

Summary
Phytoplankton and bacteria interactions have a significant role in aquatic ecosystem functioning. Associations can range from mutualistic to parasitic, shaping biogeochemical cycles and having a direct influence on phytoplankton growth. How variations in phenotype and sampling location, affect the phytoplankton microbiome is largely unknown. A high‐resolution characterization of the bacterial community in cultures of the dinoflagellate Alexandrium was performed on strains isolated from different geographical locations and at varying anthropogenic impact levels. Microbiomes of Baltic Sea Alexandrium ostenfeldii isolates were dominated by Betaproteobacteria and were consistent over phenotypic and genotypic Alexandrium strain variation, resulting in identification of an A. ostenfeldii core microbiome. Comparisons with in situ bacterial communities showed that taxa found in this A. ostenfeldii core were specifically associated to dinoflagellate dynamics in the Baltic Sea. Microbiomes of Alexandrium tamarense and minutum, isolated from the Mediterranean Sea, differed from those of A. ostenfeldii in bacterial diversity and composition but displayed high consistency, and a core set of bacterial taxa was identified. This indicates that Alexandrium isolates with diverse phenotypes host predictable, species‐specific, core microbiomes reflecting the abiotic conditions from which they were isolated. These findings enable in‐depth studies of potential interactions occurring between Alexandrium and specific bacterial taxa.
Introduction
Interactions between phytoplankton and bacteria in aquatic environments are intricate and have large impacts on ecosystem functioning (Azam and Malfatti, 2007; Buchan et al., 2014). Specifically, phytoplankton bacteria interactions, ranging from mutualism (Amin et al., 2009) to resource competition (Risgaard‐Petersen et al., 2004), have effects on the whole ecosystem in terms of the amount of carbon that is recycled or stored (Cole, 1982; Jiao et al., 2010), the level of regenerated nitrogen (Eppley and Peterson, 1979) and utilization of captured energy (Buchan et al., 2014). The diversity, functioning and efficiency of bacteria associating with phytoplankton cells (the microbiome) are thought to be determined by the biochemical composition of phytoplankton and their released organic matter (Azam and Malfatti, 2007; Buchan et al., 2014; Seymour et al., 2017). The composition of the microbiome has been shown to be host specific in association with, for example, diatoms and dinoflagellates (Schäfer et al., 2002; Guannel et al., 2011; Lawson et al., 2017; Behringer et al., 2018).
Dinoflagellates release organic matter continuously during their life cycle (Villacorte et al., 2015) affecting both composition and function of their microbiome (Sarmento et al., 2013). The role of bacteria‐dinoflagellate interactions may include growth stimuli (Sakami et al., 1999; Ferrier et al., 2002; Bolch et al., 2011), life cycle cues (Adachi et al., 2003) and regulation of toxin production (Doucette and Powell, 1997; Gallacher and Smith, 1999). Lawson et al. (2017) showed the existence of a consistent core microbiome among dinoflagellate Symbiodinium clades, pointing at taxonomically consistent interactions with significance to dinoflagellate functions. Few studies have characterized these complex interactions (Hattenrath‐Lehmann and Gobler, 2017), which is a prerequisite for understanding consequences for large scale biogeochemical processes.
Dinoflagellates of the genus Alexandrium are reported from many geographical areas in the world's oceans (Anderson et al., 2012) and are known to release bioactive compounds, potent toxins (Anderson et al., 2012) and allelochemicals that are poorly chemically characterized (Ma et al., 2009), which can interact with microbial food web dynamics (Weissbach et al., 2011). Alexandrium populations display a large intraspecific phenotypic diversity, in terms of growth rate (Suikkanen et al., 2013; Brandenburg et al., 2018), toxin production (Tillmann et al., 2009; Suikkanen et al., 2013; Martens et al., 2017; Brandenburg et al., 2018), allelochemical activity (Hakanen et al., 2014; Brandenburg et al., 2018), nutritional strategies (Glibert and Legrand, 2006) and ability to bioluminesce (Valiadi et al., 2012). Due to this large phenotypic diversity Alexandrium is an ideal model organism for investigating the consistency of microbiomes over host specific, intraspecific or environmental variations (Tahvanainen et al., 2012; Suikkanen et al., 2013; Hakanen et al., 2014). We hypothesize that the high diversity among Alexandrium isolates would result in diverse, isolate specific, microbiomes in culture.
Several earlier studies have taxonomically characterized bacteria associated with marine Alexandrium in culture, identifying Proteobacteria [mainly Roseobacter (Alphaproteobacteria) and Alteromonas (Gammaproteobacteria) clades] and Bacteroidetes as dominating bacterial phyla (Kopp, 1997; Hold et al., 2001; Biegala et al., 2002; Jasti et al., 2005). However, these studies are limited to marine conditions (salinity ~31) and low sequencing resolution and coverage, which preclude conclusions of the microbiome consistency and specific associations of individual bacterial species and Alexandrium isolates. A recent study of an Alexandrium bloom at marine conditions, using 16S amplicon sequencing, found associations with Flavobacteriia (Owenweeksia and the NS5 marine group), Alpha‐ (Rhodobacterales) and Gammaprotebacteria (Alteromonadales) (Hattenrath‐Lehmann and Gobler, 2017), showing concordance, at phylum and order level, between findings in culture with those from the environment.
In this study, we define microbiome as consisting of the whole bacterial assemblage found in the Alexandrium cultures. Our aim was to explore how host specificity, intraspecies variations and sampling location influence the diversity and consistency of the microbiome of A. ostenfeldii, A. minutum and A. tamarense isolates, and investigate the existence of a species‐specific core microbiome. In total 28 strains were included, of which 20 belong to A. ostenfeldii, isolated from four distinct locations in the brackish Baltic Sea Proper. These strains have been shown to have phenotypic (Suikkanen et al., 2013; Hakanen et al., 2014) and genotypic (Tahvanainen et al., 2012) differences. The remaining strains belonged to A. minutum (n = 7) and A. tamarense (n = 1), isolated from eight locations in the northwest Mediterranean Sea (marine conditions) with different anthropogenic impact levels, classified using the Land Uses Simplified Index, LUSI (E. Flo, unpubl.). Two in situ datasets from the Baltic Sea were used to investigate if members from the identified core microbiome of A. ostenfeldii in culture were linked to natural dinoflagellate dynamics.
Results and discussion
Identification of dinoflagellates and diversity of microbiomes
The dinoflagellates were taxonomically identified by amplification and sequencing of their ITS‐regions which corroborated identifications previously made using microscopy (S. Fraga and I. Bravo, unpubl.; Kremp et al., 2009) except for the strain AL10C, which was previously identified as A. minutum, but showed a high level of identity (99%) to an A. tamarense strain (AJ005048.1; Supporting Information Fig. S1). The Alexandrium strains were cultured using sterile methods at conditions mimicking those of their respective origins, brackish (salinity 6.5 or 7) for A. ostenfeldii and marine (salinity 31) for A. minutum/tamarense (Supporting Information Table S1). Nucleic acid samples were obtained by filtering cultures (density of A. ostenfeldii: 0.03–0.15 × 105 cells/ml and A. minutum/tamarense: 2.10–3.50 x 105 cells/ml; Supporting Information Table S1) using 0.2 μm filters, capturing the whole bacterial assemblage of the cultures at late exponential growth phase. The microbiomes were characterized using high‐throughput 16S rRNA gene amplicon sequencing, which resulted in a total of 9 776 193 sequences and 3649 operational taxonomic units (OTUs; 97% similarity clustering; Supporting Information Table S2). In this first high resolution characterization of the associated bacterial community of multiple species of Alexandrium, large differences in richness and diversity of the microbiomes were observed between the species (Supporting Information Fig. S2). At the OTU‐level, the A. ostenfeldii microbiomes showed a higher level of richness [average chao1‐index: 1081, ± 252 (SD)] and a higher diversity [average alpha Shannon index: 4.22, ± 0.67 (SD)] compared to the A. minutum/tamarense richness [average chao1‐index: 481, ± 152 (SD)] and diversity [average alpha Shannon index: 3.15, ± 0.56 (SD)] (Supporting Information Fig. S2). The differences in richness and diversity between A. ostenfeldii and A. minutum/tamarense microbiomes might reflect the longer time the latter species were kept in culture, 2 years compared to 8–16 years respectively (Supporting Information Table S1).
Similarity of microbiome composition
To study if the composition of the microbiomes associated with Alexandrium could be related to intraspecies variations of Alexandrium strains or specific conditions at the location of isolation an nMDS analysis, using Bray–Curtis dissimilarity, was performed. This analysis showed that there was a clear separation between A. ostenfeldii and the A. minutum/tamarense strains regarding frequently occurring taxa (Fig. 1 and Supporting Information Fig. S3). The A. ostenfeldii microbiomes were very homogeneous (permAnova R 2 = 0.13, P value: 0.73; Fig. 1) considering that the strains were isolated from four geographically distant locations of the Baltic Sea and the inherent intraspecific variations previously demonstrated for A. ostenfeldii (Kremp et al., 2016; Martens et al., 2017; Supporting Information Fig. S4). Consequently, our results indicate that intraspecific variations or sampling location in the Baltic Sea did not have a significant impact on the composition of the microbiomes. These findings indicate that the interactions that occur between the dinoflagellate and their microbiomes, are consistent within a species‐specific functional range of Alexandrium, disregarding phenotypic variations of individual dinoflagellate strains. In the nMDS, the A. minutum/tamarense associated microbiomes were dispersed with a spread similar to that of the A. ostenfeldii microbiomes (Fig. 1), and they did not cluster according to the anthropogenic impact level at each location (estimated using LUSI; E. Flo, unpubl.; Supporting Information Fig. S4). The results indicate a high microbiome similarity also across the A. minutum/tamarense strains. Taken together, our results show that both the environment at the sampling location, brackish or marine, and the species of Alexandrium, had important impacts on the composition of the microbiomes.
Figure 1.
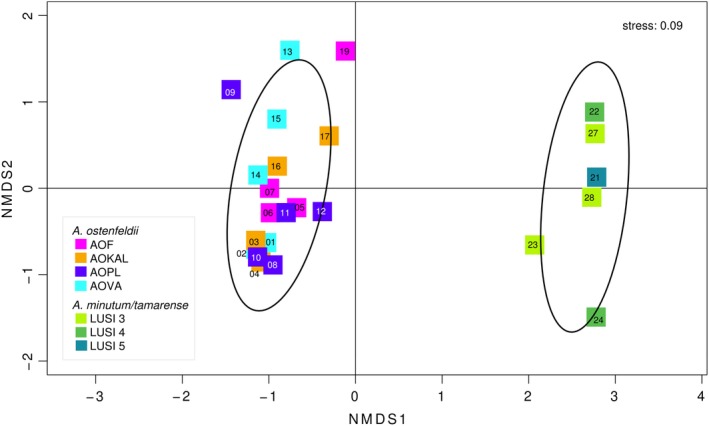
Dissimilarity analysis, nMDS, using a distance matrix with Bray Curtis dissimilarity of all strains, using R version 3.3.2, with packages Vegan (Oksanen et al., 2008) and Cluster (Maechler et al., 2016). Inherent stress: 0.09. Samples with > 162 000 read‐pairs were included in the analysis (excluding samples 18 (AOKAL), 20 (AOPL), 25 (LUSI1) and 26 (LUSI6), having < 32 000 read‐pairs; Supporting Information Table S2). The colours represent location of sampling: A. ostenfeldii – strains were isolated as resting cysts from four locations in the Baltic Sea Proper (AOF – Föglö Archipelago, Åland, AOKAL – Kalmar strait, Sweden, AOPL – Hel, Poland, AOVA – Valleviken, Sweden) (Tahvanainen et al., 2012); or level of anthropogenic impact (LUSI index): A. minutum/tamarense – strains were isolated from the north west Mediterranean Sea as vegetative cells, from locations with varying levels of anthropogenic impacts estimated using the LUSI index, 1–6 (E. Flo, unpubl.; Supporting Information Fig. S4); and sample ID for each strain (Supporting Information Fig. S4 and Table S2). The confidence limit was set at 0.80.
Microbiome of A. ostenfeldii isolates
The microbiomes of the A. ostenfeldii isolates were highly similar, forming four clusters (based on Bray–Curtis dissimilarity) of 3–7 samples each, originating from different locations (Supporting Information Fig. S3). These microbiomes included sequences affiliated with a broad range of bacterial classes: Beta‐ (58% of total sequences), Alpha‐ (14% of total sequences), Gamma‐ (3% of total sequences) and Deltaproteobacteria (6% of total sequences), Flavobacteriia (9% of total sequences) and Cytophagia (1.5% of total sequences). Represented, at family level, by Methylophilaceae (28% of total sequences), Hydrogenophilaceae (10% of total sequences), Flavobacteriaceae (7% of total sequences), Comamonadaceae (4% of total sequences), Desulfobulbaceae (4% of total sequences) and Rhodobacteraceae (3% of total sequences; Fig. 2). The high frequency of Betaproteobacteria in association with A. ostenfeldii, may be a reflection of the in situ brackish bacterial community composition of the Baltic Sea (Herlemann et al., 2011), which hosts more Betaproteobacteria compared to environments with higher salinity. The betaproteobacterial sequences, were primarily affiliated with Methylophilaceae, Burkholderiaceae/Limnobacter and Comamonadaceae (Supporting Information Fig. S5). Although Betaproteobacteria are not frequently found to dominate bacterial communities associated with phytoplankton, both Methylophilaceae and Limnobacter have previously been found in association with diatoms and coccolithophores (Amin et al., 2012; Green et al., 2015). Members of the family Methylophilaceae, are known to produce the growth promoting hormone indole‐3‐acetic acid (IAA) in association with plants (Doronina et al., 2014). Bacterial IAA has been observed to be exchanged for tryptophan in diatom cultures (Amin et al., 2015). Thus, a similar interaction may also occur between A. ostenfeldii and Methylophilaceae. Methylophilaceae prefer C1‐compounds, methanol or methylamine, as a source of carbon (Doronina et al., 2002) and they are known to degrade dimethylsulfide (DMS) (Eyice et al., 2015). The consistency of the dominating groups of the microbiome suggest that associations between these groups and A. ostenfeldii may be of importance for dinoflagellate metabolism, at brackish conditions, and are not an artefact of culturing.
Figure 2.
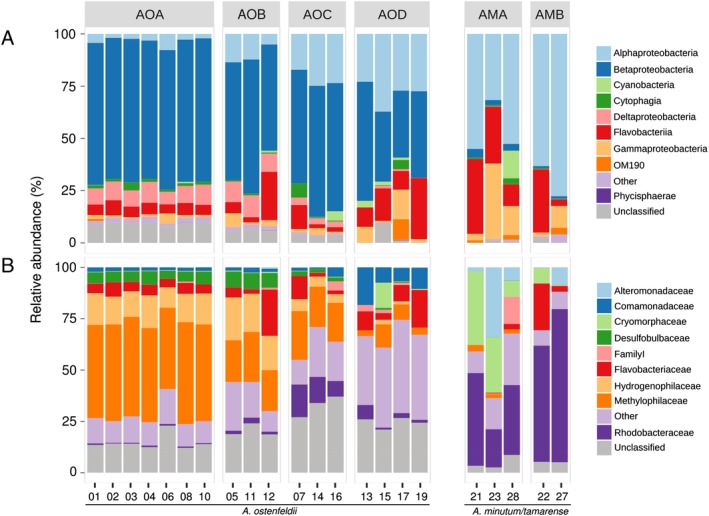
Composition of microbiomes of Alexandrium isolates at (A) Class and (B) Family level. Samples are grouped by dissimilarity (Supporting Information Fig. S3); AOA‐D: A. ostenfeldii group A–D, AMA‐B: A. minutum/tamarense group A–B. The cultures were harvested during late exponential phase using 0.2 μm, 47 mm filters and DNA was extracted (Boström et al., 2004). The V3‐V4 region of the 16S rRNA gene was amplified using primers 341F (CCTACGGGNGGCWGCAG) and 805R (GACTACHVGGGTATCTAATCC) and sequenced using Illumina MiSeq (Herlemann et al., 2011; Hugerth et al., 2014). The read‐pairs were clustered into OTUs using Usearch v8.1 (radius 1.5) (Edgar, 2013) corresponding to ~97% sequence identity and singletons were removed (Supporting Information Table S2). OTUs were classified using SILVA db 123 SSURef NR99; (Quast et al., 2013) using SINA v. 1.2.13 (Pruesse et al., 2012). The graph was constructed using ggplot2 (v. 2.2.1) (Wickham, 2009). The numbers for each sample specify the ID of each strain given in Supporting Information Fig. S4. The sequence data have been submitted to the European Nucleotide Archive (ENA) database under accession numbers ERS1617530‐ERS1617557.
Microbiome of A. minutum and A. tamarense isolates
The microbiomes of A. minutum/tamarense isolates were clustered by similarity into two groups, of 2 and 3 samples, respectively, mixing samples from different environmental conditions (estimated using LUSI; E. Flo, unpubl.; Supporting Information Figs S3 and S4). These microbiomes were dominated by sequences affiliated with Alphaproteobacteria (56% of total sequences), Flavobacteriia (21% of total sequences) and Gammaproteobacteria (13% of total sequences). At family‐level these were represented by Rhodobacteraceae (46% of total sequences), Cryomorphaceae (16% of total sequences), Alteromonadaceae (10% of total sequences) and Flavobacteriaceae (6% of total sequences; Fig. 2). The microbiomes were highly similar to what has previously been reported from cultures of marine Alexandrium species including bacterial taxa such as Rhodobacteraceae, Alteromonadaceae/Marinobacter and Cryomophaceae (Sala et al., 2005; Jasti et al., 2005). Flavobacteriia as a group are known as degraders of complex organic carbon (Kirchman, 2002), and are commonly found in close association to phytoplankton cells in culture (Grossart et al., 2005). Alphaproteobacteria belonging to the Roseobacter group can be found both attached and free‐living and exhibit a broad potential for nutrient uptake (Grossart et al., 2005; Moran et al., 2007). Gammaproteobacteria are associated with phytoplankton bloom conditions, with specialized populations responding to phytoplankton decay (Teeling et al., 2012). Taken together the results show that the microbiomes of and A. tamarense strains included in the study are representative of Alexandrium cultured at marine conditions (salinity 31).
Alexandrium core microbiome
Core microbiomes were calculated for A. ostenfeldii (Supporting Information Fig. S6) and A. minutum/tamarense (Supporting Information Fig. S7) strains, respectively, according to Lawson et al. (2017). The A. ostenfeldii core microbiome, contained five OTUs present in all 20 samples with an abundance > 0.0001%, affiliated to Limnobacter and Hoeflea (Supporting Information Fig. S6 and Table S3A). Within each group (Supporting Information Fig. S3), additional ubiquitous OTUs were identified (50), among which all four groups had OTUs that were affiliated to Hydrogenophaga, Methylothenera, Flavobacteriaceae or Comamonadaceae (Supporting Information Table S4A). These OTUs, though not identical, had the same taxonomical affiliations and were therefore considered to belong to the core A. ostenfeldii microbiome, in total 55 OTUs. For the A. minutum/tamarense isolates, 13 OTUs were present in all samples (excluding samples 25 and 26 with < 162 000 reads) (Supporting Information Fig. S7 and Table S3B). These were affiliated to Limnobacter, Pyruvatibacter, Tepidamorphus, Hoeflea, Hyphomonas, Marivita, Marinobacter, Methylophaga and Salinispirillum. Within the two groups of strains (Supporting Information Fig. S3), additional OTUs with the same taxonomical annotations were identified (39), which were affiliated to Taesokella, Fabibacter, Sphingopyxis, Spongibacter, Salinirepens and Ahrensia (Supporting Information Table S4B). Together, these 52 OTUs were considered the core microbiome of A. minutum/tamarense. The consistency of the core microbiomes indicates that associations between Alexandrium and bacteria in culture are stable and predictable. These findings enable further identification and characterization of specific Alexandrium‐bacterial interactions.
Culture conditions
There are several considerations that should be made before drawing conclusions about in situ associations from in vitro conditions (Sala et al., 2005; Garcés et al., 2007; Hattenrath‐Lehmann and Gobler, 2017; Behringer et al., 2018). For example, culture conditions and time in culture can have significant impacts as culturing can alter the bacterial community composition within hours (Sapp et al., 2007; Weissbach et al., 2010). To investigate the effect of long term culturing on the associated bacterial communities, A. ostenfeldii (strains 13–20) and A. minutum/tamarense (strains 21–28) isolates were monitored after 6 and 4 months of culturing respectively. Similar to studies of diatoms in culture (Schäfer et al., 2002; Behringer et al., 2018), the bacterial communities of the Alexandrium isolates is this study were shown to be consistent over time (Supporting Information Table S1 and Fig. S8). However, in the high‐resolution sequencing data, shifts in the relative abundance among some of the most frequent betaproteobacterial OTUs were observed in A. ostenfeldii strains 1–12, compared to strains 13–20 (Supporting Information Fig. S5). Strains 13–20 had previously been kept at the same conditions as 1–12, but were moved to another lab and sampled after 6–9 weeks of exposure to slightly different conditions with regards to light, salinity and temperature (Supporting Information Table S1). Taken together these results show that, given stable conditions, the members of the core microbiome of Alexandrium are consistent, but shifts in the relative abundance of the microbiome, including members of the core, can be expected as a reflection of changed environmental conditions.
Connections with in situ dinoflagellate populations in the Baltic Sea
In contrast to the Mediterranean Sea isolates, which were isolated as vegetative cells, the A. ostenfeldii isolates were isolated as cysts (Tahvanainen et al., 2012). To investigate if OTUs from the A. ostenfeldii core microbiome were present in the pelagic zone, and if their frequency could be related to natural dinoflagellate dynamics, two Baltic Sea microbial datasets were explored (Fig. 3A). The Prodiversa cruise took place during April 2013, representing a transect of the Baltic Sea Proper, capturing a mixed dinoflagellate spring bloom (Bunse et al., 2016; Godhe et al., 2016). The Planfish dataset was sampled during the productive season (April–October) of 2010–2011, covering a smaller geographic area west of the island of Gotland, capturing both spring and summer blooms of different dinoflagellate species (Legrand et al., 2015) (Fig. 3B). OTUs corresponding to members of the A. ostenfeldii core microbiome (OTU_000043 and OTU_000260), were found to follow the biomass dynamics of the dinoflagellate spring bloom, captured by the Prodiversa cruise (Fig. 3A). These OTUs were affiliated to Betaproteobacteria (Oxalobacteriaceae and Comamonadaceae; Supporting Information Table S4A). In the Planfish dataset, one of the same core OTUs (OTU_000260), increased in relative abundance during spring and summer dinoflagellate blooms (Fig. 3B). These results indicate that members of the core microbiome of A. ostenfeldii in culture followed in situ dinoflagellate occurrences, suggesting that they may be closely associated with Baltic Sea dinoflagellates, regardless of species.
Figure 3.
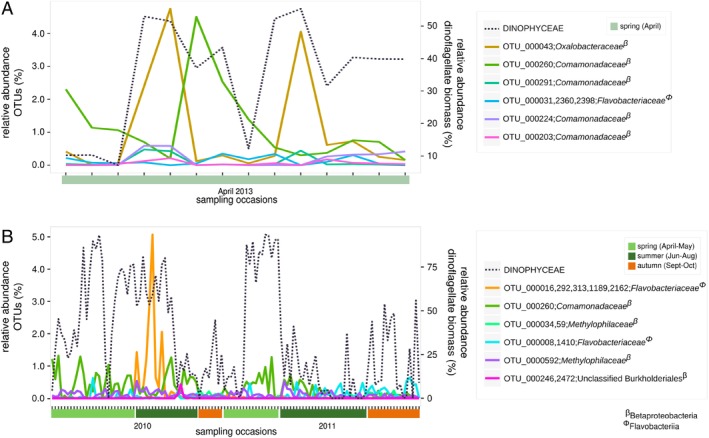
Dynamics of OTUs with a relative abundance > 0.2% and a similarity of ≥ 95% to members from the core microbiome of A. ostenfeldii, in (A) a naturally occurring spring bloom, Prodiversa, 2013 (Bunse et al., 2016; Godhe et al., 2016) and (B) a time series covering April–October in a coastal to off‐shore transect, Planfish, 2010–2011 (Legrand et al., 2015), both from the Baltic Sea Proper. Relative abundance (%) of OTUs on the left y‐axis, the dinoflagellate biomass dynamics from the corresponding datasets on the right y‐axis (%). The legend lists Planfish/Prodiversa‐OTUs named by corresponding OUTs from the A. ostenfeldii dataset, including their taxonomic affiliation at family level. The relative abundances of dinoflagellate biomass were calculated based on the carbon content of dinoflagellates per total phytoplankton carbon, including the species: Dinobryon spp, Dinophysis sp, Gymnodiniales spp, Gymnodinium spp, Gyrodinium spp, Heterocapsa rotundata, Katodinium glaucum, Peridinella catenata, Peridinella (single cell), Protoperidinium spp and Scrippsiella. Plots were made using ggplot2 (v. 2.2.1) (Wickham, 2009) in R 3.4.0.
Conclusions
This study, the first high resolution molecular characterization of the microbiome of A. ostenfeldii isolates, showed that neither intraspecific variations nor the location of sampling in the brackish Baltic Sea Proper had a significant impact on the isolate microbiome composition. Similarly, the level of anthropogenic impact at the different sites of isolation of A. minutum/tamarense strains could not be linked to the composition of the different microbiomes. Instead, the microbiomes were highly consistent among strains of A. ostenfeldii and A. minutum/tamarense, respectively, and core microbiomes were identified. These core microbiomes were likely shaped by abiotic conditions, like salinity, light and temperature as shifts in relative abundances could be seen upon changed culture conditions, and selected by the respective dinoflagellate host in a species‐specific manner. The identification of an Alexandrium core microbiome makes this model system ideal for in‐depth studies of specific interactions likely occurring between the phytoplankton and key bacterial taxa – principal for understanding the impact of interactions on the ecosystem. In future studies, core microbiomes could be identified from natural phytoplankton blooms using culture independent methods such as single cell genomics.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
Appendix S1: Supporting Information
Table S3A – Core microbiome of Alexandrium ostenfeldii, with an OTU‐abundance > 0.0001% (Lawson et al. 2017) . Specified by the Class and Family/Genus of the closest relative in GenBank (as given in Table S4A), the number of OTUs per Family and the relative abundance of those OTUs per group. Groups are specified in Fig. S4; AO – all A. ostenfeldii strains; AOA – strains 1–4, 6, 8, 10; AOB – strains 5, 11, 12; AOC – strains 7, 14, 16; AOD – strains 13, 15, 17, 19. Note that samples 18 and 20 are excluded (< 162 000 reads) and samples 9 is considered and outgroup of the A. ostenfeldii cluster (Fig. S4) and are therefore not included in any group.
Table S3B – Core microbiome of Alexandrium minutum/tamarenes, with an OTU‐abundance > 0.0001% (Lawson et al. 2017). Specified by the Class and Family/Genus of the closest relative in GenBank (as given in table S4B), the number of OTUs per Family and the relative abundance of those OTUs per group. Groups are specified in Figure S4; AM – all A. minutum/tamarense strains (excluding samples 25, 26 (< 162 000 reads); AMA – strains 21, 23, 28; AMB – strains 22, 27. Note that strain 24 is considered and outgroup of the A. minutum/tamarense cluster in Figure S4 and therefore not included in any group.
Table S4A Core microbiome (55 OTUs) of Alexandrium ostenfeldii, specifying: OTU, accession number of closest relative in GenBank, % identity, genus and family of that strain and the sequence of the OTU.
Table S4B. Core microbiome (52 OTUs) of Alexandrium minutum/tamarense, specifying: OTU, accession number of closest relative in GenBank, % identity, genus and family of that strain and the sequence of the OTU.
Acknowledgements
We thank Santiago Fraga and Isabel Bravo for isolating, identifying and providing the strains of Alexandrium minutum and A. tamarense and Carina Bunse for access to the Prodiversa dataset. Computations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) at UPPMAX (project b2016129). This project was funded by the Strategic Research Environment ECOCHANGE, Swedish Research Council Formas (CL), the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 659453) (EL) and Tryggers research Foundation (EL).
References
- Adachi, M. , Kanno, T. , Okamoto, R. , Itakura, S. , Yamaguchi, M. , and Nishijima, T. (2003) Population structure of Alexandrium (Dinophyceae) cyst formation‐promoting bacteria in Hiroshima Bay, Japan. Appl Environ Microbiol 69: 6560–6568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin, S.a. , Parker, M.S. , and Armbrust, E.V. (2012) Interactions between diatoms and bacteria. Microbiol Mol Biol Rev 76: 667–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin, S.A. , Green, D.H. , Hart, M.C. , Kupper, F.C. , Sunda, W.G. , and Carrano, C.J. (2009) Photolysis of iron‐siderophore chelates promotes bacterial‐algal mutualism. Proc Natl Acad Sci USA 106: 17071–17076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin, S.A. , Hmelo, L.R. , van Tol, H.M. , Durham, B.P. , Carlson, L.T. , Heal, K.R. , et al (2015) Interaction and signalling between a cosmopolitan phytoplankton and associated bacteria. Nature 522: 98–101. [DOI] [PubMed] [Google Scholar]
- Anderson, D.M. , Alpermann, T.J. , Cembella, A.D. , Collos, Y. , Masseret, E. , and Montresor, M. (2012) The globally distributed genus Alexandrium: multifaceted roles in marine ecosystems and impacts on human health. Harmful Algae 14: 10–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azam, F. , and Malfatti, F. (2007) Microbial structuring of marine ecosystems. Nat Rev Microbiol 5: 782–791. [DOI] [PubMed] [Google Scholar]
- Behringer, G. , Ochsenkühn, M.A. , Fei, C. , Fanning, J. , Koester, J.A. , and Amin, S.A. (2018) Bacterial communities of diatoms display strong conservation across strains and time. Front Microbiol 9: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biegala, I.C. , Kennaway, G. , Alverca, E. , Lennon, J.‐F. , Vaulot, D. , and Simon, N. (2002) Identification of bacteria associated with dinoflagellates (Dinophyceae) Alexandrium spp. using tyramide signal amplification‐fluorescent in situ hybridization and confocal microscopy. J Phycol 38: 404–411. [Google Scholar]
- Bolch, C.J.S. , Subramanian, T.A. , and Green, D.H. (2011) The toxic dinoflagellate Gymnodinium catenatum (Dinophyceae) requires marine bacteria for growth. J Phycol 47: 1009–1022. [DOI] [PubMed] [Google Scholar]
- Boström, K.H. , Simu, K. , Hagström, Å. , and Riemann, L. (2004) Optimization of DNA extraction for quantitative marine bacterioplankton community analysis. Limnol Oceanogr Methods 2: 365–373. [Google Scholar]
- Brandenburg, K.M. , Wohlrab, S. , John, U. , Kremp, A. , Jerney, J. , Krock, B. , and Van de Waal, D.B. (2018) Intraspecific trait variation and trade‐offs within and across populations of a toxic dinoflagellate. Ecol Lett 21: 1561–1571. [DOI] [PubMed] [Google Scholar]
- Buchan, A. , LeCleir, G.R. , Gulvik, C.A. , and González, J.M. (2014) Master recyclers: features and functions of bacteria associated with phytoplankton blooms. Nat Rev Microbiol 12: 686–698. [DOI] [PubMed] [Google Scholar]
- Bunse, C. , Bertos‐Fortis, M. , Sassenhagen, I. , Sildever, S. , Sjöqvist, C. , Godhe, A. , et al (2016) Spatio‐temporal interdependence of bacteria and phytoplankton during a Baltic Sea spring bloom. Front Microbiol 7: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole, J.J. (1982) Interactions between bacteria and algae in aquatic ecosystems. Annu Rev Ecol Syst 13: 291–314. [Google Scholar]
- Doronina, N. , Kaparullina, E. , and Trotsenko, Y. (2014) The family Methylophilaceae In The Prokaryotes, Alphaproteobacteria and Betaproteobacteria. Rosenberg E., DeLong E.F., Lory S., Stackebrandt E., and Thompson F. (eds). Berlin Heidelberg: Springer‐Verlag, pp. 869–880. [Google Scholar]
- Doronina, N.V. , Ivanova, E.G. , and Trotsenko, Y.A. (2002) New evidence for the ability of methylobacteria and methanotrophs to synthesize auxins. Microbiology 71: 116–118. [PubMed] [Google Scholar]
- Doucette, G.J. , and Powell, C.L. (1997) Algal‐bacterial interactions: can they determine the PSP‐related toxicity of dinoflagellates? In Harmful Algae. Reguera B., Blanco J., Fernández M.L., and Wyatt T. (eds). Grafisant: Xunta de Galicia and Inter‐ Governmental Oceanographic Commission of UNESCO, pp. 406–409. [Google Scholar]
- Edgar, R.C. (2013) UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10: 996–998. [DOI] [PubMed] [Google Scholar]
- Eppley, R.W. , and Peterson, B.J. (1979) Particulate organic matter flux and planktonic new production in the deep ocean. Nature 282: 677–680. [Google Scholar]
- Eyice, Ö. , Namura, M. , Chen, Y. , Mead, A. , Samavedam, S. , and Schäfer, H. (2015) SIP metagenomics identifies uncultivated Methylophilaceae as dimethylsulphide degrading bacteria in soil and lake sediment. ISME J 9: 2336–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrier, M. , Martin, J.L. , and Rooney‐Varga, J.N. (2002) Stimulation of Alexandrium fundyense growth by bacterial assemblages from the bay of Fundy. J Appl Microbiol 92: 706–716. [DOI] [PubMed] [Google Scholar]
- Gallacher, S. , and Smith, E.A. (1999) Bacteria and paralytic shellfish toxins. Protist 150: 245–255. [DOI] [PubMed] [Google Scholar]
- Garcés, E. , Vila, M. , Reñé, A. , Alonso‐Sáez, L. , Anglès, S. , Lugliè, A. , et al (2007) Natural bacterioplankton assemblage composition during blooms of Alexandrium spp. (Dinophyceae) in NW Mediterranean coastal waters. Aquat Microb Ecol 46: 55–70. [Google Scholar]
- Glibert, P.M. , and Legrand, C. (2006) The diverse nutrient strategies of harmful algae: focus on osmotrophy In Ecology of Harmful Algae. Granéli E., and Turner J.T. (eds). Berlin Heidelberg: Springer Verlag, pp. 163–175. [Google Scholar]
- Godhe, A. , Sjöqvist, C. , Sildever, S. , Sefbom, J. , Harardóttir, S. , Bertos‐Fortis, M. , et al (2016) Physical barriers and environmental gradients cause spatial and temporal genetic differentiation of an extensive algal bloom. J Biogeogr 43: 1130–1142. [Google Scholar]
- Green, D.H. , Echavarri‐Bravo, V. , Brennan, D. , and Hart, M.C. (2015) Bacterial diversity associated with the coccolithophorid algae Emiliania huxleyi and Coccolithus pelagicus f. braarudii . Biomed Res Int 2015: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossart, H.‐P. , Levold, F. , Allgaier, M. , Simon, M. , and Brinkhoff, T. (2005) Marine diatom species harbour distinct bacterial communities. Environ Microbiol 7: 860–873. [DOI] [PubMed] [Google Scholar]
- Guannel, M. , Horner‐Devine, M. , and Rocap, G. (2011) Bacterial community composition differs with species and toxigenicity of the diatom Pseudo‐nitzschia . Aquat Microb Ecol 64: 117–133. [Google Scholar]
- Hakanen, P. , Suikkanen, S. , and Kremp, A. (2014) Allelopathic activity of the toxic dinoflagellate Alexandrium ostenfeldii: intra‐population variability and response of co‐occurring dinoflagellates. Harmful Algae 39: 287–294. [Google Scholar]
- Hattenrath‐Lehmann, T.K. , and Gobler, C.J. (2017) Identification of unique microbiomes associated with harmful algal blooms caused by Alexandrium fundyense and Dinophysis acuminata . Harmful Algae 68: 17–30. [DOI] [PubMed] [Google Scholar]
- Herlemann, D.P. , Labrenz, M. , Jürgens, K. , Bertilsson, S. , Waniek, J.J. , and Andersson, A.F. (2011) Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J 5: 1571–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hold, G. , Smith, E.A. , Rappé, M.S. , Maas, E.W. , Moore, E.R. , Stroempl, C. , et al (2001) Characterisation of bacterial communities associated with toxic and non‐toxic dinoflagellates: Alexandrium spp. and Scrippsiella trochoidea . FEMS Microbiol Ecol 37: 161–173. [Google Scholar]
- Hugerth, L.W. , Wefer, H.A. , Lundin, S. , Jakobsson, H.E. , Lindberg, M. , Rodin, S. , et al (2014) DegePrime, a program for degenerate primer design for broad‐taxonomic‐range PCR in microbial ecology studies. Appl Environ Microbiol 80: 5116–5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasti, S. , Sieracki, M.E. , Poulton, N.J. , Giewat, M.W. , and Rooney‐Varga, J.N. (2005) Phylogenetic diversity and specificity of bacteria closely associated with Alexandrium spp. and other phytoplankton. Appl Environ Microbiol 71: 3483–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao, N. , Herndl, G.J. , Hansell, D.A. , Benner, R. , Kattner, G. , Wilhelm, S.W. , et al (2010) Microbial production of recalcitrant dissolved organic matter: long‐term carbon storage in the global ocean. Nat Rev Microbiol 8: 593–599. [DOI] [PubMed] [Google Scholar]
- Kirchman, D.L. (2002) The ecology of Cytophaga‐Flavobacteria in aquatic environments. FEMS Microbiol Ecol 39: 91–100. [DOI] [PubMed] [Google Scholar]
- Kopp, M. (1997) Phylogenetic analysis of selected toxic and non‐toxic bacterial strains isolated from the toxic dinoflagellate Alexandrium tamarense . FEMS Microbiol Ecol 24: 251–257. [Google Scholar]
- Kremp, A. , Lindholm, T. , Dreßler, N. , Erler, K. , Gerdts, G. , Eirtovaara, S. , and Leskinen, E. (2009) Bloom forming Alexandrium ostenfeldii (Dinophyceae) in shallow waters of the Åland Archipelago, Northern Baltic Sea. Harmful Algae 8: 318–328. [Google Scholar]
- Kremp, A. , Oja, J. , LeTortorec, A.H. , Hakanen, P. , Tahvanainen, P. , Tuimala, J. , and Suikkanen, S. (2016) Diverse seed banks favour adaptation of microalgal populations to future climate conditions. Environ Microbiol 18: 679–691. [DOI] [PubMed] [Google Scholar]
- Lawson, C.A. , Raina, J.B. , Kahlke, T. , Seymour, J.R. , and Suggett, D.J. (2017) Defining the core microbiome of the symbiotic dinoflagellate, Symbiodinium . Environ Microbiol Rep 10: 7–11. [DOI] [PubMed] [Google Scholar]
- Legrand, C. , Fridolfsson, E. , Bertos‐Fortis, M. , Lindehoff, E. , Larsson, P. , Pinhassi, J. , and Andersson, A. (2015) Interannual variability of phyto‐bacterioplankton biomass and production in coastal and offshore waters of the Baltic Sea. Ambio 44: 427–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, H. , Krock, B. , Tillmann, U. , and Cembella, A. (2009) Preliminary characterization of extracellular allelochemicals of the toxic marine dinoflagellate Alexandrium tamarense using a Rhodomonas salina bioassay. Mar Drugs 7: 497–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maechler, M. , Rousseeuw, P. , Struyf, A. , Hubert, M. , and Hornik, K. (2016) Cluster: Cluster Analysis Basics and Extensions. R Package Version 2.0.5. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- Martens, H. , Tillmann, U. , Harju, K. , Dell'Aversano, C. , Tartaglione, L. , and Krock, B. (2017) Toxin variability estimations of 68 Alexandrium ostenfeldii (Dinophyceae) strains from The Netherlands reveal a novel abundant gymnodimine. Microorganisms 5: 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran, M.A. , Belas, R. , Schell, M.A. , Gonzalez, J.M. , Sun, F. , Sun, S. , et al (2007) Ecological genomics of marine roseobacters. Appl Environ Microbiol 73: 4559–4569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksanen, J. , Kindt, R. , Legendre, P. , O'Hara, B. , Simpson, G.L. , Solymos, P.M. , et al (2008) The vegan package. Community Ecology Package. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- Pruesse, E. , Peplies, J. , and Glöckner, F.O. (2012) SINA: accurate high‐throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28: 1823–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast, C. , Pruesse, E. , Yilmaz, P. , Gerken, J. , Schweer, T. , Yarza, P. , et al (2013) The SILVA ribosomal RNA gene database project: improved data processing and web‐based tools. Nucleic Acids Res 41: 590–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risgaard‐Petersen, N. , Nicolaisen, M.H. , Revsbech, N.P. , and Lomstein, B.A. (2004) Competition between ammonia‐oxidizing bacteria and benthic microalgae. Appl Environ Microbiol 70: 5528–5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakami, T. , Nakahara, H. , Chinain, M. , and Ishida, Y. (1999) Effects of epiphytic bacteria on the growth of the toxic dinoflagellate Gambierdiscus toxicus (Dinophyceae). J Exp Mar Bio Ecol 233: 231–246. [Google Scholar]
- Sala, M.M. , Balagué, V. , Pedrós‐Alió, C. , Massana, R. , Felipe, J. , Arin, L. , et al (2005) Phylogenetic and functional diversity of bacterioplankton during Alexandrium spp. blooms. FEMS Microbiol Ecol 54: 257–267. [DOI] [PubMed] [Google Scholar]
- Sapp, M. , Wichels, A. , and Gerdts, G. (2007) Impacts of cultivation of marine diatoms on the associated bacterial community. Appl Environ Microbiol 73: 3117–3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarmento, H. , Romera‐Castillo, C. , Lindh, M. , Pinhassi, J. , Sala, M.M. , Gasol, J.M. , et al (2013) Phytoplankton species‐specific release of dissolved free amino acids and their selective consumption by bacteria. Limnol Oceanogr 58: 1123–1135. [Google Scholar]
- Schäfer, H. , Abbas, B. , Witte, H. , and Muyzer, G. (2002) Genetic diversity of “satellite” bacteria present in cultures of marine diatoms. FEMS Microbiol Ecol 42: 25–35. [DOI] [PubMed] [Google Scholar]
- Seymour, J.R. , Amin, S.A. , Raina, J.‐B. , and Stocker, R. (2017) Zooming in on the phycosphere: the ecological interface for phytoplankton–bacteria relationships. Nat Microbiol 2: 17065. [DOI] [PubMed] [Google Scholar]
- Suikkanen, S. , Kremp, A. , Hautala, H. , and Krock, B. (2013) Paralytic shellfish toxins or spirolides? The role of environmental and genetic factors in toxin production of the Alexandrium ostenfeldii complex. Harmful Algae 26: 52–59. [Google Scholar]
- Tahvanainen, P. , Alpermann, T.J. , Figueroa, R.I. , John, U. , Hakanen, P. , Nagai, S. , et al (2012) Patterns of post‐glacial genetic differentiation in marginal populations of a marine microalga. PLoS One 7: e53602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teeling, H. , Fuchs, B.M. , Becher, D. , Klockow, C. , Gardebrecht, A. , Bennke, C.M. , et al (2012) Substrate‐controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 336: 608–611. [DOI] [PubMed] [Google Scholar]
- Tillmann, U. , Alpermann, T.L. , da Purificação, R.C. , Krock, B. , and Cembella, A. (2009) Intra‐population clonal variability in allelochemical potency of the toxigenic dinoflagellate Alexandrium tamarense . Harmful Algae 8: 759–769. [Google Scholar]
- Valiadi, M. , Debora Iglesias‐Rodriguez, M. , and Amorim, A. (2012) Distribution and genetic diversity of the luciferase gene within marine dinoflagellates. J Phycol 48: 826–836. [DOI] [PubMed] [Google Scholar]
- Villacorte, L.O. , Ekowati, Y. , Neu, T.R. , Kleijn, J.M. , Winters, H. , Amy, G. , et al (2015) Characterisation of algal organic matter produced by bloom‐forming marine and freshwater algae. Water Res 73: 216–230. [DOI] [PubMed] [Google Scholar]
- Weissbach, A. , Rudström, M. , Olofsson, M. , Béchemin, C. , Icely, J. , Newton, A. , et al (2011) Phytoplankton allelochemical interactions change microbial food web dynamics. Limnol Oceanogr 56: 899–909. [Google Scholar]
- Weissbach, A. , Tillmann, U. , and Legrand, C. (2010) Allelopathic potential of the dinoflagellate Alexandrium tamarense on marine microbial communities. Harmful Algae 10: 9–18. [Google Scholar]
- Wickham, H. (2009) ggplot2: Elegant Graphics for Data Analysis. New York, NY: Springer. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information
Table S3A – Core microbiome of Alexandrium ostenfeldii, with an OTU‐abundance > 0.0001% (Lawson et al. 2017) . Specified by the Class and Family/Genus of the closest relative in GenBank (as given in Table S4A), the number of OTUs per Family and the relative abundance of those OTUs per group. Groups are specified in Fig. S4; AO – all A. ostenfeldii strains; AOA – strains 1–4, 6, 8, 10; AOB – strains 5, 11, 12; AOC – strains 7, 14, 16; AOD – strains 13, 15, 17, 19. Note that samples 18 and 20 are excluded (< 162 000 reads) and samples 9 is considered and outgroup of the A. ostenfeldii cluster (Fig. S4) and are therefore not included in any group.
Table S3B – Core microbiome of Alexandrium minutum/tamarenes, with an OTU‐abundance > 0.0001% (Lawson et al. 2017). Specified by the Class and Family/Genus of the closest relative in GenBank (as given in table S4B), the number of OTUs per Family and the relative abundance of those OTUs per group. Groups are specified in Figure S4; AM – all A. minutum/tamarense strains (excluding samples 25, 26 (< 162 000 reads); AMA – strains 21, 23, 28; AMB – strains 22, 27. Note that strain 24 is considered and outgroup of the A. minutum/tamarense cluster in Figure S4 and therefore not included in any group.
Table S4A Core microbiome (55 OTUs) of Alexandrium ostenfeldii, specifying: OTU, accession number of closest relative in GenBank, % identity, genus and family of that strain and the sequence of the OTU.
Table S4B. Core microbiome (52 OTUs) of Alexandrium minutum/tamarense, specifying: OTU, accession number of closest relative in GenBank, % identity, genus and family of that strain and the sequence of the OTU.