

Abstract
Chiral nanosized confinements play a major role for enantioselective recognition and reaction control in biological systems. Supramolecular self‐assembly gives access to artificial mimics with tunable sizes and properties. Herein, a new family of [Pd2L4] coordination cages based on a chiral [6]helicene backbone is introduced. A racemic mixture of the bis‐monodentate pyridyl ligand L1 selectively assembles with PdII cations under chiral self‐discrimination to an achiral meso cage, cis‐[Pd2 L1P 2 L1M 2]. Enantiopure L1 forms homochiral cages [Pd2 L1P/M 4]. A longer derivative L2 forms chiral cages [Pd2 L2P/M 4] with larger cavities, which bind optical isomers of chiral guests with different affinities. Owing to its distinct chiroptical properties, this cage can distinguish non‐chiral guests of different lengths, as they were found to squeeze or elongate the cavity under modulation of the helical pitch of the helicenes. The CD spectroscopic results were supported by ion mobility mass spectrometry.
Keywords: anion recognition, chirality, host–guest chemistry, interpenetration, supramolecular chemistry
Nanosized cages based on metallosupramolecular self‐assembly have become major players in host–guest chemistry owing to their structural and functional variability and modular composition.1 Recent design‐based approaches allow the positioning of multiple building blocks by thermodynamically controlled integrative self‐sorting.2 In biological host–guest systems, enantioselective recognition plays a pivotal role because of the inherent homochirality of most natural compounds. Hence, the formation of synthetic chiral hosts for enantioselective guest binding is not only of fundamental interest, but provides the basis for the development of selective sensors, transporters, and catalysts.3
Numerous chiral hosts based on covalent macrocyclic molecules such as cyclodextrins, cyclophanes, and calixarenes have been reported.4 Chirality has also been reported to facilitate the assembly of hydrogen‐bonded organic cages.5 More recently, chiral metallo‐supramolecular self‐assembled rings and cages have been introduced as selective receptors and enzyme‐like nanoreactors based on chiral backbones, auxiliaries, the inherent chirality of stereogenic metal centers, or the overall architecture.6 Upon metal coordination, racemic mixtures of ligands may undergo chiral self‐sorting,7 leading to homochiral8, 9, 10 or heterochiral10, 11 assemblies. Beyond their use in enantioselective recognition, chiral cages based on luminescent metal centers have been shown to exhibit unique chiroptical properties.9, 12 With respect to mechanically interlocked coordination cages,13 reports covering the implementation of homochirality are still scarce, with Hardie's dimer of cyclotriveratrylene‐based coordination cages serving as a notable example.14
Since their discovery in 1912,15 helicenes have been widely studied for properties related to their helical chirality.16 While helicenes have shown appearance in several supramolecular systems, they have never been used in the construction of coordination‐driven cages.17 We herein demonstrate that despite their highly twisted appearance, helicene‐based bis‐monodentate ligands can be used to assemble discrete [Pd2 L 4] coordination cages exhibiting chirality‐driven effects on their assembly and guest binding. We further report the first example of a homochiral interpenetrated [Pd4 L 8] dimer, comprising eight interlocked helicenes.
Ligands L1 and L2 were synthesized by Sonogashira cross‐coupling reactions from literature‐known 2,15‐dibromo[6]helicene (Figure 1) to yield racemic products, which were separated into the enantiomers by chiral HPLC (see the Supporting Information, Figure S23).18 Following our previously reported routines, the bis‐monodentate ligands were tested for the formation of self‐assembled products using [Pd(CH3CN)4](BF4)2 as the metal source in different polar organic solvents. Interestingly, in deuterated dimethyl sulfoxide (DMSO), the racemic mixture of ligand L1 was found to quantitatively assemble under chiral self‐sorting into the achiral meso cage cis‐[Pd2 L1P 2 L1M 2] (C1 meso), containing both ligand enantiomers in a 1:1 ratio, as confirmed by 1H (Figure 2 a) and NOESY NMR spectroscopy (Figure 4 b). For all herein described cages, the 1H NMR signals of the pyridine moieties (i.e., protons Ha and Hb) undergo a downfield shift upon coordination to the palladium(II) cations. The formation of the meso cage leads to splitting of all 1H NMR resonances into two sets of equal intensity. All resonances could be assigned with the help of 2D NMR techniques (COSY, NOESY, HSQC), indicating that the upper and lower halves of ligand L1 have ended up in a different surrounding upon cage formation (see the Supporting Information). The resonance splitting can be explained by symmetry considerations. The halves of the P helicenes and the halves of the M helicenes facing each other have the same chemical surrounding, which results in the same chemical shifts for the corresponding protons. Compared to this, one half of the P helicene facing the other half of the P helicene (same for M helicenes) has a different chemical surrounding, which explains the twofold splitting in the 1H NMR spectrum. A tentative trans‐configured cage would not lead to such a splitting of the NMR signals as the resulting D 2d symmetry would offer two C 2 axes perpendicular to the major C 2 axis going through both Pd centers, which would allow converting the upper half of each ligand into its lower part (Figure S7).
Figure 1.
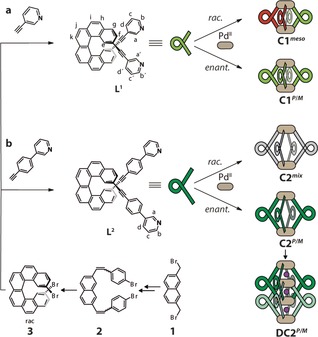
a, b) Synthesis of ligands L1 and L2 from 2,15‐dibromo[6]helicene 3 followed by separation into the P (red) and M (green) enantiomers. The addition of stoichiometric amounts of PdII leads to the quantitative formation of different coordination cages, depending on the enantiomeric composition and length of the ligand. Racemic L1 exclusively gives C1 meso, whereas racemic L2 leads to a statistical mixture of all possible stereoisomers (PPPM/MMMP/PPMM/PMPM/PPPP/MMMM, shown in gray). The enantiopure ligands give the chiral coordination cages C1 P/M and C2 P/M and the interpenetrated dimer DC2 P/M.
Figure 2.
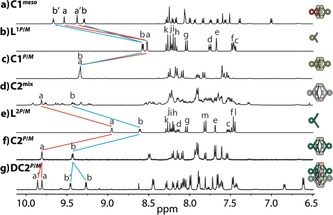
1H NMR spectra (400 MHz, [D6]DMSO, 293 K) of: a) C1 meso, b) L1P/M, c) homochiral C1 P/M, d) a statistical mixture of C2 stereoisomers, e) L2P/M, f) homochiral C2 P/M, and g) the homochiral interpenetrated cage structure DC2 P/M (here: 600 MHz, [D3]acetonitrile, 293 K).
Figure 4.
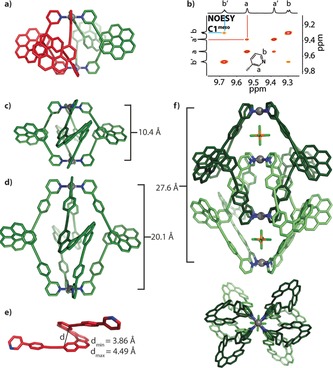
a) DFT‐calculated structure of C1 meso. b) NOESY NMR detail of the C1 meso cage supporting the cis ligand arrangement. c, d) Calculated structures of C1 M and C2 M. e) One of twelve L2P molecules in the asymmetric unit of its solid‐state structure with the found minimum/maximum helical pitches. f) X‐ray crystal structure of DC2 M, side and top view along the Pd4 axis. Pd gray, N blue, C green (M enantiomer) and red (P enantiomer), P orange, F light green).
In contrast, assembly with the enantiopure ligand L1, in either its P or M form, leads to a homochiral cage with no splitting of the 1H NMR signals (Figure 2 c). In their high‐resolution ESI mass spectra, cage C1 meso (Figure S9) and the enantiopure cages C1 P/M (Figure S11) could be identified as tetracationic [Pd2 L1 4]4+.
Next, chiral guest discrimination of C1 P was tested with (1R)‐ and (1S)‐camphorsulfonate anions (G1 R and G1 S); however, no evidence for uptake of the guests was found (Figure S21). The most probable reason is the limited size of the cavity, known as a critical factor for guest binding.19 To permit guest encapsulation, the ligand structure was extended by including 1,4‐phenylene linkers on both sides to give ligand L2. The elongation of the ligands nearly doubles the Pd–Pd distance in the modeled structures (DFT ωB97XD/def2SVP, PCM solvent: DMSO) of C2 P/M (20.1 Å; Figure 4 d) compared to C1 P/M (10.4 Å; Figure 4 c). In case of racemic L2, cage formation leads to the splitting of all 1H NMR resonances into several sets, which is indicative of a lack of chiral self‐sorting under formation of a statistical mixture of isomeric species (Figure 2 d). This picture is supported by the clean appearance of the high‐resolution ESI mass spectrum of this mixture, showing only peaks assignable to the tetracationic [Pd2 L2 4]4+ species, which is superimposable with the spectrum of the homochiral cage C2 P/M (Figure S14). The absence of chiral self‐discrimination upon cage formation from racemic L2 can be explained with the increased distance between the helicene backbones (based on the calculated structures of cages C1 and C2, the closest H–H distance between two neighboring backbones has increased from 2.39 Å to 6.20 Å).
In contrast to the results obtained in DMSO, heating the enantiopure ligand L2 with palladium(II) cations in acetonitrile was found to lead to a splitting of all NMR resonances into two sets of equal intensity, thus indicating the formation of a chiral interpenetrated cage DC2 P/M (Figure 2 g).13 In addition, the high‐resolution ESI mass spectrum contained signals for the dimeric species [3BF4@Pd4 L2 8]5+ (Figure 3 c).
Figure 3.
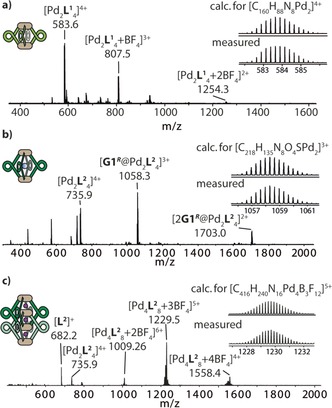
ESI mass spectra of: a) cage C1 M, b) cage C2 M after addition of (1R)‐camphorsulfonate G1 R, and c) double cage DC2 M.
Further structural insight was obtained by X‐ray diffraction methods. Crystals of enantiopure L2 (second HPLC fraction eluted from a Chiralpak IC column) suitable for X‐ray structure analysis were obtained by crystallization from DMSO (Figure 4 e). The asymmetric unit contains twelve individual helicene ligands, all of which are highly intertwined in a remarkably unordered fashion (Figure S25). The absolute configuration was unambiguously determined as the P enantiomer using the method of Parsons20 as implemented in SHELXL,21 yielding an enantiopurity‐distinguishing parameter of x=0.079(8). This assignment is in agreement with the recorded circular dichroism (CD) spectra of this compound as compared to published data on similarly substituted [6]helicenes and DFT‐calculated CD bands.22, 23
Single crystals of the dimeric cage species [2 PF6@Pd4 L2M 8] (DC2 M, based on the M ligand enantiomer eluting first from the chiral column) that were suitable for X‐ray structure analysis were obtained by slow diffusion of diethyl ether into an acetonitrile solution of the cage containing PF6 − counteranions (Figure 4 f). Synchrotron radiation was required to obtain diffraction data that could be solved with direct methods using SHELXT.24 Again, the absolute configuration could be unambiguously determined, yielding an enantiopurity‐distinguishing parameter of x=−0.02(2). The CD data were found to be in agreement with the literature‐reported absolute structure assignment of comparable helicenes.22, 23 The structure reveals that the double cage features three consecutive pockets, with the two outer ones filled with a PF6 − anion each. The Pd–Pd distances are 8.66 Å for the outer pockets and 10.33 Å for the inner cavity.
With the large cavity of monomeric C2 P/M, chiral guest discrimination could be shown for the enantiopure cages by 1H NMR titration experiments by stepwise addition of camphorsulfonates G1 R and G1 S as their tetrabutylammonium salts. Characteristic downfield shifts for the inside‐pointing proton resonance Ha were observed (Figure S22), and the results were summarized as a comparison of binding isotherms (Δδ plot; Figure 5 c). Pleasingly, both guest enantiomers showed different binding behavior when exposed to the same chiral cage; however, the combination G1 S@C2 P showed the same behavior as the enantiomeric system G1 R@C2 M, with binding constants of around 560 m −1.25 The diastereomeric combinations to this, G1 R@C2 P and G1 S@C2 M, showed a stronger extent of NMR signal shifting and a binding constant of approximately 1010 m −1. In the high‐resolution ESI mass spectra, the host–guest complexes could be identified as the triple cationic species [G1@Pd2 L2 4]3+ (Figure 3 b).
Figure 5.
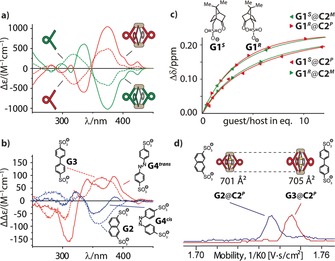
a) Circular dichroism spectra of ligands L2P/M and cages C2 P/M. b) Difference CD spectra (free host CD spectra subtracted from the host–guest CD spectra) of G2@C2 P and G3@C2 P as well as G4 trans@C2 P and G4 cis@C2 P (all in DMSO). c) Comparison of the binding isotherms for all four diastereomeric host–guest combinations G1 R/S@C2 P/M showing two “matched” and two “mismatched” cases. d) Superposition of the mobilograms obtained by trapped ion mobility ESI‐TOF mass spectrometry for host–guest complexes G2@C2P (mobility 1/K 0: 1.736 Vs cm−2, CCS: 701 Å2 at m/z 1615.4) and G3@C2P (mobility 1/K 0: 1.745 Vs cm−2, CCS: 705 Å2 at m/z 1627.9).
Furthermore, CD spectra were compared for L1P/M and C1 P/M (Figure S24) and L2P/M and C2 P/M (Figure 5 a), showing strong circular dichroism for the ligands and the cages with a positive Cotton effect for the P enantiomers. We next set out to investigate the potential utilization of the strong CD effect as an indicator for the discrimination of achiral guests. As the cages consist of four helicenes arranged like parallel springs around two connecting PdII cations, we envisioned that charged guests encapsulated between these electrostatic anchors should modulate the helical pitch of the ligand backbones. First, we compared the effect of binding short 2,7‐naphthalenedisulfonate G2 and long 4,4′‐biphenyldisulfonate G3 on the CD spectra of C2 P. Difference spectra revealed that encapsulation of the shorter guest led to a decrease in the intensity of the CD band at approximately 360 nm while binding of the longer guest increased the intensity of the same band (Figure 5 b). The assumption that such an effect is caused by tuning of the helical pitch of the helicenes was predicted by theoretical work of Mori, Inoue, and Nakai.22 We were able to confirm this hypothesis by calculating the relative CD signal intensities of unsubstituted [6]helicene under variation of its helical pitch within the limits found in the twelve individual ligands contained in the solid‐state structure of L2 (Figure S26 and Figure 4 e).
Furthermore, direct evidence for a shrinking and expansion of the cages upon addition of the short and long guests, respectively, came from trapped ion mobility ESI‐TOF mass spectrometry (timsTOF), which indicated a smaller gas phase collisional cross‐section for G2@C2 P (701 Å2) than for G3@C2 P (705 Å2), even in a mixture of both host–guest complexes (Figure 5 d and Figure S27).26 We repeated the CD experiment with azobenzene‐based guest G4,27 either in its cis or trans photoisomeric form (Figure 5 b). Remarkably, the effect of the band intensity decrease/increase could be reproduced and allows differentiation between the cis and the trans form of achiral azobenzene by CD spectroscopy, keeping in mind that the free guest itself shows no CD effect. In addition, the observed deviations from the expected band shapes were attributed to a certain degree of chirality transfer on the azobenzene chromophore, which—in contrast to guests G2 and G3—shows significant absorption around 360 nm.
In summary, a family of [Pd2 L 4] coordination cages based on a chiral helicene backbone have been developed.28 One of the cages showed integrative chiral self‐sorting, thus serving as an example of the non‐statistical formation of heteroleptic structures, while another one was found to discriminate chiral guests through different binding affinities to its enantiopure form. The strong circular dichroism of the helicene backbone could further be exploited for the size discrimination of achiral anionic guests by taking advantage of modulations of the chiroptical properties of the system upon guest‐induced changes of the helical pitch. Ion mobility mass spectrometry was employed to support these findings. In addition, the group of [Pd4L8] interpenetrated cages could be expanded by an unprecedented chiral species, as illustrated by its single‐crystal X‐ray structure. Further studies are underway to expand the guest binding and recognition features and develop a system for enantioselective catalysis inside confined environments.
Experimental Section
Cages C1 and C2 were formed by addition of [Pd(CH3CN)4](BF4)2 (0.5 equiv) to the corresponding ligands (racemic or enantiopure) in DMSO at 23 °C. Cage DC2 was formed after heating C2 in MeCN at 75 °C for 2 weeks. Single crystals suitable for X‐ray structure determination were grown for L2P from DMSO and for DC2 M by slow diffusion of Et2O into a mixture of L2M and [Pd(CH3CN)4](PF6)2 in MeCN at 7 °C. CCDC 1558206 (L2P) and 1581540 (DC2 M) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work has been supported by the Deutsche Forschungsgemeinschaft (CL 489/2‐2, RESOLV Cluster of Excellence EXC‐2033, project number 390677874) and the European Research Council through ERC Consolidator grant 683083 (RAMSES). We thank Prof. U. Diederichsen, Prof. L. Ackermann and Prof. L. Tietze (all Georg‐August University Göttingen) for access to CD and HPLC facilities. We further thank S. Löffler for support with crystallization experiments, Prof. W. Hiller (TU Dortmund), Dr. M. John (GAU Göttingen) for help with NMR spectroscopy and L. Schneider (TU Dortmund), C. Heitbrink, Dr. P. Janning (MPI Dortmund), and Dr. H. Frauendorf (GAU Göttingen) for ESI mass spectra. Diffraction data for DC2 M were collected at PETRA III, DESY, a member of the Helmholtz Association (HGF). We thank Saravanan Panneerselvam for assistance in using synchrotron beamline P11 (I‐20160736).29
T. R. Schulte, J. J. Holstein, G. H. Clever, Angew. Chem. Int. Ed. 2019, 58, 5562.
References
- 1.
- 1a. Chakrabarty R., Mukherjee P. S., Stang P. J., Chem. Rev. 2011, 111, 6810; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Han M., Engelhard D. M., Clever G. H., Chem. Soc. Rev. 2014, 43, 1848. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Mukherjee S., Mukherjee P. S., Chem. Commun. 2014, 50, 2239; [DOI] [PubMed] [Google Scholar]
- 2b. Bloch W. M., Clever G. H., Chem. Commun. 2017, 53, 8506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Castilla A. M., Ramsay W. J., Nitschke J. R., Acc. Chem. Res. 2014, 47, 2063; [DOI] [PubMed] [Google Scholar]
- 3b. Chen L.-J., Yang H.-B., Shionoya M., Chem. Soc. Rev. 2017, 46, 2555; [DOI] [PubMed] [Google Scholar]
- 3c. Pan M., Wu K., Zhang J.-H., Su C.-Y., Coord. Chem. Rev. 2019, 378, 333. [Google Scholar]
- 4.
- 4a. Tsunoda Y., Fukuta K., Imamura T., Sekiya R., Furuyama T., Kobayashi N., Haino T., Angew. Chem. Int. Ed. 2014, 53, 7243; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 7371; [Google Scholar]
- 4b. Gropp C., Trapp N., Diederich F., Angew. Chem. Int. Ed. 2016, 55, 14444; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 14659. [Google Scholar]
- 5. Beaudoin D., Rominger F., Mastalerz M., Angew. Chem. Int. Ed. 2016, 55, 15599; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 15828. [Google Scholar]
- 6.
- 6a. Carrano C. J., Raymond K. N., J. Am. Chem. Soc. 1978, 100, 5371; [Google Scholar]
- 6b. Lee S. J., Lin W., J. Am. Chem. Soc. 2002, 124, 4554; [DOI] [PubMed] [Google Scholar]
- 6c. Hembury G. A., Borovkov V. V., Inoue Y., Chem. Rev. 2008, 108, 1; [DOI] [PubMed] [Google Scholar]
- 6d. Nishioka Y., Yamaguchi T., Kawano M., Fujita M., J. Am. Chem. Soc. 2008, 130, 8160; [DOI] [PubMed] [Google Scholar]
- 6e. Siering C., Toräng J., Kruse H., Grimme S., Waldvogel S. R., Chem. Commun. 2010, 46, 1625; [DOI] [PubMed] [Google Scholar]
- 6f. Zhao C., Sun Q.-F., Hart-Cooper W. M., DiPasquale A. G., Toste F. D., Bergman R. G., Raymond K. N., J. Am. Chem. Soc. 2013, 135, 18802; [DOI] [PubMed] [Google Scholar]
- 6g. Gütz C. et al., Angew. Chem. Int. Ed. 2014, 53, 1693; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 1719. [Google Scholar]
- 7. Jędrzejewska H., Szumna A., Chem. Rev. 2017, 117, 4863. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Masood M. A., Enemark E. J., Stack T. D. P., Angew. Chem. Int. Ed. 1998, 37, 928; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1998, 110, 973; [Google Scholar]
- 8b. Lützen A., Hapke M., Griep-Raming J., Haase D., Saak W., Angew. Chem. Int. Ed. 2002, 41, 2086; [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 2190; [Google Scholar]
- 8c. Kiehne U., Weilandt T., Lützen A., Org. Lett. 2007, 9, 1283; [DOI] [PubMed] [Google Scholar]
- 8d. Maeda C., Kamada T., Aratani N., Osuka A., Coord. Chem. Rev. 2007, 251, 2743; [Google Scholar]
- 8e. Meyer-Eppler G., Topić F., Schnakenburg G., Rissanen K., Lützen A., Eur. J. Inorg. Chem. 2014, 2495; [Google Scholar]
- 8f. Boer S. A., Turner D. R., Chem. Commun. 2015, 51, 17375; [DOI] [PubMed] [Google Scholar]
- 8g. Yan L.-L., Tan C.-H., Zhang G.-L., Zhou L.-P., Bünzli J.-C., Sun Q.-F., J. Am. Chem. Soc. 2015, 137, 8550; [DOI] [PubMed] [Google Scholar]
- 8h. Pritchard V. E., Rota Martir D., Oldknow S., Kai S., Hiraoka S., Cookson N. J., Zysman-Colman E., Hardie M. J., Chem. Eur. J. 2017, 23, 6290; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8i. Tateishi T., Kojima T., Hiraoka S., Commun. Chem. 2018, 1, 727. [Google Scholar]
- 9. Yan L.-L., Tan C.-H., Zhang G.-L., Zhou L.-P., Bünzli J.-C., Sun Q.-F., J. Am. Chem. Soc. 2015, 137, 8550. [DOI] [PubMed] [Google Scholar]
- 10. Beaudoin D., Rominger F., Mastalerz M., Angew. Chem. Int. Ed. 2017, 56, 1244; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1264. [Google Scholar]
- 11.
- 11a. Kim T. W., Hong J.-I., Lah M. S., Chem. Commun. 2001, 743; [Google Scholar]
- 11b. Claessens C. G., Torres T., J. Am. Chem. Soc. 2002, 124, 14522; [DOI] [PubMed] [Google Scholar]
- 11c. Weilandt T., Kiehne U., Schnakenburg G., Lützen A., Chem. Commun. 2009, 2320; [DOI] [PubMed] [Google Scholar]
- 11d. Arribas C. S., Wendt O. F., Sundin A. P., Carling C.-J., Wang R., Lemieux R. P., Wärnmark K., Chem. Commun. 2010, 46, 4381. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Rota Martir D., Escudero D., Jacquemin D., Cordes D. B., Slawin A. M. Z., Fruchtl H. A., Warriner S. L., Zysman-Colman E., Chem. Eur. J. 2017, 23, 14358; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Li X.-Z., Zhou L.-P., Yan L.-L., Yuan D.-Q., Lin C.-S., Sun Q.-F., J. Am. Chem. Soc. 2017, 139, 8237. [DOI] [PubMed] [Google Scholar]
- 13. Frank M., Johnstone M. D., Clever G. H., Chem. Eur. J. 2016, 22, 14104. [DOI] [PubMed] [Google Scholar]
- 14. Westcott A., Fisher J., Harding L. P., Rizkallah P., Hardie M. J., J. Am. Chem. Soc. 2008, 130, 2950. [DOI] [PubMed] [Google Scholar]
- 15. Weitzenböck R., Lieb H., Monatsh. Chem. 1912, 33, 549. [Google Scholar]
- 16.
- 16a. Gingras M., Chem. Soc. Rev. 2013, 42, 968; [DOI] [PubMed] [Google Scholar]
- 16b. Saleh N., Shen C., Crassous J., Chem. Sci. 2014, 5, 3680; [Google Scholar]
- 16c. Chen C.-F., Shen Y., Helicene Chemistry, Springer, Berlin, Heidelberg, 2017; [Google Scholar]
- 16d. Mori K., Murase T., Fujita M., Angew. Chem. Int. Ed. 2015, 54, 6847; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 6951; [Google Scholar]
- 16e. Brandt J. R., Wang X., Yang Y., Campbell A. J., Fuchter M. J., J. Am. Chem. Soc. 2016, 138, 9743. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Verbiest T., Van Elshocht S., Kauranen M., Hellemans L., Snauwaert J., Nuckolls C., Katz T. J., Persoons A., Science 1998, 282, 913; [DOI] [PubMed] [Google Scholar]
- 17b. Kaseyama T., Furumi S., Zhang X., Tanaka K., Takeuchi M., Angew. Chem. Int. Ed. 2011, 50, 3684; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 3768. [Google Scholar]
- 18.
- 18a. Terfort A., Görls H., Brunner H., Synthesis 1997, 79; [Google Scholar]
- 18b. Fox J. M., Lin D., Itagaki Y., Fujita T., J. Org. Chem. 1998, 63, 2031. [Google Scholar]
- 19.
- 19a. Löffler S., Lübben J., Wuttke A., Mata R. A., John M., Dittrich B., Clever G. H., Chem. Sci. 2016, 7, 4676; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19b. Freye S., Michel R., Stalke D., Pawliczek M., Frauendorf H., Clever G. H., J. Am. Chem. Soc. 2013, 135, 8476. [DOI] [PubMed] [Google Scholar]
- 20. Parsons S., Flack H. D., Wagner T., Acta Crystallogr. Sect. B 2013, 69, 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sheldrick G. M., Acta Crystallogr. Sect. C 2015, 71, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nakai Y., Mori T., Inoue Y., J. Phys. Chem. A 2012, 116, 7372. [DOI] [PubMed] [Google Scholar]
- 23. Nakai Y., Mori T., Inoue Y., J. Phys. Chem. A 2013, 117, 83. [DOI] [PubMed] [Google Scholar]
- 24. Sheldrick G. M., Acta Crystallogr. Sect. A 2015, 71, 3. [Google Scholar]
- 25. Thordarson P., Chem. Soc. Rev. 2011, 40, 1305. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a. Jurček O., Bonakdarzadeh P., Kalenius E., Linnanto J. M., Groessl M., Knochenmuss R., Ihalainen J. A., Rissanen K., Angew. Chem. Int. Ed. 2015, 54, 15462; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 15682; [Google Scholar]
- 26b. Greisch J.-F., Chmela J., Harding M. E., Wunderlich D., Schäfer B., Ruben M., Klopper W., Schooss D., Kappes M. M., Phys. Chem. Chem. Phys. 2017, 19, 6105. [DOI] [PubMed] [Google Scholar]
- 27. Clever G. H., Tashiro S., Shionoya M., J. Am. Chem. Soc. 2010, 132, 9973. [DOI] [PubMed] [Google Scholar]
- 28.
- 28a. Malik A. U., Gan F., Shen C., Yu N., Wang R., Crassous J., Shu M., Qiu H., J. Am. Chem. Soc. 2018, 140, 2769; [DOI] [PubMed] [Google Scholar]
- 28b. Matsushima T., Kikkawa S., Azumaya I., Watanabe S., ChemistryOpen 2018, 7, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Burkhardt A., Pakendorf T., Reime B., Meyer J., Fischer P., Stübe N., Panneerselvam S., Lorbeer O., Stachnik K., Warmer M., Rödig P., Göries D., Meents A., Eur. Phys. J. Plus 2016, 131, 56. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary