

Abstract
A magnesium complex (1) featuring a bidentate aminopyridinato ligand is a remarkably selective catalyst for the dehydrocoupling of amine‐boranes. This reaction proceeds to completion with low catalyst loadings (1 mol %) under mild conditions (60 °C), exceeding previously reported s‐block systems in terms of selectivity, rate, and turnover number (TON). Mechanistic studies by in situ NMR analysis reveals the reaction to be first order in both catalyst and substrate. A reaction mechanism is proposed to account for these findings, with the high TON of the catalyst attributed to the bidentate nature of the ligand, which allows for reversible deprotonation of the substrate and regeneration of 1 as a stable resting state.
Keywords: amido ligands, dehydrocoupling, homogeneous catalysis, hydrogen storage, magnesium
Reversible coordination: A MgII complex featuring an aminopyridinato ligand is a remarkably selective catalyst for amine‐borane dehydrocoupling. The bidentate ligand allows the catalyst to reversibly regenerate the MgII complex as the resting state, rather than irreversibly forming a magnesium hydride species, leading to high efficiency and stability.
Introduction
The transition from fossil fuels to a sustainable global energy system is one of the key challenges of the 21st century. The increasing use of alternative sources has given rise to significant obstacles in storage and mobility; that is, how to utilize generated electricity for transportation or later use. Hydrogen is a promising option for a clean burning fuel, although issues remain over its storage.1, 2 Possible solutions include physical approaches such as high pressures, low temperatures and physisorption, as well as chemical storage in the form of hydrides.3, 4
Of these, amine‐boranes have emerged as lead candidates due to their high storage capacity, stability, and low environmental impact.5 These compounds have been the focus of considerable attention, with the presence of adjacent protic and hydridic hydrogens providing an excellent system for hydrogen storage and release.6, 7, 8 Hydrogen evolution from amine‐boranes may be achieved by solvolysis, thermolysis, catalysis, or some combination of the above. Of these, solvolysis creates an undesirable thermodynamic sink through the formation of very strong B−O bonds;9, 10 and thermolysis temperatures are often high, affording a complex mixture of products.11
In contrast, catalytic hydrogen release can occur at moderate temperatures, often yielding well‐defined products. Given the dominance of transition metals in catalysis, it is unsurprising that the d‐block has received the most attention in this field, with early dehydrocoupling catalysts based on rhodium and iridium.12, 13 Driven by considerations of resource availability and sustainability, recent investigations have focused on base metals such as titanium,14, 15 manganese,16, 17, 18, 19 iron,20, 21 cobalt,22, 23 and nickel.5
Looking beyond the transition metals, there has been significant work in recent years on catalysis with main group species.24, 25 This expansion has carried through to amine‐borane dehydrocoupling,26, 27, 28 with one of the most noteworthy examples being a bis‐(borane) reported by Wegner et al. capable of releasing 2.45 equivalents of H2 from NH3⋅BH3.29 Despite their ubiquitous nature, low cost, and low toxicity (barring beryllium and barium), the s‐block elements are underrepresented in this area. Predominantly investigated as storage materials rather than catalysts,30, 31, 32, 33 their lack of popularity may be due to poor selectivity and extended reaction times of, for example, 124 h (72 % conversion) with Me2NH⋅BH3 as a substrate, as reported for Group 1 bis‐(trimethylsilyl)amides.34 However, recent work is yielding more active systems.35, 36 Whilst Group 2 systems have shown greater activity towards more complex amine‐borane substrates,37, 38, 39 it should be noted that some systems studied have subsequently been shown to exhibit “spontaneous” dehydrocoupling.40 In contrast, the Me2NH⋅BH3 and iPr2NH⋅BH3 substrates investigated here showed no hydrogen release in the absence of catalyst.
Herein, we report a highly active magnesium catalyst for the dehydrocoupling of Me2NH⋅BH3 and dehydrogenation of iPr2NH⋅BH3. In addition to being readily synthesized on a multi‐gram scale, the catalyst exceeds previously published examples in terms of selectivity, substrate scope, turnover number (TON), and reaction rate. We present a detailed mechanistic investigation into this catalysis, including stoichiometric reactivity studies and kinetic measurements, and propose a catalytic cycle to account for these data.
Results and Discussion
Catalyst synthesis and investigation
Two magnesium complexes, 1 and 2, were synthesized from the reaction of the proligands (L1H or L2H)41, 42 with MeMgI⋅(OEt2)1.5 and tetramethylethylenediamine (TMEDA) in diethyl ether (Scheme 1 a and b), and obtained in high yields and purity (1: 63 % yield; 2: 86 % yield; see Supporting Information, Figures S2–S5, sections S3.2 and S4.1). The complexes were investigated as catalysts for the dehydrocoupling of Me2NH⋅BH3. While 2 was found to be unreactive, complex 1 gave excellent conversion of Me2NH⋅BH3 to the dimeric dehydrocoupling product [Me2NBH2]2 (3) with concomitant formation of H2 in C6D6 at 60 °C (Scheme 1 c, Figures S17–S18).
Scheme 1.
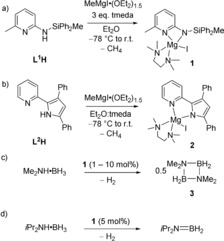
Synthesis of compounds 1 (a) and 2 (b), and catalytic dehydrocoupling/dehydrogenation of Me2NH⋅BH3 (c) and iPr2NH⋅BH3 (d) by 1.
The reaction was monitored by in situ 11B NMR spectroscopy (Figure 2 a), discontinuous 1H and 11B NMR measurements (Figure S15), and volumetrically by the measurement of hydrogen evolution (Figures S25–S26). The reaction is remarkably selective, affording [Me2NBH2]2 (3) almost exclusively (<3 % Me2N=BH2, 4, formed, Figure S18). High conversions are achieved in a relatively short time with low catalyst loadings (at 60 °C; ca. 99 % conversion after 80 mins at 10 mol % [1], 150 mins at 5 mol % [1]; Figure 1 a). High conversions were obtained even at catalyst loadings as low as 1 mol %, with >99 % in 60 h at 60 °C (implying a TON≥100; Figure S19). Increasing the temperature to 80 °C gives >95 % conversion in around 23 minutes with a catalyst loading of just 1.5 mol % (Figure S29d). The catalyst also works in THF, albeit at a significantly reduced rate (Figure S16), which is attributed to co‐ordination of the solvent to the catalyst active site.43 A similar solvent effect was seen with a lithium‐based dehydrocoupling catalyst studied by Mulvey and co‐workers.44
Figure 2.
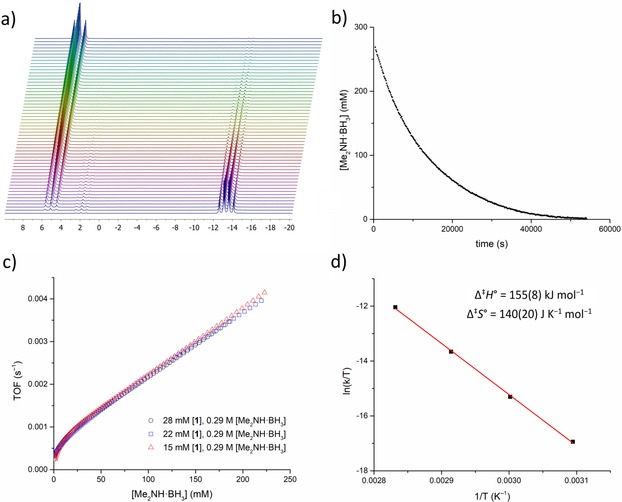
Mechanistic data from the dehydrocoupling of Me2NH⋅BH3 by 1. a) Stacked 11B NMR spectra from in situ NMR measurements. b) Plot of [Me2NH⋅BH3] (mm, as determined by integration of 11B NMR spectra) vs. time for one set of kinetic data (toluene, 60 °C, 4.5 mm 1). c) Plot of turnover frequency (TOF, υ⋅[1]−1) vs. concentration of Me2NH⋅BH3 (mm) for three experiments with different catalyst concentrations. d) Eyring plot for data collected over the temperature range 50–80 °C.
Figure 1.
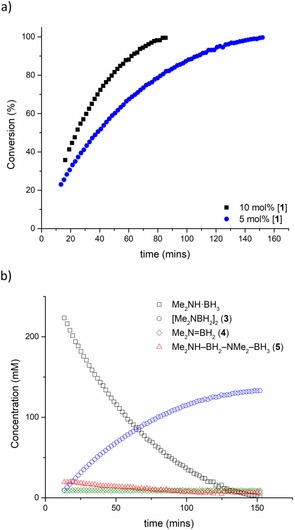
a) Conversion (mol %) vs. time (min) for the dehydrocoupling of Me2NH⋅BH3 with 5 mol % (•) and 10 mol % (▪) of 1 in C6D6 at 60 °C. Data obtained by in situ monitoring of the reaction by 11B NMR. b) Graph tracking concentration of substrate (Me2NH⋅BH3), product ([Me2NBH2]2; 3), Me2N=BH2 (4), and Me2NH‐BH2‐NMe2‐BH3 (5) over reaction course. Concentrations determined by in situ 11B NMR measurements at 60 °C in C6D6. Initial concentrations [3]=0.29 m, [1]=15 mm.
Reactions with the related amine‐borane iPr2NH⋅BH3 afforded the dehydrogenated compound iPr2N=BH2 as the dominant product (>95 %, Scheme 1 d). This reaction proceeded (albeit slowly) at room temperature (Figure S21 and S22) and increasing the temperature to 60 °C gave near total conversion in less than 1 hour (5 mol % catalyst loading, Figure S23). The formation of iPr2N=BH2 has been observed in previous work on the catalytic dehydrogenation of iPr2NH⋅BH3.45, 46, 47
To put this reactivity into context, the most effective previous alkaline earth catalyst for the reaction in Scheme 1 c was published by Hill et al. in 2010.38 This catalyst required 72 h at 60 °C to effect high (>90 %) conversions, and was less selective than 1, affording several side products including (Me2N)2BH. The same catalyst proved more effective at amine‐borane dehydrocoupling with other substrates, catalyzing the reaction between secondary and primary amines with either pinacolborane (HBPin) or 9‐borabicyclo[3.3.1]nonane (9‐BBN).48 In most cases these reactions proceeded to completion in 1 h at room temperature (10 mol % catalyst loading).48 A related magnesium complex was effective at catalyzing the dehydrogenation of iPr2NH⋅BH3 to iPr2N=BH2, but could only effect stoichiometric conversions of Me2NH⋅BH3 to 3.47 There have also been reports of magnesium and calcium bis(trimethylsilyl)amides catalyzing the dehydrocoupling of amine‐boranes with amines to afford asymmetric diaminoboranes.49 In most cases, however, these transformations required high temperatures and long reaction times (i.e. 70 °C, 48 h for coupling Me2NH⋅BH3 and tBuNH2, 2.5 mol % catalyst loading).49
Looking at Group 1 species, we can compare 1 to a recent lithium‐based catalyst published by Mulvey et al.36 Although relatively selective for the formation of [Me2NBH2]2 from Me2NH⋅BH3, the catalyst showed significantly lower activity than species 1, with 89 % conversion after 60 h at 80 °C in toluene (2.5 mol % catalyst loading). Broadening our scope to the rare‐earth metals, a lanthanum hydride complex published by Okuda et al. catalyzed the dehydrocoupling of Me2NH⋅BH3 at a comparable rate to species 1 (100 % conversion in 2 h at 60 °C, 3 mol % catalyst loading).50 However, the catalyst was less selective than 1, affording [Me2NBH2]2 and (Me2N)2BH in approximately 80:20 ratio.50 To the best of our knowledge, there are no examples of an alkaline earth catalyst (for any amine‐borane dehydrocoupling reaction) that are reported to be effective at <2.5 mol % loading,39 with 1 the first catalyst for which this has been demonstrated.51
Catalyst reusability was tested by repeated injections of Me2NH⋅BH3 to a solution of 1 in toluene, with reaction progress monitored by hydrogen evolution. At 10 mol % catalyst loading, 1 was found to tolerate two consecutive injections before a significant deterioration of activity was observed (Figure S26). This is likely due to catalyst decomposition from the gradual ingress of air and moisture with repeated injections. There are very few studies in the literature where the recyclability of main group catalysts for the dehydrocoupling of amine‐boranes has been ascertained,29 and to the best of our knowledge this is the first such determination for an s‐block metal catalyst.
Using the data gathered from in situ reaction monitoring by 11B NMR spectroscopy (Figures 1 and 2; Figures S27–S31), it is possible to gain significant mechanistic insight into this reaction. In addition to starting material, product (3), and trace amounts of Me2N=BH2 (4), the linear species Me2NH‐BH2‐NMe2‐BH3 (5) is also observed during the reaction.52 Compound 5 is a commonly observed intermediate in the dehydrocoupling of Me2NH⋅BH3,14, 16 and its concentration remains low throughout the reaction, gradually dropping to zero as the reaction nears completion (Figure 1 b). Compound 4 appears to form in the early stages of the reaction, and its concentration remains relatively constant throughout (Figure 1 b). Unlike 5, its concentration does not drop to zero. Monitoring by 1H NMR reveals that 1 is regenerated at the end of the reaction, with no appreciable change in the signals arising from the catalyst (Figure S20).
Plotting reaction rate (υ) against substrate concentration [Me2NH⋅BH3] (Figure S28) indicates that the reaction is pseudo‐first order with respect to substrate at high concentrations, with a more complex rate dependence at low substrate concentrations.53 A plot of turnover frequency (TOF=υ⋅[1]−1) against substrate concentration for three different catalyst loadings shows overlay of the data sets (Figure 2 c), indicating that the reaction is first order with respect to catalyst.53 From this, a rate equation (valid for [Me2NH⋅BH3]≫[1]) of υ=k obs[Me2NH⋅BH3][1] has been derived.
Further measurements were carried out over a range of temperatures (50–80 °C) in toluene (Supporting Information, section S4.3.2). Extraction of the pseudo‐first order rate constants for these reactions (Figure S30) allowed for the construction of an Eyring plot (Figure 2 d) and determination of the activation parameters (Δ≠ H°=+155(8) kJ mol−1 and Δ≠ S°=+140(20) J K−1 mol−1).
Kinetic isotope effect (KIE) experiments were performed using deuterated substrate analogues. Reactions with Me2ND⋅BH3 proceeded at approximately the same rate (within error) as those with Me2NH⋅BH3, suggesting that N−D bond cleavage does not feature in the rate‐limiting step of the catalytic cycle. This stands in contrast to, for example, the iron‐catalyzed dehydrocoupling of Me2NH⋅BH3 reported by Webster et al., which displayed a KIE of 2.5±0.2 with Me2ND⋅BH3.45 The distinctive 1:1:1 triplet of HD was also observed by 1H NMR as the sole by‐product in reactions with Me2ND⋅BH3 (Figure S32). It was not possible to obtain a pure sample of Me2NH⋅BD3 from the literature procedure, which afforded mixtures of Me2NH⋅BD3 and Me2NH⋅BH3 (Supporting Information, section S4.1.4). The kinetic isotope effect for B−D bonds was thus calculated from an intermolecular competition experiment with this mixed sample,54 which gave a value of k H/k D=1.6±0.1 (Supporting Information, section 4.3.3). A mixture of H2 and HD was observed by 1H NMR spectroscopy in this reaction.
Stoichiometric reactions between Me2NH⋅BH3 and 1 (Supporting Information, section S4.4.1) resulted in the slow build‐up of 3, 4, 5, and two additional signals at δ B=3.4 (t) and −14.6 (q) (Figure S35). This compound cannot be fully identified, but the signals are similar to previously reported magnesium complexes of amine‐boranes38, 47 and thus may correspond to an NMe2BH2NMe2BH3 chain bound to a magnesium atom. This compound is likely to be an intermediate of the catalytic cycle. Heating the sample to 60 °C resulted in the disappearance of these signals in the 11B NMR spectrum. Despite repeated attempts, no reaction intermediates containing coordinated magnesium could be crystallized from stoichiometric or substoichiometric experiments with Me2NH⋅BH3.
Mechanistic discussion
We propose the catalytic cycle shown in Scheme 2 to account for the observed mechanistic data. This mechanism is broadly similar to those previously proposed for previous alkaline earth metal dehydrocoupling catalysts,24 but with some key differences due to the nature of the ligand L1. It should be noted that there has been little work on the kinetics of such systems, with the mechanism being inferred from the isolation of plausible intermediates and qualitative reaction monitoring by NMR spectroscopy.38, 47 As such, the kinetic data presented in this work provides useful supporting evidence for both this mechanism and the generalized mechanism of alkaline earth dehydrocoupling catalysts.
Scheme 2.
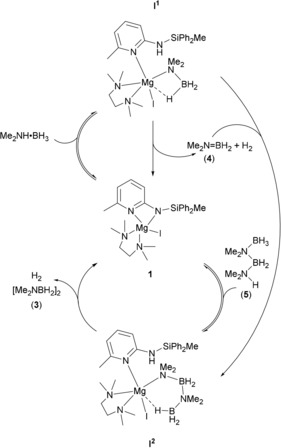
Proposed catalytic cycle for dehydrocoupling of Me2NH⋅BH3 by 1.
The first two steps involve reversible deprotonation of Me2NH⋅BH3 by the silylamide group of 1, to give species I1, followed by an irreversible β‐hydride elimination; affording hydrogen, 4, and species 1. This step likely proceeds via a short‐lived magnesium hydride, which rapidly deprotonates the pendant silyl amine to regenerate 1. This mechanism is somewhat different to previous alkaline earth catalysts, which were added as pre‐catalysts and underwent an initial irreversible deprotonation reaction, typically with elimination of an alkyl or amino species.24 Here, because of the bidentate nature of L1, the amine generated is held proximal to the magnesium and continues to take part in the reaction. This also helps explain the inactivity of 2 as a catalyst, as the less basic pyrrolide is unlikely to deprotonate Me2NH⋅BH3.
Species I1 can also undergo an insertion reaction with 4 to generate species I2. This intermediate is considered to correspond to the unknown species observed by 11B NMR in the stoichiometric experiments (Figure S35), and similar insertion reactions are commonly invoked in mechanisms of this type.24 While it has not been possible to isolate the intermediates I1 and I2, we consider them plausible based on previously proposed mechanisms for similar catalysts.24, 38 Furthermore, the silylamide is the only group on the complex sufficiently basic to deprotonate the substrate. The partial dissociation of the ligand, with the pyridine nitrogen remaining bound to the magnesium atom, seems a reasonable explanation for the regeneration of 1 at the end of the reaction with no loss of ligand. Species I2, once formed, can undergo either a reversible proton exchange (affording linear intermediate 5) or an irreversible δ‐hydride elimination to afford the major product (3) and hydrogen.
Based on the first order dependence on both 1 and Me2NH⋅BH3, the first two steps of the proposed mechanism (formation of I1 and subsequent β‐hydride elimination) must control the overall rate of the catalytic cycle. This is best modelled using the pre‐equilibrium approximation,55 in which an equilibrium is established between (Me2NH⋅BH3+1) and I1, followed by a slow β‐hydride elimination (Scheme 3). A schematic potential energy surface is shown in Figure 3 for illustrative purposes. Denoting the rate constants as k 1, k ‐1, and k 2 (Scheme 3); it can be shown that the overall rate equation for this process is given by Equation (1):55
| (1) |
Scheme 3.
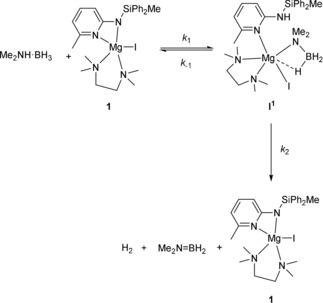
Rate controlling steps of the proposed catalytic cycle.
Figure 3.
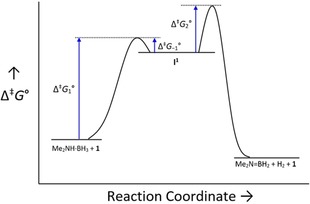
Schematic potential energy surface (PES) for the first two steps of the proposed catalytic cycle.
Use of Me2ND⋅BH3 as a substrate in this reaction will reduce the rate of both k 1 and k −1. As a result, the overall rate of reaction does not change significantly. This can also be understood as the overall rate of reaction being determined by the position of equilibrium for this deprotonation, rather than the rate of deprotonation. By contrast, use of Me2NH⋅BD3 will reduce the rate of k 2, resulting in a modest KIE for this substrate. The fact that β‐hydride elimination is the rate‐limiting step (as indicated by the KIE) suggests that the initial deprotonation must be a reversible process. If it were irreversible, one would expect intermediate I1 to accumulate in the reaction, which is not observed.
Because of the pre‐equilibrium in this reaction, the overall kinetic energy barrier for this process (Δ≠ G°) will depend on the energy barriers for k 1, k −1, and k 2 according to Equation (2):55
| (2) |
This is shown visually with the schematic potential energy surface in Figure 3. This means that the experimentally determined activation parameters for this reaction will depend on all three of these processes. As Δ≠ G 1° is the largest single term in this expression (Figure 3), the activation parameters will be dominated by the transition state for the deprotonation step, even though the β‐hydride elimination is the slowest step. This results in the large positive entropy of activation (Δ≠ S° = +140(20) J K−1 mol−1, consistent with partial dissociation of the bidentate ligand) and large enthalpy of activation (Δ≠ H° = +155(8) kJ mol−1, consistent with significant bond cleavage as we approach the transition state). It should be noted that similar activation parameters were obtained for the dehydrocoupling of 9‐BBN and N‐methylaniline by Hill's magnesium catalyst (Δ≠ H° = +125.1(3) kJ mol−1 and Δ≠ S° = +85(2) J K−1 mol−1), and that this was also attributed to deprotonation of the amine.48
The observed deviation from first‐order kinetics in Me2NH⋅BH3 can be explained by the insertion reaction between I1 and 4 being non‐rate limiting provided [Me2NH⋅BH3]≫[1], and becoming rate limiting as the reaction nears completion. The fact that compound 5 is fully consumed at the end of the reaction, while some 4 remains unreacted, can be explained by both species being steady‐state intermediates of the catalytic cycle (i.e. their rate of formation and consumption is approximately equal). This means the concentration of both 4 and 5 remains low and relatively constant throughout. As the reaction nears completion, 5 is fully converted to 3 through reaction with the catalyst. However, compound 4 can only be consumed in the presence of both Me2NH⋅BH3 and 1 (see Scheme 2). As the reaction nears completion, all the Me2NH⋅BH3 will be converted to 4 by reacting with the catalyst. This means that there is no Me2NH⋅BH3 left for 4 to react with, and thus a small amount of 4 is left unreacted once all substrate is consumed. It should be noted that the off‐metal dimerization of 4 to 3 has been observed previously, and may occur in our system given sufficient time.56, 57, 58 However, we did not observe this, and it is likely that this second‐order process is slow in the catalytic regime.47 Finally, for the reaction of iPr2NH⋅BH3 with 1, it is probable that the insertion reaction is prevented by the increased steric bulk, leading to iPr2N=BH2 as the major product. Thus, the mechanism shown in Scheme 2 accounts for all the observed mechanistic data; including the rate dependencies, kinetic isotope effects, activation parameters, and observed intermediates.
Conclusions
Magnesium complex 1 is an effective catalyst for the dehydrocoupling/dehydrogenation of Me2NH⋅BH3 and iPr2NH⋅BH3. The dehydrocoupling of Me2NH⋅BH3 afforded [Me2NBH2]2 more cleanly, rapidly, and at lower catalyst loadings than any previously reported alkaline earth metal catalyst. We propose that the bidentate ligand L1 allows the catalyst to reversibly regenerate 1 as the catalytic resting state, rather than irreversibly forming a magnesium hydride species. This distinguishes the system from previous catalysts and may account for the remarkably high efficacy and stability of 1 as a catalyst for amine‐borane dehydrocoupling.
Experimental Section
All experimental details including synthetic methods, catalytic studies, kinetic measurements, and crystallographic data are provided in the Supporting Information.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the Engineering and Physical Sciences Research Council [grant number EP/R004064/1]; The Leverhulme Trust [grant number RPG‐2014‐317]; and the University of Nottingham. We also thank Mr. Stephen Boyer (London Metropolitan University) for elemental analyses.
A. C. A. Ried, L. J. Taylor, A. M. Geer, H. E. L. Williams, W. Lewis, A. J. Blake, D. L. Kays, Chem. Eur. J. 2019, 25, 6840.
References
- 1. Marbán G., Valdés-Solís T., Int. J. Hydrogen Energy 2007, 32, 1625–1637. [Google Scholar]
- 2. Barreto L., Makihira A., Riahi K., Int. J. Hydrogen Energy 2003, 28, 267–284. [Google Scholar]
- 3. Dalebrook A. F., Gan W., Grasemann M., Moret S., Laurenczy G., Chem. Commun. 2013, 49, 8735–8751. [DOI] [PubMed] [Google Scholar]
- 4. Züttel A., Naturwissenschaften 2004, 91, 157–172. [DOI] [PubMed] [Google Scholar]
- 5. Keaton R. J., Blacquiere J. M., Baker T. R., J. Am. Chem. Soc. 2007, 129, 1844–1845. [DOI] [PubMed] [Google Scholar]
- 6. Bhunya S., Malakar T., Ganguly G., Paul A., ACS Catal. 2016, 6, 7907–7934. [Google Scholar]
- 7. Bowden M., Autrey T., Brown I., Ryan M., Curr. Appl. Phys. 2008, 8, 498–500. [Google Scholar]
- 8. Stephens F. H., Pons V., Baker R. T., Dalton Trans. 2007, 2613–2626. [DOI] [PubMed] [Google Scholar]
- 9. Graham T. W., Tsang C.-W., Chen X., Guo R., Jia W., Lu S.-M., Sui-Seng C., Ewart C. B., Lough A., Amoroso D., Abdur-Rashid K., Angew. Chem. Int. Ed. 2010, 49, 8708–8711; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 8890–8893. [Google Scholar]
- 10. Cottrell T. L., The Strengths of Chemical Bonds, Butterworths, London, 2nd ed., 1958. [Google Scholar]
- 11. Hamilton C. W., Baker R. T., Staubitz A., Manners I., Chem. Soc. Rev. 2009, 38, 279–293. [DOI] [PubMed] [Google Scholar]
- 12. Jaska C. A., Temple K., Lough A. J., Manners I., Chem. Commun. 2001, 962–963. [Google Scholar]
- 13. Denney M. C., Pons V., Hebden T. J., Heinekey D. M., Goldberg K. I., J. Am. Chem. Soc. 2006, 128, 12048–12049. [DOI] [PubMed] [Google Scholar]
- 14. Sloan M. E., Staubitz A., Clark T. J., Russell C. A., Lloyd-Jones G. C., Manners I., J. Am. Chem. Soc. 2010, 132, 3831–3841. [DOI] [PubMed] [Google Scholar]
- 15. Clark T. J., Russell C. A., Manners I., J. Am. Chem. Soc. 2006, 128, 9582–9583. [DOI] [PubMed] [Google Scholar]
- 16. Sharpe H. R., Geer A. M., Blundell T. J., Hastings F. R., Fay M. W., Rance G. A., Lewis W., Blake A. J., Kays D. L., Catal. Sci. Technol. 2018, 8, 229–235. [Google Scholar]
- 17. Gediga M., Feil C. M., Schlindwein S. H., Bender J., Nieger M., Gudat D., Chem. Eur. J. 2017, 23, 11560–11569. [DOI] [PubMed] [Google Scholar]
- 18. Muhammad S., Moncho S., Brothers E. N., Bengali A. A., Chem. Commun. 2014, 50, 5874–5877. [DOI] [PubMed] [Google Scholar]
- 19. Kakizawa T., Kawano Y., Naganeyama K., Shimoi M., Chem. Lett. 2011, 40, 171–173. [Google Scholar]
- 20. Glüer A., Förster M., Celinski V. R., Schmedt auf der Günne J., Holthausen M. C., Schneider S., ACS Catal. 2015, 5, 7214–7217. [Google Scholar]
- 21. Baker R. T., Gordon J. C., Hamilton C. W., Henson N. J., Lin P.-H., Maguire S., Murugesu M., Scott B. L., Smythe N. C., J. Am. Chem. Soc. 2012, 134, 5598–5609. [DOI] [PubMed] [Google Scholar]
- 22. Todisco S., Luconi L., Giambastiani G., Rossin A., Peruzzini M., Golub I. E., Filippov O. A., Belkova N. V., Shubina E. S., Inorg. Chem. 2017, 56, 4296–4307. [DOI] [PubMed] [Google Scholar]
- 23. Lin T.-P., Peters J. C., J. Am. Chem. Soc. 2013, 135, 15310–15313. [DOI] [PubMed] [Google Scholar]
- 24. Melen R. L., Chem. Soc. Rev. 2016, 45, 775–788. [DOI] [PubMed] [Google Scholar]
- 25. Power P. P., Nature 2010, 463, 171–177. [DOI] [PubMed] [Google Scholar]
- 26. Tang Z., Chen X., Chen H., Wu L., Yu X., Angew. Chem. Int. Ed. 2013, 52, 5832–5835; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 5944–5947. [Google Scholar]
- 27. Miller A. J. M., Bercaw J. E., Chem. Commun. 2010, 46, 1709–1711. [DOI] [PubMed] [Google Scholar]
- 28. Himmelberger D. W., Yoon C. W., Bluhm M. E., Carroll P. J., Sneddon L. G., J. Am. Chem. Soc. 2009, 131, 14101–14110. [DOI] [PubMed] [Google Scholar]
- 29.For an example of a recyclability study with a boron-based Lewis acid catalyst, Lu Z., Schweighauser L., Hausmann H., Wegner H. A., Angew. Chem. Int. Ed. 2015, 54, 15556–15559; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 15777–15780. [Google Scholar]
- 30. Stennett T. E., Harder S., Chem. Soc. Rev. 2016, 45, 1112–1128. [DOI] [PubMed] [Google Scholar]
- 31. Chua Y. S., Wu H., Zhou W., Udovic T. J., Wu G., Xiong Z., Wong M. W., Chen P., Inorg. Chem. 2012, 51, 1599–1603. [DOI] [PubMed] [Google Scholar]
- 32. Diyabalanage H. V. K., Shrestha R. P., Semelsberger T. A., Scott B. L., Bowden M. E., Davis B. L., Burrell A. K., Angew. Chem. Int. Ed. 2007, 46, 8995–8997; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 9153–9155. [Google Scholar]
- 33. Orimo S., Nakamori Y., Eliseo J. R., Züttel A., Jensen C. M., Chem. Rev. 2007, 107, 4111–4132. [DOI] [PubMed] [Google Scholar]
- 34. Bellham P., Hill M. S., Kociok-Köhn G., Dalton Trans. 2015, 44, 12078–12081. [DOI] [PubMed] [Google Scholar]
- 35. Nolla-Saltiel R., Geer A. M., Lewis W., Blake A. J., Kays D. L., Chem. Commun. 2018, 54, 1825–1828. [DOI] [PubMed] [Google Scholar]
- 36. McLellan R., Kennedy A. R., Mulvey R. E., Orr S. A., Robertson S. D., Chem. Eur. J. 2017, 23, 16853–16861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Harinath A., Anga S., Panda T. K., RSC Adv. 2016, 6, 35648–35653. [Google Scholar]
- 38. Liptrot D. J., Hill M. S., Mahon M. F., MacDougall D. J., Chem. Eur. J. 2010, 16, 8508–8515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Spielmann J., Bolte M., Harder S., Chem. Commun. 2009, 6934–6936. [DOI] [PubMed] [Google Scholar]
- 40. Helten H., Robertson A. P. M., Staubitz A., Vance J. R., Haddow M. F., Manners I., Chem. Eur. J. 2012, 18, 4665–4680. [DOI] [PubMed] [Google Scholar]
- 41. Ortu F., Moxey G. J., Blake A. J., Lewis W., Kays D. L., Inorg. Chem. 2013, 52, 12429–12439. [DOI] [PubMed] [Google Scholar]
- 42. Klappa J. J., Rich A. E., McNeill K., Org. Lett. 2002, 4, 435–437. [DOI] [PubMed] [Google Scholar]
- 43.Rates in C6D6 and THF were compared by discontinuous NMR measurements with stop-start heating (Supporting Information, section S4.2.1). It should be noted that the apparent rate when monitoring by this method (Figures S15 and S16) is substantially lower than that observed by in situ NMR measurements (Figure S27) This indicates a significant systematic error associated with stopping and starting the reaction. As such, discontinuous NMR measurements cannot be used to draw any quantitative conclusions about rates or mechanisms. They are, however, sufficient for a qualitative comparison of approximate reaction rates in different solvents.
- 44. McLellan R., Kennedy A. R., Orr S. A., Robertson S. D., Mulvey R. E., Angew. Chem. Int. Ed. 2017, 56, 1036–1041; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1056–1061. [Google Scholar]
- 45. Coles N. T., Mahon M. F., Webster R. L., Organometallics 2017, 36, 2262–2268. [Google Scholar]
- 46. Jaska C. A., Temple K., Lough A. J., Manners I., J. Am. Chem. Soc. 2003, 125, 9424–9434. [DOI] [PubMed] [Google Scholar]
- 47. Hill M. S., Hodgson M., Liptrot D. J., Mahon M. F., Dalton Trans. 2011, 40, 7783–7790. [DOI] [PubMed] [Google Scholar]
- 48. Liptrot D. J., Hill M. S., Mahon M. F., Wilson A. S. S., Angew. Chem. Int. Ed. 2015, 54, 13362–13365; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13560–13563. [Google Scholar]
- 49. Bellham P., Hill M. S., Kociok-Köhn G., Liptrot D. J., Chem. Commun. 2013, 49, 1960–1962. [DOI] [PubMed] [Google Scholar]
- 50. Cui P., Spaniol T. P., Maron L., Okuda J., Chem. Eur. J. 2013, 19, 13437–13444. [DOI] [PubMed] [Google Scholar]
- 51.To the best of our knowledge, there is only one example of an alkaline earth metal catalyst (for any transformation) that was shown to be effective at lower catalyst loadings. This was a barium species for the cross dehydrocoupling of amines and silanes. Barium is, of course, a less attractive prospect for catalysis due to its high toxicity. See Bellini C., Carpentier J.-F., Tobisch S., Sarazin Y., Angew. Chem. Int. Ed. 2015, 54, 7679–7683; [Google Scholar]; Angew. Chem. 2015, 127, 7789–7793. [Google Scholar]
- 52.Species identified by comparison with literature data, Vance J. R., Robertson A. P. M., Lee K., Manners I., Chem. Eur. J. 2011, 17, 4099–4103. [DOI] [PubMed] [Google Scholar]
- 53.We have employed the graphical techniques of Reaction Progress Kinetic Analysis (RPKA) in determining the reaction order with respect to both catalyst and substrate. For an overview of RPKA, see Blackmond D. G., Angew. Chem. Int. Ed. 2005, 44, 4302–4320; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 4374–4393. [Google Scholar]
- 54. Melander L., Saunders W. H., Reaction Rates of Isotopic Molecules, Wiley, 2nd ed., 1960. [Google Scholar]
- 55. Anslyn E. V, Dougherty D. A., Modern Physical Organic Chemistry, University Science Books, Sausalito, 1st ed., 2006. [Google Scholar]
- 56. Stevens C. J., Dallanegra R., Chaplin A. B., Weller A. S., Macgregor S. A., Ward B., McKay D., Alcaraz G., Sabo-Etienne S., Chem. Eur. J. 2011, 17, 3011–3020. [DOI] [PubMed] [Google Scholar]
- 57. Sewell L. J., Lloyd-Jones G. C., Weller A. S., J. Am. Chem. Soc. 2012, 134, 3598–3610. [DOI] [PubMed] [Google Scholar]
- 58. Robertson A. P. M., Leitao E. M., Jurca T., Haddow M. F., Helten H., Lloyd-Jones G. C., Manners I., J. Am. Chem. Soc. 2013, 135, 12670–12683. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary