

Abstract
Multiple sclerosis (MS) is an inflammatory, demyelinating, and neurodegenerative disease of the central nervous system. The complement system has an established role in the pathogenesis of MS, and evidence suggests that its components can be used as biomarkers of disease state activity and response to treatment in MS. Plasma C4a levels have been found to be significantly elevated in patients with active relapsing-remitting MS (RRMS), as compared to both controls and patients with stable RRMS. C3 levels are also significantly elevated in the cerebrospinal fluid (CSF) of patients with RRMS, and C3 levels are correlated with clinical disability. Furthermore, increased levels of factor H can predict the transition from relapsing to progressive disease, since Factor H levels have been found to increase progressively with disease progression over a 2-year period in patients transitioning from RRMS to secondary progressive (SP) MS. In addition, elevations in C3 are seen in primary progressive (PP) MS. Complement components can also differentiate RRMS from neuromyelitis optica. Response gene to complement (RGC)-32, a novel molecule induced by complement activation, is a possible biomarker of relapse and response to glatiramer acetate (GA) therapy, since RGC-32 mRNA expression is significantly decreased during relapse and increased in responders to GA treatment. The predictive accuracy of RGC-32 as a potential biomarker (by ROC analysis) is 90% for detecting relapses and 85% for detecting a response to GA treatment. Thus, complement components can serve as biomarkers of disease activity to differentiate MS subtypes and to measure response to therapy.
Keywords: Complement activation, RGC-32, Multiple Sclerosis, Glatiramer Acetate, Biomarker, Factor H
Introduction
Multiple sclerosis (MS) is an inflammatory, demyelinating, and neurodegenerative disease of the central nervous system (CNS) that particularly affects young adults. It is predominantly a T-cell-mediated disease, but innate immune system components also play an important role in its pathogenesis. There is growing pathological evidence for a significant contribution of the complement system and its activation products to neuro-inflammation in MS [1–4]. In this review, we will discuss the role of the complement system and its activation products as biomarkers of the disease state and activity as well as the response to therapy in MS.
Complement activation in MS lesions
The complement system has an established role in the pathogenesis of MS, as evidenced by deposition of complement components and activation products in the white matter plaques in brain tissue [5]. In the MS white matter lesions from MS patients, deposition of C1q, C3d, and C5b-9 are detected on and within macrophages/microglia and astrocytes and in blood vessel walls [1, 2, 6, 7]. High levels of immunoglobulins and C5b-9 deposition at sites of active myelin destruction are characteristic for pattern II MS lesions and C5–9 deposits are present only in this type of demyelinating lesion [2, 8]. In chronic MS, plaques are found to be consistently positive for complement proteins (C3, factor B, C1q), activation products (C3b, iC3b, C4d, terminal complement complex) and inhibitors (factor H, C1-inhibitor, clusterin), suggesting that there is continuing local complement synthesis, activation, and regulation despite the absence of other evidence of ongoing inflammation [9]. An initial heterogeneity of demyelinating lesions in the earliest phase of MS lesion formation [2] may disappear over time as different pathways converge in one general mechanism of demyelination. The consistent presence of complement, antibodies, and Fc receptors in phagocytic macrophages suggests that antibody- and complement-mediated myelin phagocytosis is the dominant mechanism of demyelination in established MS [3] (Figure 1).
Fig. 1. Complement system as a biomarker of disease activity and phenotype.
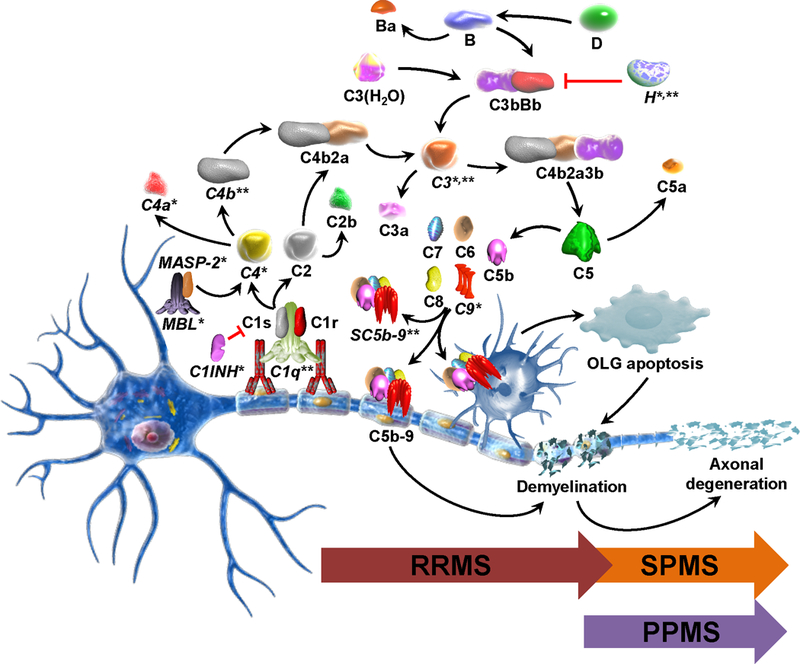
Various complement components have the potential of being biomarkers either of disease activity or of an MS phenotype. The most important complement components are marked with (*) when they are possible biomarkers in plasma and with (**) when they represent biomarkers in CSF. Complement system activation and the C5b-9 terminal complex, when present at lytic doses, lead to demyelination. In contrast, sublytic C5b-9 can protect OLG from apoptotic cell death, indicating a dual role for complement in demyelination. Human myelin and OLG are vulnerable to complement attack because they lack some of complement surface inhibitors. Classical pathway is usually activated when the C1 complex binds myelin-specific autoantibodies through its C1q component. The C1s-induced cleavage of C2 and C4 results in the generation of the classical pathway C3 convertase C4b2a. Lectin pathway is activated when MBL binds to mannose residues on the surface of a pathogen and activate MASP-2 which leads to the formation of C3 convertase. C3 convertase cleaves the C3 component and the resulting C3b participates in the formation of the C5 convertase, C4b2a3b. In the alternative pathway, factor B bound to the C3b generated through spontaneous hydrolysis of C3 (C3(H2O)) is cleaved by factor D and the resulting Bb fragment forms with C3b the alternative pathway C3 convertase, C3bBb, which amplifies the cleavage of C3. C5 convertase acts on C5 which is cleaved into C5a and C5b. C5b initiates the terminal pathway which leads to the formation of C5b-9 terminal complex
Complement staining is most apparent in plaques and peri-plaques but is also present in normal-appearing white matter and cortical areas to a greater extent than in control tissue. C1q staining is present in all plaques, suggesting a dominant role for the classical pathway. Cellular staining for complement components is largely restricted to reactive astrocytes, often adjacent to clusters of microglia that are in close apposition to complement-opsonized myelin and damaged axons [9]. We have previously shown that astrocytes in culture can secrete most of the complement proteins and expression is markedly enhanced by TNFα, IL-1β and IL-8 [10]. In addition C5b-9 deposits were found in macrophages [5], neurons [11] and OLG progenitor cells [12].
A recent study has provided evidence of complement dysregulation in MS gray matter lesions, including an association of the numerical density of C1q cells with tissue lesions. This work confirms that complement activation and dysregulation occur in all cases of MS [11]. In addition, the authors of recent study reported that C3d microglial clusters are present in chronic but not acute MS lesions. These clusters are not associated with antibody deposits or terminal complement activation. Instead, they are linked to slowly expanding lesions, localized on axons with impaired transport and associated with neuronal C3 production. C3d microglial clusters are not specific to MS, since they are also found in stroke [13]. The authors of this recent study also concluded that C3d microglial clusters in MS are not part of an acute attack against myelinated axons. Therefore, it is unlikely that they drive the formation of new lesions, but they could represent a physiological mechanism to remove irreversibly damaged axons in chronic disease [13].
Role of complement activation in demyelination and protection of oligodendrocytes from apoptosis
Complement system activation and the C5b-9 terminal complex, when present at lytic doses, lead to demyelination. In contrast, sublytic C5b-9 can protect oligodendrocytes (OLG) from apoptotic cell death, indicating a dual role for complement in demyelination [14]. The detrimental effect of the terminal complex has been shown in vitro by using myelinated CNS explant cultures treated with IgG or IgM anti-myelin antibodies and fresh serum, which induced extensive demyelination [15]. Exposure to antibody and C8-depleted serum (C8D) failed to induce demyelination, but the addition of C8 to complete the C5b-9 assembly induced extensive demyelination and eventual myelin loss [15]. Anti-myelin antibodies, including anti-galactocerebroside, are required for complement to produce significant demyelination in explants, but isolated myelin and OLG can also activate C1 and generate C5b-9 [5]. Recently, myelin-specific MS antibodies were shown to cause complement-dependent OLG loss and demyelination [16]. Human myelin and OLG are vulnerable to complement attack because they lack complement inhibitors. Human myelin lacks CD59 and CD55, and OLG lack complement receptor 1, CD46, and clusterin, whose absence prevents them from inhibiting the activation of complement [14].
Sublytic C5b-9 is able to rescue OLG from serum deprivation-induced apoptotic cell death by inhibiting cytochrome c release, and thus the activation of caspase 9 and caspase 3. Sublytic C5b-9 stimulates the phosphorylation of the pro-apoptotic factor BAD at Ser112 and Ser136 and causes dissociation of the BAD/BCL-XL complex [17]. BAD is sequestered in the cytoplasm by association with the protein 14–3-3, and the anti-apoptotic factor BCL-XL inhibits the oligomerization of the pro-apoptotic factors BAX and BAK at the outer mitochondrial membrane, which would create a channel for cytochrome c release into the cytosol and subsequent caspase activation [17]. Sublytic C5b-9 also inhibits FasL and TNF-α-induced cell death through inhibition of the cleavage of pro-apoptotic factor BID by caspase 8 [18]. Such inhibition might be achieved by increasing the expression of the caspase 8 inhibitor c-FLIP. All these effects of C5b-9 are mediated through activation of the phosphatidylinositol 3-kinase (PI3K)-Akt signaling pathway, indicating that regulation of caspase-8 and prevention of the mitochondrial insertion of the proapoptotic proteins BAD and BID are able to rescue OLG from apoptosis [18].
Given the significant involvement of complement activation in the pathogenesis of MS, numerous studies have tried to demonstrate that various complement components are associated with disease activity and phenotype, as a means of helping to stratify patients and predict outcomes that could influence therapeutic decisions.
Complement components as biomarkers in MS
Classical and lectin pathways
Studies have found no significant differences between the mean plasma and CSF levels of C1s protein in MS patients and those of control individuals [19]. However, one study has found a significant increase in the CSF levels of C1q in RRMS and SPMS patients when compared to controls [20]. In addition, plasma C1 inhibitor (C1INH) levels are significantly higher in patients with MS than in controls [19]. Moreover, mannose binding lectin (MBL) and MBL-associated serine protease −2 (MASP-2) were also significantly increased in plasma of MS patients when compared with non-MS patients [21].
Increased levels of plasma C3 and C4 components have been detected in MS patients when compared to controls, but the increase only reached statistical significance in the case of the progressive forms [19] (Table 1). C3 levels are also elevated in CSF and have been found to correlate with clinical disability as measured by the Expanded Disability Status Scale (EDSS), the number of brain MRI lesions (≥9 cerebral lesions), and the levels of neurofilament light chain (NFL), a marker for neuronal injury. The highest levels were seen in progressive MS patients, especially those with primary progressive MS (PPMS) (Table 2); in contrast, the NFL CSF levels were higher in RRMS patients. There was no difference in the C3 CSF levels between relapsing patients and patients in remission [22].
Table 1.
Plasma levels of complement components in the MS and NMOSD
| Complement components |
MS | NMOSD | References | ||
|---|---|---|---|---|---|
| RRMS | PPMS | SPMS | |||
|
Classical and lectin pathways |
C4a↑ in relapse C3↓ in stable MASP-2↑ |
C3↑ | C4↑ C3↑ MASP-2↑ |
C1s ↑ C3a ↑ C4a ↓ |
[19, 21, 24, 25, 30, 32] |
|
Terminal
pathway |
C9↓ | C9↓ | C9↓ | C5↑ SC5b-9↑ |
[19, 30, 32] |
| Inhibitors | C1INH↑ Factor H ↑ in relapse |
C1INH↑ Factor H ↑ |
C1INH↑ Factor H ↑ |
C1INH↑ Factor H ↑ |
[19, 28, 30] |
Table 2.
CSF levels of complement components in the MS and NMOSD
| Complement components |
MS | NMOSD | References | ||
|---|---|---|---|---|---|
| RRMS | PPMS | SPMS | |||
|
Classical and lectin pathways |
C1q ↑ C3 ↑ in relapse ↓ in stable C4a ↑ C4b ↑ in relapse |
C3↑ | C1q↑ C3↑ |
C3a – no difference MBL/MASP2 – no difference |
[20, 22–25, 31] |
|
Terminal pathway |
SC5b-9 ↑ | N/A | N/A | C5a ↑ in relapse SC5b-9 ↑ in relapse |
[26, 31] |
| Inhibitors | sCR2 ↑ Factor H ↓ in relapse |
Factor H↑ | sCR2↑ Factor H↑ |
N/A | [20, 23, 29] |
Another study has also found an increased expression of C3, as well as C4b (an active peptide generated by the cleavage of C4) in the CSF of patients with acute RRMS when compared to healthy controls [23]. Moreover, plasma C4a (a fragment resulting from cleavage of C4) levels were significantly elevated in patients with active RRMS when compared to both controls and patients with stable RRMS [19]. Subsequent monitoring of patients with acute RRMS in convalescence showed lower C4a levels, with the reduction being more obvious at 2–3 months post-relapse, but this trend was not maintained at 5–7 months, nor did the differences reach statistical significance. C4a levels were also weakly correlated with EDSS [24]. The CSF concentration of C4a was significantly higher in MS patients than in the control population, and it showed a moderate correlation with CSF IgG but not with CSF albumin, indicating that elevated levels of C4a most likely occur as a result of intrathecal production, as opposed to leakage from the plasma through the altered brain blood barrier (BBB), although C4a is a relatively small protein (76 amino acids) that is able to cross the intact BCB [24]. On the other hand, a study by Jongen et al. [25] has found lower C3 values in the CSF in stable RRMS patients than in controls, as well as lower plasma C3 values in stable RRMS and SPMS patients, but increased C3 index values (calculated as the CSF/serum C3 ratio multiplied by serum/CSF albumin ratio) in both stable RRMS and SPMS. This group concluded that the mean C3 index points to an increased intrathecal production of C3. The same study did not find any difference in the C4 plasma, CSF, or index values in MS patients when compared to controls [25].
In addition, significantly reduced levels of plasma C9 have been detected in MS patients when compared to controls [19]. Soluble, fluid-phase complement C5b-9 terminal complexes (SC5b-9) have been found to be increased in the CSF of patients with MS [26] (Figure 1, Table 2), and the SC5b-9 CSF concentrations correlated significantly with neurological disability, as measured by EDSS [27]. Full activation of the complement cascade during attacks of MS may be restricted to patients with more advanced disease, and it is significantly correlated with the degree of neurological disability.
Alternative pathway
No difference in mean levels of plasma factor B and in its activation product Bb was noted in MS patients when compared to controls. Elevated concentrations of factor B have been reported in acute RRMS as compared to stable RRMS [19].
Complement regulatory proteins
Factor H
Complement factor H, a major regulator of the alternative pathway, is a single-chain serum glycoprotein with the ability to recognize and bind C3b. Therefore, it regulates the enzymatic activity of C3 and C5 [4]. Factor H has the ability to differentiate self from non-self by recognizing sialic acid and glycosaminoglycan chains, thereby preventing complement activation on host surfaces. Serum factor H levels are significantly increased in PPMS and SPMS when compared to either controls or patients with RRMS [28]; this study has demonstrated that factor H can be used as an alternative biomarker to distinguish SPMS from RRMS, with a sensitivity of 89.41%, a specificity of 69.47%, and a positive predictive value of 72.38% (cutoff value >237 mg/l). Also, factor H levels were found to increase progressively with disease progression over a 2-year period in patients transitioning from relapsing to progressive disease (in contrast to patients with stable RRMS, who have constant serum factor H levels), a correlation that is independent of other phenotypic parameters such as age, disability, and disease duration. Factor H in serum was less helpful in distinguishing patients with stable RRMS from those in relapse. In patients with relapse, a small and transient, but significant, increase in factor H concentration has been detected when compared to controls [28]. In addition, quantifying factor H levels in CSF indicated no change during active disease but significantly raised levels in progressive MS [29]. However, Li et al. have found a decrease in the expression of factor H in the CSF of RRMS patients with active disease when compared to controls [23].
Tyr402His is the most common polymorphism in factor H, but the frequency of its occurrence seems to be identical in patients with MS and in controls [28].
Soluble complement receptor 2 (sCR2), clusterin, and factor I
Significant increases in sCR2 levels are evident in CSF from patients with either RRMS or SPMS when compared to controls [20] (Table 2). Furthermore, CSF sCR2 levels have been found to be significantly correlated both with CSF C3 and C1q levels as well as with a measure of disease severity. In vitro, sCR2 inhibits the cleavage and down-regulation of C3b to iC3b, suggesting that it exerts a modulatory role in complement activation downstream of C3 [20]. No significant differences in either clusterin or Factor I have been detected in the plasma and CSF levels of MS patients when compared to controls [19].
Complement activation in neuromyelitis optica (NMO)
Complement activation products are elevated in individuals with NMO spectrum disorder (NMOSD) when compared to either controls or MS patients [30–32]. The levels of four complement proteins (C1inh, C1s, C5, and factor H) are higher in patients with NMOSD than in either those with MS or controls (Table 1). A proposed model involving C1 inhibitor and C5b-9 has been able to distinguish NMOSD from MS (area under the curve [AUC]: 0.98), and the levels of C1inh and C5 can distinguish NMOSD from controls (AUC: 0.94). These findings support the contention that NMOSD can be distinguished from MS by the use of plasma complement biomarkers and that trials of anti-complement therapies may help elucidate the role of complement activation in NMOSD [30]. Recent data support a complement bystander mechanism for the assembly of C5b-9 and for early OLG injury and demyelination in NMO [33]. This publication [33] presents evidence for a complement “bystander mechanism” to account for early OLG injury in NMO, in which the activation complement following aquaporin 4 (AQP4)-IgG binding to astrocyte AQP4 results in the deposition of terminal complement C5b-9 on nearby OLG, and finally in cell lysis.
Response gene to complement-32 (RGC-32) as a biomarker of relapse and response to therapy
Response gene to complement-32 (RGC-32) was first identified in rat OLG after stimulation with sublytic C5b-9, and the gene was subsequently cloned in mice and humans [34, 35]. It is primarily involved in cell cycle regulation, and its overexpression leads to increased DNA synthesis and cell cycle progression from G1/G0 to G2/M in a number of cells, including aortic smooth muscle cells and endothelial cells [34–36]. However, RGC-32 may also behave as a tumor suppressor by inhibiting cell cycle activation, as has been demonstrated in some tumor cells [37]. Autoreactive memory T cells, which play an important role in MS pathogenesis, show impaired apoptosis in MS patients, and the number of infiltrating T cells is highly increased in the acute phase of experimental autoimmune encephalomyelitis (EAE), an animal model of MS. Remission of MS is associated with a significant reduction in the T-cell number as a result of apoptotic cell death. T-cell apoptosis is regulated in part by the Fas-FasL system. Evidence has shown that the level of FasL expression is decreased during MS relapse, a finding consistent with the increased resistance of the T cells to apoptosis [38]. When we investigated the relationship between RGC-32 and FasL in RRMS patients, we found that the levels of RGC-32 and FasL mRNAs in peripheral blood mononuclear cells (PBMCs) were significantly decreased during relapses when compared to those of stable patients, and they were significantly increased in stable patients when compared to controls [39]. Moreover, we have also found a strong direct correlation between RGC-32 and FasL mRNAs during relapses [39]. Silencing of RGC-32 expression using lentiviral shRNA RGC-32 in PBMCs from stable patients decreases the levels of FasL on the cell surfaces. Considering that RGC-32 binds to and up-regulates CDC2/cyclin B1 kinase activity, one of the regulators of FasL expression on T cells, one might speculate that RGC-32 regulates T-cell survival by modulating the expression of FasL [39].
Glatiramer acetate (GA) is one of the most common immunomodulatory therapies used for treating patients with RRMS. However, not all patients respond to treatment, and treatment failure may only be recognized after months or years of therapy. For this reason, it would be very useful to be able to predetermine patient responsiveness before starting to administer a particular therapy. There are data showing that responders to GA treatment show increased expression of RGC-32 and FasL and decreased expression of IL-21 when compared to non-responders, with these levels persisting over time. Interestingly, no changes have been seen in CDC2 or AKT expression between responders and non-responders to GA treatment [40].
The predictive accuracy of these potential biomarkers in detecting relapse and responsiveness to GA treatment has been assessed using receiver operating characteristic (ROC) analysis. The probability (area under the curve [AUC]) of detecting a relapse was 90% for RGC-32, 88% for FasL, and 75% for IL-21, and the probability of detecting a response to GA was 85% for RGC-32, 90% for FasL, and 85% for IL-21 [40] These data suggest that RGC-32, FasL, and IL-21 can serve as potential biomarkers for the detection of MS relapse and response to GA therapy [40].
RGC-32 also plays an important role in extracellular matrix production, and TGF-β is a strong inducer of RGC-32 expression in astrocytes [39]. TGF-β-induced RGC-32 expression seems to be mediated by RUNX1 and other transcription factors such as NF-κB, as detected in rat astrocytes. RGC-32 silencing leads to a significant reduction in TGF-β-induced procollagen I, fibronectin, and α-smooth muscle actin expression, the last being a marker of reactive astrocytes. These data suggest a possible role for RGC-32 in the TGF-β-mediated extracellular matrix production and gliosis seen in chronic MS and EAE lesions [41].
In conclusion, there is strong evidence for the utility of complement system levels in serum and CSF as biomarkers of tissue inflammation in MS. Complement components and activation products can serve as biomarkers of disease activity and can help differentiate the various MS subtypes as well as differentiate individuals with RRMS from healthy controls. There is strong evidence suggesting that RGC-32, FasL and IL-21 can also serve as potential biomarkers for the detection of MS relapse and response to GA therapy.
Acknowledegments:
We thank Dr. Deborah McClellan for editing this manuscript. This work was supported in part by Veterans Administration Merit Awards BX001458 and IMMB-002–065 (both to H.R.).
Funding: Veterans Administration Merit Awards BX001458 and IMMB-002–065 (both to H.R.).
Footnotes
Compliance with Ethical Standards:
Conflict of Interest: Horea Rus has received a grant from TEVA Neuroscience (CNS-2014–174). All other authors declare that they have no conflict of interest.
References
- 1.Compston DA, Morgan BP, Campbell AK, Wilkins P, Cole G, Thomas ND, Jasani B: Immunocytochemical localization of the terminal complement complex in multiple sclerosis. Neuropathol Appl Neurobiol. 1989, 15:307–316. [DOI] [PubMed] [Google Scholar]
- 2.Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H: Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. 2000, 47:707–717. [DOI] [PubMed] [Google Scholar]
- 3.Breij ECWBBP, Veerhuis R, Van den Berg C, Vloet R, Yan R, Dijkstra CD, Van der Valk P, Bö L: Homogeneity of active demyelinating lesions in established multiple sclerosis. Ann Neurol. 2008, 63:16–25. [DOI] [PubMed] [Google Scholar]
- 4.Tegla CA, Cudrici C, Patel S, Trippe R 3rd, Rus V, Niculescu F, Rus H: Membrane attack by complement: the assembly and biology of terminal complement complexes. Immunol Res. 2011, 51: 45–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rus H, Cudrici C, Niculescu F: C5b-9 complement complex in autoimmune demyelination: dual role in neuroinflammation and neuroprotection. Adv Exp Med Biol. 2006, 586:139–151. [DOI] [PubMed] [Google Scholar]
- 6.Barnett MH, Parratt JD, Cho ES, Prineas JW: Immunoglobulins and complement in postmortem multiple sclerosis tissue. Ann Neurol. 2009, 65:32–46. [DOI] [PubMed] [Google Scholar]
- 7.Barnett MH, Prineas JW: Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann Neurol 2004, 55:458–468. [DOI] [PubMed] [Google Scholar]
- 8.Lucchinetti C, Bruck W, Noseworthy J: Multiple sclerosis: recent developments in neuropathology, pathogenesis, magnetic resonance imaging studies and treatment. Curr Opin Neurol. 2001, 14:259–269. [DOI] [PubMed] [Google Scholar]
- 9.Ingram G, Loveless S, Howell OW, Hakobyan S, Dancey B, Harris CL, Robertson NP, Neal JW, Morgan BP: Complement activation in multiple sclerosis plaques: an immunohistochemical analysis. Acta Neuropathol Commun. 2014, 2:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rus HG, Kim LM, Niculescu FI, Shin ML: Induction of C3 expression in astrocytes is regulated by cytokines and Newcastle disease virus. J Immunol. 1992, 148:928–933. [PubMed] [Google Scholar]
- 11.Loveless S, Neal JW, Howell OW, Harding KE, Sarkies P, Evans R, Bevan RJ, Hakobyan S, Harris CL, Robertson NP et al. : Tissue microarray methodology identifies complement pathway activation and dysregulation in progressive multiple sclerosis. Brain Pathol. 2017: July 14. doi: 10.1111/bpa.12546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tegla CA, Cudrici C, Rozycka M, Soloviova K, Ito T, Singh AK, Khan A, Azimzadeh P, Andrian-Albescu M, Khan A et al. : C5b-9-activated, Kv1.3 channels mediate oligodendrocyte cell cycle activation and dedifferentiation. Exp Mol Pathol. 2011, 91:335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Michailidou I, Naessens DM, Hametner S, Guldenaar W, Kooi EJ, Geurts JJ, Baas F, Lassmann H, Ramaglia V: Complement C3 on microglial clusters in multiple sclerosis occur in chronic but not acute disease: Implication for disease pathogenesis. Glia. 2017, 65(2):264–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rus H, Cudrici C, Niculescu F, Shin ML: Complement activation in autoimmune demyelination: dual role in neuroinflammation and neuroprotection. J Neuroimmunol. 2006, 180:9–16. [DOI] [PubMed] [Google Scholar]
- 15.Liu WT, Vanguri P, Shin ML: Studies on demyelination in vitro: the requirement of membrane attack components of the complement system. J Immunol. 1983, 131:778–782. [PubMed] [Google Scholar]
- 16.Liu Y, Given KS, Harlow DE, Matschulat AM, Macklin WB, Bennett JL, Owens GP: Myelin-specific multiple sclerosis antibodies cause complement-dependent oligodendrocyte loss and demyelination. Acta Neuropathol Commun. 2017, 5:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soane L, Cho HJ, Niculescu F, Rus H, Shin ML: C5b-9 terminal complement complex protects oligodendrocytes from death by regulating Bad through phosphatidylinositol 3-kinase/Akt pathway. J Immunol. 2001, 167:2305–2311. [DOI] [PubMed] [Google Scholar]
- 18.Cudrici C, Niculescu F, Jensen T, Zafranskaia E, Fosbrink M, Rus V, Shin ML, Rus H: C5b-9 Terminal Complex Protects Oligodendrocytes from Apoptotic Cell Death by Inhibiting Caspase-8 Processing and Up-Regulating FLIP. J Immunol. 2006, 176:3173–3180. [DOI] [PubMed] [Google Scholar]
- 19.Ingram G, Hakobyan S, Hirst CL, Harris CL, Loveless S, Mitchell JP, Pickersgill TP, Robertson NP, Morgan BP: Systemic complement profiling in multiple sclerosis as a biomarker of disease state. Mult Scler. 2012, 18:1401–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lindblom RP, Aeinehband S, Strom M, Al Nimer F, Sandholm K, Khademi M, Nilsson B, Piehl F, Ekdahl KN: Complement Receptor 2 is increased in cerebrospinal fluid of multiple sclerosis patients and regulates C3 function. Clin Immunol. 2016, 166–167:89–95. [DOI] [PubMed] [Google Scholar]
- 21.Kwok JY, Vaida F, Augst RM, Yu DY, Singh KK: Mannose binding lectin mediated complement pathway in multiple sclerosis. J Neuroimmunol. 2011, 239:98–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aeinehband S, Lindblom RP, Al Nimer F, Vijayaraghavan S, Sandholm K, Khademi M, Olsson T, Nilsson B, Ekdahl KN, Darreh-Shori T et al. : Complement component C3 and butyrylcholinesterase activity are associated with neurodegeneration and clinical disability in multiple sclerosis. PLoS One. 2015, 10(4):e0122048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y, Qin Z, Yang M, Qin Y, Lin C, Liu S: Differential expression of complement proteins in cerebrospinal fluid from active multiple sclerosis patients. J Cell Biochem. 2011, 112:1930–1937. [DOI] [PubMed] [Google Scholar]
- 24.Ingram G, Hakobyan S, Robertson NP, Morgan BP: Elevated plasma C4a levels in multiple sclerosis correlate with disease activity. J Neuroimmunol. 2010, 223:124–127. [DOI] [PubMed] [Google Scholar]
- 25.Jongen PJ, Doesburg WH, Ibrahim-Stappers JL, Lemmens WA, Hommes OR, Lamers KJ: Cerebrospinal fluid C3 and C4 indexes in immunological disorders of the central nervous system. Acta Neurol Scand. 2000, 101:116–121. [DOI] [PubMed] [Google Scholar]
- 26.Sanders ME, Koski CL, Robbins D, Shin ML, Frank MM, Joiner KA: Activated terminal complement in cerebrospinal fluid in Guillain-Barre syndrome and multiple sclerosis. J Immunol. 1986, 136:4456–4459. [PubMed] [Google Scholar]
- 27.Sellebjerg F, Jaliashvili I, Christiansen M, Garred P: Intrathecal activation of the complement system and disability in multiple sclerosis. J Neurol Sci. 1998, 157:168–174. [DOI] [PubMed] [Google Scholar]
- 28.Ingram G, Hakobyan S, Hirst CL, Harris CL, Pickersgill TP, Cossburn MD, Loveless S, Robertson NP, Morgan BP: Complement regulator factor H as a serum biomarker of multiple sclerosis disease state. Brain. 2010, 133(Pt 6):1602–1611. [DOI] [PubMed] [Google Scholar]
- 29.Ingram G, Hakobyan S, Loveless S, Robertson N, Morgan BP: Complement regulator factor H in multiple sclerosis. J Cell Biochem. 2011, 112:2653–2654. [DOI] [PubMed] [Google Scholar]
- 30.Hakobyan S, Luppe S, Evans DR, Harding K, Loveless S, Robertson NP, Morgan BP: Plasma complement biomarkers distinguish multiple sclerosis and neuromyelitis optica spectrum disorder. Mult Scler. 2017, 23:946–955. [DOI] [PubMed] [Google Scholar]
- 31.Wang H, Wang K, Wang C, Qiu W, Lu Z, Hu X: Increased soluble C5b-9 in CSF of neuromyelitis optica. Scand J Immunol. 2014, 79:127–130. [DOI] [PubMed] [Google Scholar]
- 32.Nytrova P, Potlukova E, Kemlink D, Woodhall M, Horakova D, Waters P, Havrdova E, Zivorova D, Vincent A, Trendelenburg M: Complement activation in patients with neuromyelitis optica. J Neuroimmunol. 2014, 274:185–191. [DOI] [PubMed] [Google Scholar]
- 33.Tradtrantip L, Yao X, Su T, Smith AJ, Verkman AS: Bystander mechanism for complement-initiated early oligodendrocyte injury in neuromyelitis optica. Acta Neuropathol. 2017, 134:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Badea TC, Niculescu FI, Soane L, Shin ML, Rus H: Molecular cloning and characterization of RGC-32, a novel gene induced by complement activation in oligodendrocytes. J Biol Chem. 1998, 273:26977–26981. [DOI] [PubMed] [Google Scholar]
- 35.Badea T, Niculescu F, Soane L, Fosbrink M, Sorana H, Rus V, Shin ML, Rus H: RGC-32 increases p34CDC2 kinase activity and entry of aortic smooth muscle cells into S-phase. J Biol Chem. 2002, 277:502–508. [DOI] [PubMed] [Google Scholar]
- 36.Fosbrink M, Cudrici C, Tegla CA, Soloviova K, Ito T, Vlaicu S, Rus V, Niculescu F, Rus H: Response gene to complement 32 is required for C5b-9 induced cell cycle activation in endothelial cells. Exp Mol Pathol. 2009, 86:87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vlaicu SI, Tegla CA, Cudrici CD, Fosbrink M, Nguyen V, Azimzadeh P, Rus V, Chen H, Mircea PA, Shamsuddin A et al. : Epigenetic modifications induced by RGC-32 in colon cancer. Exp Mol Pathol. 2010, 88:67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lopatinskaya L, van Boxel-Dezaire AH, Barkhof F, Polman CH, Lucas CJ, Nagelkerken L: The development of clinical activity in relapsing-remitting MS is associated with a decrease of FasL mRNA and an increase of Fas mRNA in peripheral blood. J Neuroimmunol. 2003, 138(1–2):123–131. [DOI] [PubMed] [Google Scholar]
- 39.Tegla CA, Cudrici CD, Azimzadeh P, Singh AK, Trippe R 3rd, Khan A, Chen H, Andrian-Albescu M, Royal W 3rd, Bever C et al. : Dual role of Response gene to complement-32 in multiple sclerosis. Exp Mol Pathol. 2013, 94:17–28. [DOI] [PubMed] [Google Scholar]
- 40.Kruszewski AM, Rao G, Tatomir A, Hewes D, Tegla CA, Cudrici CD, Nguyen V, Royal W 3rd, ,Bever CT Jr., Rus V et al. : RGC-32 as a potential biomarker of relapse and response to treatment with glatiramer acetate in multiple sclerosis. Exp Mol Pathol. 2015, 99:498–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tatomir A, Cosmin T, Cudrici C, Boodhoo D, Martin A, Mekala A, Rus V, Badea C, Rus H: RGC-32 regulates TGF-β extracellular matrix production in multiple sclerosis. J Immunol. 2016, 196:189.187 [Google Scholar]