

Abstract
Despite effective surgical methods for non‐melanoma skin cancer (NMSC), patients suffer from tissue damage, scarring, or even disfigurement; thus, there is a need for chemopreventive approaches. Because of the complex interplay between glucocorticoids (GCs), inflammation, and cancer, we sought to determine the role of 11β‐hydroxysteroid dehydrogenase 1 and 2 (11βHSD1 and 2) in regulating GCs during skin cancer development and progression. 11βHSDs modulate the activation of GCs in a tissue‐specific manner and have been reported to play a role in development and progression of other types of cancer, but their role has not yet been reported in NMSC. Here, we found a significant upregulation of 11βHSD2 protein in skin cancer cells when compared to normal skin cells, suggesting a role for this enzyme in the multifactorial process of skin cancer development. In addition, inhibition of 11βHSD2 with siRNA resulted in significant reduction in colony formation in vitro. Finally, our in vivo study elucidated that inhibition of 11βHSD2 with pharmacological inhibitor, Glycyrrhetinic acid (GA) could significantly diminish tumorigenesis in a well‐studied in vivo mouse model of NMSC. Overall, these studies highlight for the first time a potential novel role for 11βHSD2 in NMSC development and may allow for new GC treatment approaches capable of avoiding deactivation by the enzyme. If 11βHSD2 can be inhibited as we have done here, or circumvented using modified GCs, this may lead to more efficacious outcomes for NMSC patients by preventing deactivation of the GC and minimizing resistance.
Keywords: 11β‐hydroxysteroid dehydrogenases, chemoprevention, glucocorticoids, glycyrrhetinic acid, non‐melanoma skin cancer, phytochemicals
Abbreviations
- 11βHSD1
11‐beta hydroxysteroid dehydrogenase 1
- 11βHSD2
11‐beta hydroxysteroid dehydrogenase 2
- DMBA
7,12‐dimethylbenz[a]anthracene
- GA
Glycyrrhetinic acid
- GC
Glucocorticoid
- GR
Glucocorticoid receptor
- MTT reagent
thiazolyl blue tetrazolium bromide
- TPA
12‐O‐Tetradecanoylphorbol‐13‐acetate
1. INTRODUCTION
Non‐melanoma skin cancer (NMSC) poses a significant public health problem as it is the most prevalent malignancy in the United States with an estimated 2‐3 million new diagnoses each year and an incidence that is continuing to rise annually.1, 2, 3, 4 Between 2002‐2006 and 2007‐2011, the average annual total cost for skin cancer increased by 126.2%, as compared to the average annual total cost for all other cancers increased only by 25.1%.5 The increasing incidence of both NMSC diagnoses and rapidly expanding treatment costs demonstrate the considerable need for prevention‐based approaches towards NMSC. Therefore, we have focused our studies on chemopreventive strategies which target the inflammation gatekeepers, GCs, to effectively diminish tumorigenesis.
The modulation of GCs at the pre‐receptor level is both necessary for their proper function and essential for tissue‐specific glucocorticoid requirements. While GCs are regulated by ligand and receptor concentrations in the plasma, an additional and vital level of pre‐receptor modulation of these critical hormones is carried out by 11β‐hydroxysteroid dehydrogenases (11βHSDS).6 The function of these enzymes is of vital importance for localized activation and deactivation of GCs.7 11βHSD1 and 11βHSD2 catalyze the interconversion of active GCs to their inactive counterparts such as the conversion of cortisol to cortisone by 11βHSD2 and, the reverse reaction whereby 11βHSD1 results in an active cortisol molecule.8 Specifically, 11βHSD2 deactivates GCs by converting the hydroxyl group on the C‐11 position to a ketone group, rendering it inactive; it does this in a tissue specific manner.9, 10 Through the enzymatic action of 11βHSD2, the amount of active GC which is available to the receptor can be carefully modulated within the designated target tissue, thus creating an advantageous target for modulating GCs to combat tumorigenesis.
Significant evidence in the literature has shown that 11βHSD2 is expressed in many different cancer tissues, however, it is not expressed in the normal tissue counterparts.11, 12, 13 Specific examples of this 11βHSD1/11βHSD2 expression “switch” as it is described in the literature include occurrences in breast cancer, colon cancer, prostate, endometrial, and ovarian cancer cell lines.12, 14, 15, 16, 17 Given the fact that GCs play an important role in skin proliferation and inflammation, it was surprising to find that 11βHSD2 characterization in NMSC has not yet been carried out. Moreover, this critical GC modulating enzyme has also not yet been evaluated for its role in transformation in NMSC, and it has been hypothesized that preferential expression of 11βHSD2 may play a role in transformation of certain types of cancer.13 Therefore, we propose that NMSC development and progression may be circumventing regulation of cellular proliferation by increasing expression of 11βHSD2 and thereby locally inactivating GCs. As the “switch” in 11βHSD1/11βHSD2 expression has been reported in various cancer types, much of the research on these enzymes as they relate to cancer has now turned towards elucidating and characterizing the mechanisms by which this change in expression is controlled.13 There is no doubt that this enzyme has a vital physiological role as it is normally responsible for inactivating intracellular GCs in typical mineralocorticoid target tissues; however, recent findings in the literature coupled with the unique role that GCs play in skin cancer development and progression suggest that this enzyme may also play a substantial role in the multifactorial process of NMSC transformation.
In this study, we sought to characterize 11βHSD2 in in vitro mouse models of NMSC and to investigate whether it plays a role in transformation. We utilized 11βHSD2 siRNA and pharmacological 11βHSD2 inhibitor, Glycyrrhetinic acid (GA) to knock down 11βHSD2 and study the effect this would have on TPA‐induced transformation of well‐established mouse models of NMSC. We also validated our 11βHSD2 characterization results from our in vitro mouse studies using a human in vitro model of NMSC to ascertain whether the same results could be achieved in a human model for clinical relevance. Finally, we performed an in vivo, two‐stage mouse carcinogenesis study over the course of 30 weeks to study the effect of 11βHSD2 inhibition by GA on DMBA/TPA‐induced tumorigenesis. Our in vitro results in both mouse and human NMSC cell models show an upregulation of 11βHSD2 in cancer cells versus normal skin cells. Moreover, upon addition of a tumor promoter to induce transformation, we see an upregulation of 11βHSD2 in pre‐neoplastic mouse keratinocytes. Corresponding with these results, our in vivo mouse studies also suggest a critical role for this enzyme in tumorigenesis as inhibition of 11βHSD2 with GA considerably delayed tumor onset and significantly decreased tumor incidence. Given the ability of GCs to alter signaling in key survival pathways and the fact that similar to many chemotherapeutic agents, skin cancer patients often develop resistance to GC therapy, an investigation into the role of 11βHSD2 in skin cancer development/progression and GC resistance is of significant relevance. As stated before, characterization of 11βHSD2 in NMSC has never been evaluated elsewhere, therefore these findings elucidate an important role for this enzyme in NMSC and may identify 11βHSD2 as a potential target for prevention and or therapy of NMSC.
1.1. Reagents
DMBA, TPA, and GA were obtained from Sigma‐Aldrich (St. Louis, MO). Stock solutions of GA (up to 20 mM) and TPA (10 mg/mL) were prepared in ethanol for in vitro studies and then diluted down for experiments. For in vivo studies, DMBA and TPA were prepared according to previously established protocol by Abel et al.18 For Western blotting analyses, primary antibodies used were for GR (Santa Cruz Biotechnology, Dallas, TX), 11βHSD1 (Santa Cruz Biotechnology), 11βHSD2 (Abcam, Cambridge, MA), and β‐actin (Abcam). Secondary antibodies for mouse and rabbit were both from Biorad (Biorad, Hercules, CA). Silencer select siRNA for 3 different regions of mouse 11βHSD2 were obtained; S67837, S67838, and S67839 (Ambion, Foster City, CA).
1.2. Cell culture
To investigate the importance of 11βHSDs in transformation using in vitro skin cancer models, we employed the use of two well‐established cell models of skin cancer; the JB6 mouse epidermal cell lines generously provided to us by the laboratory of Nancy Colburn, and a second model consisting of 3PC, MT1/2, and Ca3/7 cells which was generated in our own lab, the laboratory of Thomas J. Slaga. The JB6 model consists of clonal genetic variants that are promotion‐sensitive (P+), promotion‐resistant (P−) or transformed (Tx). The JB6 model is a widely used inducible model to study transformation, promotion, and progression at the molecular level. This model system of cell lines originated from untreated primary BALB/c mouse epidermal cell cultures that gave rise at a very low frequency to immortalized cell lines.19 Then immortalized JB6 cells underwent further change to stably acquire sensitivity to induction of anchorage independence and tumorigenicity by TPA and other tumor promoters.20 Nonselective cloning soon after observation of this change lead to the development of clonal lines that were either sensitive (P+) or resistant (P−) to tumor‐promoter‐induced neoplastic transformation.19, 20, 21 Additionally, human epidermal cell lines representing different stages of malignant transformation, PM1 and Met4 were generously provided by the laboratory of C.M. Proby. JB6 P+ and RT101 mouse epidermal keratinocytes were maintained in MEM containing 5% FBS, L‐glutamine, sodium pyruvate, 50U/mL penicillin, and 50 ng/mL streptomycin. Cells were maintained in an incubator at 5% CO2 and 37°C. 3PC, MT1/2, and Ca3/7 cells were maintained in Lonza (Houston, TX) liquid MEM Joklik (Cat no. 04‐719Q) supplemented with transferrin (10 μg/mL), FBS (8%), insulin (5 μg/mL), EGF (5 ng/mL), o‐phospoethanolamine (10 μM), 2‐amino ethanol (10 μM), gentamycin (50 μg/mL), and penicillin (50 U/mL)/streptomycin (50 ng/mL). The human epidermal cell lines were maintained in minimum essential medium supplemented with 10% FBS, 2 mM L‐glutamine, 50 U/mL penicillin, and 50 ng/mL streptomycin.
1.3. Western blotting
JB6 P+ cells or RT101 cells were plated at 2.0 × 105 cells per 100 mm culture dish. Cells were treated with 10 ng TPA or 0.1% vehicle (acetone) for 24 h. Media was aspirated and cells were rinsed twice with cold PBS before lysis in buffer containing 1% Triton X‐100, 0.5% IGEPAL, 0.05 M TrisHCl, and 0.1 M NaCl as well as protease/phosphatase inhibitors and 5 mM EDTA. Protein extraction was performed by centrifugation and quantified by the BCA protein estimation method (Pierce, Waltham, MA). Proteins were separated on 4‐12% mini‐protean gradient gels (Biorad), transferred onto nitrocellulose membrane, blocked in a 5% milk solution in PBST for 1 h prior to overnight incubation with the designated primary antibody under refrigerated conditions. Membranes were next rinsed with PBST, incubated in secondary antibody for a minimum of 1 h, and then developed using Biorad Clarity™ Western ECL blotting substrate (Biorad). Imaging of developed Western blots was performed on an EPSON scanner, followed by densitometry measurements using IMAGEJ software.
1.4. Transient knockdown of 11βHSD2 in JB6 P+ and RT101 cells
Transient knockdown of 11βHSD2 was performed using 11βHSD2 siRNA (Ambion), non‐targeted scrambled siRNA (Santa Cruz Biotechnology), and a keratinocyte nucleofection kit (Lonza). JB6 P+ and RT101 cells were transfected with 6 μL of non‐specific scramble, 11βHSD2, or GFP siRNA duplexes by electroporation at 240 V/25 ms pulse length (Amaxa, MD). After transfection, cells were plated into 100 mm dishes and allowed to grow for 48 h after which they were lysed and collected for whole cell Western blot analysis.
1.5. Anchorage independence assay of RT101 cells transiently transfected with si_HSD2
Immediately following transient knockdown of 11βHSD2, a 104 aliquot of cells were taken and directly added to a 0.33% agar medium solution with 10%FBS. The cell suspension/agar medium solution was poured as a top layer to previously prepared bottom layers of 0.5% agarose/10%FBS solution in a 6‐well plate. Cells were culture at 37°C, 5% CO2 for 10‐14 days. Where treatments were performed in soft agar, drug treatments were added to top layer prior to pouring, and final concentration was adjusted accordingly; acetone was used as a vehicle. After 10‐14 days, colonies were stained using crystal violet and quantified.
1.6. Animal model and experimental conditions
The ability of GA to inhibit tumorigenesis was examined by utilizing the two‐stage skin carcinogenesis model. Healthy female FVB mice aged 6‐7 weeks were obtained from Jackson Laboratories, Bar Harbor, ME. Upon arrival, animals were randomly segregated into eight groups: Control, DMBA/TPA, GA (low), GA (mid), GA (high), DMBA/TPA/GA(L), DMBA/TPA/GA(M), and DMBA/TPA/GA(H) (Figure 1). Each group contained 28 animals total, with eight animals whose endpoint would be the short‐term date, and 20 remaining animals whose endpoint would be at the end of the study (long term). Animals were housed in a controlled atmosphere, under hygienic conditions, with 12‐h light/dark cycles, and were provided standard animal feed. Prior to the start of the experiment, the animals were given an acclimation period of 1 week. To expose the skin of the mice for topical treatments, an approximate 2 × 3 cm portion of the dorsal region of each mouse was shaved with surgical clippers 2 days prior to the start of any topical application of compounds.
Figure 1.

Detailed animal treatment schedule to study the effect of Glycyrrhetinic acid on DMBA/TPA induced skin tumorigenesis. All treatments described were administered topically in acetone to the shaved dorsal area of mice. Group 1, the control group mice were treated with 200 μL acetone twice weekly throughout the treatment period, and at each instance where TPA was administered. In the DMBA/TPA group, DMBA administration was performed only once at week zero to groups 2, 6, 7, and 8 in a dose of 50 μg/200 μL. In each group where DMBA was administered, TPA was also given twice weekly at a dose of 2 μg/200 μL for the duration of the study. GA control, groups 3, 4, and 5, each received their respective dose of GA (at 0.25 μmol, 0.5 μmol, and 1 μmol concentrations of GA) twice weekly. For our experimental groups, 6, 7, and 8, GA was administered at the respective dose (0.25 μmol, 0.5 μmol, and 1 μmol) 30 min prior to TPA treatment, twice weekly for entire length of the study
Group 1 animals served as controls; these animals received topical application of acetone only (200 μL/mouse). To induce skin tumorigenesis, group 2 animals received a single topical application of DMBA (50 μg/200 uL of acetone) at the beginning of the study. One week later, group 2 received a topical application of TPA (2 μg/200 uL of acetone) to the same site, which continued twice weekly for 27 consecutive weeks. Groups 3, 4, and 5 served as controls for experimental groups, receiving only GA (twice weekly when experimental treatments were administered) at the following concentrations, respectively; 0.25 μmol, 0.5 μmol, and 1 μmol. To examine the effect of GA on development and progression of NMSC, animals in Groups 6, 7, and 8 were treated with the same DMBA/TPA regimen as group 2, however, they also received topical administration of GA at 30 min prior to TPA in the same doses as in groups 3, 4, and 5. The number of skin lesions >2 mm in diameter were counted and recorded each week, and general observations of the health and well‐being of the animals were recorded regularly. For further detail on treatments, a detailed schematic is shown in Figure 1.
1.7. Histopathologic examinations
Normal, tumor‐adjacent, and tumor samples were excised and collected at the end of both endpoints; therefore, samples were collected after 15 and 27 weeks. Tissues were then fixed in 10% formalin for later use. Later, the tissues were dehydrated in increasing grades of alcohol, cleared in benzene, and embedded in paraffin wax. Sections then underwent hematoxylin and eosin staining (H&E) for microscopic evaluation of characteristic markers of TPA‐induced epidermal hyperplasia, classification of papilloma, and identification of squamous cell carcinoma. H&E stained tissue sections were analyzed by a pathologist who differentiated between normal mouse skin, papilloma, and squamous cell carcinoma.
1.8. Sample preparation
Normal, tumor‐adjacent, and tumor samples were excised and collected at the end of both endpoints; samples were collected after 15 and 27 weeks. Tissues were snap frozen in liquid nitrogen until a later date upon which they were homogenized with a handheld homogenizer and lysed via sonication on ice and in RIPA lysis buffer (Pierce) containing protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific, MA). The homogenate was clarified by centrifugation at 12 000g for 15 min at 4°C. Protein lysates were stored at −80°C until needed.
1.9. Statistical analyses
For in vitro studies, data are expressed as mean ± SD. Students t‐test or ANOVA were used to calculate P values, and statistical significance was established at P < 0.05. All statistical analyses were performed using Graphpad Prism. For in vivo studies, data was collected for both tumor incidence and total number of lesions. Tumor incidence was defined as the number of animals showing carcinogenic response/total number of animals in the group. For comparisons between two groups, a Student's t‐test was used, and when comparing multiple groups, ANOVA was used. For analyzing differences between groups over time, the fisher's exact test was used to determine significance among two groups at a specific time point. For all analyses, P < 0.05 was accepted for statistical significance. All statistical analyses were performed using GraphPad Prism (GraphPad, CA).
2. RESULTS
2.1. 11βHSD2 expression is upregulated in mouse and human in vitro models of NMSC when compared to normal tissue counterparts
As upregulation of 11βHSD2 in tumors and cancer cells versus their normal counterparts has been shown in several different cancer types, but has never been evaluated in NMSC, we characterized the protein expression level of this enzyme in two well‐established mouse skin cancer models in order to determine whether 11βHSD2 was upregulated in skin cancer as it has been shown to be in other cancer types in the literature. To investigate the expression of 11βHSD2 in NMSC, we utilized the JB6 mouse epidermal keratinocyte model consisting of P+ and RT101 cells, and a second mouse NMSC model consisting of 3PC, MT1/2, and Ca3/7 cells. Our results showed that in transformed RT101 cells, basal 11βHSD2 protein levels were significantly elevated when compared with levels in untreated, pre‐neoplastic JB6 P+ cells (Figure 2A). Further, 11βHSD2 protein expression significantly increased in JB6 P+ cells after treatment with tumor promoter, TPA, when compared to acetone, the vehicle control (Figure 2A). When our second mouse NMSC model was evaluated, the same trend was observed whereby both tumorigenic cell lines, MT1/2, and Ca3/7, showed significantly elevated 11βHSD2 expression when compared to their non‐tumorigenic counterpart, 3PC cells (Figure 2A). In both models, we report for the first time in skin, that upregulation of 11βHSD2 occurs in transformed cells as compared to normal skin cells. To validate these findings in a human in vitro model, we further characterized 11βHSD2 basal expression in normal HaCaT human skin cells and compared it to highly dysplastic human skin cells (PM1) and metastatic skin cancer cells (Met4). Our results show a considerable increase in 11βHSD2 expression in PM1 highly dysplastic human skin cells and Met4 skin cancer cells when compared to HaCaT normal human skin cells (Figure 2B). These results parallel those which we obtained in our mouse in vitro studies and align with similar findings in other cancer types in the literature.
Figure 2.

A, Representative Western blot and quantification showing increased 11βHSD2 expression in TPA‐induced JB6 P+ cells as compared to basal 11βHSD2 levels in vehicle treated JB6 P+ cells; Representative Western blot and quantification showing transformed RT101 mouse epidermal keratinocyte cell lines have aberrant 11βHSD2 expression relative to normal or pre‐neoplastic cell lines (JB6 P + without TPA treatment); Representative Western blot and quantification showing increased 11βHSD2 expression in papilloma‐producing MT1/2, and carcinoma‐producing Ca3/7 cells as compared to non‐tumorigenic 3PC cell lines. B, Representative Western blot and quantification showing basal 11βHSD2 levels in PM1 dysplastic, pre‐cancerous skin cells and Met4 skin cancer cells are significantly higher than basal levels shown in normal HaCaT skin cells. C, Inhibition of 11βHSD2 via siRNA significantly decreases TPA‐induced JB6 P+ and RT101 colony formation in soft agar as determined by the anchorage independent colony formation assay. 11βHSD2 inhibition by siRNA pool of 3 different siRNA results in significantly decreased colony formation in TPA‐induced JB6 P+ cells and the same was observed in RT101 cells. These results were validated using the same 3 11βHSD2 siRNA individually in transformed RT101 cells, and the inhibition of 11βHSD2 by each siRNA resulted in significantly decreased colony formation as seen in the colony quantification of the last panel. P < 0.05 were considered statistically significant (*). Densitometry shown at right of all Western blots in this figure is the average result of 3 separate experiments. 11βHSD2 was detected using anti‐11βHSD2 antibody
2.2. 11βHSD2 transient knockdown in soft agar significantly reduces TPA‐induced colony formation of JB6 P+ cells and transformed RT101 cells
As transformed epidermal keratinocytes, RT101 cells will readily form colonies in soft agar. Thus, we sought to determine the role of 11βHSD2 in tumorigenesis by investigating whether 11βHSD2 knockdown in a transformed cell line could prevent or diminish colony formation in soft agar. As colony formation in the soft agar assay is the most efficacious correlate to tumorigenesis in vitro, determining the effect of 11βHSD2 inhibition utilizing this assay would give great insight into future in vivo tumorigenesis studies. When P+ cells were treated with TPA and 11βHSD2 was transiently knocked down, we saw a significant reduction in colony formation as compared to the scramble (Figure 2C). Further, we found that transient knockdown of 11βHSD2 in transformed RT101 cells resulted in significantly diminished colony formation over a 10‐day period when compared to the non‐specific scramble (Figure 2C). Using a pool of siRNAs for 11βHSD2 and 3 distinctly individual siRNA for 11βHSD2, we observed similar results, whereby transient knockdown of 11βHSD2 resulted in significantly reduced RT101 colony formation in soft agar with both the pooled siRNA and also with 2 individual siRNA specific for 11βHSD2 (Figure 2C).
2.3. DMBA/TPA successfully induced development of papilloma and squamous cell carcinoma, which could both be greatly inhibited by GA treatment
To determine the chemopreventive effect of GA, we performed DMBA‐initiated and TPA‐promoted mouse skin carcinogenesis in vivo. Animals from the DMBA/TPA group and the DMBA/TPA/GA groups were topically administered a single dose of DMBA at the start of our study, followed by twice weekly doses of either TPA alone or TPA 30 min after GA treatment for the duration of the study. The representative images of dorsal skin from each respective group seen in Figure 3 demonstrate that DMBA/TPA successfully induced both papilloma during the first phase of our animal study (over 15 weeks) and squamous cell carcinoma during the second phase of our animal study (26 weeks total), respectively. Moreover, representative images show that treatment of animals with DMBA/TPA/GA considerably diminished development of both papilloma and squamous cell carcinomas, with the greatest effect observed in the lowest dose GA group (Figures 3A and B).
Figure 3.

A, Representative images of papillomagenesis in the indicated groups after 15 weeks of treatment. When compared to vehicle‐treated animals (top panel), GA treatment resulted in significant inhibition of tumorigenesis as can be seen in representative images of all three DMBA/TPA/GA groups (bottom panel). B, Representative images of development of papilloma and squamous cell carcinoma in the indicated groups after 26 weeks of treatment. When compared to vehicle‐treated animals (top panel), GA treatment resulted in significant inhibition of tumorigenesis as can be seen in representative images of all three DMBA/TPA/GA groups (bottom panel)
2.4. Inhibition of 11βHSD2 by GA resulted in several anti‐tumorigenic outcomes including decreased average tumor weight, decreased tumor incidence, decreased average number of tumors, and delayed tumor onset
Our study, involving the treatment of FVB mice with GA over the length of 26 weeks provides considerable support of a chemopreventive role for this compound in DMBA/TPA‐induced skin carcinogenesis. Firstly, GA‐mediated 11βHSD2 inhibition significantly reduced average tumor weight in grams when compared to the TPA control group (Figure 4A). Compared to the DMBA/TPA‐treated group, GA significantly reduced the tumor incidence (Figure 4B) and multiplicity of skin tumor formation (Figure 4C). Tumor incidence data over both phase I and phase II of our in vivo study (26 weeks) show significant decreases in tumor incidence in TPA/GA(L) and TPA/GA(H) group for weeks 6‐16 when compared to TPA control. Although TPA/GA(L) did not reach significance, this dose was still able to reduce tumor incidence when compared to the TPA alone group. Here, tumor incidence is defined as the number of animals presenting with at least one tumor. Moreover, it substantially delayed the tumor latency period by 2 weeks in the lowest GA dose (Figure 4D). Treatment with GA in all three experimental groups (low, medium, and high [GA]) resulted in an increase in percent tumor‐free mice, with TPA/GA(L) and TPA/GA(H) showing significant increases when compared to TPA control. Even as late as 16 weeks, there is approximately a 40% difference in tumor‐free mice in the TPA/GA(L) group when compared to the TPA control group which dropped below 50% tumor‐free mice by 10 weeks.
Figure 4.
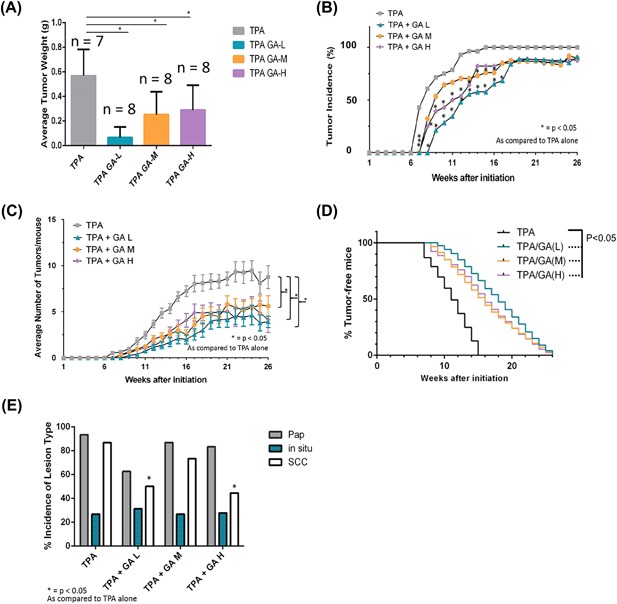
A, Chemopreventive effect of GA on DMBA/TPA‐induced skin cancer in FVB mice. GA treatment significantly reduces average tumor weight in the short‐term timeline when compared to DMBA/TPA control. Twice‐weekly topical application of GA 30 min prior to TPA‐treatment resulted in a significant decrease in average tumor weight in all three experimental DMBA/TPA/GA groups (low, medium, and high doses). B, Tumor incidence data over both phase I and phase II of our in vivo study (26 weeks) show significant decreases in tumor incidence in TPA/GA(L) and TPA/GA(H) group for weeks 6‐16 when compared to TPA control. C, Average number of tumors per mouse over both phase I and phase II of our in vivo study (26 weeks). All 3 groups elicited a significant reduction in average number of tumors/mouse when compared to the TPA alone group. D, Latency of tumors was increased in all experimental groups compared to TPA control by 2 weeks. Treatment with GA in all three experimental groups (low, medium, and high [GA]) resulted in an increase in percent tumor‐free mice, with TPA/GA(L) and TPA/GA(H) showing significant increases when compared to TPA control. E, Incidence of SCC is significantly decreased by GA treatment; Histological analysis of tissue samples after undergoing 26 weeks of TPA‐treatments reveals that treatment with GA significantly decreased the percent incidence of SCC in two out of three experimental groups (TPA/GA(L) and TPA/GA(H)). P < 0.05 were considered to be statistically significant (*)
2.5. GA significantly decreases squamous cell carcinoma incidence when compared to DMBA/TPA alone, but does not have the same effect on papilloma incidence
While GA treatment resulted in significant reduction of tumor incidence and an increase in tumor latency, it was also important to qualify whether this treatment specifically targeted the development of a particular type of lesion. Therefore, H&E stained tissues were analyzed by a pathologist to determine the specific lesion incidence in each group. As malignant conversion from papilloma to SCC is a critical step in the later stage of the DMBA/TPA two‐stage carcinogenesis method, results from this analysis offer further insight into the specific effect of GA on tumorigenesis. Our results show that GA‐mediated 11βHSD2 inhibition significantly decreased percent SCC incidence in the DMBA/TPA/GA(L) and DMBA/TPA/GA(H) groups by approximately 40% (Figure 4E). Although there was a decrease in papilloma incidence in the DMBA/TPA/GA(L) group as well, it did not reach significance. As significant differences between papilloma incidences across all groups were undetectable, but two groups showed a significant reduction in SCC as compared to DMBA/TPA control, it is possible that GA may have a stronger negative effect on conversion from papilloma to SCC rather than papillomagenesis.
2.6. GA significantly inhibits TPA‐induced 11βHSD2 expression in long‐term mouse tumor tissue
As the animal data shows a significant effect of GA‐mediated 11βHSD2 inhibition on tumorigenesis in the DMBA/TPA model of skin carcinogenesis, our next question was whether inhibition of 11βHSD2 by GA played a part in the results we observed. Therefore, we performed Western blot analyses on tumor tissues isolated from mice at both short‐term and long‐term endpoints to characterize 11βHSD1 and 11βHSD2 in both phases of the study. Our results showed that in both the short‐ and long‐term phases of the study, 11βHSD2 protein expression was significantly induced by treatment with TPA when compared to vehicle‐treated animals (Figure 5). This suggests a possible novel role for 11βHSD2 in the multifactorial process of carcinogenesis as many other studies have reported similar findings in other types of cancer. Moreover, an overall trend amongst the 11βHSDs in both short‐ and long‐term phases was observed, wherein 11βHSD1 was higher in vehicle‐treated tissue than in TPA‐treated tissue, and conversely 11βHSD2 was higher in TPA‐treated tissues and lower in vehicle‐treated tissues (Figure 5). These results suggest 11βHSD2 may play a role in tumorigenesis in NMSC, as a switch in 11βHSD1/11βHSD2 expression like the one we observed in our Western blot analyses has been shown in other cancer models. Additionally, both short‐ and long‐term studies showed a considerable reduction in TPA‐induced 11βHSD2 protein expression in the TPA/GA(L) group when compared to the TPA control (Figure 5), with the effect in the short‐term being the most dramatic. Taken together, these findings may suggest an important role for 11βHSD2 in NMSC, where it may be significantly induced by TPA‐treatment to aid in NMSC development or progression.
Figure 5.

11βHSD2 expression is significantly induced upon TPA treatment, and GA abrogates this increase in expression; (A) Representative Western blots from both short‐term and long‐term endpoints of our DMBA/TPA two stage carcinogenesis study and (B) Quantitative analysis of densitometry showing relative 11βHSD1 and 11βHSD2 expression normalized to β‐actin. P < 0.05 were considered to be statistically significant (*)
3. DISCUSSION
As GCs are known to play an essential role as regulators of cell proliferation and differentiation, modulating their activation in a tissue‐specific manner could be extremely advantageous in terms of inhibiting the induction of certain cancers. 11βHSDs are key regulators of GC activation/inactivation whose function happens at the pre‐receptor level, and therefore these enzymes could serve as potential therapeutic targets in for skin cancer. Recently, several studies have reported aberrant 11βHSD2 expression in cancer cells versus normal cell counterparts, suggested a role for 11βHSD2 in transformation, and the possibility that inhibition of this GC‐modulating enzyme could serve as a novel therapeutic target.13 For example, lung cancer studies have revealed that 11βHSD2 expression was increased in human lung cancers and experimental lung tumors.22 When 11βHSD2 was inhibited, lung tumor growth and invasion were suppressed and this correlated with increased active glucocorticoid levels in tissues.22 Other noteworthy findings occurred in colon cancer, wherein 11βHSD2 inhibition by both pharmacological inhibitor and gene silencing prevented adenoma formation, tumor growth, and metastasis in an animal model.17 Relevant findings have also been elucidated in breast cancer as well, where 11βHSD2 was observed in 66% of the breast tumor samples evaluated.12 Overexpression of 11βHSD2 in MCF‐7 cells reversed the antiproliferative effects of GCs in these cells and increased overall cell growth.23 These results suggest an increased expression of 11βHSD2 may have the ability to abrogate the antiproliferative action of GCs in certain tissues.
In these studies, we characterized 11βHSD2 in well‐established in vitro and in vivo models of NMSC, which had never been previously evaluated. We also sought to determine if 11βHSD2 plays a role in the multifactorial process of NMSC transformation. We hypothesized that 11βHSD2 contributes to malignant transformation in NMSC. We observed greater expression of 11βHSD2 in transformed RT101 cells when compared to their normal counterparts, which correlates with the “switch” observed in the literature in other types of cancer whereby normal cells will begin to preferentially express 11βHSD2 as they undergo transformation. When JB6+ epidermal keratinocytes underwent transformation by the addition of tumor promoter, TPA, we observed a significant increase in 11βHSD2 expression. Further, we tested a second well‐established in vitro mouse model of NMSC to determine if the upregulation of 11βHSD2 was prevalent in different types of mouse NMSC cells, and found 11βHSD2 was upregulated in both tumorigenic cell lines, MT1/2 and Ca3/7, but not in the non‐tumorigenic line, 3PC. These results suggest 11βHSD2 may play a role in transformation of NMSC, and may serve as a novel target for prevention. We then validated this elevated 11βHSD2 expression trend in a human in vitro skin cancer model and found that the same upregulation of 11βHSD2 evident in our mouse models was also present in human cells. After using mouse and human cell lines to show upregulation of 11βHSD2 in transformed cells as compared to their normal counterparts, we then wanted to investigate this critical enzyme's role in transformation.
To determine whether 11βHSD2 played a critical role in transformation of mouse epidermal keratinocytes, we used both pooled siRNA and individual siRNA to transiently knockdown 11βHSD2 in transformed RT101 cells. We used the anchorage independent colony formation assay to determine what effect an 11βHSD2 knockdown would have on transformed RT101's ability to grow in soft agar. Our results showed that both pooled siRNA and two of the individual siRNA were all capable of producing a knockdown which significantly reduced RT101 colony formation in soft agar over the course of 10 days. When we performed a transient 11βHSD2 knockdown in TPA‐treated JB6 P+ cells, we saw a significant decrease in colony formation when compared to the scramble TPA‐treated P+ cells. Our in vitro results suggest 11βHSD2 may serve as a novel and critical target for inhibition of NMSC development. These data showed for the first time in detail, increased 11βHSD2 expression in two well‐established mouse models of NMSC and also a well‐established human model of NMSC; these findings provide substantial support of a possible role for this GC‐regulating enzyme in transformation in a cancer where it had previously not been evaluated. As the anchorage independent colony formation assay is typically considered to be an in vitro indicator of tumorigenicity, these results suggest that when 11βHSD2 is inhibited in an animal model, we may be able to achieve minimized tumor size or diminished tumor formation. Therefore, we next sought to test this hypothesis in an in vivo mouse NMSC model.
We evaluated the effect of 11βHSD2 expression inhibition in a well‐established in vivo model of NMSC using phytonutrient, GA as an inhibitor of the enzyme. We chose GA because phytonutrients are naturally derived compounds isolated from plants, which can modulate disease progression and have been widely studied for their efficacious use in cancer and other diseases.24 Pentacyclic triterpenoids have been shown to be efficacious in inhibition of skin tumorigenesis, however, the role of 11βHSD2 as a target in this chemopreventive process has not been evaluated. In our in vivo studies, we observed a significant decrease in tumor incidence, a significant decrease in squamous cell carcinoma incidence, and an increase in tumor latency in the DMBA/TPA/GA experimental groups as compared to the DMBA/TPA group. Histopathological evaluation of tissues showed that topical administration of GA did not have any adverse effects on the epidermis or mouse weight over 26 weeks. Histological analysis of the DMBA/TPA group also show a characteristically thickened epidermis, that is, absent in GA control groups and diminished in DMBA/TPA/GA experimental groups. To investigate whether inhibition of 11βHSD2 played a role in the decrease in tumors exerted by GA treatment, we evaluated 11βHSD2 protein expression in tissues from both phase 1 (short‐term) and phase 2 (long‐term) animals. Our results show a significant induction of 11βHSD2 protein expression in in TPA‐treated tissues when compared to vehicle‐treated tissues from both short‐ and long‐term phases. An overall trend amongst the 11βHSDs in both short‐ and long‐term phases was observed, wherein 11βHSD1 was higher in vehicle‐treated tissue than in TPA‐treated tissue, and conversely 11βHSD2 was higher in TPA‐treated tissues and lower in vehicle‐treated tissues. These results correlate with the “switch” reported in the literature in other models which we mentioned earlier in the introduction. These findings further validate our earlier in vitro studies, where we observed a significant induction of 11βHSD2 protein expression upon TPA treatment, and saw the same inverse expression trend between 11βHSD1 and 11βHSD2. Finally, GA abrogated TPA‐induced 11βHSD2 protein expression in both short‐ and long‐term phases. Because GA‐mediated 11βHSD2 inhibition resulted in significantly reduced tumor incidence as well as a delay of tumor onset in our in vivo work, this may elucidate a possible therapeutic approach for NMSC by modulating the 11βHSD2 enzyme in a tissue‐specific manner. If GA is used to locally inhibit 11βHSD2 rather than systemic inhibition, adverse effects caused by 11βHSD2 inhibition should not be expected.
Given the rise of NMSC incidence, high rate of lesion recurrence, and serious tissue damage that can occur from lesion development, elucidating novel chemopreventive approaches to NMSC and a greater understanding of the mechanisms by which they occur are of critical importance.2, 25, 26, 27 The goal of this study, was to investigate whether 11βHSDs, pre‐receptor regulators of GC activation, played a role in NMSC development and/or progression; this was achieved by using pharmacological 11βHSD2 inhibitor, GA, in conjunction with tumor promoter, TPA, to determine if 11βHSD2 inhibition could significantly abrogate tumorigenesis. Major findings of this study are that 11βHSD2 expression is upregulated in TPA‐treated in vitro and in vivo models of NMSC, and both genetic knockdown and pharmacological inhibition of 11βHSD2 result in significantly reduced soft agar colony formation and tumorigenesis in cell and animal models respectively. Results of this study are significant because the specific role of 11βHSDs in NMSC have not been well‐studied, and many others have suggested 11βHSD2 may be a pro‐proliferative force because of its ability to inactivate glucocorticoids in a tissue‐specific manner.12, 13, 14, 23 The role of 11βHSD2 in tumorigenesis has already been elucidated in colorectal cancer, and its inhibition has suppressed colon carcinogenesis in both mouse and human models.17, 28 As GCs are well‐known inhibitors of cell proliferation and inducers of cell differentiation via GRs, pre‐receptor regulation of the availability of active GCs to the receptor is vital for tightly maintaining the proper function of GCs in specific situations. It is highly disadvantageous for the tumor environment to have the capability to increase 11βHSD2 expression in a tissue‐specific manner to achieve a decrease in the functionality of GCs; such an environment would result in GC inactivation, reduced anti‐inflammatory signals, and a setting that favors tumorigenesis. Further investigation into understanding the mechanistic players involved in GA‐mediated 11βHSD2 inhibition is certainly warranted to provide a more thorough understanding of the impact of these findings. In conclusion, this work has characterized the 11βHSD enzymes in well‐established human and mouse in vitro and mouse in vivo models of skin cancer, shown a significant role for 11βHSD2 in transformation of NMSC in mouse in vitro models, and elucidated GA‐mediated 11βHSD2 inhibition to be a potential chemopreventive target for prevention tumorigenesis in a well‐studied mouse model of NMSC.
CONFLICTS OF INTEREST
The authors declare that they have no conflict of interest.
ACKNOWLEDGMENTS
Histological evaluations of samples from in vivo studies were performed by Dr. Martha Hanes of the UT Health Science Center San Antonio. Financial Support was provided by the National Institutes of Health (R01 CA164159) Diversity Supplement and a Cancer Prevention and Research Institute of Texas training grant (RP140105).
Mancha‐Ramirez AM, Yang X, Liang H, et al. Harnessing the gatekeepers of glucocorticoids for chemoprevention of non‐melanoma skin cancer. Molecular Carcinogenesis. 2019;58:102–112. 10.1002/mc.22912
REFERENCES
- 1. Farzan SF, Karagas MR, Christensen BC, Li Z, Kuriger JK, Nelson HH. RNASEL and MIR146A SNP‐SNP interaction as a susceptibility factor for non‐melanoma skin cancer. PLoS ONE. 2014;9:e93602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Eisemann N, Waldmann A, Geller AC, et al. Non‐melanoma skin cancer incidence and impact of skin cancer screening on incidence. J Invest Dermatol. 2014;134:43–50. [DOI] [PubMed] [Google Scholar]
- 3. Schulze HJ, Cribier B, Requena L, et al. Imiquimod 5% cream for the treatment of superficial basal cell carcinoma: results from a randomized vehicle‐controlled phase III study in Europe. Br J Dermatol. 2005;152:939–947. [DOI] [PubMed] [Google Scholar]
- 4. Samarasinghe V, Madan V. Nonmelanoma skin cancer. J Cutan Aesthet Surg. 2012;5:3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Guy GP, Machlin SR, Ekwueme DU, et al. Prevalence and costs of skin cancer treatment in the U.S., 2002‐2006 and 2007‐2011. Am J Prev Med. 2015;48:183–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rabbitt EH, Lavery GG, Walker EA, Cooper MS, Steward PM, Hewison M. Prereceptor regulation of glucocorticoid action by 11beta‐hydroxysteroid dehydrogenase: a novel determinant of cell proliferation. FASEB J. 2002;16:36–44. [DOI] [PubMed] [Google Scholar]
- 7. Seckl JR, Walker BR. Minireview: 11beta‐hydroxysteroid dehydrogenase type 1‐ a tissue‐specific amplifier of glucocorticoid action. Endocrinology. 2001;142:1371–1376. [DOI] [PubMed] [Google Scholar]
- 8. Stewart PM, Prescott SM. Can licorice lick colon cancer? J Clin Invest. 2009;119:760–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Seckl JR, Walker BR. 11beta‐hydroxysteroid dehydrogenase type 1 as a modulator of glucocorticoid action: from metabolism to memory. Trends Endocrinol Metab. 2004;15:418–424. [DOI] [PubMed] [Google Scholar]
- 10. Chapman KE, Coutinho AE, Zhang Z, Kipari T, Savill JS, Seckl JR. Changing glucocorticoid action: 11β‐Hydroxysteroid dehydrogenase type 1 in acute and chronic inflammation. J Steroid Biochem Mol Biol. 2013;137:82–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Coulter CL, Smith RE, Stowasser M, Sasano H, Krozowski ZS, Gordon RD. Expression of 11beta‐hydroxysteroid dehydrogenase type 2 (11betaHSD‐2) in the developing human adrenal gland and human adrenal cortical carcinoma and adenoma. Mol Cell Endocrinol. 1999;154:71–77. [DOI] [PubMed] [Google Scholar]
- 12. Koyama K, Myles K, Smith R, Krozowski Z. Expression of the 11beta‐hydroxysteroid dehydrogenase type II enzyme in breast tumors and modulation of activity and cell growth in PMC42 cells. J Steroid Biochem Mol Biol. 2001;76:153–159. [DOI] [PubMed] [Google Scholar]
- 13. Rabbitt EH, Gittoes NJ, Steward PM, Hewison M. 11beta‐hydroxysteroid dehydrogenases, cell proliferation and malignancy. J Steroid Biochem Mol Biol. 2003;85:415–421. [DOI] [PubMed] [Google Scholar]
- 14. Hundertmark S, Buhler H, Rudolf M, Weitzel HK, Ragosh V. Inhibition of 11 beta‐hydroxysteroid dehydrogenase activity enhances the antiproliferative effect of glucocorticosteroids on MCF‐7 and ZR‐75‐1 breast cancer cells. J Endocrinol. 1997;155:171–180. [DOI] [PubMed] [Google Scholar]
- 15. Manning JR, Bailey MA, Soares DC, Dunbar DR, Mullins JJ. In silico structure‐function analysis of pathological variation in the HSD11B2 gene sequence. Physiol Genomics. 2010;42:319–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Manolis T, Lee YC, Temkin S, Hellman M, Nacharaju VL, Abulafia O. NAD dependent 11beta‐hydroxysteroid dehydrogenase activity in human endometrium and endometrial tumors. Gynecol Obstet Invest. 2006;62:103–107. [DOI] [PubMed] [Google Scholar]
- 17. Zhang MZ, Xu J, Yao B, et al. Inhibition of 11beta‐hydroxysteroid dehydrogenase type II selectively blocks the tumor COX‐2 pathway and suppresses colon carcinogenesis in mice and humans. J Clin Invest. 2009;119:876–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Abel E, Angel JM, Kiguchi K, DiGiovanni J. Multi‐stage chemical carcinogenesis in mouse skin: Fundamentals and applications. Nat Protoc. 2009;4:1350–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Colburn NH, Vorder Bruegge WF, Bates JR, et al. Correlation of anchorage‐independent growth with tumorigenicity of chemically transformed mouse epidermal cells. Cancer Res. 1978;38:624–634. [PubMed] [Google Scholar]
- 20. Colburn NH, Wendel EJ, Abruzzo G. Dissociation of mitogenesis and late‐stage promotion of tumor cell phenotype by phorbol esters: mitogen‐resistant variants are sensitive to promotion. Proc Natl Acad Sci USA. 1981;78:6912–6916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Srinivas L, Gindhart TD, Colburn NH. Tumor‐promoter‐resistant cells lack trisialoganglioside response. Proc Natl Acad Sci. 1982;79:4988–4991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chang J, Xue M, Yang S, et al. Inhibition of 11β‐Hydroxysteroid dehydrogenase type II suppresses lung carcinogenesis by blocking tumor COX‐2 expression as well as the ERK and mTOR signaling pathways. PLoS ONE. 2015;10:e0127030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lipka C, Mankertz J, Fromm M, et al. Impairment of the antiproliferative effect of glucocorticosteroids by 11beta‐hydroxysteroid dehydrogenase type 2 overexpression in MCF‐7 breast‐cancer cells. Horm Metab Res. 2004;36:437–444. [DOI] [PubMed] [Google Scholar]
- 24. Calhoun LN, Kwon YM. Structure, function and regulation of the DNA‐binding protein Dps and its role in acid and oxidative stress resistance in Escherichia coli: a review. J Appl Microbiol. 2011;110:375–386. [DOI] [PubMed] [Google Scholar]
- 25. Chen AC, Halliday GM, Damian DL. Non‐melanoma skin cancer: carcinogenesis and chemoprevention. Pathology. 2013;45:331–341. [DOI] [PubMed] [Google Scholar]
- 26. Gordon R. Skin cancer: an overview of epidemiology and risk factors. Semin Oncol Nurs. 2013;29:160–169. [DOI] [PubMed] [Google Scholar]
- 27. Boukamp P. Non‐melanoma skin cancer: what drives tumor development and progression? Carcinogenesis. 2005;26:1657–1667. [DOI] [PubMed] [Google Scholar]
- 28. Yang HS, Knies JL, Stark C, Colburn NH. Pdcd4 suppresses tumor phenotype in JB6 cells by inhibiting AP‐1 transactivation. Oncogene. 2003;22:3712–3720. [DOI] [PubMed] [Google Scholar]