

Abstract
Background
The prognosis of idiopathic pulmonary fibrosis (IPF) is the worst among all interstitial lung diseases, and is related to the disease itself. Comorbidities or complications can worsen IPF. We assessed the effect of comorbidities on the survival of IPF patients. A retrospective review of patients with IPF was completed.
Material/Methods
Information on demographic features, clinical examination, and comorbidities at baseline were obtained. Then, median, 1-year, and 5-year survival was calculated. A total of 380 patients with IPF admitted to Beijing Chao-Yang Hospital from 1 April 2002 to 31 March 2015 were followed up until December 2016.
Results
Of these 380 patients, 71.9% died during the study period. Median survival was 2.25 years and overall 5-year survival was 28.5%. Also, 86.3% of patients were males. A total of 248 cases underwent lung function tests, and 178 patients underwent bronchoalveolar lavage (BAL). Multivariate analyses showed that forced expiratory volume in 1 second/forced vital capacity (FVC), diffusing capacity of the lungs for carbon monoxide percent predicted, FVC% predicted, the number of macrophages, neutrophils, and lymphocytes in BAL fluid, pulmonary hypertension, hypoxemia, and hydropower disorder were independent prognostic indicators of IPF, GAP gender (G), age (A), and 2 pulmonary physiological parameters (P) model can help to predict prognosis of IPF.
Conclusions
Spirometry, GAP model, and BAL are helpful to forecast the prognosis of IPF. IPF patients also suffering from pulmonary arterial hypertension, hypoxemia, and hydropower disorder have a poor prognosis.
MeSH Keywords: Idiopathic Pulmonary Fibrosis, Risk Factors, Survival Rate
Background
Idiopathic pulmonary fibrosis (IPF) is a devastating lung disease, the incidence of which increases markedly with suboptimal treatment [1]. The median survival of IPF patients is 2–3 years [2–5]. Symptoms are non-specific at IPF onset, with the most prominent being dyspnea upon exertion and non-productive cough. Hence, the diagnosis and prognosis of IPF can be challenging.
Several national recommendations/guidelines from European countries have provided new guidance on the diagnosis and treatment of IPF. IPF guidelines from 2011 and 2015 revealed an increasing focus on the diagnosis and treatment of IPF. In addition, other IPF-related questions have not been answered, and there is a lack of management guidance.
In addition, based on multiple genetic variants and population-based investigations, the prevalence of IPF in white populations has been shown to differ from that from other ethnicities [6]. That is, there are differences between ethnicities with respect to IPF. Hence, the characteristics of IPF in China must be clarified to provide a more personalized and effective approach to treat IPF patients. Therefore, we undertook a retrospective study to ascertain the epidemiology, clinical examination, and comorbid features of IPF. In this way, we wished to provide a basis for prediction of the prognosis of IPF.
Material and Methods
Patient selection
The Human Ethics Review Committee of Beijing Chao-Yang Hospital (Beijing, China) approved our study protocol. All of the subjects included in the present work provided written informed consent.
Consecutive patients with IPF admitted between April 2002 and March 2015 to Beijing Chao-Yang Hospital formed the study cohort. All IPF patients were diagnosed based on criteria set by the American Thoracic Society/European Respiratory Society [7]. For study inclusion, patients had to fulfill the following criteria: (i) manifestations of progressive dyspnea and bilateral (predominantly) basal crackles; (ii) restricted lung function and impairment of gas exchange; (iii) typical abnormalities indicative of usual interstitial pneumonia (UIP) on high-resolution computed tomography (HRCT) of the chest, including bilateral reticular abnormalities of the lung with predominantly basal/sub-pleural honeycombing and/or traction bronchiectasis, Figure 1; (iv) bronchoalveolar lavage fluid (BALF) profiles and/or pathologic features indicative of UIP on surgical lung biopsy (Figure 2); and (v) no evidence of known causes of pulmonary fibrosis.
Figure 1.

HRCT scan of the IPF patients demonstrated the grid changes near the pleura (red arrow).
Figure 2.

Pathology features in the patients with IPF, the lung tissue was obtained through transbronchial lung biopsy. [(A) Masson staining (collagen fiber) showed disorder alveolar structure and interstitial fibrosis in IPF patients (×100/HP); (B) α-SAM (fibrinogen specific antibody) staining (×100/HP); (C) The characteristic pathological changes of terminal alveolar bronchopathy with submucosal gland structure in terminal alveolar structure. MUC5B stain was positive by immunohistochemistry assay (×100/HP); (D) Masson staining (collagen fiber) showed that the alveolar structure was disorder and interstitial fibrosis was seen in IPF patients; (E) α-SAM (fibrinogen specific antibody) staining (×100/HP); (F) Terminal alveolar bronchopathy with submucosal gland structure with MUC5B stain positive (×100/HP); (G) Masson staining (×200/HP); (H) α-SAM (fibrinogen specific antibody) staining (×100/HP); (I) MUC5B positive expression (×100/HP)].
Patients were excluded if they had indications of other types of idiopathic interstitial pneumonias, connective tissue-diseases, chronic hypersensitivity pneumonitis, or drug/occupational/environmental exposure-related interstitial lung disease. Patients were also excluded if there was a lack of HRCT images of the chest or results of clinical follow-up. Such investigations were carried out during regular visits to our unit. These were done every 3–6 months or at time intervals determined by clinical requirements, or by telephone calls for patients unable to visit our clinic. Survival data were obtained from medical records, telephone interviews, and a database (Social Security Death Index). Finally, 380 patients with IPF were enrolled into our study.
Data collection
A standard form was used to collect information on demographics (age, sex, race, smoking status), comorbidities (pulmonary hypertension, hypoxemia, hydropower disorder (i.e., low protein, low potassium, low chlorine, low sodium and other pathologic conditions), gastroesophageal reflux, pulmonary arterial hypertension (PAH), and diabetes mellitus (DM)), BALF (percentage of macrophages, neutrophils, eosinophils and lymphocytes), and pulmonary function tests (PFTs).
Spirometry data, such as total lung capacity (TLC), forced vital capacity percent predicted (FVC% predicted), diffusing capacity of the lungs for carbon monoxide (DLCO), diffusing capacity of the lungs for carbon monoxide percent predicted (DLCO% predicted), and forced expiratory volume in 1 second (FEV1), were obtained in accordance with American Thoracic Society guidelines [8]. We then used the GAP system, which include gender (G), age (A), and 2 pulmonary physiological parameters(P)-percentage predicted forced vital capacity (FVC[%]), and percentage predicted diffusion capacity of the lungs for carbon monoxide (DLco[%]) for staging IPF [9]. The survival time and survival status were confirmed until the last telephone follow-up (December 2016). Comorbidities were assessed in relation to the prognosis.
Statistical analyses
Continuous data are given as the mean ±SD. Categorical data are shown as frequencies. Unless specified otherwise, the number of patients with available data (n) was used for the calculation of summary statistics. The chi-squared test was used to compare categorical variables. The Kolmogorov-Smirnov test was employed to compare continuous variables. Survival was evaluated using survival analyses provided by SPSS tables (SPSS v22.0, IBM, Armonk, NY, USA) and the log-rank test was used to determine significance. Kaplan-Meier analysis was undertaken to estimate cumulative survival. Differences in the hazard ratio (HR) for death were evaluated using Cox proportional hazards regression. Unadjusted and adjusted Cox proportional hazards regression were undertaken, and HRs are presented along with 95% confidence intervals. All statistical analyses were done using SPSS v22.0, and P<0.05 was considered significant.
Results
Demographic data
A total of 380 IPF patients formed the study cohort. We found that 71.9% of these 380 IPF patients died during the study period. Also, 22.6% patients were still being followed up at our institution at the end of the study period. One-year survival was 53% and 5-year survival was 28.5%. In addition, 86.3% of IPF patients were male. The mean age at which any symptom or sign of IPF appeared was 63±9.25 years.
For survival analyses, patients were classified into 3 groups: I (<50 years), II (50–75 years), and III (>75years). Kaplan-Meier analyses revealed the median survival for these 3 groups to be 65.044, 48.320, and 35.176 months, respectively. Significant differences were not observed between the age groups when they were used as predictors of survival (P=0.268) (Table 1). The median survival for females was 45.005 months, and for males it was 46.623months (P=0.887) (Table 1). More than half of the patients were smokers, but smoking was not associated with worse survival (HR=0.19). Multivariate analyses revealed age, sex, and smoking status were not independent predictors of survival in IPF patients (Table 2).
Table 1.
Survival time of various strata in IPF patients.
| Variables | Num. | Survival time (mean, month) | P values |
|---|---|---|---|
| Age | 0.268 | ||
| <50 years | 22 | 65.044 | |
| 50–75 years | 325 | 48.320 | |
| >75 years | 57 | 35.176 | |
| Gender | 0.887 | ||
| Male | 347 | 46.623 | |
| Female | 55 | 45.005 | |
| FVC (%pred) | 0.048 | ||
| <45 | 17 | 25.388 | |
| ≥45 | 208 | 47.070 | |
| DLCO(%pred) | 0.000 | ||
| >60% | 46 | 51.038 | |
| ≤60%, >45% | 91 | 46.956 | |
| ≤45%, >30%) | 66 | 25.427 | |
| ≤30% | 22 | 19.182 | |
| GAP stage | 0.041 | ||
| Stage I | 107 | 64.78 | |
| Stage II | 96 | 35.33 | |
| Stage III | 22 | 31.60 | |
| Hypoxemia | 0.002 | ||
| Yes | 210 | 41.172 | |
| No | 192 | 52.083 | |
| Hydropower disorder | 0.004 | ||
| Yes | 76 | 31.683 | |
| No | 326 | 49.349 | |
| Pulmonary hypertension | 0.024 | ||
| Yes | 78 | 32.198 | |
| No | 324 | 51.263 | |
Table 2.
Univariate regression analysis of overall survival for IPF patients.
| Variables | B value | SE | P values | Relative risk (95%CI) |
|---|---|---|---|---|
| Age | 0.011 | 0.007 | 0.084 | 1.011 (0.998–1.025) |
| Gender | 0.023 | 0.166 | 0.889 | 1.024 (0.740–1.417) |
| Smoking | 0.003 | 0.137 | 0.056 | 1.021 (1.012–1.117) |
| FEV1 (%pred) | 0.006 | 0.008 | 0.449 | 1.006 (0.991–1.021) |
| FVC (%pred) | −0.327 | 0.131 | 0.012 | 0.721 (0.558–0.932) |
| TLC (%pred) | −0.007 | 0.004 | 0.110 | 0.993 (0.985–1.002) |
| DLCO (%pred) | 0.008 | 0.003 | 0.009 | 1.008 (1.002–1.014) |
| FEV1/FVC (%) | 0.025 | 0.007 | 0.002 | 0.846 (1.023–1.108) |
| BAL macrophage | −0.028 | 0.008 | 0.021 | 0.997 (0.959–0.989) |
| BAL neutrophil | −0.009 | 0.004 | 0.016 | 0.991 (0.983–0.998) |
| BAL eosinophil | −0.016 | 0.019 | 0.391 | 0.984 (0.947–1.021) |
| BAL lymphocyte | 0.024 | 0.010 | 0.015 | 1.024 (1.005–1.044) |
| PAH | 0.355 | 0.160 | 0.027 | 1.426 (1.042–1.951) |
| Hypoxemia | −0.356 | 0.119 | 0.003 | 0.7 (0.555–0.884) |
| Hydropower | 0.459 | 0.162 | 0.005 | 1.582 (1.152–2.173) |
| GER | 0.251 | 0.188 | 0.182 | 1.285 (0.889–1.857) |
| Arterial hypertension | 0.014 | 0.145 | 0.923 | 1.014 (0.763–1.348) |
| Diabetes | 0.070 | 0.146 | 0.632 | 1.072 (0.806–1.427) |
Baseline lung function and prediction for survival from IPF
A total of 236 cases of the 380 IPF patients completed LFTs. Multivariate analyses of prognostic factors using a Cox proportional hazards model showed that the FVC% predicted and DLCO% predicted were highly correlated with survival from IPF (P=0.012; P=0.009) (Table 2). Kaplan-Meyer survival curves demonstrated that FVC% predicted and DLCO% predicted were significantly associated with disease severity. Furthermore, higher value for FEV1/FVC was associated with worse survival (Table 2). Survival was poor among patients with FVC% predicted <45 (P=0.048) (Figure 3). DLCO% predicted could be divided into 4 stages: I (>60%), II (≤60%, >45%), III (≤45%, >30%), and IV (≤30%). Stage IV of DLCO% predicted was strongly correlated with survival in IPF patients (P<0.001, Figure 4).
Figure 3.
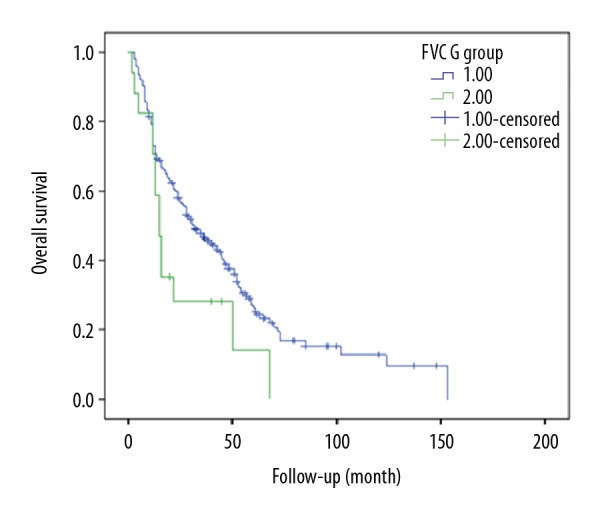
Survival curve of the IPF patients in different FVC groups (1: FVC ≥50%; 2: FVC% <50%).
Figure 4.
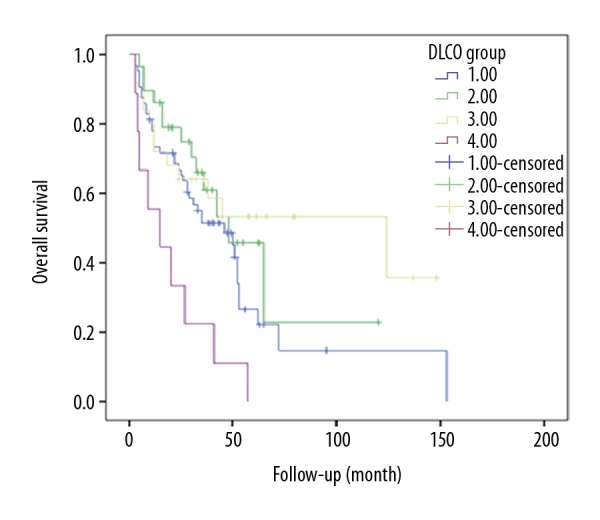
Survival curve of the IPF patients in different DLCO groups (1: >60%; 2: ≤60%, >45%; 3: ≤45%, >30%; 4: ≤30%).
GAP model and prediction of survival from IPF
Total GAP score was calculated using the method suggested by Ley et al. [12]. All 4 clinical variables were examined: gender (female: 0 points, male: 1 point), age (0–2 points), FVC (%) (0–2 points), and DLCO (%) (0–2 points). Ultimately, we divided the patients on the basis of GAP score (0–7) into 3 stages: I (0–3 points), II (4–5 points), and III (6–8 points) (Table 3). Stage III of GAP was highly correlated with survival in IPF patients (P=0.041, Figure 5).
Table 3.
GAP data summary.
| Variable | GAP points | No. of patients |
|---|---|---|
| Gender | ||
| Female | 0 | 27 (13.0) |
| Male | 1 | 180 (87.0) |
| Age, years | ||
| ≤60 | 0 | 71 (34.3) |
| 61–65 | 1 | 33 (15.9) |
| >65 | 2 | 103 (49.8) |
| Physiology | ||
| FVC, %predicted | ||
| >75 | 0 | 105 (50.7) |
| 50–75 | 1 | 80 (38.6) |
| <50 | 2 | 22 (10.6) |
| DLco, %predicted | ||
| >55 | 0 | 86 (41.5) |
| 36–55 | 1 | 55 (26.6) |
| <35 | 2 | 65 (31.4) |
| GAP stage | ||
| Stage I | 0–3 | 107 (47.5) |
| Stage II | 4–5 | 96 (42.6) |
| Stage III | 6–8 | 22 (9.9) |
Figure 5.
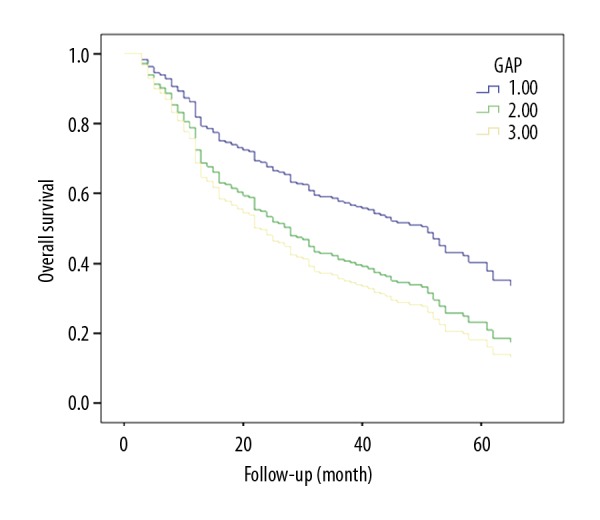
Survival curve of the IPF patients in different GAP groups (1: 0–3 points; 2: 4–5 points; 3: 6–8 points).
Baseline cell profiles in BALF and prediction of survival from IPF
Evaluation of cell profiles in BALF can be beneficial for the diagnosis of IPF. Few studies have used this method to predict the prognosis of IPF.
A total of 176 patients out of the 380IPFcases underwent BAL. The percentage of lymphocytes was 4.9±5.29, and that of eosinophils was 2.78±6.0, macrophages was 45.08±24.36, and neutrophils was 46.95±23.49. Multivariate analyses of prognostic factors using a Cox proportional hazards model showed that the percentage of macrophages, neutrophils, and lymphocytes was highly correlated with survival from IPF (P=0.021, P=0.016, and P=0.015, respectively), but there was no correlation with the percentage of eosinophils (Table 2).
Comorbidities and prediction of survival from IPF
We focused on 6 common IPF comorbidities (pulmonary hypertension, hypoxemia, hydropower disorder, gastroesophageal reflux, PAH, and DM) and the clinical syndrome known as “combined pulmonary fibrosis”.
The most prevalent comorbidities among IPF patients are listed in Table 2. Analyses of the Cox proportional hazards model of the prognostic comorbidities of IPF showed that PAH, hypoxemia, and hydropower disorder were highly correlated with survival from IPF (P=0.027, P=0.003, and P=0.005, respectively) (Table 2). IPF comorbid with hypoxemia, hydropower disorder, and PAH showed significant differences in survival from IPF (P=0.024, P=0.002, and P=0.004, respectively) (Table 1).
Discussion
IPF is a heterogeneous illness, and predicting survival for individual patients can be challenging. Contrary to some reports [2,8], we found that age, sex, and smoking status were not independent predictors of survival in multivariate analyses, but these findings are similar to data from our previous studies [10]. Nadrous and coworkers determined the impact on survival in IPF patients, and reported that age and sex were not predictors of survival from IPF, findings that are in accordance with our data [11].Olson and colleagues analyzed IPF data from 1992 to 2003 from the National Center for Health Statistics [12].They concluded that IPF was more common in men than in women but, over the study period, the prevalence of mortality accelerated more steeply in women than in men [12].In our study, 86.3% were males and the median age was 63±9.25 years. Hence, IPF is predominantly a disease of elderly men, and increasing age and being male are powerful predictors for IPF. That is, age and sex are risk factors for the development of IPF but are less predictive of survival from IPF.
The direct cause of death due to idiopathic pulmonary fibrosis was respiratory failure. FEV1, FVC, and DLCO are most common clinically used parameters for evaluation the pulmonary function. Therefore, FEV1, FVC, and DLCO were assessed for association with survival. In IPF, lung function is restricted, and values for FVC, TLC, and DLCO are decreased. Spirometry is used for IPF diagnosis, and can be used to predict survival. Our study showed that DLCO% predicted and FVC% predicted were highly correlated with survival in IPF patients, observations that are consistent with our previous study and with those of Schmidt et al. [13]. Also, stratified analyses demonstrated that a combination of DLCO% predicted <30% and FVC% predicted <45% was associated with significantly worse survival in IPF patients. Recently, Nathan et al. showed that categorization according to the baseline values of FVC% predicted and DLCO% predicted can be effectively used to identify long-term outcomes in IPF. Thus, DLCO% predicted and FVC% predicted could be good predictors of survival in patients with IPF [14]. In this part of our study there were 2 limitations: we did not analyze peripheral oxygen saturation and lung function was not monitored.
Morbidity and mortality are high in IPF and the clinical course and prognosis vary widely among individual patients [15]. Some studies have attempted to predict clinical course and prognosis using age, gender, lung function change, radiological pattern, clinical symptom, and so on. However, none of these predictive models have been widely adopted. The GAP model uses 4 variables: gender (G), age (A), FVC%] FVC% predicted, and DLCO% predicted; these 4 variables are commonly measured and the GAP system are commonly used in asthma, chronic obstructive pulmonary disease (COPD), and lung cancer [9]. The purpose of our study was to predict the prognosis the IPF by using the GAP model. We found that GAP stages differed in terms of survival, with survival in stage III patients significantly lower than in the other stages (P=0.041<0.05 (Table 3, Figure 3), and these results are consistent with other studies [16,17]. A limitation of our study was that GAP scores should be analyzed with the other common comorbidities and examinations.
Another common examination used for the diagnosis of IPF is bronchoalveolar lavage (BAL). Multivariate analyses of prognostic factors showed that the percentage of macrophages, neutrophils, and lymphocytes was highly correlated with survival from IPF, but that the percentage of eosinophils was not. These findings are similar to those observed in other studies [18–22]. In our study, the median percentage of lymphocytes was 4.9±5.29, which is lower than the normal range of lymphocytes found in BALF [18–20]. Other studies investigating the cell profiles of BALF in IPF patients have shown that a paucity of lymphocytes reflects the histologic UIP pattern [19], and is associated with a poor outcome [24–28]. The proposed response of epithelial cells/fibroblasts to IPF is not suggestive of lung inflammation [29–31], but lack of an inflammatory stimulus in the fibro-proliferative model cannot be expected to lead to an increase in the number of lymphocytes in BALF. This hypothesis may explain the poor response of IPF/UIP patients to anti-inflammatory agents such as corticosteroids. Overall, the clinical utility of BAL at the time of prognosis prediction of IPF should be reconsidered.
IPF is a complex disease associated with various respiratory and non-respiratory comorbidities [32]. The presence of comorbidities with IPF can significantly influence the prognosis and inform management strategies. Hence, clinicians must recognize the potential for these concurrent conditions and be able to identify and manage them. Our study showed that PAH was associated with a poor outcome, an observation that is consistent with our former study [13] and other reports [33]. Also, DM and gastroesophageal reflux are likely to affect IPF. Contrary to previous reports [34], we found that hypoxemia and hydropower disorder were independent predictors of survival in multivariate analyses and were associated with a significantly worse prognosis. Hence, IPF patients suffering from hypoxemia and hydropower disorder may be suffering from decompensation. Hypoxemia in IPF patients leads to a further decline in lung function and gas diffusion, so the body cannot compensate for hypoxia. We showed that hypoxia is a risk factor for IPF prognosis. A potential limitation of our study was that we did not analyze the correlation between lung function and hypoxia. Predicting survival in IPF patients has been the focus of many studies over the last 30 years [35]. In agreement with other studies, our study showed that IPF is predominantly a disease of elderly people, and several important findings emerged from our study. IPF is associated with a poor prognosis, but survival is difficult to predict for individual patients [7,36].
Conclusions
Spirometry, GAP model and BAL are useful to predict the prognosis of IPF. IPF patients suffering from pulmonary arterial hypertension, hypoxemia, and hydropower disease have a poor prognosis. We hope that the results of the present study will lead to a more personalized and elective approach to treating IPF patients.
Footnotes
Source of support: The Key Program of National Natural Science Foundation of China (No. 81430001); National Key Technologies R & D Program Precision Medicine Research (No.2016YFC0901101), and the Inner Mongolia Autonomous Region Natural Science Foundation Project (No. 2017MS (LH) 0853)
Conflict of interest
None.
References
- 1.Noth I, Martinez FJ. Recent advances in idiopathic pulmonary fibrosis. Chest. 2007;132:637–50. doi: 10.1378/chest.06-1927. [DOI] [PubMed] [Google Scholar]
- 2.Kim HJ, Perlman D, Tomic R. Natural history of idiopathic pulmonary fibrosis. Respir Med. 2015;109:661–70. doi: 10.1016/j.rmed.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 3.Ley B, Collard HR, King TE., Jr Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;183:431–40. doi: 10.1164/rccm.201006-0894CI. [DOI] [PubMed] [Google Scholar]
- 4.Barlo NP, van Moorsel CH, van den Bosch JM, Grutters JC. Predicting prognosis in idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis. 2010;27:85–95. [PubMed] [Google Scholar]
- 5.Luppi F. Predicting the future of patients with idiopathic pulmonary fibrosis: Another step forward. Lancet Respir Med. 2017;14:30436–38. doi: 10.1016/S2213-2600(17)30436-8. [DOI] [PubMed] [Google Scholar]
- 6.Noth I, Zhang Y, Ma SF, et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: A genome-wide association study. Lancet Respir Med. 2013;1:309–17. doi: 10.1016/S2213-2600(13)70045-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.American Thoracic Society. Idiopathic pulmonary fibrosis: Diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS) Am J Respir Crit Care Med. 2000;161:646–64. doi: 10.1164/ajrccm.161.2.ats3-00. [DOI] [PubMed] [Google Scholar]
- 8.Beydon N, Davis SD, Lombardi E, et al. American Thoracic Society/European Respiratory Society Working Group on Infant and Young Children Pulmonary Function Testing. An official American Thoracic Society/European Respiratory Society statement: Pulmonary function testing in preschool children. Am J Respir Crit Care Med. 2007;175:1304–45. doi: 10.1164/rccm.200605-642ST. [DOI] [PubMed] [Google Scholar]
- 9.Ley B, Ryerson CJ, Vittinghoff E, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med. 2012;156:684–91. doi: 10.7326/0003-4819-156-10-201205150-00004. [DOI] [PubMed] [Google Scholar]
- 10.Nadrous HF, Ryu JH, Douglas WW, et al. Impact of angiotensin-converting enzyme inhibitors and statins on survival in idiopathic pulmonary fibrosis. Chest. 2004;126:438–46. doi: 10.1378/chest.126.2.438. [DOI] [PubMed] [Google Scholar]
- 11.Olson AL, Swigris JJ, Lezotte DC, et al. Mortality from pulmonary fibrosis increased in the United States from 1992 to 2003. Am J Respir Crit Care Med. 2007;176:277–84. doi: 10.1164/rccm.200701-044OC. [DOI] [PubMed] [Google Scholar]
- 12.Schmidt SL, Nambiar AM, Tayob N, et al. Pulmonary function measures predict mortality differently in IPF versus combined pulmonary fibrosis and emphysema. Eur Respir J. 2011;38:176–83. doi: 10.1183/09031936.00114010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cai M, Zhu M, Ban C, et al. Clinical features and outcomes of 210 patients with idiopathic pulmonary fibrosis. Chin Med J (Engl) 2014;127:1868–73. [PubMed] [Google Scholar]
- 14.Nathan SD, Shlobin OA, Ahmad S, et al. Pulmonary hypertension and pulmonary function testing in idiopathic pulmonary fibrosis. Chest. 2007;131:657–63. doi: 10.1378/chest.06-2485. [DOI] [PubMed] [Google Scholar]
- 15.Ley B, Collard HR, King TE. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;183:431–40. doi: 10.1164/rccm.201006-0894CI. [DOI] [PubMed] [Google Scholar]
- 16.Salisbury ML, Xia M, Zhou Y, et al. Idiopathic pulmonary fibrosis: Gender-age-physiology index stage for predicting future lung function decline. Chest. 2016;149:491–98. doi: 10.1378/chest.15-0530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee SH, Kim SY, Kim DS, et al. Predicting survival of patients with idiopathic pulmonary fibrosis using GAP score: A nationwide cohort study. Respir Res. 2016;17:131. doi: 10.1186/s12931-016-0454-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boomars KA, Wagenaar SS, Mulder PG, et al. Relationship between cells obtained by bronchoalveolar lavage and survival in idiopathic pulmonary fibrosis. Thorax. 1995;50:1087–92. doi: 10.1136/thx.50.10.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tabuena RP, Nagai S, Tsutsumi T, et al. Cell profiles of bronchoalveolar lavage fluid as prognosticators of idiopathic pulmonary fibrosis/usual interstitial pneumonia among Japanese Patients. Respiration. 2005;72:490–98. doi: 10.1159/000087673. [DOI] [PubMed] [Google Scholar]
- 20.Nagai S, Kitaichi M, Itoh H, et al. Idiopathic nonspecific interstitial pneumonia/fibrosis: Comparison with idiopathic pulmonary fibrosis and BOOP. Eur Respir J. 1998;12:1010–19. doi: 10.1183/09031936.98.12051010. [DOI] [PubMed] [Google Scholar]
- 21.Pesci A, Ricchiuti E, Ruggiero R, De Micheli A. Bronchoalveolar lavage in idiopathic pulmonary fibrosis: What does it tell us? Respir Med. 2010;104:S70–73. doi: 10.1016/j.rmed.2010.03.019. [DOI] [PubMed] [Google Scholar]
- 22.Papanikolaou IC, Drakopanagiotakis F. Polychronopoulos vs. acute exacerbations of interstitial lung diseases. Curr Opin Pulm Med. 2010;16:480–86. doi: 10.1097/MCP.0b013e32833ae49d. [DOI] [PubMed] [Google Scholar]
- 23.Wuyts WA, Thomeer M, Dupont LJ, Verleden GM. An algorithm to tackle acute exacerbations in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;177:1397. doi: 10.1164/ajrccm.177.12.1397. author reply 1398. [DOI] [PubMed] [Google Scholar]
- 24.Turner-Warwick M, Haslam PL. The value of serial bronchoalveolar lavage in assessing progress of patients with cryptogenic fibrosing alveolitis. Am Rev Respir Dis. 1987;135:26–34. doi: 10.1164/arrd.1987.135.1.26. [DOI] [PubMed] [Google Scholar]
- 25.Rudd RM, Haslam PL, Turner-Warwick M. Cryptogenic fibrosing alveolitis: Relationships of pulmonary physiology and bronchoalveolar lavage to response to treatment and prognosis. Am Rev Respir Dis. 1981;124:1–8. doi: 10.1164/arrd.1981.124.1.1. [DOI] [PubMed] [Google Scholar]
- 26.Inage M, Nakamura H, Kato S, et al. Levels of cytokeratin 19 fragments in bronchoalveolar lavage fluid correlate to the intensity of neutrophil and eosinophil-alveolitis in patients with idiopathic pulmonary fibrosis. Respir Med. 2000;94:155–60. doi: 10.1053/rmed.1999.0712. [DOI] [PubMed] [Google Scholar]
- 27.Wells AU, Hansell DM, Haslam PL. Bronchoalveolar lavage cellularity: Lone cryptogenic fibrosing alveolitis compared with the fibrosing alveolitis of systemic sclerosis. Am J Respir Crit Care Med. 1998;157:1474–82. doi: 10.1164/ajrccm.157.5.9609096. [DOI] [PubMed] [Google Scholar]
- 28.Peterson MW, Monick M, Hunninghake GW. Prognostic role of eosinophils in pulmonary fibrosis. Chest. 1992;1:51–56. doi: 10.1378/chest.92.1.51. [DOI] [PubMed] [Google Scholar]
- 29.Harada T, Watanabe K, Nabeshima K, et al. Prognostic significance of fibroblastic foci in usual interstitial pneumonia and non-specific interstitial pneumonia. Respirology. 2013;18:278–83. doi: 10.1111/j.1440-1843.2012.02272.x. [DOI] [PubMed] [Google Scholar]
- 30.Quadrelli S, Molinari L, Ciallella L, et al. Radiological versus histopathological diagnosis of usual interstitial pneumonia in the clinical practice: Does it have any survival difference? Respiration. 2010;79:32–37. doi: 10.1159/000225987. [DOI] [PubMed] [Google Scholar]
- 31.Lazor R, Letovanec I, Beigelman C. Idiopathic interstitial pneumonias. Rev Prat. 2014;64:933–37. 939–40. [PubMed] [Google Scholar]
- 32.Vancheri C, Cottin V, Kreuter M, Hilberg O. IPF, comorbidities and management implications. Sarcoidosis Vasc Diffuse Lung Dis. 2015;32:17–23. [PubMed] [Google Scholar]
- 33.Mannino DM, Etzel RA, Parrish RG. Pulmonary fibrosis deaths in the United States, 1979–1991. An analysis of multiple-cause mortality data. Am J Respir Crit Care Med. 1996;153:1548–52. doi: 10.1164/ajrccm.153.5.8630600. [DOI] [PubMed] [Google Scholar]
- 34.Novelli F, Tavanti L, Cini S, et al. Determinants of the prognosis of idiopathic pulmonary fibrosis. Eur Rev Med Pharmacol Sci. 2014;18:880–86. [PubMed] [Google Scholar]
- 35.Raghu G, Weycker D, Edelsberg J, et al. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2006;174:810–16. doi: 10.1164/rccm.200602-163OC. [DOI] [PubMed] [Google Scholar]
- 36.Nair GB, Matela A, Kurbanov D, Raghu G. Newer developments in idiopathic pulmonary fibrosis in the era of anti-fibrotic medications. Expert Rev Respir Med. 2016;10:699–711. doi: 10.1080/17476348.2016.1177461. [DOI] [PubMed] [Google Scholar]