Our effort nominated mitochondrial pyruvate carrier subunit 2 (MPC2), a component of the mitochondrial pyruvate carrier (MPC), as a putative enabling component of PCa metabolism. The MPC is a hetero-oligomeric complex made up of co-stabilizing proteins MPC1 and MPC211,12. The carrier assembles on the inner mitochondrial membrane and imports the metabolic end product of glycolysis, pyruvate, into the mitochondrial matrix for incorporation into intermediary metabolism in the citric acid cycle (TCA). The MPC has been characterized as a Warburg-suppressive complex in highly glycolytic models of colon cancer13, but the contrasting metabolic features characterizing AR-driven PCa position the MPC to fuel, rather than suppress, oncogenic growth.
The metabolism operant in AR-driven PCa is thought to be unique because primary PCa is highly lipogenic, less glycolytic, and more reliant on oxidative phosphorylation (OxPhos) than most other solid tumors14,15. Therefore, we hypothesized AR-driven PCa models would funnel pyruvate into the mitochondria via the MPC to fuel OxPhos, lipogenesis, and other biosynthetic processes originating from TCA metabolism that are necessary for proliferation. In line with this expectation, experimental MPC inhibition in AR-driven PCa models restricts proliferation, OxPhos, and lipogenesis while activating the integrated stress response (ISR). ISR activation triggers the G1/S cell cycle checkpoint and promotes glutamine assimilation in an attempt to salvage TCA function and regain metabolic homeostasis. It follows that experimental glutamine restriction greatly amplifies the effects of MPC inhibition. Last, MPC function is conserved in preclinical PCa models in-vivo, and MPC inhibition in this setting activates ISR signaling while suppressing tumor growth. Together, our findings define the MPC as an enabling component of AR-driven PCa metabolism and suggest inhibition of this complex may have therapeutic potential for the treatment of lethal castrate-resistant PCa.
Results
MPC Subunits are Increased in PCa
To nominate AR target genes involved in metabolism, we accessed PCa mRNA expression data from the Cancer Genome Atlas (TCGA)16 through the cBioPortal17 and calculated a Spearman score for all annotated genes based on co-expression with the AR/luminal marker genes KRT8 and KRT18 as well as the direct AR target gene PSA (KLK3). We rank-ordered each gene list and identified genes present in the top 5% of every list to nominate 483 preliminary candidate genes. Next, to identify genes involved in central metabolic pathways, candidate genes were keyword-screened by their annotated RefSeq function and NCBI GeneRif summary. Final candidate genes were rank-ordered by expression fold change in benign prostate tissue vs. PCa specimens in the TCGA (Fig. 1a, Supplementary Data 1).
Figure 1. The MPC is increased in PCa specimens and associated with poor clinical outcomes.
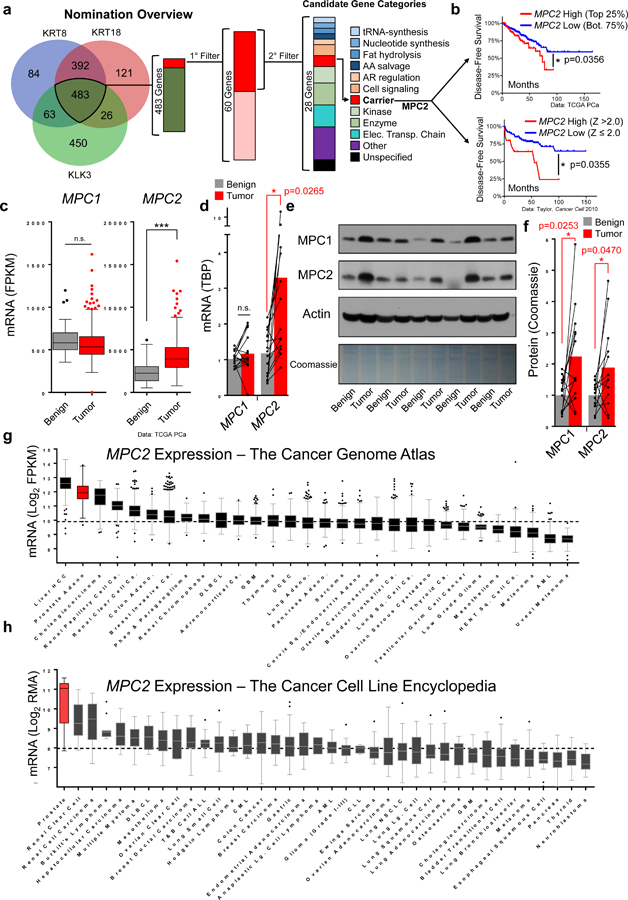
a, Nomination overview to identify candidate AR target genes involved in metabolism. b, Kaplan-Meier plot with primary prostate tumor cohorts defined by high or low MPC2 expression based on Z scores or quartiles. Data from the Cancer Genome Atlas (TCGA) PCa (n=497) and Taylor 2010 (n=131). c, RNA-sequencing comparing MPC1 and MPC2 expression in benign prostate tissue (n=52) and prostate tumor (n=497) specimens Data from TCGA PCa. d, MPC1 and MPC2 mRNA expression measured by qPCR and normalized to Tata-binding protein (TBP) in benign prostate tissue and matched adjacent primary prostate tumors (n=15 independent pairs). e, MPC1 and MPC2 protein expression measured by immunoblotting in 5 representative benign prostate tissue and matched adjacent primary prostate tumors. f, Densitometry quantification of 13 benign prostate and adjacent prostate tumor pairs normalized to total protein loading (Coomassie). Plot includes 5 pairs in (d) and 8 additional pairs in Supp. Fig 1a. g, MPC2 mRNA expression in all available TCGA RNA-seq datasets ordered by median expression; n=9,121 samples across 30 tumor types. h, MPC2 mRNA expression across all cell line models in the online Cancer Cell Line Encyclopedia ordered from high to low MPC2 expression; n=865 samples across 38 tumor types. Data in panel b were statistically analyzed using a two-sided log-rank test *p < 0.05. Data in panel c, g, and h are represented as box and whisker plots using the Tukey method in Graphpad Prism: The box extends from the 25th to the 75th percentile, the line in the middle of the box represents the median, whiskers represent the inter-quartile distance multiplied by 1.5, and data points outside of this range are plotted individually. Data in panel d and f are represented as a bar designating the mean combined with a scatter plot with individual pairs connected by a line. Statistical analysis was performed using a two-tailed Student’s t-test (panel c) or a two-tailed paired Student’s t-test (panel d and f): n.s. not significant, *p < 0.05, ***p < 0.001.
Our efforts nominated MPC2 as a putative AR-regulated gene with a critical role in metabolism. Patients with high levels of MPC2 tumor mRNA expression suffer decreased disease-free survival (Fig. 1b) and MPC2 mRNA is significantly increased in primary prostate tumors relative to benign prostate tissue (Fig. 1c). Consistent with TCGA data, MPC1 mRNA expression was not altered and MPC2 mRNA expression was significantly increased in an independent validation cohort of primary prostate tumors compared to matched adjacent benign tissue in radical prostatectomy specimens (Fig. 1d, Supplementary Fig. 1b). In contrast to mRNA expression, both MPC1 and MPC2 protein were increased in prostate tumors relative to adjacent matched benign tissue (Fig. 1e, f, Supplementary Fig. 1a, c). This finding may derive from the co-stabilizing nature of the MPC subunits11–13 and suggests increased MPC2 mRNA expression drives stabilization of an intact and functional MPC in prostate tumors. In line with this idea, castrate-resistant PCa specimens18 exhibit MPC2 mRNA upregulation and locus amplification (Supplementary Fig. 1d).
To place these findings into a broader context, we queried all mRNA expression data sets available in the TCGA and found median expression of MPC2 in PCa was second highest among all profiled tumor types (Fig. 1g). In contrast, MPC1 expression was not elevated in PCa relative to other tumor types (Supplementary Fig. 1e). Similarly, in the Cancer Cell Line Encyclopedia (CCLE)19, median MPC2 expression in PCa models was approximately two log2-fold greater than any other cancer type (Fig. 1h) while MPC1 expression was not elevated (Supplementary Fig. 1f). Together, these findings demonstrate MPC2 expression is uniquely increased in PCa and suggest increased MPC2 expression may drive MPC complex stabilization and function in prostate tumors.
MPC2 Transcription is Regulated by AR
Similar to human tumor specimens, protein expression of both MPC subunits was elevated in hormonally responsive AR-driven PCa models compared to non-transformed RWPE1 prostate cells (Fig. 2a). MPC subunit expression was greatest in AR positive castrate-resistant PCa models but was virtually absent in AR negative models. Our initial nomination predicted MPC2 as an AR-regulated gene, and the correlation between AR and MPC expression in the cell line models likewise suggested a regulatory relationship. Similar to the canonical AR target gene KLK3 (PSA) (Supplementary Fig. 2a), MPC2 mRNA was increased by androgens (Dihydrotestosterone (DHT) or Metribolone (R1881)) and this induction was blocked by the anti-androgen enzalutamide (Enz) in multiple hormone-responsive PCa cell lines (Fig. 2b). Though MPC2 ranks among the most hormone-responsive genes in independent PCa RNA-sequencing datasets20 (Supplementary Fig. 2b), MPC1 mRNA expression was not altered in response to hormonal manipulations (Supplementary Fig. 2a). We hypothesized the androgen-driven increase in MPC2 mRNA would drive the accumulation of both MPC1 and MPC2 proteins as observed in human tumor specimens. However, in tissue culture cells, while MPC2 protein increased in response to 72-hour androgen stimulation, MPC1 protein was unchanged (Fig. 2c). To examine MPC regulation in a more physiologic setting, we implanted VCaP xenografts into mice, allowed four weeks for tumor establishment, then paired the mice by tumor volume and randomized each pair to sham surgery or castration. Tumors were collected one week after surgery and tumors from castrated mice had significantly less MPC1 and MPC2 protein with the exception of one pair (arrow) in which the castrate tumor harbored increased expression of MPC subunits concomitant with the emergence of the recognized constitutively active AR splice variant, AR-V721 (Fig. 2d). To examine MPC expression during castrate-resistant outgrowth, we implanted intact mice with VCaP tumors as before, but allowed tumors to grow after castration. Castrate-resistant VCaP tumors regained MPC expression (Fig. 2e). Next, we examined MPC expression in the AR-driven castrate-resistant cell line, LNCaP-androgen ABLation (ABL)22. In contrast to hormone-responsive LNCaP cells, ABL cells maintained proliferation (Supplementary Fig. 2e) and MPC expression (Fig. 2f) during hormonal manipulations. Interestingly, though MPC protein was not altered in response to hormonal manipulations in ABL cells, AR is required for transcriptional induction of MPC2 in response to androgens in these cells (Supplementary Fig. 2f). These data demonstrate AR regulates the MPC and MPC expression re-emerges and is maintained during castrate-resistant growth in AR-driven PCa.
Figure 2. AR controls the MPC in PCa through transcriptional regulation of MPC2.
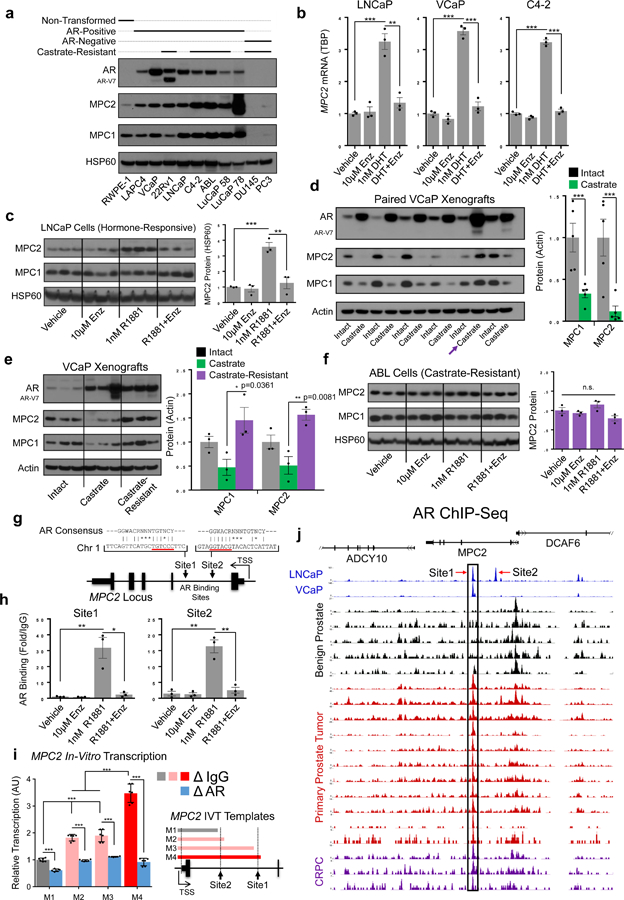
a, MPC1 and MPC2 protein measured by immunoblotting across prostate cancer models. HSP60 is used as a mitochondrial loading control. Untreated protein lysates collected during routine culturing (cell lines) or growth (PDX models) and the immunoblot is one representative result from 3 independent experiments b, MPC2 mRNA was assessed in hormonally responsive PCa cell line models were treated as indicated for 48h and MPC2 mRNA expression was measured by qPCR. c, LNCaP cells were treated for 72h and protein expression was assessed by immunoblotting; n=3 independent cultures per treatment (1 per lane). d, Mice bearing established VCaP xenografts were paired by tumor volume and randomized to sham or castration surgery. Protein expression was assessed by immunoblotting one week after surgery. Pair 5 (arrow) was excluded from quantification at right due to the emergence of AR-V7. e, Mice bearing established VCaP xenografts were randomized to sham (n=3) or castration (n=6) surgery. 3 sham and 3 castration xenografts were collected one week after surgery. The remaining 3 castrate xenografts were allowed to establish castrate-resistant growth and were collected 9 weeks after castration. Protein expression was measured by immunoblotting. f, Castrate-resistant ABL cells were treated for 72h and protein expression was assessed by immunoblotting; n=3 independent cultures per treatment (1 per lane). g, The MPC2 locus with putative AR binding sites indicated. h, AR binding at the MPC2 locus was assessed by ChIP-qPCR in LNCaP cells 16 hours after treatment. i, in-vitro transcription to assess the functional relevance of the AREs in the MPC2 locus (n=2 pooled experiments measured in technical triplicates). j, ChIP-Seq data from LNCaP cells, VCaP cells, benign human prostate, primary human prostate tumors, and CRPC specimens. Data from Stelloo25 and Pomerantz24. n =3 independent cultures per treatment for qPCR and ChIP experiments in panels b and h, respectively. Data in bar graphs are represented as the mean ± SEM. Statistical analysis was performed using a two-tailed Student’s t-test: n.s. not significant, *p < 0.05, **p < 0.01, ***p < 0.001.
To determine if AR mediates direct transcriptional control of MPC2, we applied transcription factor binding motif analysis which identified two putative androgen response element half-sites located in the first intron of the MPC2 locus (Fig. 2g). AR chromatin immunoprecipitation (ChIP) experiments confirmed androgen-dependent AR recruitment to both MPC2 sites that was blocked by the anti-androgen enzalutamide (Fig. 2h). To assess the functional relevance of the AR binding sites in the MPC 2 locus, we performed in-vitro transcription (IVT) using chromatinized IVT templates23 (Supplementary Fig. 2h–j). MPC2 transcription increased with the addition of each AR binding site on the IVT templates and AR immunodepletion abrogated MPC2 transcription (Fig. 2i). As before, PSA was used as a positive control in these experiments (Supplementary Fig. 2c, d, g). Last, AR binding at the MPC2 locus was conserved in primary prostate tumors and CRPC specimens from published ChIP-Seq data24,25 (Fig. 2j). These data demonstrate AR regulates the MPC through direct transcriptional control of MPC2 in PCa models and suggest this relationship is conserved in human PCa.
MPC Inhibition Disrupts Metabolism in AR-Driven PCa
The TCA is repurposed into a biosynthetic hub to support the demands of uncontrolled proliferation during oncogenesis26. Key TCA outputs in this context include citrate for lipogenesis, reducing equivalents for OxPhos, and intermediates for amino acid synthesis (Fig. 3A). To examine the consequences of MPC inhibition, we treated AR-dependent PCa cell line models with the established MPC inhibitor, UK509927. In basal culturing conditions, MPC inhibition resulted in a significant, dose-responsive decrease in proliferation in hormone responsive PCa cell line models (Fig. 3b). To confirm the specificity of this effect, we applied the same treatment to two AR-negative PCa cell lines lacking MPC expression and a colon cancer cell line where the MPC has been reported as a Warburg repressor13. In contrast to AR-dependent cell lines, AR negative cells showed little or no decrease in proliferation in response to equivalent doses of UK5099 (Supplementary Fig. 3a). To experimentally isolate the role of AR signaling in these processes, LNCaP cells were cultured in hormone-depleted charcoal stripped serum (CSS) and androgens were added to specifically induce AR-driven proliferation, OxPhos, and lipogenesis. MPC inhibition restricted AR-driven cellular proliferation (Fig. 3c), maximal (uncoupled) OxPhos capacity (Fig. 3d), and lipogenesis (Fig. 3e, f). Further, in addition to decreased lipid content, TEM also revealed MPC inhibition resulted in swelling of mitochondrial cristae, an observation consistent with reduced oxygen consumption and decreased ATP production.
Figure 3. MPC inhibition delays proliferation and disrupts TCA outputs in AR-driven PCa models.
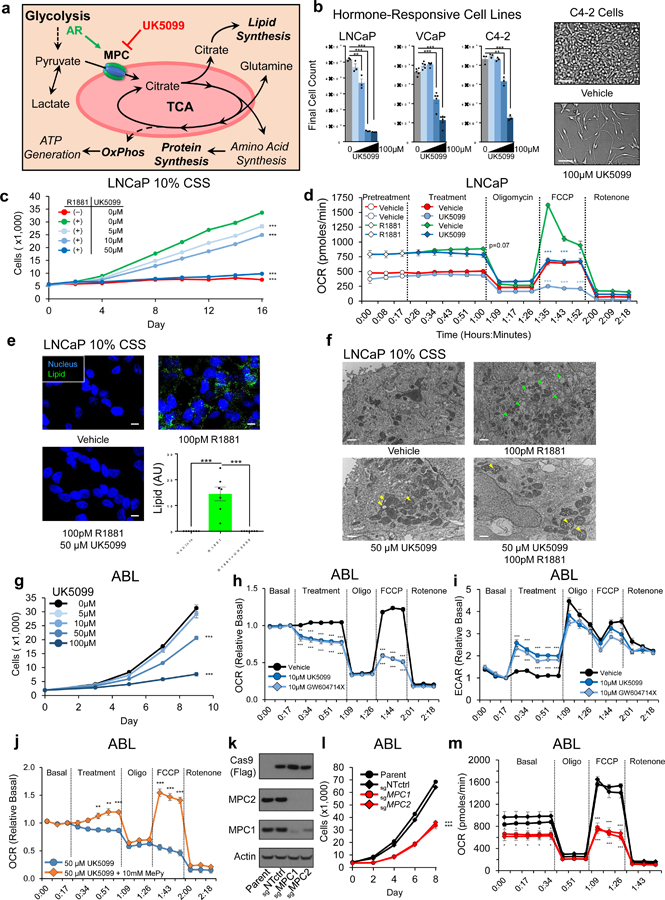
a, Model depicts metabolic and biosynthetic outputs of the TCA in relation to glycolysis and the MPC. b, Hormone-responsive AR-driven cell lines were treated with 0, 5µM, 10µM, 50µM, or 100µM of MPC inhibitor UK5099. Cells proliferation was quantified using in-situ image-based analysis on the final day of the experiment. Scale bar is 100 µm; n=3 (LNCaP, C4–2) or n=5 (VCaP) independent wells per treatment. c, LNCaP cells in 10% hormone-depleted CSS growth media were treated with 100pM of synthetic androgen R1881 with or without UK5099. Cell proliferation was assessed using image-based cell counting; n=5 independent wells per treatment. d, Oxygen consumption rate (OCR) was assessed in LNCaP cells pretreated for 72h; vehicle or 5µM UK5099 was injected at the indicated timepoint. e, LNCaP cells in 10% CSS were treated for 96h and then stained for neutral lipids (LipidTOX). Scale bar is 10µm. f, LNCaP cells were treated for 96h and then imaged using TEM. Green arrowheads specify lipids, yellow arrowheads specify swollen mitochondrial cristae. Scale bar is 1 µm; n=3 independent experiments per treatment and approximately 15 images from each treatment were collected from different grids with representative images shown g, Proliferation during MPC inhibition in castrate-resistant ABL cells was quantified as in (c); n=6 independent wells per treatment. h,i, Metabolic potentials in ABL cells were assessed by measuring OCR (h) and the rate of extracellular acidification (ECAR) (i). j, OCR was assessed in ABL cells supplemented with membrane-bypassing methyl-pyruvate (MePy) during UK5099 treatment. k, Immunoblot of ABL cells with Cas9-mediated genetic disruption of the MPC1 or MPC2 locus. sgNTctrl is a non-targeting sgRNA and parent is unmodified cells. l,m, Proliferation (l) and OCR (m) were assessed in the cells described in (k); n=5 independent wells per treatment for proliferation experiment in panel l. n=7 fields from 3 independent cover slips per treatment for lipid staining in e. n=5 independent wells per treatment for ECAR & OCR experiments in panel d, h, i, j, and m. Data in bar and line graphs are represented as the mean ± SEM. Statistical analysis was performed using a two-tailed Student’s t-test *p < 0.05, **p < 0.01, ***p < 0.001.
AR reactivation during androgen deprivation therapy drives castration resistance and direct AR targeting in this setting is challenging28. However, continued reliance on AR-driven programs may impose metabolic dependencies concomitant with disease progression. To model this disease, we pursued experiments in the castrate-resistant ABL model22, which proliferates and maintains MPC protein expression during treatment with androgens and anti-androgens (Fig. 2f, Supplementary Fig. 2e). However, in contrast to hormone-responsive LNCaP cells, AR knockdown does not decrease baseline levels of MPC2 transcription, suggesting additional factors maintain MPC2 transcription in the hormone-free culturing conditions in which ABL proliferates (Supplementary Fig. 3f). Regardless, MPC function is required in this AR-dependent model, as ABL cells treated with UK5099 or a thiazolidine-class MPC inhibitor, GW604714X29, exhibited a dose-dependent decrease in proliferation (Fig. 3g, Supplementary Fig. 3b) and restricted basal and maximal OCR concomitant with an increased rate of extracellular acidification (ECAR) resulting from lactic acid secretion (Fig. 3h, i). In contrast, OxPhos in DU145 cells with low MPC expression was not impacted by UK5099 but was markedly restricted by glutamine withdrawal (Supplementary Fig. 3c), suggesting these cells oxidize glutamine in the absence of the MPC while ABL cells oxidize MPC-imported pyruvate.
Compounds that suppress the MPC may inhibit MCT1 (SLC16A1), a plasma membrane lactate transporter30. To examine this possibility, we treated ABL cells with UK5099 and the MCT1 inhibitor AZD396531. UK5099-mediated increases in lactate secretion were blocked when MCT1-mediated lactate export was inhibited using AZD3965, suggesting UK5099 does not meaningfully impact MCT1 in these conditions (Supplementary Fig. 3d). Further, in contrast to MPC inhibition, maximal MCT1 inhibition did not inhibit proliferation in ABL cells (Supplementary Fig. 3e). Last, the constitutively high rate of lactate secretion in MCT1-expressing DU145 cells with low MPC expression is not impacted by UK5099 (Supplementary Fig. 3f), and UK5099-mediated OCR restriction is rescued by membrane-bypassing methyl pyruvate (Fig. 3j). These results suggest phenotypes resulting from MPC inhibition using UK5099 are not attributable to off-target effects on MCT1. However, we noted a discrepancy in the concentration of UK5099 required for growth inhibition (~50 µM) compared to OxPhos restriction (~10µM) and hypothesized albumin in the serum present in growth media may sequester UK5099 and prevent its action. In line with this idea, cells were markedly sensitized to UK5099 in low serum conditions, but the addition of albumin reduced the effectiveness of UK5099 (Supplementary Fig. 3g). Conversely, the addition of serum to assay media during OCR measurements blunted cellular responses to UK5099 (Supplementary Fig. 3h).
To examine the effect of genetic MPC disruption, we generated single guide RNAs targeting the first exon of MPC1 or MPC2. Immunoblotting confirmed Cas9-mediated disruption of these proteins and, as expected, genetic disruption of either MPC subunit resulted in depletion of the complex (Fig. 3K). Similar to pharmacologic MPC inhibition, MPC KO cells exhibited a decreased rate of cellular proliferation as well as decreased basal and maximal OCR (Fig. 3L&M). Collectively, these data characterize the MPC as a required metabolic component of AR-driven proliferation that is operant in hormone-responsive PCa and maintained in the setting of castrate-resistant disease.
MPC Flux is Required in Castrate-Resistant PCa
To examine specific metabolic impacts of MPC inhibition on TCA function, we began by assessing the relative steady-state levels of metabolic intermediates during MPC inhibition. MPC inhibition did not alter the levels of early glycolytic intermediates, but pyruvate and lactate began to accumulate immediately upstream of the pharmacological MPC blockade (Fig. 4a). Downstream, TCA intermediates and anaplerotic amino acid pools were depleted, suggesting MPC-trafficked pyruvate constitutes a major TCA input in these cells. To gauge the impact of MPC inhibition on TCA function, we measured the NAD+/NADH ratio and cellular ATP content. The ratio of NAD+/NADH was increased while ATP was decreased (Fig. 4b, c), suggesting a decrease in cellular reducing potential contributes to OxPhos disruption and prevents efficient ATP generation during MPC inhibition. In agreement, we found phosphorylation of the central energy sensor AMP-activated protein kinase (AMPK) and its substrate, acetyl-CoA carboxylase (ACC), were increased during MPC inhibition (Fig. 4f). AMPK activation likely contributes to our previous observation that lipogenesis is restricted during MPC inhibition (Fig. 3e, f) because ACC is the rate-limiting lipogenic enzyme and ACC phosphorylation is inhibitory. Last, we measured reduced glutathione to gauge the impact of MPC inhibition on cellular anti-oxidant capacity. We found reduced glutathione content was decreased (Fig. 4d) concomitant with increased cellular reactive oxygen species (ROS) (Fig. 4e). In agreement with increased ROS, immunoblotting demonstrated increased content of NRF2, a master regulator of the oxidative stress response (Fig. 4f). In summary, MPC inhibition results in profound disruption of metabolic homeostasis with resultant impacts on intracellular metabolite pools, reducing potential, ATP content, and anti-oxidant capacity.
Figure 4. MPC inhibition disrupts TCA function and prevents cell cycle progression.
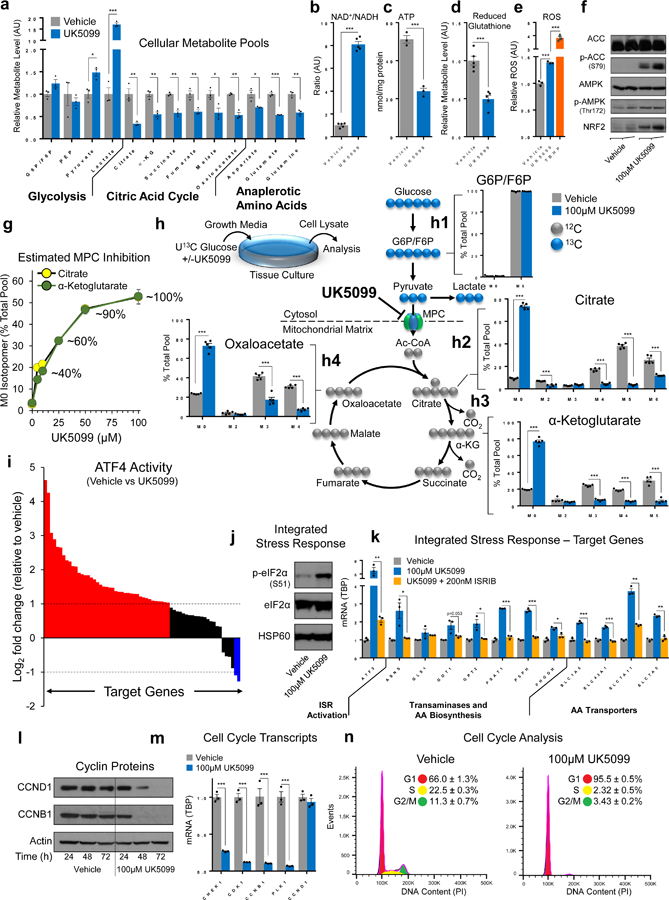
a, Metabolites were quantified in ABL cells treated for 2h. b-e, NAD+/NADH (b), ATP (c), reduced glutathione (d), and ROS (e) were quantified in ABL cells; treatment was applied for 6h (b), 48h (c), 2h (d), or 1h (e). f, p-ACC, p-AMPK, and NRF2 were assessed via immunoblot in ABL cells treated for 72h; AMPK and ACC are loading controls. g, Functional UK5099-mediated MPC inhibition was assessed by incubating ABL cells with U13C glucose and measuring the M0 isotopomer of citrate and α-ketoglutarate after 48h. h, Glucose incorporation into TCA metabolite pools was quantified by pretreating ABL cells for 2h (vehicle or UK5099), then adding U13C glucose for 48h. i, ATF4 activity was assessed by RNA-sequencing in ABL cells treated for 72h (see supplementary fig. 4c). Average log2 fold change (FC) of known ATF4 target genes is plotted; FC>1 are red, FC<1 are blue, and all others are black. j, ISR activity was assessed via immunoblotting in ABL cells treated for 24h. k, Transcriptional activation of the ISR was assessed via qPCR in ABL cells treated for 6h. ISRIB is a small molecule that inhibits the ISR. l, Cyclin content was assessed via immunoblot in ABL cells. m, Cell cycle-dependent mRNA transcripts were assessed in ABL cells treated for 48h. n, Cell cycle distribution was assessed using flow cytometry in ABL cells treated for 48h. n =3 independent cultures per treatment for metabolite measurements in a, c, e, and g. n=5 independent cultures per treatment for metabolite measurements in b, d, and h. For immunoblots, the experiment was performed 2 (l) or 3 (f, j) independent times with similar results and a representative blot with one biological replicate per lane is illustrated. n=3 biological replicates for RNA sequencing (i), qPCR (k and m), and cell cycle analysis (n). 100µM UK5099 used in all experiments in this figure unless otherwise indicated. Data in bar and line graphs are represented as the mean ± SEM. Statistical analysis was performed using a two-tailed Student’s t-test *p < 0.05, **p < 0.01, ***p < 0.001.
Most cultured cells convert glucose to lactate and utilize glutamine as the major TCA carbon source32. In sharp contrast, our results suggest PCa metabolism relies on MPC flux, which implies glucose-derived pyruvate is the major TCA carbon source in these models. To directly assess the carbon source supplying TCA metabolism in our models, we performed isotopomeric metabolic flux analysis using uniformly labelled 13C glucose. We began with a dose-response experiment to empirically determine the concentration of UK5099 required to achieve maximal MPC blockade by measuring the content of unlabeled (M0) citrate and α-ketoglutarate in cells cultured in U13C glucose during MPC inhibition (Fig. 4g, Supplementary Fig. 4a). Based on the results of this experiment, we pursued subsequent tracing experiments using 100 µM UK5099 to achieve near-maximal MPC blockade. In vehicle treated cells, labelled glucose is taken up and incorporated via glycolysis, making up virtually all (~98.8%) of the glucose-6-phosphate / fructose-6-phosphate (G6P/F6P) pool (Fig. 4h1). Turning next to mitochondrial metabolite pools, the isotopomeric distribution of citrate indicates virtually all (~91%) citrate molecules contained glucose-derived carbon (M2, M3, M4, M5, M6 isotopomers) (Fig. 4h2). In agreement, 13C labeled α-ketoglutarate (α-kg) and oxaloacetate species made up the majority (81% and 76%, respectively) of their total respective metabolite pools (Fig. 4h3, h4). Next, we blocked MPC flux using UK5099. MPC inhibition had no impact on the isotopomeric distribution of upstream G6P/F6P (Fig. 4h1). In contrast, downstream of the MPC blockade, unlabeled M0 isotopomers of citrate, α-kg, and oxaloacetate were dramatically increased (73%, 79%, and 73%, respectively) (Fig. 4h2–4). This dramatic shift in isotopomeric distribution occurred concomitant with a decrease in metabolite pool size (Fig. 4a). Together, these data confirm glucose as the primary TCA carbon source in these cells and support a critical role for the MPC in maintaining metabolic outputs of the TCA. Likewise, the dramatic increase in M0 isotopomers suggests alternative carbon sources supply the TCA during MPC inhibition.
MPC inhibition in AR-driven PCa models restricts cell proliferation and the constellation of phenotypic effects resulting from MPC inhibition suggests global shifts in cell signaling and metabolism. To identify the predominant cell signaling events underpinning these responses to MPC inhibition, we applied a reverse phase protein array (RPPA). RPPA analysis indicated MPC suppression precipitates a multi-factorial stress response with elements of bioenergetic stress (AMPK, ACC), cytoprotective heat shock protein activation (HSP27, BiP-GRP78), and enhanced anti-oxidant capacity (SOD1) (Supplementary Data 2, Supplementary Fig. 4b). RPPA analysis likewise indicated MPC inhibition broadly suppresses cell-cycle checkpoint machinery (CHK1, ATM, CDC25C, CDK1, CYCLIN-B1, PLK1) while inhibiting protein translation (S6 pS235/pS236). Though these findings were congruent with characterized phenotypes, the specific mechanism(s) underpinning these responses were not immediately clear. Therefore, we pursued RNA sequencing to characterize the global transcriptional response to MPC inhibition (Supplementary Data 3, Supplementary Fig. 4c). Gene Set Enrichment Analysis (GSEA)33 indicated MPC inhibition results in protein misfolding and ER stress concomitant with suppression of DNA replication and progression through mitosis (Supplementary Fig. 4d). Consistent with this finding, master transcription factor regulators of the integrated stress response (ISR), including ATF4, XBP1, and CHOP as well as their target genes were prominently increased during MPC inhibition (Fig. 4i, Supplementary Fig. 4c). Together, RPPA and RNA-Seq analyses favor a model in which MPC inhibition precipitates activation of the ISR which in turn delays cell cycle progression.
Mechanistically, ISR activation occurs through phosphorylation of eIF2α and subsequent translation of ATF4, which mediates transcription of target genes to resolve the ISR and regain homeostasis34. In line with this idea, MPC suppression results in eIF2α phosphorylation and transcription of ATF4 target genes (Fig. 4j, k). To confirm ISR signaling is required for the transcriptional response to MPC inhibition, we suppressed ISR signaling during MPC inhibition. Pharmacologic and siRNA-mediated ISR suppression rescued transcriptional changes measured during MPC inhibition (Fig. 4k, Supplementary Fig. 4e). ISR activation can prevent G1/S cell cycle progression through depletion of Cyclin D135. In agreement, MPC suppression results in cyclin depletion concomitant with dramatic reduction of a battery of G2/M-phase dependent cell cycle mRNA transcripts and activation of the G1/S checkpoint as assessed by flow cytometry (Fig. 4l–n). To determine if MPC inhibition resulted in similar effects in other cell line models of prostate cancer, we applied UK5099 to a battery of AR positive and AR negative cell lines and found the majority of AR positive cell lines demonstrated ISR activation while AR negative cell lines exhibited little or no response (Supplementary Fig. 4f). Importantly, while ABL cells demonstrate a dose-responsive increase in ISR signaling during MPC inhibition (Supplementary Fig. 4g), AR negative DU145 cells do not respond to equivalent doses of UK5099 as measured by ATF4 translation, cyclin depletion, or loss of G2/M-dependent mRNA transcripts (Supplementary Fig. 4h–j). Collectively, these experiments demonstrate MPC inhibition activates the ISR which in turn delays cell cycle progression by activating the G1/S checkpoint.
MPC Suppression Increases Glutamine Reliance
ATF4 action is shaped by the initial ISR stimulus and coordinates efforts to regain homeostasis34. However, ISR action may mask MPC reliance in the engineered metabolic conditions present in-vitro. Therefore, to establish experimental conditions designed to isolate the cellular requirement for MPC activity, we sought to identify and disrupt relevant ATF4-mediated processes. Our isotopomeric flux analysis using labelled glucose during MPC suppression suggests alternative carbon sources supply the TCA during MPC inhibition (Fig. 4h) and glutamine oxidation can maintain the TCA during impaired mitochondrial pyruvate transport36. Therefore, we examined RNA-sequencing data generated during MPC suppression (Supplementary Table 3) to identify biologically cohesive metabolic programs that may enable increased glutamine uptake and TCA assimilation. These efforts uncovered a clear pathway made up of the plasma membrane glutamine transporter SLC1A5, Asparagine Synthetase (ASNS), and Phosphoserine Aminotransferase (PSAT1), which were coordinately upregulated by ATF4 during MPC inhibition (Fig. 4k, Fig. 5a, b).
Figure 5. ISR signaling coordinates glutamine uptake and incorporation during MPC suppression.
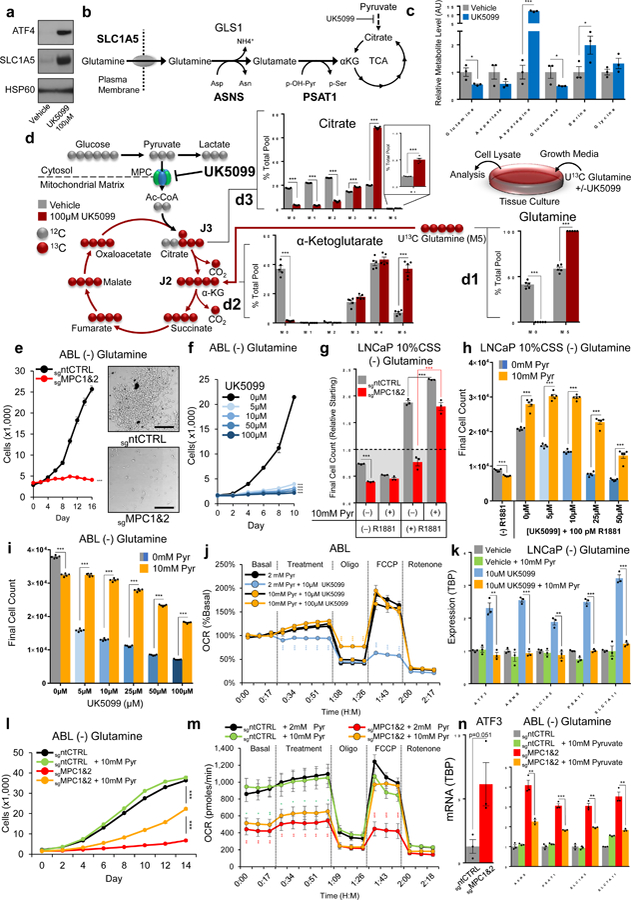
a, Glutamine uptake capacity (SLC1A5) and ISR activation (ATF4) were assessed by immunoblotting ABL cells after 72 hours of MPC inhibition; experiment performed 3 independent times with similar results and a representative blot with one biological replicate per lane is illustrated. b, Model depicts glutamine uptake and subsequent transamination reactions to incorporate glutamine into the TCA as α-ketoglutarate. c, Flux along the proposed pathway in (b) was examined by treating ABL cells (vehicle or 100µM UK5099) in amino-acid free HBSS for two hours and assessing selected metabolites using mass spectroscopy. d, Glutamine incorporation into TCA metabolite pools was assessed by pretreating ABL cells with 100µM UK5099 or vehicle 2 hours prior to the addition of U13C glutamine. Isotopomeric distribution for indicated metabolites was measured after 48 hours using mass spectroscopy. e,f, Cellular proliferation in glutamine-restricted conditions was assessed in Cas9-modified ABL cells (e) or during pharmacologic MPC inhibition (f). Scale bar is 250µm. g,h, Androgen-mediated cellular proliferation in glutamine-restricted conditions was assessed in Cas9-modified LNCaP cells (g) or during pharmacologic MPC inhibition (h). i-k, Cellular proliferation (i), maximal oxidative capacity (j), and ISR induction (k) were assessed in ABL cells following the addition of sodium pyruvate during pharmacological MPC inhibition. l-n, Cellular proliferation (l), maximal oxidative capacity (m), and ISR induction (n) were assessed in Cas9-modified ABL cells following the addition of sodium pyruvate. n=3 independent wells for measurements in c, g, k, and n. n=5 independent wells for measurements in d-f, h-j, l, and m. Data in bar and line graphs are represented as the mean ± SEM. Statistical analysis was performed using a two-tailed Student’s t-test *p < 0.05, **p < 0.01, ***p < 0.001.
To examine flux along this pathway during MPC inhibition, we placed cells in amino-acid free HBSS and measured relevant metabolite levels (Fig 5c). Consistent with increased pathway flux, MPC inhibition increased asparagine and serine content while decreasing glutamine and glutamate content. To directly examine glutamine content in the TCA during MPC inhibition, we turned to isotopomeric metabolic flux analysis using U13C-labelled glutamine. In vehicle-treated cells, about half (41%) of the glutamine pool was detected as the M0 isotopomer, indicating this glutamine was synthesized from endogenous processes rather than exogenous uptake (Fig. 5d1). Likewise, 35% of the α-kg pool and 17% of the citrate pool were endogenously synthesized and contained no carbon from exogenous 13C glutamine (Fig. 5d2–3). In agreement, M5 α-kg derived directly from exogenous glutamine made up 7% of the α-kg pool, and M4 citrate made up 20% of the citrate pool. Collectively, these measurements demonstrate limited incorporation of exogenous glutamine into the TCA in basal growth conditions. In contrast, during MPC inhibition, endogenously synthesized glutamine (M0) was undetectable while exogenously-derived M5 α-kg increased to 37% and M4 citrate increased to 68%. Overall, these isotopomeric distributions are consistent with a continuous influx of exogenous glutamine into the TCA during MPC inhibition and suggests enhanced glutamine reliance in this setting.
In line with the idea that MPC suppression increases glutamine reliance, we found glutamine restriction during MPC inhibition amplified TCA metabolite depletion (Supplementary Fig. 5a) and ATF4 activation (Supplementary Fig. 5b, c). Likewise, cells grown in the absence of glutamine or in the presence of a glutaminase inhibitor were dramatically sensitized to MPC inhibition (Fig. 5e, f, Supplementary Fig. 5d–f). Notably, AR positive models that express the MPC were able to grow in the absence of glutamine while AR negative models lacking significant MPC expression suffered proliferative arrest (Supplementary Fig. 5g). The addition of 2mM of each TCA intermediate to growth cultures failed to rescue MPC inhibition, suggesting these cells are not equipped to import and assimilate exogenous TCA intermediates (Supplementary Fig. 5h). However, the addition of 2mM glutamate or alanine resulted in a partial and near-complete rescue, respectively (Supplementary Fig. 5i) Of critical importance, metabolic bypass of the MPC using supraphysiologic pyruvate supplementation (Fig. 5g, h) augments AR-driven proliferation and rescues characterized phenotypes resulting from experimental MPC inhibition (Fig. 5i–n, Sup. Fig. 5j–l). Indeed, pyruvate supplementation not only rescues, but increases proliferation, positioning the MPC as a bona-fide rate limiting component of AR-driven metabolism (Fig 5g–i). Last, accumulating evidence suggests lactate is an important carbon source for tumor TCA metabolism in-vivo37,38. Exogenous lactate must be converted to pyruvate prior to oxidation in the TCA, but it is not clear if pyruvate is converted in the cytosol or the mitochondrial matrix and there is evidence for both39,40. However, in agreement with a recent report41, we found MPC inhibition in our models prevented lactate oxidation (Supplementary Fig. 5m, n), suggesting MPC activity is required for TCA incorporation of exogenous lactate following cytosolic lactate-to-pyruvate conversion.
Pharmacologic MPC Inhibition Suppresses AR-Driven PCa Growth
In contrast to other solid malignancies, human prostate tumors are not glucose avid and may yet rely on mitochondrial OxPhos14,15. Therefore, while fluorodeoxyglucose positron emission tomography (FDG-PET) studies are not useful for primary disease detection or monitoring42, new clinical imaging approaches, specifically hyperpolarized [1-13C] pyruvate imaging43, are emerging that can provide new insight into the unique metabolic properties of PCa. To assess the pyruvate avidity of our models in-vivo, we implanted mice with VCaP or ABL xenografts and examined metabolic characteristics of tumors in real time using hyperpolarized [1-13C] pyruvate imaging. Like human PCa, we found these tumors were pyruvate avid with similar pyruvate to lactate conversion (Fig. 6a). While hyperpolarized [1-13C] pyruvate imaging allowed us to assess tumor pyruvate uptake, the 1-13C label on pyruvate is lost as CO2 following mitochondrial import, preventing subsequent TCA assessment using this method. Therefore, to confirm MPC activity was operant and targetable in our tumor models in-vivo, we implanted tumor bearing mice with jugular venous catheters and, following a 6 hour fast, infused U13C glucose for 6 hours with or without UK5099. Mice maintained similar blood glucose levels during the infusion (Supplementary Fig. 6a), and, consistent with MPC inhibition, tumors from mice infused with UK5099 contained significantly more M0 citrate and less higher order citrate isotopomers despite similar G6P/F6P labelling (Fig. 6b1, 2). These results suggest these xenograft models display similar metabolic characteristics as human PCa and confirm MPC activity is conserved and targetable in models of PCa in-vivo.
Figure 6. MPC inhibition suppresses tumor growth in preclinical models of AR-driven PCa.
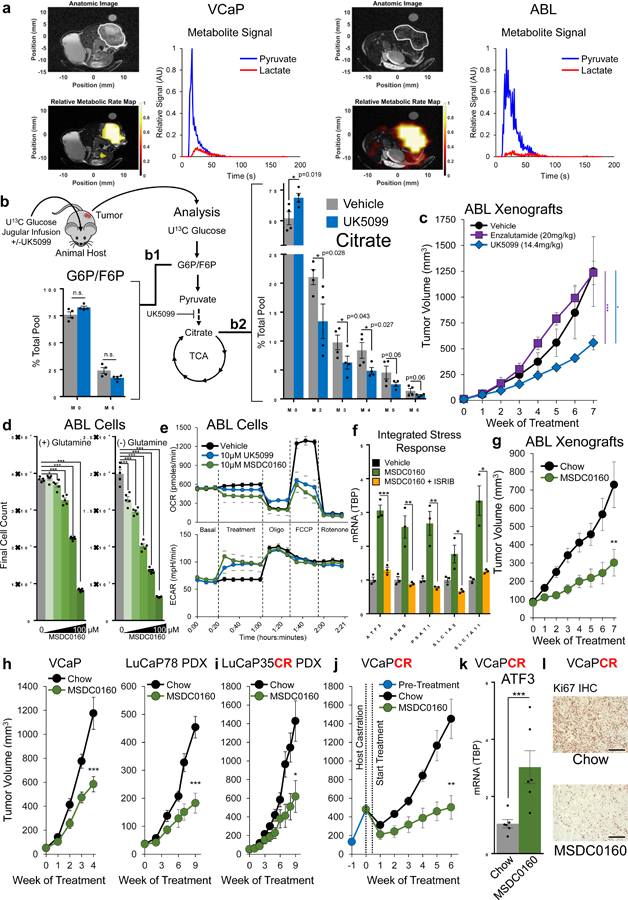
a, In-vivo metabolism of VCaP and ABL xenografts was assessed using 1-13C Hyperpolarized pyruvate imaging; n=2 tumors of each model with one representative imaging experiment shown. b, Mice harboring VCaP xenografts were infused with U13C glucose (30mg/kg/min) mixed with vehicle or UK5099 (3mg/kg/h) for 6 hours. Metabolites were analyzed using mass spectroscopy. n=4 tumors per treatment. c, Mice harboring ABL xenografts were treated via I.P. injection every other day and tumor growth was monitored; n=4 (enzalutamide) or n=5 (vehicle, UK5099) tumors per treatment. d, Proliferation of ABL cells in response to 0, 5, 10, 25, 50, or 100µM of the MPC inhibitor MSDC0160 with or without glutamine. n=5 independent cultures per treatment. e, Extracellular metabolic fluxes in ABL cells were assessed in response to treatment with MSDC0160 or UK5099. n=5 independent wells per treatment. f, ISR activation was assessed via qPCR in ABL cells treated for 6 hours. n=3 independent cultures per treatment. g, Castrated mice harboring ABL xenografts were fed a diet milled with MSDC0160 designed to deliver 30mg/kg/day or a matched control diet and tumor growth was monitored. n=12 chow and 13 MSDC0160 animals. h, Intact mice harboring the indicated tumor were fed the control or MSDC0160 diet and tumor growth was monitored. VCaP n=9 chow, 11 MSDC0160. LuCAP78 n=8 per treatment. i, Castrate mice harboring LuCAP35CR PDX xenografts were fed the control or MSDC0160 diet and tumor growth was monitored. n=7 mice per treatment. j, VCaP tumors were implanted in intact mice and grown for 3 weeks, at which time mice were castrated and randomized to the control or MSDC0160 diet and tumor growth was monitored. n=6 animals per treatment arm. k, ISR activation in the tumors from (j) was assessed using qPCR to measure ATF3 mRNA. n=6 animals per treatment arm. l, Representative Ki67 staining of VCaPCR tumors in (j) Scale bar is 200 µm. Ki67 staining is quantified in Sup. Fig. 6f. Data in bar and line graphs are represented as the mean ± SEM. Statistical analysis was performed using a one-tailed (panel b) or two-tailed (all other panels) Student’s t-test: n.s. not significant, *p < 0.05, **p < 0.01, ***p < 0.001.
To examine the impact of MPC suppression on tumor growth, we treated mice harboring ABL tumor xenografts with UK5099 or the antiandrogen enzalutamide. While enzalutamide treatment did not impact growth of this castrate-resistant xenograft, UK5099 treatment resulted in a significant reduction in tumor volume (Fig. 6c). UK5099 treatment was well-tolerated, and animals treated with this drug maintained weight and did not display any obvious abnormalities in feeding or behavior (Supplementary Fig. 6b). In contrast to mice treated with enzalutamide, UK5099-treated animals did not display prostate regression, suggesting MPC inhibition is not intrinsically deleterious to normal prostate tissue (Supplementary Fig. 5c). However, recognizing the limited translational potential implicit in the use of a tool compound such as UK5099, we transitioned subsequent in-vivo experiments to a recently developed clinically viable small molecule MPC inhibitor, MSDC0160.
MSDC0160 is a PPAR-γ-sparing thiazolidinedione (TZD) in clinical development for Alzheimer’s disease44 and type 2 diabetes45 with therapeutic promise in models of Parkinson’s disease46. Similar to UK5099, MSDC0160 inhibits PCa cell growth (Fig. 6d), restricts basal and maximal OCR, increases ECAR (Fig. 6e), and elicits the ISR (Fig. 6f). MSDC0160 is orally bioavailable and the compound itself is not taste aversive to mice, allowing us to deliver MSDC0160 milled into an animal diet. Similar to the results observed with UK5099 treatment, ABL tumor growth in castrate mice maintained on an MSDC0160 diet was suppressed compared to mice maintained on a matched chow diet (Fig. 6g). We applied this experimental approach to hormone-responsive, AR-driven VCaP and LuCaP78 PDX xenografts and again found the MSDC0160 diet inhibited xenograft growth (Fig. 6h). Similarly, MSDC0160 inhibited tumor growth in AR positive, castrate-resistant LuCaP35CR PDXs (Fig. 6i) Last, we implanted VCaP xenografts into a cohort of intact animals, allowed tumor establishment, then castrated the cohort and randomized animals to MSDC0160 or a matched control diet. Castrate-resistant outgrowth was disrupted in animals maintained on the MSDC0160 diet (Fig. 6j, Supplementary Fig. 6d). Similar to in-vitro findings, we found evidence for activation of the ISR in MSDC0160-treated tumors compared to control tumors (Fig. 6k, Supplementary Fig. 6e). Likewise, Ki67 staining was markedly decreased in tumors from mice maintained on the MSDC0160 diet (Fig. 6l, Supplementary Fig. 6f), suggesting delayed cell cycle progression resulting from ISR activation. We found no evidence for overt treatment-associated toxicity, as animals fed the MSDC0160 diet maintained weight (Supplementary Fig. 6g), and a pathological review of vital organs and the urogenital tract at the conclusion of the experiment revealed no obvious abnormalities (Supplementary. Fig. 6h). Overall, these experiments demonstrate MPC suppression using a clinically viable small molecule suppresses tumor growth in several preclinical models of hormone-responsive and castrate-resistant PCa.
Discussion
The metabolic properties of the prostate gland and PCa15 position the MPC to facilitate oncogenic metabolism. In contrast to all other tissues, normal prostate epithelium produces and secretes citrate through a physiologic truncation of the TCA at the level of aconitase14. Because citrate is produced from the condensation of oxaloacetate and pyruvate-derived acetyl-CoA in the mitochondrial matrix, mitochondrial pyruvate import is critical to ensure an abundant supply of pyruvate to fuel citrate production. Thus, AR’s regulation of the MPC in the setting of PCa may stem from AR’s regulation of citrate biosynthesis in normal prostate tissue. During oncogenic transformation, zinc depletion de-represses aconitase and enzymatically unifies the TCA47, enabling AR-dependent metabolic reprogramming to fuel tumor growth and progression7. The sum of our data suggest MPC activity is a necessary component of the AR-driven metabolic program that enables the growth of PCa in the hormone-responsive and castrate-resistant stages of the disease. In contrast, AR negative prostate cancer models lack MPC expression, are unresponsive to pharmacologic MPC inhibition, and require glutamine for proliferation in-vitro. These observations suggest fundamental differences in the metabolic underpinnings of AR positive and AR negative prostate cancer. The mechanisms responsible for the apparent loss of MPC expression in AR negative models remain to be clarified, but may relate to loss of AR-dependent transcriptional programs that normally drive tissue differentiation. This model reconciles our findings with principles set forth by Rutter and colleagues, who have reported MPC expression is maintained in differentiated epithelia but decreases during oncogenic transformation13,48. Regardless, our observations fill a critical conceptual gap in the understanding of the metabolic underpinnings of AR-driven PCa, suggesting AR regulation of the MPC enables glycolytic flux to be funneled directly into mitochondria to fuel the TCA metabolism that gives rise to the increased OxPhos and lipogenesis characteristic of PCa.
Acute disruption of MPC flux interrupts TCA outputs, resulting in a multi-faceted stress response that delays cell cycle progression and attempts to salvage TCA metabolism by coordinating increased uptake and assimilation of glutamine. Previous work established glutamine oxidation maintains TCA metabolism during MPC suppression36, and the current study identifies the predominant cell signaling mechanisms likely underpinning this process. Functional glutamine restriction markedly enhances the effect of MPC disruption and future work will be aimed at identifying productive metabolic inhibitor combinations for therapy. Of particular note, glutaminase inhibitors including CB839 are under clinical investigation and a recent report described a promising new SLC1A5 inhibitor, V-9302, with single agent activity in a variety of preclinical tumor models49. In our studies, experimental MPC inhibition using a clinically viable MPC inhibitor, MSDC0160, suppressed tumor growth in a variety of hormone-responsive and castrate-resistant AR-driven models of PCa. These results add to the accumulating evidence suggesting inhibition of the MPC may confer therapeutic benefit in neurodegenerative and metabolic diseases46,50–52 as well as cancer41,53. Moreover, with the recent discovery that TZD-class compounds (e.g. PPAR-γ-sparing MSDC0160) directly inhibit the MPC54, our findings partially reconcile long-standing observations that TZDs can inhibit PCa growth through PPAR-γ-independent mechanisms55–57.
Our metabolic tracing studies suggest MPC activity fuels TCA metabolism in prostate cancer cells in-vitro, and this metabolic dependency is likely maintained in-vivo. These results are aligned with increasing evidence that metabolites derived from glycolytic metabolism, rather than glutamine, often supply TCA metabolism in-vivo32,58. Recent evidence suggests glucose can feed the TCA via circulating lactate37,38, and while our in-vivo experiments do not allow us to differentiate between glucose and lactate as the carbon source that is actually entering the tumor, the distinction may become inconsequential because glucose and lactate are primarily converted to pyruvate prior to mitochondrial entry via the MPC39. Indeed, MPC inhibition in PCa cells suppressed lactate oxidation in our models and is known to interrupt lactate uptake in models of cervical, pharyngeal, and breast cancer41. Therefore, MPC blockade may, in theory, prevent mitochondrial utilization of exogenously-derived glucose, pyruvate, and lactate in PCa as well as other solid tumors.
In summary, AR regulates the MPC in prostate adenocarcinoma, and MPC inhibition disrupts metabolism and inhibits growth of hormone-dependent and castrate-resistant models of PCa. While our current data suggests additional AR-independent transcriptional inputs to the MPC2 locus in castrate-resistant disease, these findings begin to address a critical unmet clinical need for treating the most common lethal form of prostate cancer. Future efforts will be focused on designing rational combinatorial therapies to maximize the therapeutic effect of MPC suppression and identifying patients that are most likely to benefit from these approaches.
Experimental Procedures
Ethical Compliance Statement.
Studies involving human specimens were approved by the Institutional Review Board at Baylor College of Medicine. Human prostatectomy specimens were collected after obtaining informed consent and specimens were acquired through the Human Tissue Acquisition and Pathology Core of the Dan L. Duncan Cancer Center at Baylor College of Medicine. All experiments using animals were approved by the Baylor College of Medicine Institutional Animal Care and Use Committee (IACUC), an Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) International-approved committee.
Clinical datasets and bioinformatic nomination.
Publicly available tumor data sets were accessed from the TCGA data portal (https://portal.gdc.cancer.gov/) and the cBioPortal for cancer genomics (http://www.cbioportal.org/). Spearman scores were generated using KRT8, KRT18, and KLK3 in the TCGA prostate cancer RNA-Seq database. Next, gene lists were rank-ordered and candidate genes were identified as those present in the top 5% of each Spearman correlation list. Ref-Seq gene summaries and Gene Rifs were collected from the National Center for Biotechnology Information (NCBI, https://www.ncbi.nlm.nih.gov/) for keyword screening. Next, gene lists were filtered using two keyword screens. The first keyword screen included the terms “Metabolism”, “Metabolic,” “Transporter,” “Carrier,” and “Enzyme.” The second keyword screen included the terms “Glucose,” “Glycolysis,” “Pyruvate,” “Lactate,” “Mitochondria,” “Citric Acid Cycle,” “Fatty Acid,” “Beta Oxidation,” “Amino Acid,” “Plasma Membrane,” and “Mitochondrial Membrane.” Genes with all or any part of these terms were selected. Last, remaining genes were rank-ordered based on their mRNA expression fold-change in benign prostate specimens compared to prostate tumors in the TCGA dataset. MPC2 was our top candidate with associated Keyword hits of “Carrier” and “Mitochondria”, a relative fold-change of 1.73 in prostate tumors compared to benign prostate tissue in the TCGA dataset, and a statistically significant disease-free survival association from a well-characterized clinical cohort59 as well as the TCGA dataset. Other nominated genes included PRDX4, ACY1, SMPD2, FAAH, and PNKP. The full list is available in Supplementary Data 1. Data from the Cancer Cell Line Encyclopedia for MPC2 (previously BRP44) and MPC1 (previously BRP44L) was collected from the CCLE data portal at: https://portals.broadinstitute.org/ccle/home.
Data set accession numbers from specific publications are as follows:
GSE21032 (Taylor et. al. 2010) (Fig. 1b, bottom)
GSE36139 (Barretina et. al. 2012) (Fig. 1h, Supplementary Fig. 1f)
GSE65478 (Stelloo et. al. 2015) & GSE70079 (Pomerantz et. al 2015) (Fig. 2j)
Prostate cancer models, specimens, and tissue culture.
RWPE1, 22RV1, LNCaP, DU145, and PC3 cell lines were acquired from American Type Culture Collection (ATCC). LAPC4, originally generated by R. Reiter et. al., and ABL, originally generated by Z. Culig et. al., were a kind gift from Nicholas Mitsiades, who also provided protein lysates from LuCaP58 and LuCaP78 PDX tumors. LuCaP35CR and LuCaP78 models were acquired from Eva Corey at the University of Washington. VCaP, originally generated by K. Pienta et. al., and C4–2, originally generated by G. Thalmann et. al. cells were a kind gift from Nancy Weigel at Baylor College of Medicine. Fresh human radical prostatectomy specimens were collected intra-operatively and immediately flash frozen in liquid nitrogen. Human cancer specimens contained a minimum of 70% cancer and benign tissues were free of cancer on pathological examination. The identity of all models was confirmed using short tandem repeat (STR) profiling at the M.D. Anderson CCSG-Characterized Cell Line Core (see Supplementary Data 4). All models were routinely screened for mycoplasma by the Baylor College of Medicine Tissue Culture Core (see Supplementary Data 4). Cell lines were maintained in ATCC-recommended growth media in a humidified 5% CO2 tissue culture incubator at 37°C. Cell lines without ATCC growth media recommendations were cultured as follows: LAPC4 – Iscove’s modified Dulbecco’s medium supplemented with 15% FBS and an additional 2mM glutamine. ABL – phenol-red-free RPMI supplemented with 10% CSS. C4–2 – RPMI supplemented with 10% FBS. No antibiotics or antimycotics were included in any growth media. No cell lines in this study are present in the database of commonly misidentified cell lines that is maintained by ICLAC and NCBI Biosample.
Small molecules.
Enzalutamide and AZD3965 were purchased from Selleck Chemicals. Dihydrotestosterone (DHT) was purchased from Cayman Chemical. R1881, UK5099, ISRIB, and BPTES were purchased from Sigma Aldrich. GW604714X was synthesized by David Gooden at Duke University. Oligomycin, FCCP, and Rotenone were purchased from Seahorse Biosciences. MSDC0160 was kindly provided under an MTA from Jerry Colca at the Metabolic Solutions Development Company.
Quantitative PCR.
Total mRNA from cells and tissue specimens was isolated using the RNeasy kit (Qiagen). mRNA was reverse transcribed to cDNA using the high capacity cDNA reverse transcription kit (Thermo Fisher). qPCR was performed using the StepOne Real-Time PCR system (Applied Biosystems) and qPCR master mix (Kapa Biosystems) with standard cycling parameters. TaqMan qPCR primer sets for human MPC1, MPC2, and KLK3 (PSA) were purchased from Thermo Fisher. Other qPCR Taqman sets were designed using the Universal Probe Library System (Roche) and are available in Supplementary Data 4.
Western blotting.
Total protein was extracted from cells and tissue specimens using modified NP40 lysis buffer (50mM Tris, 150mM NaCl, 1% NP40, 0.1% SDS, 5% Glycerol supplemented with cOmplete EDTA-free protease inhibitor and Phos-Stop tablets (Roche)). Tissue specimens were mechanically processed at cryogenic temperatures using a micro mortar and pestle. Lysates were then sonicated using a probe sonicator to shear genomic DNA and release lipid-membrane-bound proteins. The resulting specimen was centrifuged (>10,000 RPM) for 10 minutes at 4°C in a microfuge and supernatant was collected. These total protein lysates were quantified (Thermo-Pierce BCA protein assay) for equal loading. Lysates were run on NuPAGE Novex Bis-Tris 4–12% polyacrylamide gels (Thermo Fisher) with MES or SDS running buffer (Thermo Fisher) and transferred to 0.22 micron PVDF (Millipore). Blots were incubated overnight in primary antibody in 5% w/v blotting-grade blocker dissolved in PBS-T and developed using HRP-labelled secondary antibodies and Amersham ECL or Amersham ECL prime chemiluminescent detection reagent. A list of antibodies used in this manuscript is available in Supplementary Data 4 and includes links to manufacturer and external antibody validation data.
Chromatin immunoprecipitation.
LNCaP cells were plated in 10% FBS RPMI and allowed to grow for 48 hours, at which point growth media was replaced with 10% CSS RPMI for 48 hours. Next, cells were treated as indicated for 16 hours (enzalutamide treatment groups were pre-treated with enzalutamide for two hours to ensure androgen blockade was in place prior to the addition of R1881). Chromatin was isolated (Active Motif high sensitivity ChIP kit) and sheared (Diagenode Bioruptor bath sonicator) using 20 cycles (30 seconds on, 30 seconds off). Chromatin immunoprecipitation was performed using anti-AR (active motif) or normal rabbit IgG (Millipore). Immunoprecipitated DNA was amplified using PCR or qPCR and primer sets are available in Supplementary Data 4. Data are reported as fold-change of AR binding normalized to input and IgG control immunoprecipitation.
In-Vitro Transcription (IVT)
Generation of IVT templates.
The MPC2 and PSA/KLK3 fragments were PCR-amplified from LNCaP genomic DNA with Phusion high-fidelity DNA polymerase (New England Biolabs). The primer sequences used to generate the fragments are available in Supplementary Data 4. Each fragment was gel-purified and was used to further PCR amplification in bulk using OneTaq DNA polymerase (NewEngland Biolabs), precipitated, and reconstituted into chromatin using H1-depleted core histones purified from HeLa cells. The chromatinized templates were used in IVT reactions.
Chromatin reconstitution.
8 μg of each OneTaq-amplified fragment was mixed with 12 μg core histones in the presence of 2M NaCl in 1x CRB (10 mM Tris-HCl (pH 7.5), 1mM EDTA, 0.05% Igepal) in final 15 μl, and let stand at room temperature (RT) for 20 minutes. Thereafter, the nucleohistone mixture was diluted by adding 1xCRB every 20 minutes in the following sequence: 5, 5, 5, 7.5, 12.5, 25 μl, and finally 75 μl (with 0.5mg/ml BSA, to final 0.25mg/ml). The chromatin thus prepared is stable for years at 4oC. The chromatin preparation was verified by limited micrococcal nuclease (MNase) digestion. 0.5 μg of chromatin was digested with 25U and 50U of MNase (Worthington) in final 2mM CaCl2 for 1 minute at RT. Digestion was stopped by adding EDTA to final 10 mM. The digests were deproteinized for a 15-minute digestion with Proteinase K followed by phenol-chloroform extraction and ethanol precipitation. The digests were run on a 2.5% agarose gel in 1xTG buffer at 4oC, stained with EtBr.
Nuclear Extract (NE) preparation.
NE was prepared as follows. LNCaP cells were grown in 10% FBS RPMI to near-confluency. Cells were scraped off the plates (6 × 15cm plates) and were washed twice with 10 ml of cold phosphate buffered saline (PBS), and swollen in 10 ml of Buffer A (10 mM HEPES, pH7.9 supplemented with fresh 0.5 mM DTT/0.5 mM PMSF) for 20 minutes on ice. The swollen cells were snap-frozen in liq. N2 in 1 ml aliquots, and stored at −80oC. To prepare NE, the aliquots were thawed on ice and nuclei were pelleted at 3500xg for 10 min at 4oC. The pellet was resuspended in ½ packed cell volume (pcv) of LSB (20mM KCl, 20 mM HEPES, pH7.9 and 0.5 mM DTT/0.5 mM PMSF), to which ½ pcv of HSB (1.2M KCl, 20 mM HEPES, pH7.9 and 0.5 mM DTT/0.5 mM PMSF) was added dropwise. The Lysate was rotated in the cold room for 30 minutes, and then clarified by centrifugation at 15,000xg for 10 min at 4oC. The supernatant was diluted with equal volume of no-salt Buffer D (20 mM Hepes-KOH (pH 7.9), 20% glycerol, 0.2 mM EDTA). Precipitated proteins were removed by centrifugation at was collected after centrifugation 15,000xg for 10 min at 4oC, and the supernatant was collected as NE.
Immunodepletion.
Pierce Protein A/G magnetic beads (88802; 3μl slurry per μg antibody) were washed and blocked with 1mg/ml BSA in 1x Buffer D (with 100 mM KCl) for 30 minutes in cold room. The beads were collected, resuspended in 200 μl Buffer D, and antibody was added (5 μg per mg NE). Anti-AR antibody (ActiveMotif 39781) and control Rabbit IgG (sc-2027) were used. The antibodies were bound to the beads overnight in the cold room. Next day, the beads were collected and resuspended in 0.25mg NE and rotated in the cold room for 2hrs. The NE was collected and added to fresh antibody-bound beads for another 2 hrs. The final NE thus immunodepleted was used in IVT reactions. Extent of immunodepletion was examined by immunoblotting.
In-Vitro Transcription.
IVT conditions were as in Panigrahi et al.23. 0.2 pmole of the chromatinized template was incubated with 40 µg of NE in a final buffer condition of 12 mM Hepes-KOH (pH 7.9), 12% glycerol, 60 mM KCl, 12 mM MgCl2, 0.12 mM EDTA, 0.3 mM DTT, 1 mM ATP, 0.9 mM acetyl CoA, and 5nM R1881 in a final volume of 45 μl at room temperature. After 25 min, 5 µl of 5 mM NTP mixture (GTP, CTP, TTP) was added to the reactions and shifted to 37 oC for 45 minutes. 250 ul of Tri-Reagent was added to each reaction to extract RNA. 15 μl of BAN (4-bromoanisole) was added for efficient phase separation. Aqueous phase (RNA) was collected and precipitated with 150 µl of isopropanol for 20 min at RT; 20 µg of glycogen was added to aid precipitation. RNA precipitates were collected at 15,000 rpm in a table-top microcentrifuge for 15 min at 4oC; washed with 75% ethanol (prepared with DEPC-treated H2O; Ambion) at RT, air-dried, and dissolved in 35 µl DEPC-treated H2O at 55oC for 10 min. The RNA samples were digested with 1 µl of DNase (Turbo DNA-free kit; Ambion) along with 4 µl of DNase buffer as recommended by the manufacturer. DNase digestion was carried out for 1 hr at 37oC, and the reactions were stopped by adding the DNase inactivation reagent. Two µl of the resultant RNA sample was used in each One-step RT-qPCR reaction using primer pairs downstream of the TSS of MPC2 (For: GAGAATTGTGCGGCATCATCTTTA, Rev: CATGTTGGAGAAGGGAAAGTGAAG) or PSA/KLK3 (For: CTATCCCAGAGACCTTGATGCTTG, Rev: CATTTGTTGTCTCAGGCCAGATAG). To verify that no significant “carry-over” DNA contamination remained after DNase digestion, each sample was also subjected to parallel qPCR reactions without reverse transcriptase. The CT values were normalized to an amplicon from 10 fmoles of template DNA, which is equivalent to the template amount in the RNA sample (if extracted alongside RNA, and was not destroyed by DNase treatment). The transcript levels were further normalized to the ΔIgG reaction with template M1 (or P1, the templates with no enhancers). The resultant values were expressed as “relative transcription”.
Proliferation assays.
Depending on the experiment, between 2,500 and 10,000 cells per well were plated in flat-bottom, optically pure 96 well plates (Greiner bio-one 655090). Cells were allowed to attach for 12–24 hours, then treatment was applied. Plates were serially imaged during the experiment (beginning immediately after treatment) using a Celigo S Image Cytometer (Nexcelom Bioscience) and Celigo image analysis software was used to count cells. For each experiment, the same image analysis algorithm was applied to all wells for all time points.
Fluorescence microscopy.
LNCaP cells were plated in 10% CSS onto acid-etched, poly-D-lysine coated coverslips and allowed to attach for 24 hours before treatments. Cells were treated for 96 hours, then washed with ice cold PBS and fixed with 4% paraformaldehyde. Sodium borohydride was used to quench autofluorescence derived from residual paraformaldehyde. Cells were stained using DAPI (Sigma) for nuclei and LipidTOX green (ThermoFisher) for neutral lipids. Imaging was performed with the GE Deltavision deconvolution microscope. Lipid staining was quantified using ImageJ.
Transmission Electron Microscopy.
Cells were fixed in plastic petri plates in Modified Karnovshy’s fixative60, washed in 0.1M cacodylate, stained with 0.1% tannic acid and postfixed in 1% OsO4 + 0.8% potassium ferricyanide. After an en bloc stain with saturated aqueous uranyl acetate, cells were dehydrated through a gradient series of ethanol to 100% then infiltrated with a gradient series of resin to 100% LX112. Cells were embedded in the plates using LX112 resin, and polymerized at 45°C overnight, then 60°C for 2 days. Cells were easily separated from the warm plastic plates and mounted for ultra-thin sectioning. Ultra-thin sections were cut at 55–60nm using a Diatome Ultra45 knife on a Leica UC-7 ultra-microtome. The sections were viewed on a Hitachi H7500 transmission electron microscope and images were captured using an AMT XR-16 digital camera and AMT Image Capture, v602.600.51 software.
Extracellular flux analysis.
Cells were plated on XF24 well cell culture microplates (Agilent) to form a consistent and confluent monolayer at the time of experimental measurements. Extracellular flux analysis was performed using the Seahorse XF cell mitochondrial stress test kit (Seahorse Bioscience P/N 103015–100), with UK5099 injection sequenced into the manufacturer’s protocol. Experimental media was XF DMEM supplemented with glucose (10mM), pyruvate (1mM), and glutamine (2mM) except as noted. Final concentration of oligomycin used in the experiment was 2µM, FCCP was 2µM and Rotenone/antimycin was 0.5µM. Extracellular flux experiments were performed on a Seahorse XF24 Analyzer.
Lactate Secretion:
Lactate content in growth media was assessed using an enzyme-based colorimetric kit (Biovision) according to the manufacturer’s protocol.
Histology and Immunohistochemistry.
Tissue specimens were fixed in neutral formalin buffered saline (10%) and embedded in paraffin. Hematoxylin and eosin staining was performed using standard methods and tissue specimens from experimental animals were reviewed in a blinded fashion by a clinical pathologist (M. M. I.) For Ki67 staining, 3–4 micron tissue sections were cut from paraffin blocks and baked overnight in a dry slide incubator, then deparaffinized on a Shandon-Lipshaw Varistain using a series of incubations in xylene, ethanol, then water. Antigen retrieval was achieved by incubating slides in Tris-HCL 9.0 AR buffer in a T-FAL OPTIMA pressure cooker. Slides were rinsed, then endogenous peroxidase was blocked by immersing slides in 3% hydrogen peroxide for 5 minutes. Following washes in nanopure water and TBS-20, primary antibody was applied at a dilution of 1:200 (Ki67, MIB-1 Clone, Dako). Following primary antibody incubation, slides were washed and then incubated with envision-labelled polymer-HRP Anti-Mouse (Dako). Slides were washed, then DAB+ solution (DakoCytomation) was added for a 15-minute incubation, after which slides were rinsed with nanopure water. Chromogen signal was enhanced using DAB Sparkle Enhancer (Biocare). Slides were washed, then counterstained with Harris Hematoxylin, dehydrated, cleared, then mounted using Cytoseal (VWR).
Reactive Oxygen Species Measurement:
ROS content was assessed using 2’,7’-dichlorodihydrofluorescein diacetate (H2DCFDA) following the manufacturer’s protocol (abcam). Data was collected using a plate reader (Biotek). Cell-free wells containing equivalent concentrations of UK5099 were used for background correction. tert-Butyl hydroperoxide (Sigma) served as a positive control for ROS.
Flow Cytometry.
Following treatment, cells were fixed, stained and permeabilized using a propidium-iodide based method61. DNA content was assessed using an Attune NxT Acoustic Focusing Cytometer and data was analyzed using the FlowJo software package.
RNA-Sequencing.
Total RNA was extracted using a TRIzol based kit (Zymo) and Poly-A RNA was purified from total RNA using Dynabeads Oligo dT25 (Invitrogen). The RNA-Seq library was generated using KAPA strand RNA-Seq library prep kit (KR-0934) and sequenced on a Hiseq2500 sequencer. Data was mapped using TopHat2 onto the human genome UCSC hg19, and quantified using Cufflinks and the GENCODE gene model. Pathway enrichment analysis was carried out using Gene Set Enrichment Analysis (GSEA) against the Molecular Signature Database (MSigDB) compiled pathway compendium. Analyzed data are available in Supplementary Data 3. Raw data are available under accession number GSE114708 on the NCBI GEO database.
Reverse-Phase Protein Array.
Cellular proteins were denatured by 1% SDS (with Beta‐mercaptoethanol) and diluted in five 2‐fold serial dilutions in dilution lysis buffer. Serial diluted lysates were arrayed on nitrocellulose‐coated slides (Grace Bio Lab) by Aushon 2470 Arrayer (Aushon BioSystems). Total 5808 array spots were arranged on each slide including the spots corresponding to serial diluted: 1) “Standard Lysates”; 2) positive and negative controls prepared from mixed cell lysates or dilution buffer, respectively.
Each slide was probed with a validated primary antibody plus a biotin‐conjugated secondary antibody. Only antibodies with a Pearson correlation coefficient between RPPA and western blotting of greater than 0.7 were used for RPPA. Antibodies with a single or dominant band on western blotting were further assessed by direct comparison to RPPA using cell lines with differential protein expression or modulated with ligands/inhibitors or siRNA for phospho‐ or structural proteins, respectively.
The signal obtained was amplified using a Dako Cytomation–Catalyzed system (Dako) and visualized by DAB colorimetric reaction. The slides were scanned, analyzed, and quantified using a customized software to generate spot intensity. Each dilution curve was fitted with a logistic model (“Supercurve Fitting” developed by the Department of Bioinformatics and Computational Biology in MD Anderson Cancer Center, “http://bioinformatics.mdanderson.org/OOMPA”). This fits a single curve using all the samples (i.e., dilution series) on a slide with the signal intensity as the response variable and the dilution steps are independent variable. The fitted curve is plotted with the signal intensities – both observed and fitted ‐ on the y ‐ axis and the log2 ‐ concentration of proteins on the x‐axis for diagnostic purposes. The protein concentrations of each set of slides were then normalized for protein loading. Correction factor was calculated by: 1) median ‐ centering across samples of all antibody experiments; and 2) median-centering across antibodies for each sample. RPPA Data is available in Supplementary Data 2.
Cas9 MPC knockout.
sgRNA targeting sequences in the first exon of MPC1 and MPC2 were designed using the online tool from Feng Zhang’s lab (http://crispr.mit.edu). A non-mammalian targeting control sgRNA sequence with similar GC content was generated by scrambling the sequence of the MPC2 guide and confirming no mammalian recognition sites (≥4 mismatches) using Cas-OFFinder62. Guide sequences are available in Supplementary Data 4. Guide sequences were cloned into the lentiCRISPR v2 plasmid (Feng Zhang, Addgene plasmid #52961) and lentiviral particles were generated in 293t cells (ATCC) using packaging plasmids pCMV-VSV-G (Bob Weinberg, Addgene plasmid #8454) and psPAX2 (Didier Trono, Addgene plasmid #12260). ABL cells were selected for vector incorporation using 1 µg/mL puromycin (Gibco). Cas9 expression and MPC disruption were confirmed via Western blotting.
Metabolite measurements.
Metabolites from cell lines and quality control standards were extracted as follows. Briefly, cell pellets were thawed at 4°C and subjected to at least three freeze-thaw cycles in liquid nitrogen and over ice to disrupt the plasma membrane. Next, 750 µL of ice-cold methanol:water (4:1) containing 20 µL of spiked internal standards were added to each cell and tissue extract. This was followed by sequential addition of ice cold chloroform and water in a 3:1 ratio to make the final ratio of water, methanol, and chloroform 1:4:3:1 (water:methanol:chloroform:water). Both organic (methanol and chloroform) and aqueous layers were separated individually and combined to remove cellular debris. Next, the extract was deproteinized using a 3 KDa molecular filter (Amicon Ultracel −3K Membrane, Millipore Corporation, Billerica, MA) and the filtrate containing metabolites was dried under vacuum (Genevac EZ-2plus, Gardiner, NY). Prior to mass spectrometry, the dried extracts were resuspended in identical volumes of injection solvent composed of water: methanol (50:50).Glycolytic intermediates, TCA intermediates, NAD+/NADH, reduced glutathione, and selected amino acids were measured using liquid chromatography coupled to mass spectroscopy (LC/MS) as described previously63,64.
For extraction of tumor metabolites, 750 μl of water/methanol (1:4) was added to 50 mg of snap-frozen tumor and samples were homogenized, then mixed with 450 μl ice-cold chloroform. The resulting solution was mixed with 150 μl ice-cold water and vortexed again for 2 minutes. The solution was incubated at –20°C for 20 minutes and centrifuged at 4°C for 10 minutes to partition the aqueous and organic layers. The aqueous and organic layers were combined and dried at 37°C for 45 minutes in an automatic Environmental Speed-Vac system (Thermo Fisher Scientific). The extract was reconstituted in a 500-μl solution of ice-cold methanol/water (1:1) and filtered through a 3-kDa molecular filter (Amicon Ultracel 3-kDa Membrane) at 4°C for 90 minutes to remove proteins. The filtrate was dried at 37°C for 45 minutes in a speed vacuum and stored at –80°C until MS analysis. Prior to MS analysis, the dried extract was resuspended in a 50-μl solution of methanol/water (1:1) containing 0.1% formic acid and then analyzed using multiple reaction monitoring (MRM). Ten microliters were injected and analyzed using a 6490 QQQ triple quadrupole mass spectrometer (Agilent Technologies) coupled to a 1290 Series HPLC system via selected reaction monitoring (SRM).
Separation of glycolytic and TCA intermediates.
Briefly, aqueous phase chromatographic separation was achieved using three solvents: water (solvent A), water with 5mM ammonium acetate (pH 9.9), and100% acetonitrile (ACN) (solvent B). The binary pump flow rate was 0.2 ml/min with a gradient spanning 80% B to 2% B over a 20 minute period followed by 2% B to 80% B for a 5 min period and followed by 80% B for 13 minute time period. The flow rate was gradually increased during the separation from 0.2 mL/min (0–20 mins), 0.3 mL/min (20–25 min), 0.35 mL/min (25–30 min), 0.4 mL/min (30–37.99 min), and finally set at 0.2 mL/min (5 min). Glycolytic and TCA intermediates were separated on a Luna Amino (NH2) column (3 µm, 100A 2 × 150 mm, Phenomenex), that was maintained in a temperature-controlled chamber (37°C).
Separation of selected amino acids, NAD+/NADH, and reduced glutathione.
Briefly, samples were delivered to the mass spectrometer via normal phase chromatography using either a 4.6mm x 10cm Amide XBridge HILIC column (Waters) or a Luna 3µm NH2 100A (Phenomenex) at 300µL/min. Separation was achieved beginning with 85% solvent B (HPLC-grade ACN with or without 0.1% formic acid) to 35% B from 0–3.5 minutes, 35% B to 2% B from 3.5–11.5 minutes, 2% B from 11.5 to 16.5 minutes, and 2% B to 85% B from 16.5–17.5 minutes to complete the separation. 85% B was held for 7 minutes to re-equilibrate the column at the end of the run and all columns used in this study were washed and reconditioned after every 50 injections.
Glycolytic and TCA intermediates were measured using negative ionization mode with an ESI voltage of −3500ev. Approximately 9–12 data points were acquired per detected metabolite. Selected amino acids, NAD+/NADH, and glutathione were measured using positive ionization mode with an ESI voltage of −4000ev and approximately 9–12 data points were acquired per detected metabolite. For all samples, ten microliters of sample were injected and analyzed using a 6495 QQQ triple quadrupole mass spectrometer (Agilent) coupled to a 1290 series HPLC system via Selected Reaction Monitoring (SRM). Data was analyzed using the MassHunter Workstation software package (Agilent).
ATP was measured from cell pellets using an enzyme-based colorimetric kit (Biovision) per the manufacturer’s protocol.
13C tracing experiments were performed with glucose- or glutamine-free, phenol-red-free RPMI containing 10% dialyzed FBS (Gibco). For glucose tracing, cells were plated and allowed to attach in basal growth media. Once cells had attached (~24–48 hours), cells were rinsed twice with PBS and growth media was replaced with glucose-free media for 12 hours. After 12 hours, glucose-free media was replaced with the equivalent media supplemented with 10mM U-13C glucose with or without UK5099. Cells were incubated for 48 hours, then washed twice with ice cold PBS and snap frozen in-situ by floating the tissue culture vessel on liquid nitrogen. Metabolites were extracted and measured as above. For glutamine tracing, the same procedure was followed except cells were starved for glutamine for 12 hours, and glutamine-free media was replaced with the equivalent media supplemented with 2mM U-13C glutamine.
Animal procedures.
All experiments used 6–8-week-old male Fox Chase SCID Beige mice (CB17.Cg-PrkdcscidLystbg-J/Crl) (Charles River). For tumor growth experiments, a time to event power analysis was conducted when possible prior to initiation of the experiment based on available in-vitro and in-vivo data. Mice were observed daily to screen for behavioral changes resulting from tumor burden or experimental treatment. Tumor growth was formally assessed once per week using a digital caliper and tumor volume was estimated using the formula (L2*W)/2. Per institutional guidelines, the sum of the two greatest tumor diameter measurements was not allowed to exceed 1.5cm. Tumors from all mice were included in the final analysis with the exception of one mouse in the enzalutamide treatment arm of Fig. 6C, which was found moribund and was euthanized prior to the experimental endpoint.
In the ABL xenograft experiment described in Fig. 6C, as well as PDX model experiments, we implanted tumor pieces PDX style. Briefly, Beige-SCID mice harboring established ABL flank tumors of similar volume were sacrificed and ABL tumors were collected and cut into small pieces (~20–25 mg). Regions of necrosis were removed, and pieces were then implanted into the flanks of recipient Beige-SCID mice and allowed to engraft for 3 weeks at which point they were randomized by tumor volume to treatment arms. For UK5099 and enzalutamide IP injections, the treatment solution contained the treatment compound(s) dissolved in 200 µL sterile phosphate-buffered saline with 2.5% 2-hydroxypropyl beta cyclodextrin (ChemCruz) and 9% DMSO (Sigma). Treatment was unblinded as each prepared drug solution had a characteristic appearance. Treatment was delivered every other day via intraperitoneal injection. Mice were sacrificed 48 hours after the final treatment dose. In selected mice, the prostate gland and seminal vesicles were microdissected and massed to confirm prostate regression in response to enzalutamide treatment. To generate ABL and VCaP xenografts, 2–5 million cells mixed 1:1 with high protein content Matrigel (Corning) were injected into the flanks of mice. Tumors were allowed to establish for 2 weeks, at which point animals were randomized to treatment arms. For MSDC0160 delivery, animals were offered a diet containing MSDC0160 designed to deliver 30mg/kg/day or a matched control diet. Tumor growth was monitored using digital calipers and tumor volume was calculated using the formula (L2*W)/2. Castration surgery in selected experiments was performed via an abdominal approach.
For tumor infusion experiments, venous catheters were surgically implanted into the jugular veins of mice harboring established VCaP tumors 3–4 days prior to U13C glucose infusions. Infusions were performed as in Davidson et. al.32 with minor modifications. Briefly, animals were fasted 6 hours prior to infusions, which began at 10:00am for all studies to minimize confounding variables related to circadian rhythms. Infusions were performed in free-moving conscious animals. U13C glucose was delivered at a constant infusion rate of 30mg/kg/min with or without UK5099, which was infused at 3mg/kg/hr for a total of 6 hours. At the end of the infusion, animals were briefly anesthetized with isoflurane then euthanized by cervical dislocation. Tissues were collected within 2–3 minutes and snap frozen using a BioSqueezer (Biospec) precooled with liquid nitrogen to rapidly quench metabolism. Specimens were stored at −80°C prior to metabolite extraction and analysis.
Dynamic Imaging of Hyperpolarized Pyruvate.
Twenty-six mg of [1-13-C]-pyruvic acid (Cambridge Isotopes) containing 1.5 mM Gadoteridol (Bracco Diagnostics) and 15 mM OX063 (GE Healthcare) were polarized using a HyperSense dissolution DNP system (Oxford Instruments). The polarized substrate was rapidly dissolved in 4 mL of 80mM NaOH, 50 mM NaCl, 0.1 g/L EDTA and 40 mM Trizma pH 7.6 (Sigma Aldrich) to yield a neutral, isotonic, 37°C solution of 80mM hyperpolarized [1-13C] pyruvate. Two hundred μL of the hyperpolarized pyruvate solution was administered to the animals via a tail vein catheter.
Imaging was performed on a 7T Biospec MR system with a 1H/13C dual-tuned volume coil (Bruker Biospin MRI) and a custom built 13C surface coil. Anatomic imaging was performed using a fast spin-echo (FSE) sequence. Dynamic 13C data was acquired using a previously described radial multi-band frequency encoding sequence65 and a constrained reconstruction algorithm66. The constrained reconstruction results were normalized and relative signal curves for hyperpolarized pyruvate and lactate were generated as well as apparent relative metabolic conversion rate maps which were co-registered with the anatomic images.
Statistical analyses.
Statistical analyses are described in each figure and were performed using GraphPad Prism or R. All experimental design and analysis is done in collaboration with our biostatistics team in the Dan L. Duncan Cancer Center, who assisted with statistical test selection and assured all data met the assumptions of the statistical test with regard to distribution and variance. When possible, sample sizes were selected based on estimates of effect size derived from available preliminary data. Differences between experimental groups were designated as significant at the 95% confidence level.
Reporting Summary.
Experimental design details (sample size determination, data exclusions, replication, randomization, etc.) are described in the associated Nature Research Life Sciences Reporting Summary.
Data Availability.
Analyzed RPPA data is available in Supplementary Data 2, analyzed RNA-sequencing data is available in Supplementary Data 3, and raw RNA-sequencing data is available under accession number GSE114708 on the NCBI GEO database. All other data described, analyzed, and represented in the figures present in this study are available from the corresponding authors upon reasonable request.
Supplementary Material
Acknowledgements
This study was supported by grants from the US National Institutes of Health (NIH): grant no. R21CA205257 (S.E.M.), F30CA196108 (D.A.B.), K01DK096093, R03DK105006, R01DK114356 (S.M.H.), R01CA211150 (J.A.B.), R01CA220297 (N.P.), F30AG050412 (H.M.S.), U01CA167234 (A.S.), PO1DK113954 and HD08818 (B.W.O.), the Prostate Cancer Foundation (S.E.M.), The Caroline Weiss Law Scholar Foundation (S.E.M.), The M.D. Anderson Physician Scientist Development Program (S.E.M.), and the Cancer Research and Prevention Institute of Texas (RP140021-P5 (J.A.B.) and RP140106 (MDACC)). D.A.B. is a C. Thomas Caskey Scholar. D.A.B., H.M.S., and M.P.H. are supported by the Baylor College of Medicine (BCM) Medical Scientist Training Program. S.M.H. is supported by the BCM Bridge to Independence Program and the Alkek Center for Molecular Discovery. M.P.H. is supported by the B.R.A.S.S. program, and the Robert and Janice McNair Foundation. V.P., N.P., A.S., K.R., and C.C. are supported by the CPRIT Core Facility Support Award RP120092, the NCI Cancer Center Support Grant P30CA125123, intramural funds from the Dan L. Duncan Cancer Center, and Alkek Center for Molecular Discovery. Additional support is from the American Cancer Society (127430-RSG-15–105-01-CNE to N.P.), CPRIT (RP150451 to A.S.) and GE Healthcare (J.A.B and C.W). The BCM Metabolomics Core is a designated Agilent Technologies Center for Excellence in Mass Spectrometry.
We thank the following BCM Cores: Human Tissue Acquisition and Pathology (NCI-CA125123), Mouse Metabolism (Dir. P. Saha), Integrated Microscopy (NCI-CA125123, NIDDK-56338–13/15, CPRIT RP150578, and the John S. Dunn Gulf Coast Consortium for Chemical Genomics), Cytometry and Cell Sorting (Dir. J. Sederstrom, NIAID P30AI036211, NCI P30CA125123, and NCRR S10RR024574). We thank the M.D.A.C.C. Characterized Cell Line Core and Small Animal Imaging Facility (P30-CA016672). We thank J. Colca and the Metabolic Solutions Development Company for generously providing MSDC0160. We thank D. Townley for TEM imaging and B. Krishnan, M. Finegold, and G. Stoica for reviewing TEM images. We thank J. Rutter, D. Moore, and A. Means for insightful conversations related to this manuscript. We acknowledge the joint participation by the Diana Helis Henry Medical Research Foundation through its direct engagement in the continuous active conduct of medical research (S.E.M. and A.S.).
Footnotes
Competing Interests
The authors declare no competing interests.
References
- 1.Pavlova NN & Thompson CB The Emerging Hallmarks of Cancer Metabolism. Cell Metab 23, 27–47 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DeBerardinis RJ & Chandel NS Fundamentals of cancer metabolism. Sci. Adv 2, e1600200 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Keibler MA et al. Metabolic requirements for cancer cell proliferation. Cancer Metab 4, 16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vander Heiden MG & DeBerardinis RJ Understanding the Intersections between Metabolism and Cancer Biology. Cell 168, 657–669 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Q et al. Androgen receptor and nutrient signaling pathways coordinate the demand for increased amino acid transport during prostate cancer progression. Cancer Res 71, 7525–7536 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Deblois G & Giguère V Oestrogen-related receptors in breast cancer: control of cellular metabolism and beyond. Nat. Rev. Cancer 13, 27–36 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Massie CE et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J 30, 2719–33 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mohler JL et al. Prostate Cancer, Version 1.2016. J. Natl. Compr. Canc. Netw 14, 19–30 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Mitsiades N A road map to comprehensive androgen receptor axis targeting for castration-resistant prostate cancer. Cancer Res 73, 4599–605 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Scher HI & Sawyers CL Biology of progressive, castration-resistant prostate cancer: Directed therapies targeting the androgen-receptor signaling axis. J. Clin. Oncol 23, 8253–8261 (2005). [DOI] [PubMed] [Google Scholar]
- 11.Herzig S et al. Identification and functional expression of the mitochondrial pyruvate carrier. Science 337, 93–6 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Bricker DK et al. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science 337, 96–100 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schell JC et al. A Role for the Mitochondrial Pyruvate Carrier as a Repressor of the Warburg Effect and Colon Cancer Cell Growth. Mol. Cell 1–14 (2014). doi: 10.1016/j.molcel.2014.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Costello LC & Franklin RB The clinical relevance of the metabolism of prostate cancer; zinc and tumor suppression: connecting the dots. Mol. Cancer 5, 17 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zadra G, Photopoulos C & Loda M The fat side of prostate cancer. Biochim. Biophys. Acta (2013). doi: 10.1016/j.bbalip.2013.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abeshouse A et al. The Molecular Taxonomy of Primary Prostate Cancer. Cell 163, 1011–1025 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cerami E et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2, 401–4 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beltran H et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med 1–12 (2016). doi: 10.1038/nm.4045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barretina J et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chattopadhyay I et al. Src promotes castration-recurrent prostate cancer through androgen receptor-dependent canonical and non-canonical transcriptional signatures. Oncotarget 8, 10324–10347 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu R et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res 69, 16–22 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Culig Z et al. Switch from antagonist to agonist of the androgen receptor bicalutamide is associated with prostate tumour progression in a new model system. Br. J. Cancer 81, 242–51 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Panigrahi AK et al. SRC-3 Coactivator Governs Dynamic Estrogen-Induced Chromatin Looping Interactions during Transcription. Mol. Cell 70, 679–694.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pomerantz MM et al. The androgen receptor cistrome is extensively reprogrammed in human prostate tumorigenesis. Nat. Genet 47, 1346–1351 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stelloo S et al. Androgen receptor profiling predicts prostate cancer outcome. EMBO Mol Med 7, 1450–1464 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sullivan LB, Gui DY & Heiden MG Vander. Altered metabolite levels in cancer: implications for tumour biology and cancer therapy. Nat. Rev. Cancer 16, 680–693 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Halestrap AP The mitochondrial pyruvate carrier. Kinetics and specificity for substrates and inhibitors. Biochem. J 148, 85–96 (1975). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anantharaman A & Friedlander TW Targeting the androgen receptor in metastatic castrate-resistant prostate cancer: A review. Urol. Oncol. Semin. Orig. Investig 34, 356–367 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Hildyard JCW, Ammälä C, Dukes ID, Thomson S. a & Halestrap AP Identification and characterisation of a new class of highly specific and potent inhibitors of the mitochondrial pyruvate carrier. Biochim. Biophys. Acta 1707, 221–30 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Halestrap AP The monocarboxylate transporter family-Structure and functional characterization. IUBMB Life 64, 1–9 (2012). [DOI] [PubMed] [Google Scholar]
- 31.Polański R et al. Activity of the monocarboxylate transporter 1 inhibitor AZD3965 in small cell lung cancer. Clin. Cancer Res 20, 926–37 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davidson SM et al. Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab 23, 517–528 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A 102, 15545–50 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pakos-Zebrucka K et al. The integrated stress response. EMBO Rep 17, 1374–1395 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hamanaka RB, Bennett BS, Cullinan SB & Diehl JA PERK and GCN2 contribute to eIF2alpha phosphorylation and cell cycle arrest after activation of the unfolded protein response pathway. Mol. Biol. Cell 16, 5493–501 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang C et al. Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Mol. Cell 56, 414–24 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Faubert B et al. Lactate Metabolism in Human Lung Tumors. Cell 171, 358–371.e9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hui S et al. Glucose feeds the TCA cycle via circulating lactate. Nature (2017). doi: 10.1038/nature24057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Herbst EAF, George MAJ, Brebner K, Holloway GP & Kane DA Lactate is Oxidized Outside of the Mitochondrial Matrix in Rodent Brain. Appl. Physiol. Nutr. Metab 2–4 (2017). doi: 10.1139/apnm-2017-0450 [DOI] [PubMed] [Google Scholar]
- 40.Chen YJ et al. Lactate metabolism is associated with mammalian mitochondria. Nat. Chem. Biol 12, 937–943 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Corbet C et al. Interruption of lactate uptake by inhibiting mitochondrial pyruvate transport unravels direct antitumor and radiosensitizing effects. Nat. Commun 9, 1208 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu IJ, Zafar MBMB, Lai Y-HY, Segall GMGM & Terris MKMK Fluorodeoxyglucose positron emission tomography studies in diagnosis and staging of clinically organ-confined prostate cancer. Urology 57, 108–111 (2001). [DOI] [PubMed] [Google Scholar]
- 43.Nelson SJ et al. Metabolic imaging of patients with prostate cancer using hyperpolarized [1-13C]pyruvate. Sci. Transl. Med 5, 198ra108 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shah R et al. An Evaluation of MSDC-0160, A Prototype mTOT Modulating Insulin Sensitizer, in Patients with Mild Alzheimer’s Disease. Curr. Alzheimer Res 11, 564–573 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Colca JR et al. Clinical proof-of-concept study with MSDC-0160, a prototype mTOT-modulating insulin sensitizer. Clin. Pharmacol. Ther 93, 352–359 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ghosh A et al. Mitochondrial pyruvate carrier regulates autophagy, inflammation, and neurodegeneration in experimental models of Parkinsons disease. Sci. Transl. Med 8, 368ra174–368ra174 (2016). [DOI] [PubMed] [Google Scholar]
- 47.Kolenko V, Teper E, Kutikov A & Uzzo R Zinc and zinc transporters in prostate carcinogenesis. Nat. Rev. Urol 10, 219–26 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schell JC et al. Control of intestinal stem cell function and proliferation by mitochondrial pyruvate metabolism. Nat. Cell Biol 19, 1027–1036 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schulte ML et al. Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat. Med 24, 194–202 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McCommis KS et al. Targeting the mitochondrial pyruvate carrier attenuates fibrosis in a mouse model of nonalcoholic steatohepatitis. Hepatology 65, 1543–1556 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Divakaruni AS et al. Inhibition of the mitochondrial pyruvate carrier protects from excitotoxic neuronal death. J. Cell Biol 216, 1091–1105 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rauckhorst AJ et al. The mitochondrial pyruvate carrier mediates high fat diet-induced increases in hepatic TCA cycle capacity. Mol. Metab 6, 1468–1479 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nancolas B et al. The anti-tumour agent lonidamine is a potent inhibitor of the mitochondrial pyruvate carrier and plasma membrane monocarboxylate transporters. Biochem. J 473, 929–36 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Divakaruni AS et al. Thiazolidinediones are acute, specific inhibitors of the mitochondrial pyruvate carrier. Proc. Natl. Acad. Sci. U. S. A 110, 5422–7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lyles BE, Akinyeke TO, Moss PE & Stewart LV Thiazolidinediones regulate expression of cell cycle proteins in human prostate cancer cells via PPARg-dependent and PPARg-independent pathways. Cell Cycle 8, 268–277 (2009). [DOI] [PubMed] [Google Scholar]
- 56.Fröhlich E & Wahl R Chemotherapy and chemoprevention by thiazolidinediones. Biomed Res. Int 2015, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Suzuki S et al. Pioglitazone, a peroxisome proliferator-activated receptor γ agonist, suppresses rat prostate carcinogenesis. Int. J. Mol. Sci 17, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hensley CTT et al. Metabolic Heterogeneity in Human Lung Tumors. Cell 164, 681–694 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Taylor BS et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 18, 11–22 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Morris JK A formaldehyde glutaraldehyde fixative of high osmolality for use in electron microscopy. J. Cell Biol 27, 1A–149A (1965).5857256 [Google Scholar]
- 61.Darzynkiewicz Z & Juan G DNA content measurement for DNA ploidy and cell cycle analysis. Curr. Protoc. Cytom Chapter 7, Unit 7.5 (2001). [DOI] [PubMed] [Google Scholar]
- 62.Bae S, Park J & Kim JS Cas-OFFinder: A fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30, 1473–1475 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Putluri N et al. Metabolomic profiling reveals a role for androgen in activating amino acid metabolism and methylation in prostate cancer cells. PLoS One 6, e21417 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kettner NM et al. Circadian Homeostasis of Liver Metabolism Suppresses Hepatocarcinogenesis Article Circadian Homeostasis of Liver Metabolism Suppresses Hepatocarcinogenesis. Cancer Cell 1–16 (2016). doi: 10.1016/j.ccell.2016.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ramirez MS et al. Radial spectroscopic MRI of hyperpolarized [1-13C] pyruvate at 7 tesla. Magn. Reson. Med 72, 986–995 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bankson JA et al. Kinetic modeling and constrained reconstruction of hyperpolarized [1–13C]-pyruvate offers improved metabolic imaging of tumors. Cancer Res 75, 4708–4717 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Analyzed RPPA data is available in Supplementary Data 2, analyzed RNA-sequencing data is available in Supplementary Data 3, and raw RNA-sequencing data is available under accession number GSE114708 on the NCBI GEO database. All other data described, analyzed, and represented in the figures present in this study are available from the corresponding authors upon reasonable request.