

Abstract
An increasing number of biocatalytic oxidation reactions rely on H2O2 as a clean oxidant. The poor robustness of most enzymes towards H2O2, however, necessitates more efficient systems for in situ H2O2 generation. In analogy to the well‐known formate dehydrogenase to promote NADH‐dependent reactions, we here propose employing formate oxidase (FOx) to promote H2O2‐dependent enzymatic oxidation reactions. Even under non‐optimised conditions, high turnover numbers for coupled FOx/peroxygenase catalysis were achieved.
Keywords: biocatalysis, formate oxidase, hydrogen peroxide, oxidation, oxyfunctionalisation
Enzymatic oxidation and oxyfunctionalisation reactions are currently receiving tremendous interest in the context of preparative organic chemistry.1 Especially if selectivity is desired, enzymatic reactions often excel over the chemical counterparts. Amongst available biocatalysts, monooxygenases are of particular interest.2 Monooxygenases, however, rely on molecular oxygen, which is reductively activated at the enzyme active site. The reducing equivalents required are mostly derived more or less directly from reduced nicotinamide cofactors (NAD(P)H). While issues regarding the in situ regeneration of NAD(P)H have largely been solved, so that it can be used catalytic amounts,3 the so‐called oxygen dilemma poses a more severe challenge:4 many monooxygenases cannot utilise NAD(P)H directly but depend on single‐electron mediators to transform the hydride transfer from NAD(P)H into two successive single‐electron transport events. The reduced mediators, however, also directly interact with dissolved molecular oxygen and are re‐oxidised (Scheme 1 a). As a consequence, reactive oxygen species are formed in a futile cycle that uncouples the regeneration reaction from the oxygenation reaction. In extreme cases, up to 95 % of the reducing equivalents provided by the co‐substrate are wasted.4
Scheme 1.

Biocatalytic oxyfunctionalisations using monooxygenases (a) or peroxizymes (b). Monooxygenases often are prone to the oxygen dilemma while peroxizymes productively make use of the oxygen dilemma.
A viable solution of the challenge outlined above is to make use of H2O2‐dependent “Peroxizymes”.5 By using H2O2, Peroxizymes actually make use of the oxygen dilemma instead of being hampered by it (Scheme 1 b).
A range of enzymes are able to use H2O2. Peroxidases for example, represent model enzymes for H2O2‐dependent oxidation,6 polymerization,7 or halogenation reactions.8 More recently, peroxygenases have received a lot of attention for selective oxyfunctionalisation reactions.9 In addition to these reactions, hydrolase‐catalysed formation of peracids for H2O2‐driven epoxidation10 and Baeyer–Villiger oxidations11 are becoming popular.
Enzymes, however, are also prone to oxidative inactivation by H2O2 12 which is why a broad range of in situ H2O2 generation methods have been investigated in recent years (Table S2 compares some established systems with respect to efficiency and waste generation). The goal is to provide the production enzymes with H2O2 at rates that allow high catalytic turnover while minimising the undesired oxidative inactivation by excess H2O2.13 Today, glucose oxidase (GOx) is the catalyst of choice for in situ H2O2 generation.14 It couples the oxidation of glucose to the reductive activation of O2 to form H2O2 in a highly efficient and robust fashion. The GOx system, however, suffers from high levels of waste generation (196 g of gluconate waste per mol H2O2 equivalent are generated).15 Additionally, practical issues such as the high viscosity of the reaction medium have to be dealt with at larger reaction scales. Formate would be a more suitable reductant for the reductive activation of O2 (generating only 44 g of volatile and therefore not accumulating CO2 waste per mol H2O2 equivalent). The systems available today, however, either rely on bioincompatible transition‐metal catalysts,16 or are too complex15, 17 or too elaborate18 to be practical.
Recently, a formic acid oxidase from Aspergillus oryzae (AoFOx) has been reported as the first member of the glucose‐methanol‐choline (GMC) oxidoreductase superfamily that oxidizes formic acid instead of simple alcohols.19 This enzyme features an optimum pH range from 2.8–6.8 and a k cat value of 82 s−1 over that range. It contains an unusual 8‐formyl flavin adenine dinucleotide (FAD) cofactor, which is formed in situ from FAD through self‐oxidation. Its unique catalytic properties render AoFOx a promising candidate for H2O2‐dependent enzymatic reactions. We therefore set out to evaluate the potential of AoFOx as a catalyst to promote H2O2‐dependent biocatalytic oxidation reactions (Scheme 2).
Scheme 2.

The formate oxidase from Aspergillus oryzae (AoFOx) enables in situ H2O2 generation from formate and ambient oxygen to promote a broad range of biocatalytic oxidation/oxyfunctionalisation reactions.
AoFOx was prepared according to a previously published procedure.19 In short, AoFOx was expressed in recombinant Escherichia coli and partially purified to remove catalase. Overall, from 1 L culture broth, 38 mg of purified enzyme were obtained within 1 day (Figure S1 in the Supporting Information).
Having AoFOx in hand, we decided to first apply this enzyme for some selective oxyfunctionalisation reactions catalysed by the recombinant evolved unspecific peroxygenase from Agrocybe aegerita (rAaeUPO) heterologously expressed in Pichia pastoris.20 As model reaction, we first focused on the selective hydroxylation of ethyl benzene into (R)‐1‐phenylethanol. A preliminary optimisation of the reaction conditions (Figure 1) revealed that the bienzymatic cascade operates optimally in slightly acidic reaction media (pH 6, Figure 1 a, Figure S2), which is in line with the reported preferences of the enzymes.21 An apparent optimal temperature of 25 °C was determined (Figure 1 b, Figure S3). Between 20 and 35 °C, the initial rates of the overall system were largely temperature‐independent (Figure S2) but the reaction ceased sooner at elevated temperatures. At 40 °C for example, no further product formation was observed after 2 h. In contrast, steady product accumulation occurred at 30 °C or lower. This behaviour can be attributed to the comparably poor thermal robustness of wt‐AoFOx.
Figure 1.

Characterisation of the reaction parameters that influence the efficiency of the bienzymatic hydroxylation of ethyl benzene. Individual reaction conditions are given in the captions of Figures S2–8.
We determined an apparent optimal formate concentration of 200 mm (Figure 1 d, Figure S4), which represents a compromise between the relatively high K M value of wt‐AoFOx at this pH19d–19f and the decreasing peroxygenative activity of UPOs at higher formate concentrations.22
The relative ratio of (H2O2‐generating) AoFOx and (H2O2‐consuming) rAaeUPO had a very pronounced effect on the efficiency of the overall reaction system (Figure 1 c, Figure S5). The highest initial rate was observed at an equimolar ratio of the two enzymes, albeit at the expense of poor long‐term stability of the overall system (after 5 h, no further product formation was observed; Figure S5). Lower ratios of AoFOx to rAaeUPO gave lower productivity but significantly greater robustness. At a ratio of 1:5, stable product formation for at least 24 h was observed.
The availability of molecular oxygen had a significant influence on the overall reaction (Figure 1 e). Under ambient atmosphere without stirring, an O2 transfer rate of 0.84±0.03 mm h−1 was estimated (Figure S7), which limits the productivity of the overall system. Increasing the O2 availability by increasing the O2 partial pressure in the headspace of the reaction dramatically increased the productivity of the overall reaction more than ten‐fold (Figure 1 e, Figure S8).
It is worth mentioning that appropriate negative controls (i.e. reactions leaving out either one of the enzymes or reactions in the absence of formate) were performed for all of the reactions reported. With the sole exception of CytC‐catalysed sulfoxidation, where traces of sulfoxide were also observed in the absence of CytC, the control reactions gave no product formation.
Next, we explored the enzyme and product scope of the AoFOx‐catalysed H2O2 generation system to promote various H2O2‐dependent biocatalytic oxidation reactions (Figure 2).
Figure 2.
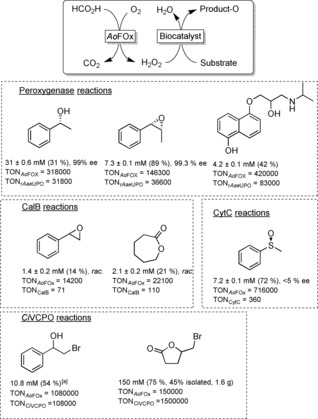
The scope of AoFOx‐driven peroxizyme reactions: The peroxygenase from Agrocybe aegerita (rAaeUPO) enabled selective hydroxylations and epoxidations; lipase B from Candida antarctica (CalB) mediated chemoenzymatic epoxidation and Baeyer–Villiger oxidations; cytochrome C (CytC, a heme‐containing electron‐transport protein) was applied to the sulfoxidation of thioanisole; and V‐dependent chloroperoxidase from Curcuvaria inaequalis (CiVCPO)‐initiated hydroxyhalogenation and halolactonisation reactions. For details about the reaction schemes and experimental results, please refer to the respective section in the Supporting Information. Yields shown are calculated from the product concentration divided by the initial starting material concentration.
First, we investigated some peroxygenase‐catalysed hydroxylation and epoxidation reactions. The proposed H2O2‐generation system enabled excellent catalytic performance of the peroxygenase used. Both product concentrations and rAaeUPO‐turnover numbers were at least as high as for previous methods using more complicated H2O2 generation systems.15, 18, 20d, 23
The stereospecific hydroxylation of ethyl benzene was performed on a semi‐preparative scale, yielding 434 mg of (R)‐1‐phenylethanol (>99 % ee, see the Supporting Information for further details). A very satisfactory turnover number for the AoFOx of more than 300 000 was achieved, which suggests that this in situ H2O2 generation system is economically feasible. It is also worth mentioning that up to 31±3 mm (R)‐1‐phenylethanol was produced (Figure S6), which is one of the highest numbers observed so far using rAaeUPO.15 It should be mentioned here that in case of volatile reagents, imperfect mass balances were observed upon prolonged reaction times. We believe that this is a technical issue that will be overcome in future scale‐up experiments.
Cytochrome C (CytC), another heme‐containing protein capable of catalysing H2O2‐driven oxygen transfer reactions, especially sulfoxidation,16 was evaluated next. Compared to the turnover numbers observed with rAaeUPO, the numbers achieved with CytC appear rather low. However, these numbers are still significantly higher than those achieved previously using other H2O2‐generation systems.16 The lack of enantioselectivity in the sulfoxidation of thioanisol is in accordance with previous reports.16 It should be kept in mind here that the natural role of CytC is not that of an enzyme but rather that of an electron‐transport protein.
Another important H2O2‐driven reaction is the so‐called perhydrolase reaction of lipases.1b, 24 In short, a lipase catalyses the perhydrolysis of carboxylic (esters) to yield a reactive peracid, which in turn can undergo Baeyer–Villiger oxidations of ketones or Prilezhaev oxidations of C=C‐double bonds. Our proposed AoFOx H2O2‐generation system proved to be applicable in principle to drive these reactions (Figure 2). Using the lipase B from Candida antarctica, CalB) together with octanoic acid as cocatalyst gave catalytic turnover in the chemoenzymatic Baeyer–Villiger oxidation of cyclohexanone as well as the chemoenzymatic epoxidation of styrene. However, compared to the other systems investigated here, rather low turnover numbers for the biocatalyst were observed. This can be attributed to the low affinity of CalB towards H2O2 in aqueous systems25 resulting in low CalB activity under the conditions chosen. Further investigations aiming at higher in situ H2O2 concentrations are currently ongoing.
Finally, we evaluated AoFOx to promote halogenation reactions catalysed by the V‐dependent haloperoxidase from Curvularia inaequalis.8b,8c, 26 The hydroxyhalogenation of styrene gave acceptable results in terms of product yield and catalyst performance. Again, the volatility of the reagents impaired the final product concentration and thereby the catalytic numbers. A completely different picture evolved, however, when using 4‐pentenoic acid as starting material. Here, a perfect mass balance was observed and full conversion of the starting material into the desired bromolactone was observed. We also scaled up this reaction to the gram scale: Starting from 200 mm 4‐pentenoic acid, 150 mm of the desired bromolactone was obtained, which could be separated from the reaction mixture by simple extraction (Figure S14). Thus, 1.6 g of the pure product was obtained.27
In conclusion, we present herein the proof‐of‐concept for a simple H2O2 generation system based on formate oxidases such as the FOx from Aspergillus oryzae (AoFOx). This system stands out in terms of practical simplicity and excellent performance, even at this early stage of development. Furthermore, the turnover numbers achieved with AoFOx exceed those of established systems by orders of magnitude (Table S2).
Three decades ago, the introduction of formate dehydrogenases as NADH regeneration catalysts ushered in a new era in bioreduction catalysis.28 We are convinced that formate oxidases will have a similar impact for biooxidation/functionalisation catalysis. Further developments in our laboratories will focus on further engineering AoFOx (in particular, a lower K M value towards formate is highly desirable) and further characterisation and optimisation of the synthetic schemes to fully explore its synthetic potential.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors gratefully acknowledge funding by the European Research Commission (ERC consolidator grant, No. 648026), the European Union (H2020‐BBI‐PPP‐2015‐2‐1‐720297), the Netherlands Organisation for Scientific Research (VICI grant No. 724.014.003), the National Science Foundation (NSF) of the United States (grant IIP‐1540017) and the Comunidad de Madrid Synergy CAM Project Y2018/BIO‐4738‐EVOCHIMERA‐CM.
F. Tieves, S. J.-P. Willot, M. M. C. H. van Schie, M. C. R. Rauch, S. H. H. Younes, W. Zhang, J. Dong, P. Gomez de Santos, J. M. Robbins, B. Bommarius, M. Alcalde, A. S. Bommarius, F. Hollmann, Angew. Chem. Int. Ed. 2019, 58, 7873.
References
- 1.
- 1a. Liang Y., Wei J., Qiu X., Jiao N., Chem. Rev. 2018, 118, 4912–4945; [DOI] [PubMed] [Google Scholar]
- 1b. Dong J., Fernández-Fueyo E., Hollmann F., Paul C., Pesic M., Schmidt S., Wang Y., Younes S., Zhang W., Angew. Chem. Int. Ed. 2018, 57, 9238–9261; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 9380–9404; [Google Scholar]
- 1c. Fessner N. D., ChemCatChem 2019, 10.1002/cctc.201801829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Roduner E., Kaim W., Sarkar B., Urlacher V. B., Pleiss J., Gläser R., Einicke W.-D., Sprenger G. A., Beifuß U., Klemm E., Liebner C., Hieronymus H., Hsu S.-F., Plietker B., Laschat S., ChemCatChem 2013, 5, 82–112; [Google Scholar]
- 2b. Urlacher V. B., Girhard M., Trends Biotechnol. 2012, 30, 26–36; [DOI] [PubMed] [Google Scholar]
- 2c. Nestl B. M., Hammer S. C., Nebel B. A., Hauer B., Angew. Chem. Int. Ed. 2014, 53, 3070–3095; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 3132–3158; [Google Scholar]
- 2d. Bernhardt R., Urlacher V. B., Appl. Microbiol. Biotechnol. 2014, 98, 6185–6203; [DOI] [PubMed] [Google Scholar]
- 2e. Fasan R., ACS Catal. 2012, 2, 647–666; [Google Scholar]
- 2f. Jung S. T., Lauchli R., Arnold F. H., Curr. Opin. Biotechnol. 2011, 22, 809–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Kara S., Schrittwieser J. H., Hollmann F., Ansorge-Schumacher M. B., Appl. Microbiol. Biotechnol. 2014, 98, 1517–1529; [DOI] [PubMed] [Google Scholar]
- 3b. Weckbecker A., Gröger H., Hummel W. in Biosystems Engineering I: Creating Superior Biocatalysts, Vol. 120, Springer-Verlag Berlin, Berlin, 2010, pp. 195–242. [Google Scholar]
- 4. Holtmann D., Hollmann F., ChemBioChem 2016, 17, 1391–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.As Peroxizymes, we define all enzymes that (in principle) can use H2O2 as stoichiometric oxidant. Examples are the classical peroxidases and peroxygenases but also other heme-containing biocatalysts such as P450 monooxygenases and also hydrolases.
- 6. van Rantwijk F., Sheldon R. A., Curr. Opin. Biotechnol. 2000, 11, 554–564. [DOI] [PubMed] [Google Scholar]
- 7. Hollmann F., Arends I. W. C. E., Polymers 2012, 4, 759–793. [Google Scholar]
- 8.
- 8a. Seel C. J., Králík A., Hacker M., Frank A., König B., Gulder T., ChemCatChem 2018, 10, 3960–3963; [Google Scholar]
- 8b. Dong J. J., Fernandez-Fueyo E., Li J., Guo Z., Renirie R., Wever R., Hollmann F., Chem. Commun. 2017, 53, 6207–6210; [DOI] [PubMed] [Google Scholar]
- 8c. Fernández-Fueyo E., van Wingerden M., Renirie R., Wever R., Ni Y., Holtmann D., Hollmann F., ChemCatChem 2015, 7, 4035–4038; [Google Scholar]
- 8d. Wischang D., Hartung J., Tetrahedron 2012, 68, 9456–9463. [Google Scholar]
- 9.
- 9a. Hofrichter M., Ullrich R., Curr. Opin. Chem. Biol. 2014, 19, 116–125; [DOI] [PubMed] [Google Scholar]
- 9b. Wang Y., Lan D., Durrani R., Hollmann F., Curr. Opin. Chem. Biol. 2017, 37, 1–9. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Meyer-Wassewitz J., Hohmann D., Ansorge-Schumacher M. B., Kraume M., Drews A., Biochem. Eng. J. 2017, 126, 68–77; [Google Scholar]
- 10b. Zhou P., Wang X., Zeng C., Wang W., Yang B., Hollmann F., Wang Y., ChemCatChem 2017, 9, 934–936. [Google Scholar]
- 11.
- 11a. Markiton M., Boncel S., Janas D., Chrobok A., ACS Sustainable Chem. Eng. 2017, 5, 1685–1691; [Google Scholar]
- 11b. Teixeira A. R. S., Flourat A., Peru A. M., Brunissen F., Allais F., Front. Chem. 2016, 4, 11; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11c. Drozdz A., Chrobok A., Chem. Commun. 2016, 52, 1230–1233. [DOI] [PubMed] [Google Scholar]
- 12. Valderrama B., Ayala M., Vazquez-Duhalt R., Chem. Biol. 2002, 9, 555–565. [DOI] [PubMed] [Google Scholar]
- 13. Sabuzi F., Churakova E., Galloni P., Wever R., Hollmann F., Floris B., Conte V., Eur. J. Inorg. Chem. 2015, 3519–3525. [Google Scholar]
- 14. Sheldon R. A., Pereira P. C., Chem. Soc. Rev. 2017, 46, 2678–2691. [DOI] [PubMed] [Google Scholar]
- 15. Ni Y., Fernández-Fueyo E., Baraibar A. G., Ullrich R., Hofrichter M., Yanase H., Alcalde M., van Berkel W. J. H., Hollmann F., Angew. Chem. Int. Ed. 2016, 55, 798–801; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 809–812. [Google Scholar]
- 16. Hollmann F., Schmid A., J. Inorg. Biochem. 2009, 103, 313–315. [DOI] [PubMed] [Google Scholar]
- 17. Rocha-Martin J., Velasco-Lozano S., Guisan J. M., Lopez-Gallego F., Green Chem. 2014, 16, 303–311. [Google Scholar]
- 18.
- 18a. Zhang W., Fernández-Fueyo E., Ni Y., van Schie M., Gacs J., Renirie R., Wever R., Mutti F. G., Rother D., Alcalde M., Hollmann F., Nat. Catal. 2018, 1, 55–62; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18b. Zhang W., Burek B. O., Fernández-Fueyo E., Alcalde M., Bloh J. Z., Hollmann F., Angew. Chem. Int. Ed. 2017, 56, 15451–15455; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 15654–15658. [Google Scholar]
- 19.
- 19a. Doubayashi D., Ootake T., Maeda Y., Oki M., Tokunaga Y., Sakurai A., Nagaosa Y., Mikami B., Uchida H., Biosci. Biotechnol. Biochem. 2011, 75, 1662–1667; [DOI] [PubMed] [Google Scholar]
- 19b. Maeda Y., Doubayashi D., Oki M., Nose H., Sakurai A., Isa K., Fujii Y., Uchida H., Biosci. Biotechnol. Biochem. 2009, 73, 2645–2649; [DOI] [PubMed] [Google Scholar]
- 19c. Maeda Y., Doubayashi D., Oki M., Nose H., Fujii Y., Uchida H., J. Biosci. Bioeng. 2009, 108, S106-S106; [DOI] [PubMed] [Google Scholar]
- 19d. Robbins J. M., Bommarius A. S., Gadda G., Arch. Biochem. Biophys. 2018, 643, 24–31; [DOI] [PubMed] [Google Scholar]
- 19e. Robbins J. M., Souffrant M. G., Hamelberg D., Gadda G., Bommarius A. S., Biochemistry 2017, 56, 3800–3807; [DOI] [PubMed] [Google Scholar]
- 19f. Robbins J. M., Geng J., Barry B. A., Gadda G., Bommarius A. S., Biochemistry 2018, 57, 5818–5826. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Molina-Espeja P., Garcia-Ruiz E., Gonzalez-Perez D., Ullrich R., Hofrichter M., Alcalde M., Appl. Environ. Microbiol. 2014, 80, 3496–3507; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20b. Molina-Espeja P., de Santos P. G., Alcalde M. in Directed Enzyme Evolution: Advances and Applications (Ed.: M. Alcalde), Springer International Publishing, Cham, 2017, pp. 127–143; [Google Scholar]
- 20c. Molina-Espeja P., Ma S., Mate D. M., Ludwig R., Alcalde M., Enzyme Microb. Technol. 2015, 73–74, 29–33; [DOI] [PubMed] [Google Scholar]
- 20d. Gomez de Santos P., Canellas M., Tieves F., Younes S. H. H., Molina-Espeja P., Hofrichter M., Hollmann F., Guallar V., Alcalde M., ACS Catal. 2018, 8, 4789–4799. [Google Scholar]
- 21. Ullrich R., Nüske J., Scheibner K., Spantzel J., Hofrichter M., Appl. Environ. Microbiol. 2004, 70, 4575–4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Perez D. I., Mifsud Grau M., Arends I. W. C. E., Hollmann F., Chem. Commun. 2009, 6848–6850. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Peter S., Kinne M., Ullrich R., Kayser G., Hofrichter M., Enzyme Microb. Technol. 2013, 52, 370–376; [DOI] [PubMed] [Google Scholar]
- 23b. Kluge M., Ullrich R., Scheibner K., Hofrichter M., Green Chem. 2012, 14, 440–446. [Google Scholar]
- 24.
- 24a. Björkling F., Frykman H., Godtfredsen S. E., Kirk O., Tetrahedron 1992, 48, 4587–4592; [Google Scholar]
- 24b. Björkling F., Godtfredsen S. E., Kirk O., J. Chem. Soc. Chem. Commun. 1990, 1301–1303. [Google Scholar]
- 25.
- 25a. Ma Y., Li P., Willot S. J.-P., Zhang W., Ribitsch D., Choi Y. H., Zhang T., Verpoort R., Hollmann F., Wang Y., ChemSusChem 2019, 10.1002/cssc.201900043; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25b. Zhou P. F., Lan D. M., Popowicz G. M., Wang X. P., Yang B., Wang Y. H., Appl. Microbiol. Biotechnol. 2017, 101, 5689–5697; [DOI] [PubMed] [Google Scholar]
- 25c. Zhou P., Wang X., Yang B., Hollmann F., Wang Y., RSC Adv. 2017, 7, 12518–12523. [Google Scholar]
- 26.
- 26a. Wever R., van der Horst M. A., Dalton Trans. 2013, 42, 11778–11786; [DOI] [PubMed] [Google Scholar]
- 26b. Wever R., Renirie R., Peroxidases and Catalases: Biochemistry Biophysics, Biotechnology, and Physiology, Wiley, Hoboken, 2010, pp. 363–385. [Google Scholar]
- 27.This corresponds to a 45 % yield of isolated product. However, upon performing a second extraction, more product (albeit contaminated with the starting material) was obtained.
- 28.
- 28a. Shaked Z., Whitesides G. M., J. Am. Chem. Soc. 1980, 102, 7104–7105; [Google Scholar]
- 28b. Popov V. O., Lamzin V. S., Biochem. J. 1994, 301, 625–643; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28c. Tishkov V. I., Popov V. O., Biochemistry 2004, 69, 1252–1253; [DOI] [PubMed] [Google Scholar]
- 28d. Bommarius A. S., Schwarm M., Stingl K., Kottenhahn M., Huthmacher K., Drauz K., Tetrahedron: Asymmetry 1995, 6, 2851–2888. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary