

Abstract
The preparation of the first stable diylide‐substituted stannylene and germylene (Y2E, with E=Ge, Sn and Y=[PPh3‐C‐SO2Tol]−) is reported. The synthesis is easily accomplished in one step from the sulfonyl‐substituted metalated ylide YNa and the corresponding ECl2 precursors. Y2Ge and Y2Sn exhibit unusual structures in the solid state and in solution, in which the three adjacent lone pairs in the C‐E‐C linkage are arranged coplanar to each other. As shown by DFT studies, this bonding situation is preferred over the typical π‐donation from the ligands into the empty p‐orbital at the metal due to the strong anion‐stabilizing ability of the sulfonyl groups in the ylide backbone and their additional coordination to the metal. The alignment of the three lone pairs leads to a remarkable boost of the HOMO energy and thus of the donor strengths of the tetrylenes. Hence, Y2Ge and Y2Sn become stronger donors than their diamino or diaryl congeners and comparable to cyclic alkyl(amino)carbenes. First reactivity studies confirm the high reactivity of Y2Ge and Y2Sn, which for example undergo an intramolecular C−H activation reaction via metal–ligand cooperation.
Keywords: germylenes, main group elements, stannylenes, subvalent compounds, ylides
Compounds with Group 14 elements in low oxidation states have been the subject of intense research interest in the past years due to their applicability in element–hydrogen bond activations, which are important processes in many catalytic cycles normally only enabled by toxic transition metals.1 The ability of tetrylenes to undergo bond activations is largely determined by the singlet–triplet gap, which in turn can be manipulated by the choice of substituents. Classical substituents that allow the stabilization and isolation of these usually reactive compounds are amino or bulky aryl groups.2, 3 However, several other functional groups have been employed successfully in this chemistry and have led to a further tunability of the orbital setting at the central element and the propensity to undergo bond activation reactions.4 For example, N‐heterocyclic olefins5 and imines,6 boryl,7 and even metallo substituents8 have been reported for germylenes.9
More recently, the introduction of ylide substituents has also been recognized as a means for the stabilization and electronic manipulation of tetrylenes.10 Comparable to amino groups, ylide substituents can act as strong π‐donors. However, because carbon has lower electronegativity than nitrogen, ylide functionalization should result in more electron‐rich tetrylenes with enhanced nucleophilicity. Despite significant effort, the number of isolated ylide‐substituted carbenes and carbene analogues is extremely limited. No acyclic system has been reported to date and most synthetic efforts addressed mixed amino(ylide) tetrylenes to tame the expected high donor strength. Although the isolation of amino(ylide) carbenes (e.g. A) has been attempted, no stable system has been reported as yet (Figure 1).11, 12 In the case of the heavier analogues, Driess and co‐workers succeeded in the isolation of cyclic silylenes of type B, which are the only diylide‐substituted tetrylenes isolated so far.13 The cyclic amino(ylide)silylene C 14 and germylenes D 15 reported by Kato and Baceiredo are the only isolated and also structurally characterized ylide‐functionalized tetrylenes. These compounds exhibited strong donor properties, thus proving the strong donation from the ylide substituent.
Figure 1.
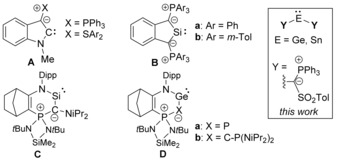
Ylide‐stabilized tetrylenes reported so far (Dipp=2,6‐iPr2C6H3).
Recently, we reported on the ready isolation of metalated ylides and their use in ylide functionalization,16 which, for example, was used for the synthesis of stable boron cations and highly electron‐rich phosphines.17 We envisioned that metalated ylides should also be ideal reagents for the stabilization of tetrylenes via simple salt metathesis reactions. Thus, also the synthesis of the first acyclic ylide‐substituted system and hence heavier tetrylenes with high nucleophilicity and stronger donor properties should be accessible. To test this hypothesis, we set out to isolate the diylidestannylene and germylene Y2Sn and Y2Ge based on the metalated ylide YNa (Scheme 1).16a Indeed, treatment of 2 equiv of YNa with GeCl2⋅dioxane or SnCl2 selectively provided the corresponding tetrylenes Y2Sn and Y2Ge, which were isolated as pale‐yellow solids in 68 and 63 % yield, respectively. Y2Sn and Y2Ge are characterized by singlets in the 31P{1H} NMR spectrum at δ P=6.88 ppm and 7.98 ppm, respectively, and a doublet for the ylide carbon atom at approx. δ C=52 ppm in the 13C{1H} NMR spectrum. In the case of the stannylene, the 117Sn NMR signal appears at δ Sn=−122.2 ppm. This highfield‐shifted signal indicates additional coordination of the sulfonyl groups in solution.18
Scheme 1.
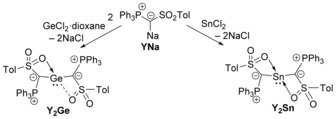
Preparation of the diylide‐substituted tetrylenes, Y2Sn and Y2Ge.
Single crystals of Y2Ge and Y2Sn were prepared to unambiguously confirm the nature of the compounds (see the Supporting Information). The structures prove the coordination of the two ylide groups as well as the additional coordination of the sulfonyl moieties to the tetrel (Figure 2 a).19 While both sulfonyl groups symmetrically bind to the tin center in Y2Sn, only one of the sulfonyl groups strongly interacts with the germanium center in Y2Ge (d Ge‐O=2.299(1) and 3.269(4) Å), probably due to the increased ring strain. The most interesting feature, however, concerns the arrangement of the ylide groups relative to the central C‐E‐C moiety. In contrast to typical π‐donor substituents, the ylide groups (P‐C‐S plane) in both tetrylenes arrange perpendicularly to the C‐E‐C linkage (Figure 2 b, conformer 1). This suggests that no π‐donation from the ylide substituents into the empty p‐orbital at Ge/Sn is possible and that the lone pairs remain localized at the ylide carbon atoms. This results in an unusual bonding situation, in which three lone pairs of electrons are in plane and located next to each other (canonical structure a, Figure 3 a). This is in clear contrast to typical diaminotetrylenes, in which the nitrogen atoms donate electron density into the empty p‐orbital at the central atom (conformer 2). To the best of our knowledge, such an electronic situation has never been observed for any tetrylene. Due to the lack of π‐donation, the Sn−C and Ge−C bond lengths (2.23 and 2.04 Å, respectively) are in the range of single bonds20 and the P–C and C–S distances in the ylide groups are comparable to those in the free ylide YH (P–C: 1.646(2) Å and S–C: 1.626(2) Å).16a The C‐E‐C angles of 103.05(7) (for Sn) and 105.94(7)° (for Ge) are comparable to those of other germylenes and stannylenes.5, 6, 7, 8, 9, 21
Figure 2.
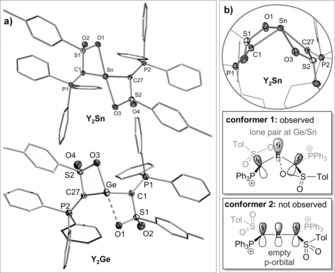
a) Molecular structures of Y2Sn and Y2Ge (ellipsoids at 50 % probability level, hydrogens and solvent molecules omitted for clarity). b) View on the C‐E‐C plane. Selected bond lengths [Å] and angles [°]: Y2Sn: Sn–C1 2.224(2), Sn–C27 2.238(2), Sn–O1 2.428(1), Sn–O3 2.479(1), S2–C1 1.663(2), P2–C27 1.701(2), P1–C1 1.692(2), S2–C27 1.658(2); C1‐Sn‐C27 103.05(7), S1‐C1‐P1 120.8(1), S2‐C27‐P2 121.2(1); Y2Ge: Ge–C1 2.035(2), Ge–C27 2.049(2), Ge–O3 2.299(1), S1–C1 1.662(2), S2–C27 1.662(2), P1–C1 1.688(2), P2–C27 1.687(2); C1‐Ge‐C27 105.94(7), S1‐C1‐P1 123.3(1), S2‐C27‐P2 123.5(1).
Figure 3.
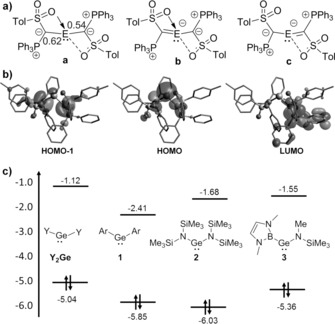
a) Possible canonical structures of Y2E and calculated Wiberg bond indices for Y2Ge, b) Kohn–Sham orbitals of Y2Ge (isosurface value=0.030 e Å−3) and c) comparison of the HOMO–LUMO energies in eV of different germylenes (Ar=2,6‐dimethylphenyl).
The unsymmetrical coordination of both ylide substituents in Y2Ge is inconsistent with the single signal observed in the 31P{1H} NMR spectrum at room temperature, thus indicating a fluxional behavior in solution. VT‐NMR studies in [D8]toluene revealed a broadening (no splitting) of the signal upon cooling, which is in line with a fast exchange process (see the Supporting Information). For Y2Sn, which exhibits a sharp singlet in its 31P{1H} NMR spectrum, no broadening was observed. This agrees well with a symmetrical coordination by the two ylide substituents also in solution (cf. 119Sn NMR shift). Thus, the NMR studies indicate that the structures are also retained in solution.
To gain further insight into the electronic properties of the tetrylenes, DFT calculations were performed at the PBE0‐D3/def2‐tzvp level of theory. The structural parameters closely matched those found in the crystal structures of Y2Ge and Y2Sn (e.g. different Ge–O distances in Y2Ge). The Wiberg bond indices (Figure 3 a) of the Ge−C bonds amount to only 0.54 and 0.62 (0.44 for Y2Sn) and are thus lower than those calculated for Ar2Ge 1 (Ar=2,6‐dimethylphenyl; WBI=0.73). Natural bond orbital (NBO) analysis also only shows a single bond between Ge and C and two lone pairs residing at the ylidic carbon atoms. Thus, in line with the experimental data, the calculations show that the canonical structure a (Figure 3 a) reflects best the electronic situation in Y2E. This is in contrast to silylene C, in which strong π‐interaction from the ylide to the silicon center was observed.14 This difference probably arises from the different substituents in the ylide backbone. In contrast to the amino substituent in C, the sulfonyl group considerably stabilizes the negative charge at the ylidic carbon atoms in Y2E and thus favors electrostatic and negative hyperconjugation effects over π‐interactions in the Ge–C linkage. This demonstrates the dramatic impact of the substituents in the ylide backbone on the electronics and thus on the reactivity of the tetrylene.
To further study the electronic situation, the hypothetical conformer 2 was calculated, in which the ylide substituents are forced into a coplanar arrangement with the C‐Ge‐C linkage to allow for π‐delocalization with the p‐orbital at germanium. Accordingly, the WBIs of the Ge−C bond increase to approximately 0.8, which is comparable to the Ge−N bond in the amino/boryl germylene 3 reported by Aldridge,4b but still is considerably smaller than that found in the phosphanylide‐stabilized germylene Da,15b thus further proving the tunability of the donor properties by the ylide substituents. It is also important to note that conformer 2 is clearly energetically disfavored over conformer 1 (ΔG=35 kJ mol−1), thus confirming the experimental observations.
We hypothesized that the unique electronic structure of Y2E decisively impacts the orbital energies and thus the donor properties of the tetrylenes. The calculated HOMO of Y2Ge and Y2Sn mainly resides on the metal center, while the LUMO is distributed over one of the ylide ligands (Figure 3 b).22 The HOMO‐1 is greatly localized at the ylidic carbon atoms, thus reflecting the two lone pairs at the C atoms. A comparison of the HOMO–LUMO energies with those of other acyclic germylenes showed that Y2Ge is indeed a stronger donor and weaker acceptor than diaryl, diamino, or even the amino/boryl germylene 3 (Figure 3 c). This is a consequence of the coplanar arrangement of the three lone pairs which raises the HOMO energy (by 0.17 eV relative to conformer 2). Thus, the HOMO energy of Y2Ge is boosted to the level of alkyl(amino)carbenes (see the Supporting Information). This is also confirmed by the pyramidalization of the GaCl3 moiety in the energy‐optimized Y2Ge⋅GaCl3 complex, which was shown to correlate with the donor strength of a given ligand.23, 24 The calculated sum of Cl‐Ga‐Cl angles in Y2Ge⋅GaCl3 amounts to 327°, which corresponds to a Tolman electronic parameter of 2032.3 cm−1. Thus, Y2Ge is a considerably stronger donor than germylenes 1–3 and comparable to cyclic alkyl(amino)carbenes. This makes Y2Ge the germylene with the highest donor capacity known so far. The same holds true for stannylene Y2Sn.
While the stannylene is stable in solution at room temperature for several days without showing evidence of decomposition reactions, the germylene decomposed slowly in THF in the course of one week to form a new product along with an equivalent amount of ylide YH. The same product is formed within 1 h when a toluene solution of Y2Ge is heated to 90 °C. XRD analysis revealed the new compound to be cyclotetragermane 4. Compound 4 is presumably formed by C−H activation of one of the PPh3 phenyl groups, thus generating the cyclometalated germylene 4‐Int, which eliminates the ylide ligand and tetramerizes to 4 (Scheme 2). DFT studies suggest that the C−H activation does not occur at the germanium center, but via addition across the Ge−C bond, which is in line with the lone pairs at the carbon atoms. This indicates that ylide functionalization does not only increase the donor strength of the germylene but may also result in reactivities via metal–ligand cooperation. Compound 4 could be isolated as a yellow solid in 33 % yield. The tetragermane is characterized by a signal at δ P=15.7 ppm in the 31P{1H} NMR spectrum and a doublet at δ C=37.5 ppm in the 13C{1H} NMR spectrum for the ylide carbon atom (1 J PC=81.3 Hz). In the solid state, 4 shows a fully planar Ge4 core, at which the ylide substituents bind in an alternating fashion to minimize steric repulsion between the PPh3 moieties. The Ge–Ge bonds (2.480(1)–2.493(1) Å) are in the range of other tetragermanes.25
Scheme 2.
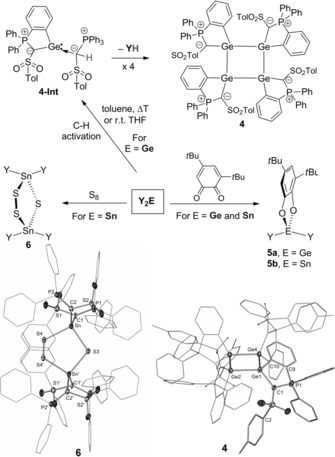
Reactivity studies of Y2Ge and Y2Sn and molecular structures of compound 4 and 6 (thermal ellipsoids at the 50 % probability level).
Despite the instability of Y2Ge, it can be applied in further transformations. Y2Ge readily reacts within a couple of minutes with 3,5‐di‐tert‐butyl‐o‐benzoquinone to form 5 a. The same [4+1] cycloaddition reaction was observed with Y2Sn to generate 5 b. Both compounds could be isolated as colorless solids and were fully characterized. While the germylene shows no selective reaction with elemental sulfur, Y2Sn selectively generates compound 6 with a nonplanar, C 2‐symmetric Sn2S3 five‐membered ring. To the best of our knowledge, formation of such a five‐membered Sn‐S cycle has never been observed with any other stannylene.26 In contrast to the structures of the tetrylenes, no coordination of the tin and germanium center, respectively, by the sulfonyl group is observed in the molecular structures of 4–5 b and only a weak interaction by one of the sulfonyl groups in 6. This shows that—although important for the stabilization of Y2Ge and Y2Sn—the sulfonyl coordination is easily opened to facilitate substrate coordination and/or attack.
In conclusion, we have isolated the first diylide‐stabilized germylene and stannylene synthesized via simple salt metathesis reactions using an α‐metalated ylide. These tetrylenes feature an unusual electronic structure with three lone pairs arranged in a coplanar fashion. This arrangement results in a boost of the HOMO and LUMO energy levels and in a remarkable increase of the donor strength, thus making Y2Ge a stronger donor than classical germylenes and comparable to cyclic alkyl(amino)carbenes. This electronic structure also leads to novel reactivity patterns, such as an intramolecular C−H activation by the Ge–C linkage. Thus, ylide substituents may be used as a tool to impart unique properties to low‐valent main group compounds which were so far not accessible with other substituents. The forced alignment of lone pairs through a sophisticated molecular design may also be used to enhance the donor strengths of other ligands.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This project has received funding from the European Research Council under the European Union's Horizon 2020 research and innovation program (No 677749) and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC‐2033 – project number 390677874.
C. Mohapatra, L. T. Scharf, T. Scherpf, B. Mallick, K.-S. Feichtner, C. Schwarz, V. H. Gessner, Angew. Chem. Int. Ed. 2019, 58, 7459.
References
- 1.
- 1a. Mizuhata Y., Sasamori T., Tokitoh N., Chem. Rev. 2009, 109, 3479; [DOI] [PubMed] [Google Scholar]
- 1b. Asay M., Jones C., Driess M., Chem. Rev. 2011, 111, 354; [DOI] [PubMed] [Google Scholar]
- 1c. Power P. P., Nature 2010, 463, 171; [DOI] [PubMed] [Google Scholar]
- 1d. Yao S., Xiong Y., Driess M., Organometallics 2011, 30, 1748. [Google Scholar]
- 2.
- 2a. Li L., Fukawa T., Matsuo T., Hashizume D., Fueno H., Tanka K., Tamao K., Nat. Chem. 2012, 4, 361; [DOI] [PubMed] [Google Scholar]
- 2b. Merrill W. A., Wright R. J., Stanciu C. S., Olmstead M. M., Fettinger J. C., Power P. P., Inorg. Chem. 2010, 49, 7097; [DOI] [PubMed] [Google Scholar]
- 2c. Hadlington T. J., Hermann M., Frenking G., Jones C., Chem. Sci. 2015, 6, 7249; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2d. Woodul W. D., Carter E., Mìller R., Richards A. F., Stasch A., Kaupp M., Murphy D. M., Driess M., Jones C., J. Am. Chem. Soc. 2011, 133, 10074; [DOI] [PubMed] [Google Scholar]
- 2e. Spikes G. H., Peng Y., Fettinger J. C., Steiner J., Power P. P., Chem. Commun. 2005, 6041; [DOI] [PubMed] [Google Scholar]
- 2f. Schneider J., Sindlinger C. P., Freitag S. M., Schubert H., Wesemann L., Angew. Chem. Int. Ed. 2017, 56, 333; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 339; [Google Scholar]
- 2g. Walz F., Moos E., Garnier D., Köppe R., Anson C. E., Breher F., Chem. Eur. J. 2017, 23, 1173; [DOI] [PubMed] [Google Scholar]
- 2h. Veith M., Grosser M., Z. Naturforsch. B 1982, 37, 1375; [Google Scholar]
- 2i. Oetzel J., Weyer N., Bruhn C., Leibold M., Gerke B., Pöttgen R., Maier M., Winter R. F., Holthausen M. C., Siemeling U., Chem. Eur. J. 2017, 23, 1187; [DOI] [PubMed] [Google Scholar]
- 2j. Guthardt R., Oetzel J., Schweizer J. I., Bruhn C., Langer R., Maurer M., Vícha J., Shestakova P., Holthausen M. C., Siemeling U., Angew. Chem. Int. Ed. 2019, 58, 1387; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1401. [Google Scholar]
- 3.
- 3a. Davidson P. J., Lappert M. F., J. Chem. Soc. Chem. Commun. 1973, 317; [Google Scholar]
- 3b. Harris D. H., Lappert M. F., J. Chem. Soc. Chem. Commun. 1974, 895. [Google Scholar]
- 4.
- 4a. Peng Y., Guo J.-D., Ellis B. D., Zhu Z., Fettinger J. C., Nagase S., Power P. P., J. Am. Chem. Soc. 2009, 131, 16272; [DOI] [PubMed] [Google Scholar]
- 4b. Usher M., Protchenko A. V., Rit A., Campos J., Kolychev E. L., Tirfoin R., Aldridge S., Chem. Eur. J. 2016, 22, 11685. [DOI] [PubMed] [Google Scholar]
- 5. Hering-Junghans C., Andreiuk P., Ferguson M. J., McDonald R., Rivard E., Angew. Chem. Int. Ed. 2017, 56, 6272; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 6368. [Google Scholar]
- 6.
- 6a. Inoue S., Leszczynska K., Angew. Chem. Int. Ed. 2012, 51, 8589; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 8717; [Google Scholar]
- 6b. Ochiai T., Szilvási T., Franz D., Irran E., Inoue S., Angew. Chem. Int. Ed. 2016, 55, 11619; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 11791. [Google Scholar]
- 7. Protchenko A. V., Birjkumar K. H., Dange D., Schwarz A. D., Vidovic D., Jones C., Kaltsoyannis N., Mountford P., Aldridge S., J. Am. Chem. Soc. 2012, 134, 6500. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Juckel M. M., Hicks J., Jiang D., Zhao L., Frenking G., Jones C., Chem. Commun. 2017, 53, 12692; [DOI] [PubMed] [Google Scholar]
- 8b. Inomata K., Watanabe T., Miyazaki Y., Tobita H., J. Am. Chem. Soc. 2015, 137, 11935. [DOI] [PubMed] [Google Scholar]
- 9.Further examples:
- 9a. Dong Z., Reinhold C. R. W., Schmidtmann M., Müller T., Angew. Chem. Int. Ed. 2016, 55, 15899; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 16131; [Google Scholar]
- 9b. Dong Z., Bedbur K., Schmidtmann M., Müller T., J. Am. Chem. Soc. 2018, 140, 3052; [DOI] [PubMed] [Google Scholar]
- 9c. Izod K., Rayner D. G., El-Hamruni S. M., Harrington R. W., Baisch U., Angew. Chem. Int. Ed. 2014, 53, 3636; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 3710; [Google Scholar]
- 9d. Suzuki Y., Sasamori T., Guo J.-D., Tokitoh N., Chem. Eur. J. 2018, 24, 364; [DOI] [PubMed] [Google Scholar]
- 9e. Jana A., Huch V., Scheschkewitz D., Angew. Chem. Int. Ed. 2013, 52, 12179; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 12401; [Google Scholar]
- 9f. Pelzer S., Neumann B., Stammler H.-G., Ignat'ev N., Hoge B., Angew. Chem. Int. Ed. 2016, 55, 6088; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 6192; [Google Scholar]
- 9g. Nieder D., Huch V., Yildiz C. B., Scheschkewitz D., J. Am. Chem. Soc. 2016, 138, 13996; [DOI] [PubMed] [Google Scholar]
- 9h. Yao S., Xiong Y., Szilvási T., Grützmacher H., Driess M., Angew. Chem. Int. Ed. 2016, 55, 4781; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 4859. [Google Scholar]
- 10.
- 10a. Borthakur B., Phukan A. K., Chem. Eur. J. 2015, 21, 11603; [DOI] [PubMed] [Google Scholar]
- 10b. Guha A. K., Gogoi U., Phukan A. K., Int. J. Quantum Chem. 2013, 113, 2471. [Google Scholar]
- 11.
- 11a. Fürstner A., Alcarazo M., Radkowski K., Lehmann C. W., Angew. Chem. Int. Ed. 2008, 47, 8302; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 8426; [Google Scholar]
- 11b. Nakafuji S., Kobayashi J., Kawashima T., Angew. Chem. Int. Ed. 2008, 47, 1141; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 1157. [Google Scholar]
- 12.Carbenes with “inverse” ylide substituents:
- 12a. Lavigne F., El Kazzi A., Escudie Y., Maerten E., Kato T., Saffon-Merceron N., Branchadell V., Cosso F. P., Baceiredo A., Chem. Eur. J. 2014, 20, 12528; [DOI] [PubMed] [Google Scholar]
- 12b. Asay M., Kato T., Saffon-Merceron N., Cossio F., Baceiredo A., Bertrand G., Angew. Chem. Int. Ed. 2008, 47, 7530; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 7640. [Google Scholar]
- 13. Asay M., Inoue S., Driess M., Angew. Chem. Int. Ed. 2011, 50, 9589; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 9763. [Google Scholar]
- 14. Alvarado-Beltran I., Baceiredo A., Saffon-Merceron N., Branchadell V., Kato T., Angew. Chem. Int. Ed. 2016, 55, 16141; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 16375. [Google Scholar]
- 15.
- 15a. Berthe J., Garcia J. M., Ocando E., Kato T., Saffon-Merceron N., De Cózar A., Cossío F. P., Baceiredo A., J. Am. Chem. Soc. 2011, 133, 15930; [DOI] [PubMed] [Google Scholar]
- 15b. Del Rio N., Baceiredo A., Saffon-Merceron N., Hashizume D., Lutters D., Müller T., Kato T., Angew. Chem. Int. Ed. 2016, 55, 4753; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 4831; [Google Scholar]
- 15c. Del Rio N., Lopez-Reyes M., Baceiredo A., Saffon-Merceron N., Hashizume D., Lutters D., Müller T., Kato T., Angew. Chem. Int. Ed. 2017, 56, 1365; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1385. [Google Scholar]
- 16.
- 16a. Scherpf T., Wirth R., Molitor S., Feichtner K.-S., Gessner V. H., Angew. Chem. Int. Ed. 2015, 54, 8542; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 8662; [Google Scholar]
- 16b. Scharf L. T., Gessner V. H., Inorg. Chem. 2017, 56, 8599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.
- 17a. Scherpf T., Schwarz C., Scharf L. T., Zur J.-A., Helbig A., Gessner V. H., Angew. Chem. Int. Ed. 2018, 57, 12859; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 13041; [Google Scholar]
- 17b. Weber P., Scherpf T., Rodstein I., Lichte D., Scharf L. T., Gooßen L. J., Gessner V. H., Angew. Chem. Int. Ed. 2019, 58, 3203–3207; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 3235–3239. [Google Scholar]
- 18. Mansell S. M., Russell C. A., Wass D. F., Inorg. Chem. 2008, 47, 11367. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. El Ezzi M., Lenk R., Madec D., Sotiropoulos J.-M., Mallet-Ladeira S., Castel A., Angew. Chem. Int. Ed. 2015, 54, 805; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 819; [Google Scholar]
- 19b. Mehring M., Löw C., Schürmann M., Uhlig F., Jurkschat K., Mahieu B., Organometallics 2000, 19, 4613. [Google Scholar]
- 20.
- 20a. Escudié J., Couret C., Ranaivonjatovo H., Satge J., Coord. Chem. Rev. 1994, 130, 427; [Google Scholar]
- 20b. Meyer H., Baum G., Massa W., Berndt A., Angew. Chem. Int. Ed. Engl. 1987, 26, 798; [Google Scholar]; Angew. Chem. 1987, 99, 790; [Google Scholar]
- 20c. Lazraq M., Escudié J., Couret C., Satgé J., Dräger M., Dammel R., Angew. Chem. Int. Ed. Engl. 1988, 27, 828; [Google Scholar]; Angew. Chem. 1988, 100, 885; [Google Scholar]
- 20d. Tokitoh N., Kishikawa K., Okazaki R., J. Chem. Soc. Chem. Commun. 1995, 1425; [Google Scholar]
- 20e. Ghereg D., Gornitzka H., Ranaivonjatovo H., Escudié J., Dalton Trans. 2010, 39, 2016. [DOI] [PubMed] [Google Scholar]
- 21. Grützmacher H., Pritzkow H., Edelman F. T., Organometallics 1991, 10, 23. [Google Scholar]
- 22.Note that the orbital reflecting the empty p-orbital at Ge is higher in energy (E=0.73 eV) due to the weak donor ability of the sulfonyl group.
- 23. El-Hellani A., Monot J., Tang S., Guillot R., Bour C., Gandon V., Inorg. Chem. 2013, 52, 11493. [DOI] [PubMed] [Google Scholar]
- 24.Attempts to experimentally determine the TEP failed. Reactions with Ni(CO)4, Rh(acac)CO2, or GaCl3 led to no selective formation of a single species due side reactions such as chloride elimination or further CO loss, which is in line with the donor properties of the compound.
- 25.
- 25a. Rupar P. A., Jennings M. C., Baines K. M., Organometallics 2008, 27, 5043; [Google Scholar]
- 25b. Zirngast M., Flock M., Baumgartner J., Marschner C., J. Am. Chem. Soc. 2009, 131, 15952; [DOI] [PubMed] [Google Scholar]
- 25c. Richards A. F., Brynda M., Olmstead M. M., Power P. P., Organometallics 2004, 23, 2841. [Google Scholar]
- 26.
- 26a. Hitchcock P. B., Jang E., Lappert M. F., J. Chem. Soc. Dalton Trans. 1995, 3179–3187; [Google Scholar]
- 26b. Thompson J. R., Ahmet I. Y., Johnson A. L., Kociok-Köhn G., Eur. J. Inorg. Chem. 2016, 4711; [Google Scholar]
- 26c. Foley S. R., Yap G. P. A., Richeson D. S., Organometallics 1999, 18, 4700. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary