

Abstract
Alloimmune risk stratification in renal transplantation has lacked the necessary prognostic biomarkers to personalize recipient care or optimize clinical trials. HLA molecular mismatch improves precision compared to traditional antigen mismatch but has not been studied in detail at the individual molecule level. This study evaluated 664 renal transplant recipients and correlated HLA‐DR/DQ single molecule eplet mismatch with serologic, histologic, and clinical outcomes. Compared to traditional HLA‐DR/DQ whole antigen mismatch, HLA‐DR/DQ single molecule eplet mismatch improved the correlation with de novo donor‐specific antibody development (area under the curve 0.54 vs 0.84) and allowed recipients to be stratified into low, intermediate, and high alloimmune risk categories. These risk categories were significantly correlated with primary alloimmune events including Banff ≥1A T cell–mediated rejection (P = .0006), HLA‐DR/DQ de novo donor‐specific antibody development (P < .0001), antibody‐mediated rejection (P < .0001), as well as all‐cause graft loss (P = .0012) and each of these correlations persisted in multivariate models. Thus, HLA‐DR/DQ single molecule eplet mismatch may represent a precise, reproducible, and widely available prognostic biomarker that can be applied to tailor immunosuppression or design clinical trials based on individual patient risk.
Keywords: clinical research/practice, clinical trial design, histocompatibility, kidney transplantation/nephrology, major histocompatibility complex (MHC), rejection: antibody‐mediated (ABMR), rejection: T cell mediated (TCMR), risk assessment/risk stratification
Short abstract
Primary alloimmune risk precisely determined using HLA class II molecular mismatch may facilitate personalized treatment and monitoring strategies for kidney transplant recipients.
Abbreviations
- ABMR
antibody‐mediated rejection
- dnDSA
de novo donor‐specific antibody
- PRA
panel reactive antibody
- TCMR
T cell–mediated rejection
1. INTRODUCTION
In a 2016 US Food and Drug Administration public meeting on patient‐focused drug development, transplant recipients voiced their desire to have immunosuppressive therapy individualized and simplified to avoid side effects while ensuring efficacy.1 Unfortunately, when induction therapy is selected for renal transplant recipients, current evidence shows that an individual's clinical risk factors only account for 10%‐33% of the observed variation in practice, while transplant center effect was responsible for the majority (51%‐61%) of the variation.2 Compounding the problem, randomized controlled trials (RCTs) of immunosuppression minimization that have attempted to identify “low risk” recipients using clinical, serologic, and histologic criteria have been unsuccessful, suggesting that traditional risk factors hold little utility to personalize patient care.3, 4, 5 Thus, an unmet need in transplantation is the accurate definition of an individual's alloimmune risk for a given donor at the time of transplant: a fundamental requirement if the field is to move to precision medicine.
In 2017, the American Society of Transplantation and the American Society of Histocompatibility and Immunogenetics established the Sensitization in Transplantation: Assessment of Risk (STAR) Working Group. The goal of this expert panel is to conduct critical reviews of the pretransplant diagnostics literature and make recommendations for alloimmune risk assessment building on the 2009 Kidney Disease: Improving Global Outcomes (KDIGO) and the 2013 Transplant Society clinical practice guidelines.6, 7, 8 The STAR 2017 report created a framework that recommended 2 independent risk assessments: 1 related to the risk of immunologic memory and a second related to the risk of a primary (ie, de novo) alloimmune response posttransplant. In this context, the STAR Working Group identified that the HLA molecular mismatch was a key determinant of an individual's primary alloimmune risk and called for research to determine optimal approaches to define HLA molecular mismatch risk categories.
In this study, building on our prior work using HLAMatchmaker as a computational tool to assess donor‐recipient HLA relatedness,9, 10 we evaluated a novel approach to quantify HLA molecular mismatch allowing us to more precisely classify individuals into low, intermediate, or high alloimmune risk categories at the time of kidney transplant. These risk categories correlated with primary alloimmune events (ie, T cell–mediated rejection [TCMR], de novo donor‐specific antibody [dnDSA] development, antibody‐mediated rejection [ABMR]), as well as all‐cause graft loss.
2. CONCISE METHODS
2.1. Study population
Approval was obtained from the institutional review board (H2011:211) and was in adherence with the declaration of Helsinki. Seven hundred twenty‐four adult and pediatric consecutive renal transplants between January 1999 and July 2016 were considered for inclusion. Patients with primary nonfunction (n = 17), or pretransplant donor‐specific antibody (DSA) (n = 43) were excluded, leaving 664 recipients (adult n = 606, pediatric n = 58) for analysis. Median follow‐up was 91 months (range 18‐226). Recipients who moved (n = 33) or died with a functioning graft (n = 112) were censored at last follow‐up. Standard immunosuppression consisted of a calcineurin inhibitor (tacrolimus [87%] or cyclosporin [13%]), mycophenolate mofetil, and prednisone. Induction therapy with thymoglobulin (21%) or basiliximab (18%) was used in 39% of patients. Details on clinical, serologic, and histologic monitoring posttransplant have been reported previously and can be found in the Supplemental Methods.9, 11
2.2. HLA typing and epitope mismatch identification
High‐resolution Class II HLA typing (HLA‐DRβ1/3/4/5 and HLA‐DQα1/β1) was performed using sequence‐specific oligonucleotide probes or sequence‐specific primer technology (LABType® HD SSO, Micro SSP™; One Lambda, Los Angeles, CA). HLAMatchmaker software (HLA DRDQDP Matching version 2.0) was used to define Class II eplet mismatches between donors and recipients.
HLA eplet identification is based on 2 underlying principles: (a) the immune system recognizes and develops antibodies against nonself‐antigens, or more specifically the epitopes on those antigens, while ignoring self‐antigens/epitopes; and (b) epitope binding affinity is largely determined by a small number of polymorphic amino acids near the center of the epitope.12 An eplet is defined as a single polymorphic amino acid or a small patch of polymorphic amino acids within a 3 angstrom (0.3 nm) radius (Figure 1) on or near the surface of an HLA molecule. An eplet represents the smallest functional unit of an epitope‐paratope interface, which may drive antibody specificity through interactions with the central complementary‐determining regions of the antibody paratope. An epitope is defined by the complete antigen‐antibody interface (≈15 angstrom [1.5 nm]) made up of amino acids essential for specificity as well as those that affect affinity but not specificity.
Figure 1.
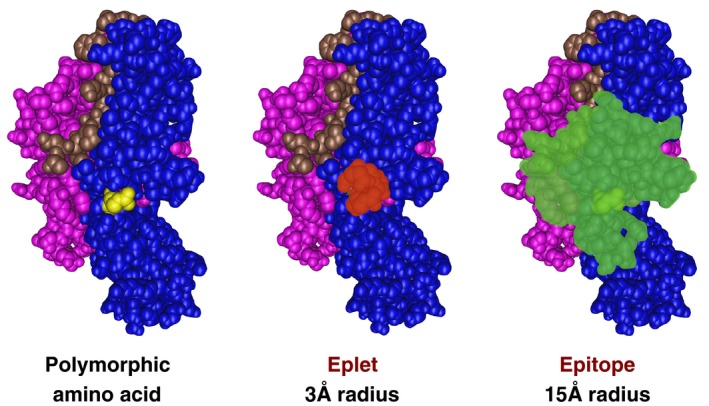
HLA donor‐recipient mismatch drives allorecognition. An amino acid polymorphism (yellow) present in the donor and not present in the recipient is the most basic unit of mismatch. An eplet is defined as a single polymorphic amino acid or a small patch of polymorphic amino acids within a 3 angstrom (0.3 nm) radius on or near the surface of an HLA molecule. An eplet represents the smallest functional unit of an epitope‐paratope interface, which may drive antibody specificity through interactions with the central complementary determining regions of the antibody paratope. The complete epitope (green) represents all amino acids within a 15 angstrom (1.5 nm) radius typical for an antibody paratope
2.3. Traditional HLA mismatch vs molecular HLA mismatch assessment
Class II HLA‐DR/DQ donor‐recipient mismatch was evaluated by 3 different methods in this study (Figure 2). First is the traditional whole antigen method where each HLA‐DRβ1 or HLA‐DQβ1 donor antigen is assigned 1 mismatch if different from either recipient antigen, resulting in a sum score of 0, 1, or 2 at each locus. The second method, published previously,9 uses HLAMatchmaker DRDQDP (version 2) to determine the eplet mismatches for each of the HLA‐DRβ1/3/4/5, HLA‐DQα1, and HLA‐DQβ1 alleles, which are summed for each locus. In both of the first 2 methods the total score for that HLA locus is correlated with dnDSA development at that locus. The third method used HLAMatchmaker to determine the eplet mismatch for each HLA‐DR or HLA‐DQ molecule individually and correlated the single molecule eplet mismatch with dnDSA development against that molecule specifically (Table 1). Thresholds were then developed by receiver operating characteristic (ROC) curve analysis (see section 2.1) so that recipients could be categorized by whether any of their individual HLA‐DR or DQ molecules had eplet mismatch loads above or below the thresholds. For HLA‐DR each maternal and parenteral HLA‐DRβ1 (n = 1328), HLA‐DRβ3 (n = 481), HLA‐DRβ4 (n = 392), HLA‐DRβ5 (n = 231) donor alleles were considered. Donor null alleles at HLA‐DRβ3/4/5 (n = 224) did not count toward the total. For HLA‐DQ, α and β alleles inherited as a haplotype were considered as 1 HLA‐DQα1β1 molecule (n = 1328).
Figure 2.
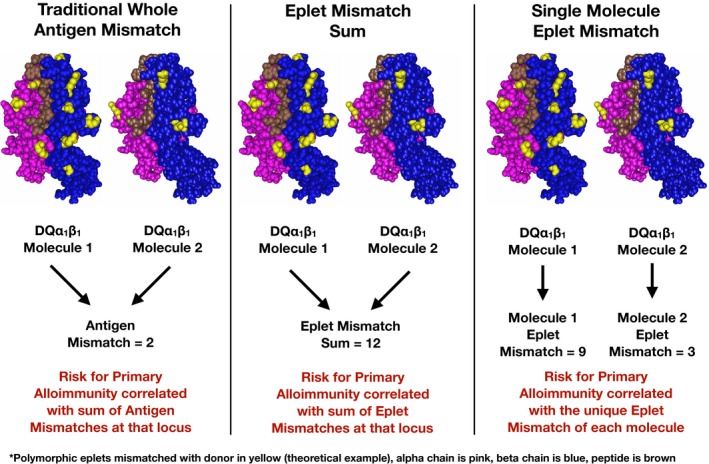
Comparing HLA mismatch methods to define low risk for dnDSA. Methods of quantifying HLA mismatches are compared using a theoretical example at the HLA‐DQ locus. dnDSA, de novo donor‐specific antibody
Table 1.
Comparison of HLA‐DR/DQ mismatch quantification methods
| Description | Calculation of HLA‐DR mismatch score | HLA‐DR dnDSA AUCa | Calculation of HLA‐DQ mismatch score | HLA‐DQ dnDSA AUCa | Benefits | Drawbacks | |
|---|---|---|---|---|---|---|---|
| Traditional whole antigen mismatch | The sum of mismatched donor antigens for a given locus | DRβ1 allele 1 + DRβ1 allele 2 | 0.58 | DQβ1 allele 1 + DQβ1 allele 2 | 0.54 | Historical. Understood by all | Imprecise. No information about the relative similarity or difference between donor and recipient antigens |
| Eplet mismatch sum | The sum of mismatched donor eplets for a given locus | DRβ1 allele 1 + DRβ3/4/5 allele 1 + DRβ1 allele 2 + DRβ3/4/5 allele 2 | 0.72 | DQα1 allele 1 + DQβ1 allele 1 + DQα1 allele 2 + DQβ1 allele 2 | 0.72 | Increased precision compared to antigen matching. One mismatch score for each HLA locus | Composite score of all molecules within a locus less precise in recipients with 1 antigen matched and 1 mismatched |
| Single molecule eplet mismatch | The number of eplet mismatches for each specific molecule within each locus | DRβ1 allele 1 or DRβ1 allele 2 or DRβ3/4/5 allele 1 or DRβ3/4/5 allele 2 | 0.84 | DQα1 allele 1 + DQβ1 allele 1 or DQα1 allele 2 + DQβ1 allele 2 | 0.84 | Improved precision, particularly for recipients with 1 antigen matched and 1 mismatched | Antibody specificity must be known. Specificities may overlap across molecules |
AUC, area under the curve as a correlate with de novo donor‐specific antibody (dnDSA) development.
2.4. Statistics
Comparisons between baseline predictors and clinical outcomes were done using Student t test for parametric continuous variables and Wilcoxon‐rank test for nonparametric data. Chi‐squared or Fisher exact tests were used to test categorical variables. Comparisons across multiple groups were done using Kruskal‐Wallis test for nonparametric data and analysis of variance for parametric variables. Survival analysis was done by the Kaplan‐Meier method using the log‐rank test for significance. ROC analysis was used to identify HLA‐DR or DQ molecule specific thresholds best associated with dnDSA development. Cox proportional hazards model was used to evaluate predictors of Banff ≥1A TCMR‐free survival, dnDSA free‐survival, ABMR‐free survival, and all‐cause graft loss. Variables for multivariate regression were selected based on bivariate screening, with P values ≤.2 used to identify candidates for inclusion in the final model. Statistical software used was JMP (version 14.0; SAS Inc., Cary, NC).
3. RESULTS
This consecutive cohort (n = 664) had a median follow‐up of 91 months (range 18‐227 months) and a median 10‐year all‐cause graft survival of 74% (death‐censored graft survival of 87%). Screening serial sera, HLA dnDSA developed in 82 recipients (12%) at an average of 5.9 (range 0.5‐17.0) years posttransplant. De novo DSA developed against Class I alone (n = 10), Class II alone (n = 50), or Class I and II (n = 22). Two recipients with Class I dnDSA alone went on to graft loss at a median 11 years (range 9‐13 years) after dnDSA development. Graft survival in the Class I dnDSA alone group was not different from the no dnDSA group (P = .39, Figure S1). Recipients who developed Class II dnDSA alone, or Class I and II dnDSA had decreased graft survival compared with those who did not develop dnDSA (P < .0001).
3.1. Defining low risk for primary alloimmunity by HLA molecular mismatch
The range of HLA‐DRβ1/3/4/5 single molecule eplet mismatches was 0‐22 and the range of HLA‐DQα1β1 single molecule eplet mismatches was 0‐31. Each HLA‐DRβ1/3/4/5 and HLA‐DQα1β1 molecule was analyzed individually for the risk of dnDSA development against that molecule. Using this approach, a ROC analysis identified a molecule specific threshold of ≥7 HLA‐DR eplet mismatches and ≥9 HLA‐DQ eplet mismatches associated with an area under the curve (AUC) of 0.84 and 0.84 for HLA‐DR and HLA‐DQ dnDSA development, respectively (sensitivity, 81% and 90%, specificity 75% and 64%, respectively, Table 1, Figure S2). By comparison using the traditional HLA whole antigen mismatch as a correlate with dnDSA development, the AUC was only 0.54 and 0.58 for HLA‐DR, and DQ, respectively.
Using the HLA‐DR/DQ molecule‐specific mismatch thresholds, recipients were stratified into 3 groups: Group A (n = 93) HLA‐DR = 0 and HLA‐DQ = 0; Group B (n = 73) HLA‐DR = 1‐6 and/or HLA‐DQ = 1‐8; and Group C (n = 498) HLA‐DR ≥7 or HLA‐DQ ≥9. Baseline demographics within these groups are compared in Table 2. Recipients in Group B were more likely to have a repeat transplant (P < .01), younger age (P = .03), and were less likely to have received induction therapy (P < .01) compared to Group C. Of note, risk factors known to influence dnDSA development such as nonadherence, maintenance immunosuppression, and tacrolimus coefficient of variation were similar among the 3 groups.
Table 2.
HLA‐DR/DQ single molecule eplet mismatch subgroup demographics
| Group A | Group B | Group C | P value | P value | |
|---|---|---|---|---|---|
| DR=0 and DQ=0 | DR=1‐6 and/or DQ=1‐8 | DR≥7 or DQ≥9 | |||
| n = 93 | n = 73 | n = 498 | All groups | Group B vs C | |
| First transplant | 93% | 89% | 97% | .0118 | .0075 |
| Recipient age (y) | 40.4 ± 14.1 | 41.0 ± 15.0 | 44.9 ± 16.5 | .0028 | .0269 |
| Donor age (y) | 38.7 ± 12.8 | 40.9 ± 16.0 | 40.6 ± 15.0 | .4386 | .7205 |
| Living donor | 77% | 45% | 44% | <.0001 | .8183 |
| Ethnicity (white vs other) | 79% | 62% | 65% | .0186 | .6172 |
| Cold ischemic time (h) | 4.2 ± 3.6 | 6.9 ± 5.6 | 7.3 ± 5.5 | <.0001 | .7962 |
| Delayed graft function | 7% | 19% | 14% | .0333 | .2845 |
| Induction therapy | 20% | 27% | 44% | <.0001 | .0056 |
| Basiliximab | 14% | 15% | 20% | ||
| Thymoglobulin | 7% | 12% | 24% | ||
| Tacrolimus vs cyclosporin | 90% | 88% | 87% | .6485 | .8627 |
| Tacrolimus CV 0‐12 mo (n = 582) | 34.2 ± 9.4 | 39.1 ± 13.9 | 36.3 ± 12.1 | .1728 | .1459 |
| Mycophenolate | 100% | 100% | 100% | ns | ns |
| Nonadherence | 14% | 12% | 16% | .6458 | .3987 |
CV, coefficient of variation; ns, not significant.
3.2. Comparison of HLA‐DR/DQ mismatch quantification methods
Traditional HLA‐DR/DQ whole antigen mismatch, HLA‐DR/DQ eplet mismatch sum thresholds (previously published HLA‐DR and DQ thresholds each ≤11),9 and HLA‐DR/DQ single molecule eplet mismatch thresholds were compared as correlates for HLA‐DR/DQ dnDSA‐free survival (Figure 3) and Banff ≥1A TCMR‐free survival (Figure 4). Table 1 and Figures 1 and 2 outline the key differences between these 3 methods.
Figure 3.
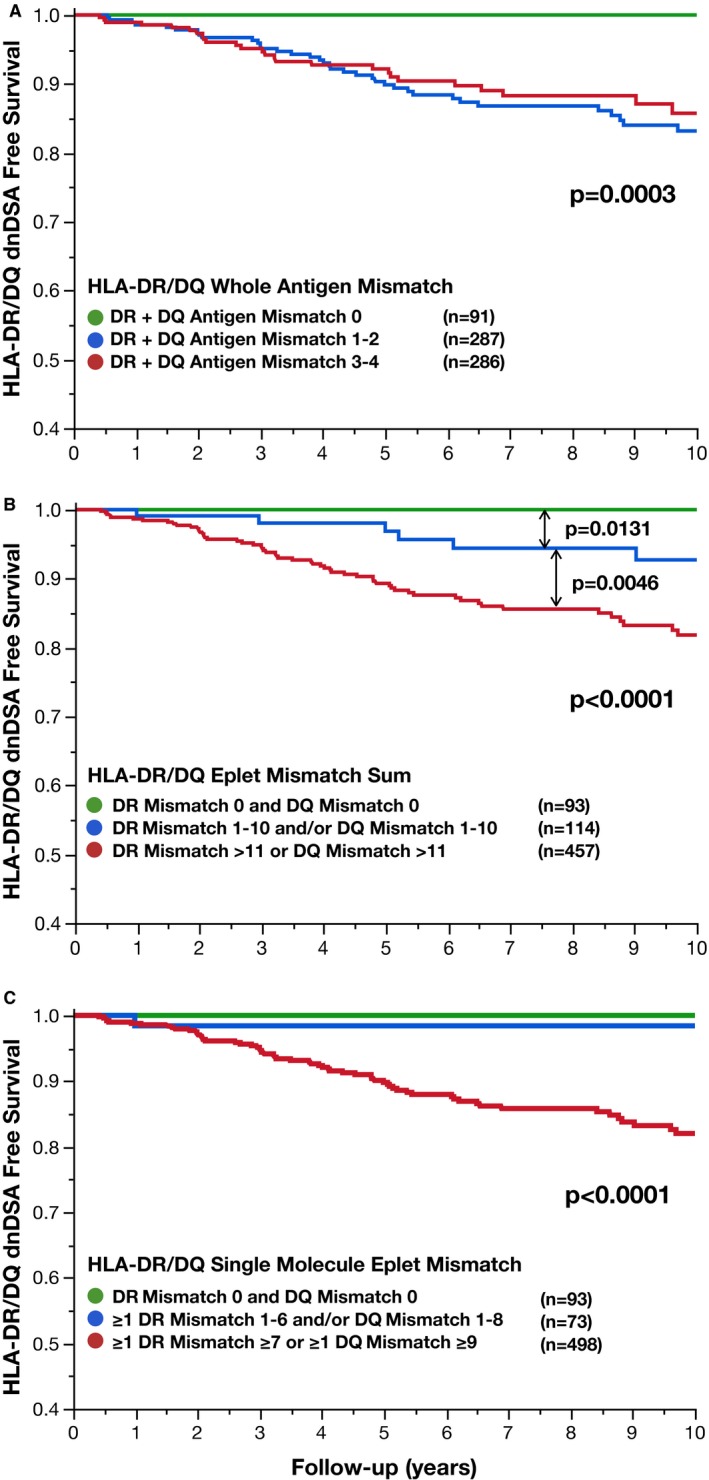
Comparing HLA mismatch methods to define low risk for dnDSA. Traditional HLA‐DR/DQ whole antigen mismatch (A), HLA‐DQ/DQ eplet mismatch sum (B), and HLA‐DR/DQ single molecule eplet mismatch (C) are correlated with de novo donor‐specific antibody‐free survival
Figure 4.
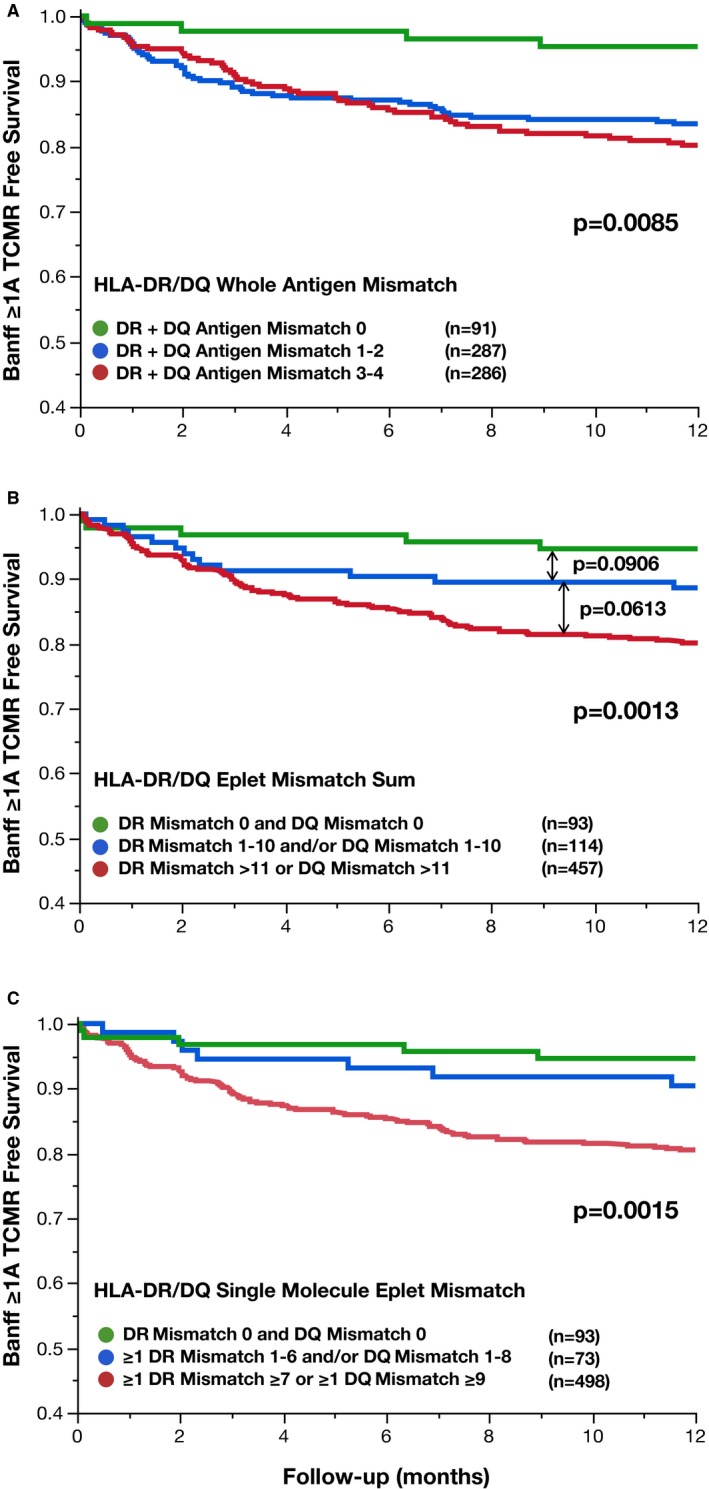
Comparing HLA mismatch methods to define low risk for TCMR. Traditional HLA‐DR/DQ whole antigen mismatch (A), HLA‐DQ/DQ eplet mismatch sum (B), and HLA‐DR/DQ single molecule eplet mismatch (C) are correlated with Banff ≥1A T cell–mediated rejection‐free survival. TCMR, T cell–mediated rejection
Traditional HLA‐DR/DQ whole antigen mismatch greater than zero was associated with significantly lower HLA‐DR/DQ dnDSA‐free survival (P = .0003). However, there was no statistical difference in HLA‐DR/DQ dnDSA‐free survival between HLA‐DR/DQ whole antigen risk groups other than zero (P = .48, Figure 3A). This was also true in a locus‐specific analysis of HLA‐DR or HLA‐DQ dnDSA development (Figure S3). HLA‐DR/DQ eplet mismatch sum also was associated with HLA‐DR/DQ dnDSA‐free survival (Figure 3B). Using the previously published thresholds of 11 HLA‐DR or DQ eplet mismatches to risk stratify recipients, a significant dose‐response relationship was demonstrated (P < .0001, Figure 3B).9 By comparison, HLA‐DR/DQ single molecule eplet mismatch risk groups with HLA‐DR <7 and HLA‐DQ <9 mismatches (Groups A and B, n = 166, 25% of the cohort) developed HLA‐DR/DQ dnDSA in only 2 recipients (1.2%) in the first 10 years posttransplant. However, recipients with single molecule HLA‐DR ≥7 or HLA‐DQ ≥9 eplet mismatches (Group C, n = 498) had significantly increased risk of HLA‐DR/DQ dnDSA development (P < .0001, Figure 3C).
At least 1 surveillance or for‐cause biopsy was performed in the first‐year posttransplant in 522/664 (79%) of patients. Traditional HLA‐DR/DQ whole antigen mismatch greater than zero was associated with significantly decreased Banff ≥1A TCMR‐free survival in the first year posttransplant (P = .0085, Figure 4A). However, there was no difference in Banff ≥1A TCMR‐free survival between HLA‐DR/DQ whole antigen risk groups other than zero (P = .49, Figure 4A). HLA‐DR/DQ eplet mismatch sum was also associated with Banff ≥1A TCMR‐free survival (P = .0013, Figure 4B). Using the thresholds of ≥11 HLA‐DR or DQ eplet mismatches, a trend of increased risk of Banff ≥1A TCMR across eplet mismatch groups was evident (Figure 4B). Using HLA‐DR/DQ single molecule eplet mismatch risk groups, recipients with HLA‐DR <7 and HLA‐DQ <9 (Groups A and B, n = 166, 25% of the cohort) developed Banff ≥1A TCMR in ≤10% of recipients in the first year posttransplant. However, recipients with HLA‐DR ≥7 or HLA‐DQ ≥9 single molecule eplet mismatches (Group C, n = 498) had significantly increased risk of Banff ≥1A TCMR (20% at 12 months, P = .0018, Figure 4C).
3.3. Defining intermediate and high risk for primary alloimmunity by HLA molecular mismatch
Groups A and B were combined into a single Low Risk category based on the prior analysis (HLA‐DR <7 and HLA‐DQ <9). Because Group C recipients represented 75% of the cohort, we sought to stratify these recipients further by repeating the ROC analysis at the HLA‐DR and DQ loci after exclusion of recipients in the Low Risk category. For HLA‐DR, no additional cutoff could be identified; however, a single molecule threshold of ≥15 HLA‐DQ eplet mismatches was identified. When Group C recipients were split into groups of above or below 15 HLA‐DQ eplets, 2 alloimmune risk categories were identified: Intermediate Risk (HLA‐DR ≥7 and HLA‐DQ ≤14, or HLA‐DR 0‐6 and HLA‐DQ 9‐14), and High Risk (HLA‐DR 0‐22 and HLA‐DQ 15‐31). Recipient demographics of the low, intermediate, and high HLA molecular mismatch risk categories are shown in Table S1. Differences between groups were found for primary transplant, recipient age, living donor, cold ischemic time, and induction therapy. There were no significant differences in baseline demographic between intermediate‐ and high‐risk groups.
Low, Intermediate, and High HLA molecular mismatch risk categories were significantly associated with Banff ≥1A TCMR (P = .0006, Figure 5A). In a Cox model, Banff ≥1A TCMR‐free survival was significantly less for recipients in the Intermediate (hazard ratio [HR] 2.02, 95% confidence interval [CI] 1.1‐3.9, P = .0230) and High (HR 3.33, 95% CI 1.9‐6.3, P < .0001) risk categories compared to the Low Risk category. Correlates associated with Banff ≥1A TCMR are shown in Table S2. Significant multivariate correlates of Banff ≥1A TCMR‐free survival were recipient age (HR 0.98, 95% CI 0.97‐0.99, P = .0037), cyclosporin vs tacrolimus (HR 5.39, 95% CI 3.6‐8.0, P < .0001), delayed graft function (HR 2.04, 95% CI 1.3‐3.1, P = .0035), and HLA molecular mismatch risk category (HR High vs Low 3.94, 95% CI 2.2‐7.5, P < .0001; HR High vs Intermediate 1.6, 95% CI 1.1‐2.4, P = .0233; HR Intermediate vs Low 2.28, 95% CI 1.2‐4.5, P = .0075).
Figure 5.
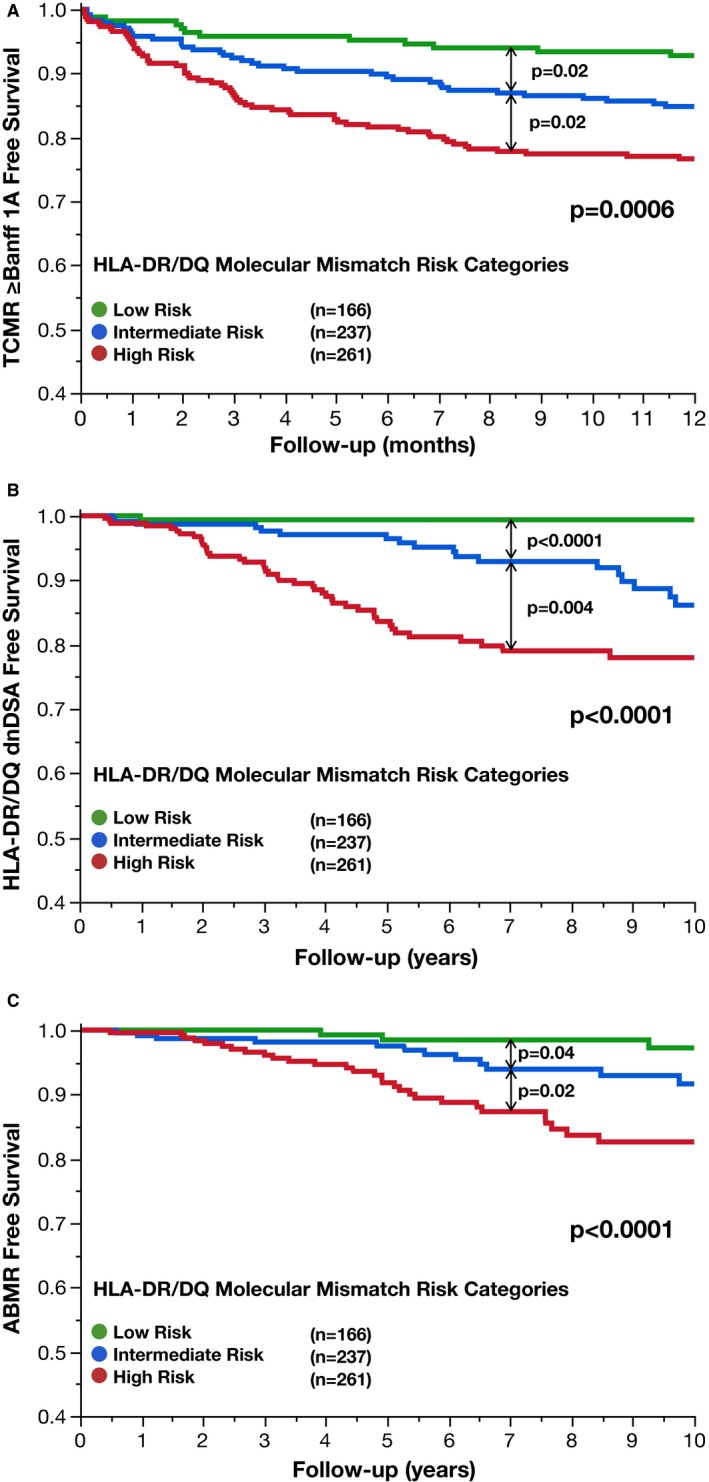
HLA molecular mismatch category correlates with Banff ≥1A TCMR, dnDSA development, and ABMR. HLA‐DR/DQ molecular mismatch categories (low, intermediate, and high) were correlated with Banff ≥1A T cell–mediated rejection‐free survival (A), de novo donor‐specific antibody‐free survival (B), and antibody‐mediated rejection‐free survival (C). ABMR, antibody‐mediated rejection; dnDSA, de novo donor‐specific antibody; TCMR, T cell–mediated rejection
Low, Intermediate, and High HLA molecular mismatch risk categories were significantly associated with dnDSA development (P < .0001, Figure 5B). In a Cox model, dnDSA‐free survival was significantly less for recipients in the Intermediate (HR 10.18, 95% CI 3.0‐63.5, P < .0001), and High (HR 20.8, 95% CI 6.4‐127.4, P < .0001) risk categories compared to the Low Risk category. Correlates associated with dnDSA‐free survival are shown in Table S3. Significant multivariate correlates of dnDSA development were recipient age (HR 0.97, 95% CI 0.96‐0.99, P = .0010), cyclosporin vs tacrolimus (HR 2.11, 95% CI 1.3‐3.4, P = .0043), nonadherence (HR 2.83, 95% CI 1.7‐4.6, P < .0001), and HLA molecular mismatch risk category (HR High vs Low 18.31, 95% CI 5.6‐11.4, P < .0001; HR High vs Intermediate 1.84, 95% CI 1.1‐3.1, P = .0149; HR Intermediate vs Low 9.96, 95% CI 2.9‐62.1, P < .0001).
At least 1 renal biopsy was available in 57/72 (79%) of recipients post‐dnDSA. ABMR developed in 40/57 (70%) of recipients post‐dnDSA development. ABMR‐free survival was significantly associated with HLA molecular mismatch risk categories (P < .0001, Figure 5C). Correlates associated with ABMR‐free survival are shown in Table S4. Significant multivariate correlates of ABMR were recipient age (HR 0.98, 95% CI 0.96‐0.99, P = .0116), nonadherence (HR 3.52, 95% CI 2.0‐6.3, P < .0001), and HLA molecular mismatch risk category (HR High vs Low 5.06, 95% CI 2.1‐14.9, P < .0001; HR High vs Intermediate 1.87, 95% CI 1.0‐3.6, P = .0420; HR Intermediate vs Low 2.71, 95% CI 1.0‐8.3, P = .0394).
3.4. HLA molecular mismatch correlates with all‐cause graft loss
Low, Intermediate, and High HLA molecular mismatch risk categories were significantly associated with all‐cause graft loss (P = .0003, Figure S4). Correlates associated with all‐cause graft loss are shown in Table S5. In a multivariate analysis, covariates significantly associated with all‐cause graft loss were recipient age (HR 1.03, 95% CI 1.01‐1.04, P < .0001), delayed graft function (HR 2.56, 95% CI 1.80‐3.58, P < .0001), nonadherence (HR 2.00, 95% CI 1.35‐2.87, P = .0006), and alloimmune risk category (HR High vs Low 1.66, 95% CI 1.12‐2.53, P = .0120; HR High vs Intermediate 1.20, 95% CI 0.9‐1.7, P = .3; HR Intermediate vs Low 1.39, 95% CI 0.9‐1.2, P = .1).
4. DISCUSSION
The key finding in this study is that in the absence of donor‐specific memory (ie, no preformed DSA by solid phase single antigen bead assessment), quantifying the differences of HLA‐DR/DQ mismatches at the molecular level can improve precision in primary alloimmune risk categorization. As a prognostic biomarker available pretransplant, its potential application includes the development of personalized immunosuppression protocols, as well as being a drug development tool for enrichment/stratification in clinical trial design to improve trial efficiency. These applications would address major unmet needs in transplantation from both the perspective of the patient and the pharmaceutical industry/academia.13, 14
Traditional alloimmune risk factors reported by KDIGO and recently enumerated in the 2017 Consensus on Managing Modifiable Risk in Transplantation report were pretransplant DSA, panel reactive antibody (PRA) >0%, younger recipient age, African American ethnicity, and HLA‐DR mismatch.7, 15 Pretransplant DSA, a measure of alloimmune memory, correlates with ABMR, transplant glomerulopathy, and graft loss.16 However, as pretransplant DSA is avoided in most kidney transplants, methods for primary alloimmune risk stratification are needed. Although elevated PRA has been correlated with allograft outcomes, recent work using state‐of‐the‐art antibody assessment in combination with more complete HLA typing (ie, HLA‐C, HLA‐DQ, and HLA‐DP) has shown that when preformed DSA are ruled out, calculated PRA alone is not prognostic of graft outcomes.17, 18, 19 Younger recipient age is a well‐known correlate of alloimmune risk in transplantation, likely as a result of a more robust immune system, even after adjustment for the higher prevalence of nonadherence.10 Unfortunately, there is a lack of studies to define what age cutoff may be important, and how recipient age might be used in the precision medicine context. Although certain ethnic minorities have been associated with worse outcomes, these data are confounded by socioeconomic status, HLA mismatch, and differences in drug metabolism.20 Moreover, population migration and genetic admixture makes self‐reported ethnicity increasingly imprecise such that ethnicity may be prognostic at a population level but is unlikely to have any prognostic utility at an individual level.21, 22
Since the 1950s, HLA mismatch has been known to correlate with transplant outcome.23 Unfortunately, while traditional HLA whole antigen mismatch, especially Class II, correlates with outcomes at the population level, the lack of precision limits its utility at the individual level. Traditional HLA whole antigen mismatches only evaluates whether the donor and recipient molecules are the same or different. The issue is that some mismatched HLA molecules are nearly identical while others may be very disparate—information ignored with traditional HLA mismatch assessment. Fortunately, this relative difference can be captured and quantified by HLA molecular mismatch comparisons24, 25 such as the HLAMatchmaker eplet mismatch analysis used in this study. Because the range of eplet mismatches is wide, it is logical to ask what cutoff might best correlate with a primary alloimmune response. De novo DSA development is a useful outcome for model development because it can be detected noninvasively, its onset can be determined by serial screening, and specificity can be assigned to the single molecule mismatch level. Previously, it was reported that the sum of adding the eplet mismatches within the same locus correlated with dnDSA development at that locus.9 However, since single molecule specificity of dnDSA can be determined in most cases, the analysis can be refined further to ask if the eplet mismatch for each individual molecule correlates with the development of dnDSA to that molecule.26 The single molecule approach would be expected to be more precise, especially for patients who have only 1 of the 2 molecules mismatched.
Using the traditional HLA whole antigen mismatch as a correlate with dnDSA development, the AUC was only 0.54 and 0.58 for HLA‐DR and DQ, respectively. When applying the sum of the eplet mismatch within a locus, the AUC improved to 0.72 for both HLA‐DR and DQ.9 This was enhanced further in the current study using the single molecule eplet analysis to an AUC of 0.84 for HLA‐DR and DQ. Notably, results of the single molecule method provide multiple individual scores within each locus for each recipient. Thus, to bring this evaluation to the individual recipient level, thresholds were determined and used to categorize each recipient as low, intermediate, or high primary alloimmune risk based on the molecular mismatch scores across all HLA‐DRβ1/3/4/5 and HLA‐DQα1/β1 molecules. These risk categories were highly correlated with dnDSA development, ABMR, Banff ≥1A TCMR, and all‐cause graft loss.
The need for reliable prognostic and predictive biomarkers at the time of transplant to allow individualization of immunosuppression in patients without alloimmune memory has been recognized.1 A prognostic biomarker is one that indicates an increased (or decreased) likelihood of a future clinical event.27 HLA‐DR/DQ molecular mismatch has consistently been shown to correlate with a significantly increased risk of dnDSA development, ABMR, transplant glomerulopathy, and graft loss after adjustment for other risk factors.9, 10, 11, 26, 28, 29 A predictive biomarker is used to identify individuals who are more likely to respond after exposure to a particular medical product or environmental agent.27 In this regard, Class II HLA eplet mismatch has been shown in 2 observational cohort studies and 1 RCT to identify high alloimmune risk patients who have increased rates of dnDSA development, rejection, and graft loss when immunosuppression is minimized through protocol‐driven withdrawal or recipient nonadherence.3, 9, 28 Conversely, and of equal importance, these studies also identified a subset of recipients with low alloimmune risk who tolerated immunosuppression minimization. If validated in prospective clinical trials, this would provide the evidence that HLA‐DR/DQ single molecule mismatch can act as both a prognostic and a predictive biomarker capable of identifying which individuals require more or less immunosuppression to control their primary alloimmune response.21
As a prognostic biomarker, HLA‐DR/DQ molecular mismatch has several favorable characteristics. It is available at the time of transplant, modern HLA typing methods already provide the inputs necessary for its evaluation, and analysis software is free and is already being incorporated into HLA typing software from vendors. Thus, HLA‐DR/DQ molecular mismatch is cost effective, reproducible, and will be widely available in histocompatibility laboratories.
At present, given excellent short‐term clinical outcomes, the evaluation of novel drugs focuses on the average treatment effect in the overall kidney transplant population requiring Phase 3 clinical trials lasting ≥5 years with relatively large sample sizes to demonstrate superiority to standard of care.13, 14, 15, 30 This inefficient trial design is in part due to the lack of robust and precise pretransplant risk assessment for primarily alloimmune events resulting in the inclusion of heterogeneous populations. Using the HLA‐DR/DQ molecular mismatch score as a prognostic biomarker could significantly address this issue by enriching Phase 2 and 3 clinical trials with patients based upon risk categorization in studies evaluating novel drugs.
4.1. Limitations
Due to the relatively small sample size and the associated risk of type II error, risk quantification should be interpreted with caution, and should be validated in an independent cohort. Histology was available in 79% of recipients in the first year posttransplant; however, 97% of the death‐censored graft loss occurred in the cohort with at least 1 biopsy. Methods of risk stratification will need to be tested prospectively and in independent cohorts with varying ethnicities to confirm their general applicability.
5. CONCLUSIONS
A prerequisite to precision medicine is a prognostic biomarker that correlates with clinical outcomes, is reproducible, and ideally is widely available and cost effective. Using thresholds identified for dnDSA development as an outcome, we show that the HLA‐DR/DQ molecular mismatch score allows for low‐, intermediate‐, and high‐risk stratification for Banff ≥1A TCMR, dnDSA development, ABMR, and all‐cause graft loss. Given that all transplant programs worldwide are supported by accredited histocompatibility laboratories, this biomarker could be readily applied at little additional cost. Once validated, the HLA‐DR/DQ molecular mismatch score could be used to tailor immunosuppression based on individual patient risk, as well as in the design of clinical trials.
DISCLOSURE
The authors of this manuscript have conflicts of interest to disclose as described by the American Journal of Transplantation. D.N.R. is a consultant with Astellas Pharma, and P.W.N. is a consultant with Astellas Pharma and Vitaeris Inc. The other authors have no conflicts of interest to disclose.
Supporting information
ACKNOWLEDGMENTS
C.W. received funding from a Research Manitoba operating grant. C.W., J.H., I.W.G, D.N.R, and P.W.N. are funded by the Canadian Institutes for Health Research. J.H. holds a Canadian Institutes for Health Research New Investigator award. P.W.N. holds the Flynn Family Chair in Renal Transplantation. V.K. was supported by the National Institute for Health Research Blood and Transplant Research Unit in Organ Donation and Transplantation at the University of Cambridge in collaboration with Newcastle University and in partnership with National Health Service Blood and Transplant. The views expressed are those of the authors and not necessarily those of the National Health Service, the National Institute for Health Research, the Department of Health, or National Health Service Blood and Transplant.
Wiebe C, Kosmoliaptsis V, Pochinco D, et al. HLA‐DR/DQ molecular mismatch: A prognostic biomarker for primary alloimmunity. Am J Transplant. 2019;19:1708–1719. 10.1111/ajt.15177
REFERENCES
- 1. Ettenger R, Albrecht R, Alloway R, et al. Meeting report: FDA public meeting on patient‐focused drug development and medication adherence in solid organ transplant patients. Am J Transplant. 2018;18(3):564‐573. [DOI] [PubMed] [Google Scholar]
- 2. Dharnidharka VR, Naik AS, Axelrod DA, et al. Center practice drives variation in choice of US kidney transplant induction therapy: a retrospective analysis of contemporary practice. Transpl Int. 2017;31(2):198‐211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hricik DE, Formica RN, Nickerson P, et al. Adverse outcomes of tacrolimus withdrawal in immune‐quiescent kidney transplant recipients. J Am Soc Nephrol. 2015;26(12):3114‐3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dugast E, Soulillou JP, Foucher Y, et al. Failure of calcineurin inhibitor (tacrolimus) weaning randomized trial in long‐term stable kidney transplant recipients. Am J Transplant. 2016;16(11):3255‐3261. [DOI] [PubMed] [Google Scholar]
- 5. Gatault P, Kamar N, Büchler M, et al. Reduction of extended‐release tacrolimus dose in low‐immunological‐risk kidney transplant recipients increases risk of rejection and appearance of donor‐specific antibodies: a randomized study. Am J Transplant. 2017;17(5):1370‐1379. [DOI] [PubMed] [Google Scholar]
- 6. Tambur AR, Campbell P, Claas FH, et al. Sensitization in Transplantation: Assessment of Risk (STAR) 2017 working group meeting report. Am J Transplant. 2018;15(4):462‐511. [DOI] [PubMed] [Google Scholar]
- 7. Special issue: KDIGO clinical practice guideline for the care of kidney transplant recipients. Am J Transplant. 2009;9(suppl. 3):S1‐S155. [DOI] [PubMed] [Google Scholar]
- 8. Tait BD, Süsal C, Gebel HM, et al. Consensus guidelines on the testing and clinical management issues associated with HLA and non‐HLA antibodies in transplantation. Transplantation. 2013;95(1):19‐47. [DOI] [PubMed] [Google Scholar]
- 9. Wiebe C, Rush DN, Nevins TE, et al. Class II eplet mismatch modulates tacrolimus trough levels required to prevent donor‐specific antibody development. Am Soc Nephrol. 2017;28(11):3353‐3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wiebe C, Pochinco D, Blydt‐Hansen TD, et al. Class II HLA epitope matching‐a strategy to minimize de novo donor‐specific antibody development and improve outcomes. Am J Transplant. 2013;13(12):3114‐3122. [DOI] [PubMed] [Google Scholar]
- 11. Wiebe C, Gibson IW, Blydt‐Hansen TD, et al. Rates and determinants of progression to graft failure in kidney allograft recipients with de novo donor‐specific antibody. Am J Transplant. 2015;15(11):2921‐2930. [DOI] [PubMed] [Google Scholar]
- 12. Duquesnoy RJ, Askar M. HLAMatchmaker: a molecularly based algorithm for histocompatibility determination. V. Eplet matching for HLA‐DR, HLA‐DQ and HLA‐DP. Hum Immunol. 2007;68:12‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stegall MD, Morris RE, Alloway RR, Mannon RB. Developing new immunosuppression for the next generation of transplant recipients: the path forward. Am J Transplant. 2016;16(4):1094‐1101. [DOI] [PubMed] [Google Scholar]
- 14. OʼConnell PJ, Kuypers DR, Mannon RB, et al. Clinical trials for immunosuppression in transplantation: the case for reform and change in direction. Transplantation. 2017;101(7):1527‐1534. [DOI] [PubMed] [Google Scholar]
- 15. Neuberger JM, Bechstein WO, Kuypers DRJ, et al. Practical recommendations for long‐term management of modifiable risks in kidney and liver transplant recipients: a guidance report and clinical checklist by the Consensus on Managing Modifiable Risk in Transplantation (COMMIT) group. Transplantation. 2017;101:S1‐S56. [DOI] [PubMed] [Google Scholar]
- 16. Djamali A, Kaufman DB, Ellis TM, Zhong W, Matas A, Samaniego M. Diagnosis and management of antibody‐mediated rejection: current status and novel approaches. Am J Transplant. 2014;14(2):255‐271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wiebe C, Gibson IW, Blydt‐Hansen TD, et al. Evolution and clinical pathologic correlations of de novo donor‐specific HLA antibody post kidney transplant. Am J Transplant. 2012;12(5):1157‐1167. [DOI] [PubMed] [Google Scholar]
- 18. Bray RA, Nolen JDL, Larsen C, et al. Transplanting the highly sensitized patient: the emory algorithm. Am J Transplant. 2006;6(10):2307‐2315. [DOI] [PubMed] [Google Scholar]
- 19. Wehmeier C, Hönger G, Cun H, et al. Donor specificity but not broadness of sensitization is associated with antibody‐mediated rejection and graft loss in renal allograft recipients. Am J Transplant. 2017;17(8):2092‐2102. [DOI] [PubMed] [Google Scholar]
- 20. Williams RC, Opelz G, McGarvey CJ, Weil EJ, Chakkera HA. The risk of transplant failure with HLA mismatch in first adult kidney allografts from deceased donors. Transplantation. 2016;100(5):1094‐1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wiebe C, Ho J, Gibson IW, Rush DN, Nickerson PW. Carpe diem‐time to transition from empiric to precision medicine in kidney transplantation. Am J Transplant. 2018;18(7):1615‐1625. [DOI] [PubMed] [Google Scholar]
- 22. Mannon RB, Askar M, Jackson AM, Newell K, Mengel M. Meeting report of the STAR‐Sensitization in Transplantation Assessment of Risk: naïve abdominal transplant organ subgroup focus on kidney transplantation. Am J Transplant. 2018; 18;(7):1604. [DOI] [PubMed] [Google Scholar]
- 23. Harrison JH, Merrill JP, Murray JE. Renal homotransplantation in identical twins. Surg Forum. 1956;6:432‐436. [PubMed] [Google Scholar]
- 24. Kosmoliaptsis V, Dafforn TR, Chaudhry AN, Halsall DJ, Bradley JA, Taylor CJ. High‐resolution, three‐dimensional modeling of human leukocyte antigen class I structure and surface electrostatic potential reveals the molecular basis for alloantibody binding epitopes. Hum Immunol. 2011;72(11):1049‐1059. [DOI] [PubMed] [Google Scholar]
- 25. Wiebe C, Kosmoliaptsis V, Pochinco D, Taylor C, Nickerson P. A comparison of HLA molecular mismatch methods to determine HLA immunogenicity. Transplantation. 2018;102:1338‐1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kosmoliaptsis V, Mallon DH, Chen Y, Bolton EM, Bradley JA, Taylor CJ. Alloantibody responses after renal transplant failure can be better predicted by donor‐recipient HLA amino acid sequence and physicochemical disparities than conventional HLA matching. Am J Transplant. 2016;16(7):2139‐2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. FDA‐NIH Biomarker Working Group . BEST (Biomarkers, EndpointS, and other Tools) Resource. Silver Spring, MD: Food and Drug Administration (US); 2016. [PubMed] [Google Scholar]
- 28. Wiebe C, Nevins TE, Robiner WN, Thomas W, Matas AJ, Nickerson PW. The synergistic effect of class II HLA epitope‐mismatch and nonadherence on acute rejection and graft survival. Am J Transplant. 2015;15(8):2197‐2202. [DOI] [PubMed] [Google Scholar]
- 29. Sapir‐Pichhadze R, Tinckam K, Quach K, Laupacis A. HLA‐DR and ‐DQ eplet mismatches and transplant glomerulopathy: a nested case‐control study. Am J Transplant. 2015;15(1):137‐148. [DOI] [PubMed] [Google Scholar]
- 30. Freidlin B, Korn EL. Biomarker enrichment strategies: matching trial design to biomarker credentials. Nat Rev Clin Oncol. 2014;11(2):81‐90. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials